

The plasma potassium level is normally maintained within narrow limits (typically, 3.5 to 5.0 mmol per liter) by multiple mechanisms that collectively make up potassium homeostasis. Such strict regulation is essential for a broad array of vital physiologic processes, including the resting cellular-membrane potential and the propagation of action potentials in neuronal, muscular, and cardiac tissue, along with hormone secretion and action, vascular tone, systemic blood-pressure control, gastrointestinal motility, acid–base homeostasis, glucose and insulin metabolism, mineralocorticoid action, renal concentrating ability, and fluid and electrolyte balance.1-3
The importance of potassium homeostasis is underscored by the well-recognized finding that patients with hypokalemia or hyperkalemia have an increased rate of death from any cause.4,5 In addition, derangements of potassium homeostasis have been associated with pathophysiologic processes, such as progression of cardiac and kidney disease and interstitial fibrosis.1,3,6
The need for tight regulation of the extracellular level of potassium is illustrated by the potential for derangements in the level during the ingestion of a normal meal. An average adult has approximate levels of 60 to 80 mmol of total extracellular potassium and levels of 20 to 25 mmol of total plasma potassium. Meals may contain more potassium than the total plasma potassium content, but because of rapid clearance by renal and extrarenal mechanisms, the variations in the plasma potassium level during the course of a day are commonly no greater than 10%.7 Renal potassium excretion also has a circadian rhythm independent of food intake and modulates other mechanisms that control potassium excretion. Here we review the mechanisms that regulate potassium homeostasis and describe the important role that the circadian clock exerts on these processes.
From a clinical perspective, the importance of the circadian clock is illustrated by the benefits of timed drug administration. For example, the time of drug administration can affect the therapeutic benefit.8,9 Aldosterone and cortisol have an endogenous circadian secretion pattern, so sampling at specific times will reduce variability and improve clinical assessment. Moreover, the action of these hormones is influenced by the circadian clock.10 The substantial daily variation in urinary potassium excretion justifies caution in the use of random urine sampling to evaluate hypokalemia or hyperkalemia. Without consideration of the time of collection, random measurement of urinary potassium may either underestimate or overestimate the 24-hour rate of potassium excretion. Finally, the time of day affects the adaptation to a potassium load and can be important in emergency potassium-replacement therapy.11
Potassium Homeostasis
Potassium homeostasis denotes the maintenance of the total body potassium content and plasma potassium level within narrow limits in the face of potentially wide variations in dietary potassium intake. It involves two concurrent processes — external and internal. External potassium homeostasis regulates renal potassium excretion to balance potassium intake, minus extrarenal potassium loss and correction for any potassium deficits. Internal potassium regulation controls the asymmetric distribution of total body potassium with the greater part (approximately 98%) intracellular and only a small fraction (approximately 2%) extracellular.2 Much evidence supports the role of the circadian clock in external homeostasis, and some evidence indicates a role in internal homeostasis.7,12-14
External Potassium Balance
External potassium balance involves three control systems (Fig. 1A). Two systems can be categorized as “reactive,” whereas a third system is considered to be “predictive.” A negative-feedback system reacts to changes in the plasma potassium level and regulates the potassium balance. Potassium excretion increases in response to increases in the plasma potassium level, leading to a decrease in the plasma level. A reactive feed-forward system that responds to potassium intake in a manner that is independent of changes in the systemic plasma potassium level has been recognized.2,15 Currently, the component mechanisms remain under study and are incompletely delineated. Because oral potassium intake was seen to produce a marked kaliuresis in the absence of effective increases in the plasma potassium level, investigators postulated that potassium receptors reside in the gut, hepatic portal vein, or liver.2,15 Experiments with the use of vagotomy and hypophysectomy support the role of vagal afferents and the pituitary as components of this system.16,17 Evidence in animal models shows that an oral potassium load leads to kaliuresis, but aldosterone, vasopressin, α-melanocyte–stimulating hormone (α-MSH), γ-MSH, and peptides such as glucagon-like peptide 1, guanylin, uroguanylin, and other candidate substances do not appear to be responsible.18 The nature of the kaliuretic factors requires further investigation.
Figure 1. Overview of Potassium Homeostasis.
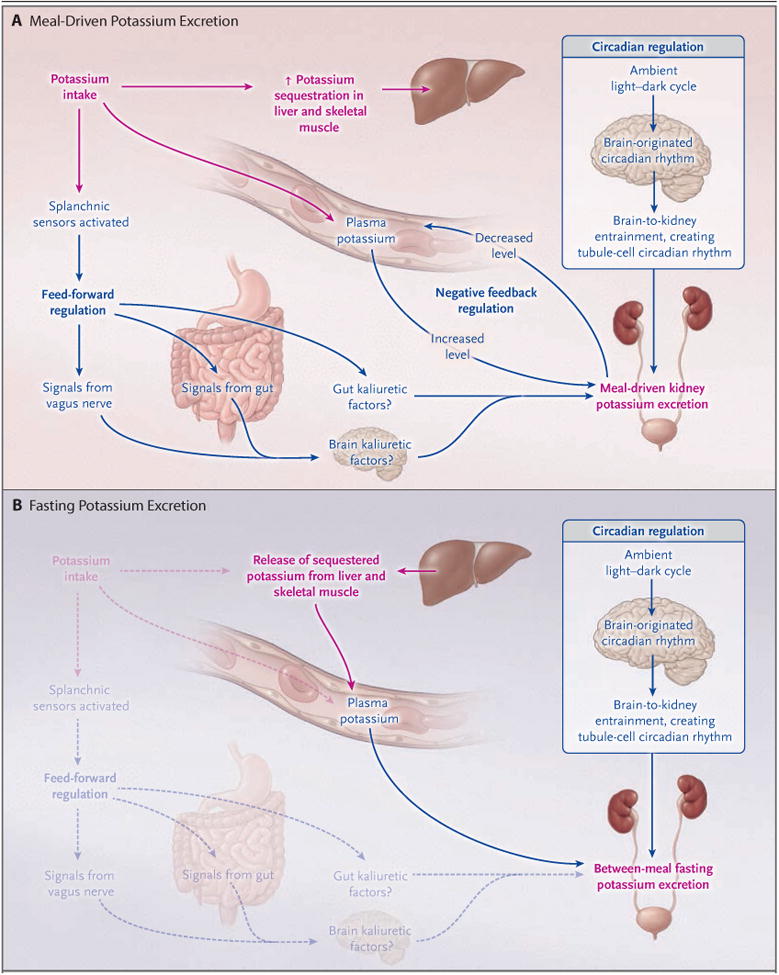
Shown are the known mechanisms that regulate external potassium balance and the pathways for net potassium movement associated with meal-driven potassium intake (Panel A) and with between-meal fasting (Panel B). Meal-driven potassium intake initiates both an increase in potassium excretion and sequestration of potassium in liver and skeletal muscle. Increased excretion is driven by reactive mechanisms, which can be either dependent on the plasma potassium level (negative-feedback regulation) or independent of the plasma potassium level (feed-forward regulation) initiated at splanchnic receptors. The circadian rhythm drives a predictive regulation of the tubule mechanisms responsible for potassium excretion, generated by a central clock and transmitted to circadian clocks in the tubule cells responsible for variations in potassium excretion. This rhythm enhances excretion during the active daylight phase and diminishes it during the inactive nighttime phase. In combination, these components provide maintenance of total body potassium levels within narrow limits without appreciable changes in the plasma potassium level. Between periods of meal intake, potassium is released from intracellular stores (primarily liver and skeletal muscle) for excretion.
A predictive system appears to modulate the effect of reactive systems, enhancing physiologic mechanisms at the time of day when food intake characteristically occurs — typically, during the day in humans and at night in nocturnal rodents.7 This predictive system is driven by a circadian oscillator in the suprachiasmatic nucleus of the brain and is entrained to the ambient light–dark cycle. The central oscillator (clock) entrains intracellular clocks in the kidney that generate the cyclic changes in excretion. When food intake is evenly distributed over 24 hours, and physical activity and ambient light are held constant, this system produces a cyclic variation in potassium excretion.7,13,19
After the ingestion of a meal, the feed-forward system induces kaliuresis.20-22 If the quantity of potassium is sufficient to increase the plasma potassium level, the feedback system is activated.20-22 Intake can vary widely throughout the day. Although the circadian clock and potassium intake from meals alter potassium excretion rapidly (≤24 hours), potassium excretion responds appropriately with intake. For example, under normal conditions in persons who consume four equal meals at 6-hour intervals, meal-induced kaliuresis is greater during the day than at night.23 However, balance studies show that with large, prolonged step increases or decreases in potassium intake, potassium balance may not be fully achieved for several days.
Internal Potassium Homeostasis
Internal potassium homeostasis is the maintenance of an asymmetric distribution of total body potassium between the intracellular and extracellular fluid. This occurs by the balance of active cellular uptake by sodium–potassium adenosine triphosphatase, an enzyme that pumps sodium out of cells while pumping potassium into cells (called the sodium–potassium pump rate), and passive potassium efflux (called the leak rate). Case 1 illustrates the dramatic effect of derangements in the proper coupling between potassium pump and leak rates (Box 1).
Box 1. Case 1.
Case 1 illustrates the dramatic effect of derangements in the proper coupling between potassium-pump rates and potassium-leak rates
An 11-year-old boy was admitted for evaluation of hypokalemia and muscle weakness. He awoke at 3:00 a.m. with paralysis of the lower limbs and severe weakness in both upper limbs, with a muscle-strength grade of 1, on a scale of 0 (lowest) to 5 (highest). His mother called for an ambulance. The patient had a history of having similar episodes approximately three to four times per year, frequently in the early morning. Most attacks were less severe than this one, but a recent episode had prompted admission for near paralysis and hypokalemia. The attacks were characterized by weakness, principally in the thighs with occasional involvement of the upper limbs. None were associated with loss of consciousness.
On admission (5:00 a.m., 2 hours after the beginning of the episode), the patient's plasma potassium level was 1.6 mmol per liter, and he promptly received 40 mmol of potassium chloride by mouth. After approximately 90 minutes without relief of the paralysis, 40 mmol per liter of potassium chloride was infused in 0.9% saline intravenously over 2 hours. However, this treatment did not substantially increase the plasma potassium level (1.7 mmol per liter). The patient received an additional 100 mmol of potassium chloride by mouth at the completion of the intravenous potassium chloride infusion. His muscle strength improved, and the plasma potassium level increased to 2.2 mmol per liter by 11:45 a.m. His strength improved progressively. The plasma potassium level increased to 5.4 mmol per liter at 2:40 p.m. but decreased to 4.0 mmol per liter by 4:15 p.m. Subsequent plasma potassium values remained between 3.7 and 4.8 mmol per liter, and by 11:00 p.m. (20 hours after the beginning of the episode), the patient had recovered most of his strength and his symptoms had largely abated. The results of subsequent laboratory testing for levels of triiodothyronine and free thyroxine were normal. The patient was prescribed acetazolamide and potassium chloride tablets and a high-potassium diet.
This case illustrates one form of periodic paralysis with profound hypokalemia. Although quite rare, this disease provides substantial insight into the principal factors that dictate the plasma potassium level.24 In the absence of exogenous potassium administration, the plasma potassium level fell precipitously during an attack as cellular potassium uptake exceeded potassium leak from cells, principally skeletal muscle. In such cases, cautious use of potassium is generally recommended because the hypokalemia does not reflect potassium deficiency but rather transcellular potassium redistribution.25 During attacks, the balance between cellular pump potassium uptake and potassium efflux is shifted to an increase in cellular potassium uptake relative to potassium efflux. As this case illustrates, during the peak of the attack (between 3:00 a.m. and 8:00 a.m.), plasma potassium values may remain depressed despite substantial potassium administration.
Periodic paralysis encompasses cases associated with episodic muscle weakness or paralysis.26,27 Many of these cases are hereditary, typically with an autosomal dominant inheritance pattern — hence the designation of familial periodic paralysis. Both hypokalemic and hyperkalemic forms of familial periodic paralysis exist, although the increase in the plasma potassium level is typically small and may not exceed the normal range in the latter.
Hypokalemic familial periodic paralysis typically presents in the first two decades of life, with attacks typically lasting several hours; the attacks may be brief or last for several days. Factors that are implicated in the precipitation of attacks include stress, strenuous exercise, and carbohydrate-rich meals. Two genetically distinct mutations account for the majority of the familial cases and are due to a mutation in the gene encoding either skeletal muscle calcium channel α1 subunit (CACNA1) or skeletal muscle sodium channel voltage-gated type IV α subunit (SCN4A).
Hyperkalemic familial periodic paralysis usually presents earlier in life than hypokalemic familial periodic paralysis, frequently in infancy or early childhood, with episodes of transient paralysis. Factors that are implicated in the precipitation of attacks include exposure to cold temperature, fasting, rest after exercise, and potassium ingestion. Mutations in SCN4A are known to produce this condition.
Thyrotoxic hypokalemic periodic paralysis is an uncommon manifestation of thyrotoxicosis that is characterized by abrupt development of hypokalemia and episodes of muscular weakness. Its incidence is substantially greater in Asians than in non-Asians, and most patients present in their 20s or 30s.28 Although there is a higher incidence of hyperthyroidism in women than in men, the development of periodic paralysis associated with hyperthyroidism is more frequent in men. A recent study indicates that loss of function of the skeletal muscle–specific potassium channel Kir2.6 may contribute to this disorder.29
The differential diagnosis of hypokalemic paralysis should include nonperiodic paralysis and periodic paralysis that can be familial or sporadic. Other more common conditions should be considered in patients presenting with hypokalemia and paralysis, including renal tubular acidosis. In one series, renal tubular acidosis was the most frequent cause of hypokalemia with paralysis.30,31 This is particularly the case if there is substantial potassium depletion or if provoked by high-carbohydrate caloric sources.32 Autoimmune disorders such as Sjögren's syndrome and pernicious anemia should also alert the clinician to the possibility of renal tubular acidosis and potassium depletion as potential causes of hypokalemic paralysis.31,33
Little increase in the plasma potassium level occurs during potassium absorption from the gut in normal persons owing to potassium excretion by the kidney and potassium sequestration by the liver and muscle (Fig. 1A). Between meals, the plasma potassium level is nearly constant, as potassium excretion is balanced by the release of sequestered intracellular potassium (Fig. 1B). Potassium depletion primarily involves a loss of potassium from muscle, although it may be reflected in reductions in the plasma potassium level. When the potassium loss is corrected, potassium retention from intake replaces the deficit.2,15
Insulin, catecholamines, and mineralocorticoids stimulate potassium uptake into muscle and other tissues. Absorption of meal-derived glucose stimulates insulin secretion with a consequent insulin-driven potassium uptake in muscle. The effectiveness of insulin in the treatment of hyperkalemia depends on its capacity to drive potassium into skeletal muscle, thereby decreasing the plasma potassium level. In the absence of a change in the total body potassium content, severe hypokalemia may result from a minor increase in intracellular potassium as a result of a resetting of pump–leak kinetics.34 The pump–leak kinetics are not altered by short-term elevations in aldosterone but are reset by chronic mineralocorticoid stimulation, which reduces the plasma potassium level in the absence of discernable changes in the total body potassium content.34-36 Such actions contribute largely to the reductions in plasma potassium associated with increased secretion or administration of aldosterone. Nevertheless, supraphysiologic rates of aldosterone secretion, as in primary hyperaldosteronism, may be associated with potassium depletion. Case 2 illustrates the importance of extrarenal potassium homeostasis to the maintenance of the plasma potassium level (Box 2).
Box 2. Case 2.
Case 2 illustrates the importance of glucose and insulin to extrarenal potassium homeostasis and to maintenance of the plasma potassium level
A 35-year-old woman presented with nausea, vomiting, and muscle weakness for the past several days. Before this episode, she had had a good appetite. Her medical history was unremarkable except for a previous diagnosis of nephrolithiasis. She had a 15-year pack-history of smoking tobacco and reported taking no prescription medicines, diuretics, or nonprescription or other drugs, including laxatives.
Her blood pressure was 108/88 mm Hg, and the heart rate was 110 beats per minute; respirations were unlabored, and she was afebrile. The chest and cardiovascular examination was normal. Muscle strength was initially judged to be modestly reduced, with a score of 3 out of 5. The results of initial laboratory tests were as follows: a normal differential blood count; sodium, 137 mmol per liter; potassium, 1.6 mmol per liter; chloride, 108 mmol per liter; bicarbonate, 16 mmol per liter; anion gap, 13; blood urea nitrogen, 10 mg per deciliter (3.6 mmol per liter); creatinine, 0.8 mg per deciliter (71 μmol per liter); arterial blood pH, 7.32; and partial pressure of carbon dioxide, 25 mm Hg. On urinalysis, the urine pH was 7.5, specific gravity 1.005, with no leukocytes, protein, blood, or nitrites. Electrocardiography revealed prominent U waves. Nephrocalcinosis was confirmed on renal ultrasonography and intravenous pyelography.
She was treated with intravenous potassium (40 mmol per liter) in 5% glucose and oral potassium (40 mmol). Severe muscle weakness (grade 1 out of 5) ensued, and a repeat plasma potassium level was 1.2 mmol per liter. The patient was admitted to the intensive care unit for close observation and electrolyte replacement. Potassium replacement was continued intravenously as 40 to 60 mmol of potassium chloride per liter in normal saline, and the plasma potassium level increased to 3.6 mmol per liter. The patient's muscle strength rapidly returned to normal. Subsequent evaluation confirmed the diagnosis of hypokalemic (type I) distal renal tubular acidosis. On hospital discharge, she was prescribed 25 ml of potassium citrate and citric acid oral solution four times daily. At a follow-up visit 2 weeks later, she had no symptoms and had normal plasma electrolytes.
This case illustrates a potentially life-threatening complication that can ensue from administration of intravenous glucose solutions (despite potassium-replacement therapy) to patients with potassium depletion.32 The glucose-enhanced insulin secretion results in rapid stimulation of cellular potassium uptake. Life-threatening cardiac arrhythmias and worsening muscle weakness can develop during the infusion of potassium with glucose in the treatment of hypokalemia. The infusion of glucose reduces the plasma potassium level in both healthy persons and in those with hypokalemia, but the complications can be more serious when hypokalemia or potassium depletion is present. In the correction of potassium depletion, oral administration of potassium is recommended if practicable. If intravenous potassium repletion is used, it should be in glucose-free solutions, to avoid neuromuscular paralysis or cardiac arrhythmias.
Aberrant Potassium Homeostasis
The concurrent activities of the external and internal systems act to maintain the plasma potassium level within narrow limits. However, in clinical practice, clinicians often encounter deviations from normal levels when potassium intake is greatly altered. Hypokalemia and hyperkalemia frequently occur as the result of nonhomeostatic processes that are not regulated by changes in the potassium balance. These processes increase or decrease potassium excretion but not in response to changes in potassium intake (e.g., action of diuretics, alterations in acid–base balance, or impaired kidney function) or limit the capacity of the kidney to compensate (e.g., in chronic kidney disease).
Renal Potassium Handling
The healthy kidney has a robust capacity to excrete potassium, and under normal conditions, most persons can ingest very large quantities of potassium (400 mmol per day or more) without clinically significant hyperkalemia.23,37 Potassium that is filtered at the glomerulus is largely re-absorbed in the proximal tubule and the loop of Henle. Consequently, the rate of renal potassium excretion is determined mainly by the difference between potassium secretion and potassium re-absorption in the cortical distal nephron and collecting duct. Both of these processes are regulated — potassium ingestion stimulates potassium secretion and inhibits potassium re-absorption.2,38 Factors that regulate potassium secretion and reabsorption can be divided into those that serve to preserve potassium balance (homeostatic) and those that affect potassium excretion without intrinsically acting to preserve potassium balance (contra-homeostatic) (Table 1). Examples of the latter include flow rate in the renal tubular lumen and the luminal sodium level. The acid–base balance also affects potassium excretion. The predominant effect of acidosis is to inhibit potassium clearance, whereas the predominant effect of alkalosis is to stimulate potassium clearance. Cases 3 and 4 illustrate the evaluation of hypokalemia from renal and extrarenal causes (Box 3).
Table 1. Factors Regulating Potassium Secretion and Potassium Reabsorption.
| Change | Potassium Secretion | Potassium Reabsorption | ||
|---|---|---|---|---|
| Homeostatic | Contra-homeostatic | Homeostatic | Contra-homeostatic | |
| Increases effect | Potassium loading Aldosterone in the presence of hyperkalemia |
Increased luminal flow rate Increased luminal sodium delivery Decreased luminal chloride Exogenous mineralocorticoid agonists, fludrocortisone, diuretics Metabolic alkalosis |
Potassium restriction and depletion Progesterone |
Acidosis Exogenous mineralocorticoid agonists (e.g., fludrocortisone) |
|
| ||||
| Decreases effect | Potassium restriction and depletion | Decreased luminal flow rate Decreased luminal sodium delivery Drugs that inhibit sodium absorption (e.g., amiloride, triamterene, trimethoprim, pentamidine, digitalis) Inhibitors of renin–angiotensin– aldosterone system (RAAS)* Potassium-channel inhibitors and other mechanisms, including metabolic acidosis, cyclooxygenase inhibitors (nonsteroidal antiinflammatory drugs), and calcineurin inhibitors |
Potassium loading Tissue kallikrein |
Inhibitors of renin–angio-tensin–aldosterone system (RAAS) |
RAAS inhibitors include those that affect aldosterone synthesis (e.g., heparin); those that affect aldosterone regulation, including the inhibition of renin secretion (e.g., beta-blockers, cyclooxygenase inhibitors), direct renin inhibitors (e.g., aliskiren), angiotensin-converting–enzyme inhibitors (e.g., captopril), and angiotensin II–receptor blockers (e.g., losartan); and those that affect aldosterone action, including mineralocorticoid receptor blockers (e.g., spironolactone, eplerenone).
Box 3. Cases 3 and 4.
Cases 3 and 4 illustrate the approach to hypokalemia of renal and extrarenal origin and the response to therapy
In Case 3, a 32-year-old man was referred to the nephrology clinic because of persistent hypokalemia. The patient was a construction worker who had no known medical illnesses until a job-related laceration required medical attention. In the emergency department, blood was obtained for type and cross-matching, complete blood count, and measurement of electrolytes. Hemostasis was obtained, but the laboratory reported a panic value for the plasma potassium level of 1.8 mmol per liter, and the patient was transferred to the intensive care unit for intravenous potassium replacement.
Over the next 72 hours, the patient received approximately 500 mmol of potassium. The plasma potassium level increased initially to 2.1 to 2.2 mmol per liter and remained essentially constant thereafter at 2.5 to 2.7 mmol per liter, despite intravenous infusion of more than 180 mmol of potassium chloride for 72 hours. The failure to correct the plasma potassium level suggested a renal cause of the hypokalemia. The patient was prescribed 80 mmol of potassium chloride per day and a high-potassium diet and was referred to the nephrology unit.
The patient had minimal symptoms and reported having only mild muscle cramps and weakness during the summer months, which usually did not interfere with his work or activities. The blood pressure was 108/72 mm Hg, the pulse 68 beats per minutes and regular, and the respiratory rate 14 breaths per minute and unlabored; the patient was afebrile. The patient was a muscular man with no abnormalities on physical examination. Electrolytes values were as follows: sodium, 141 mmol per liter; potassium, 2.8 mmol per liter; chloride, 95 mmol per liter; bicarbonate, 31 mmol per liter; creatinine, 0.7 mg per deciliter (62 μmol per liter); blood urea nitrogen, 14 mg per deciliter (5.0 mmol per liter); glucose, 92 mg per deciliter (5.1 mmol per liter); calcium, 9.2 mg per deciliter (2.3 mmol per liter); phosphorus, 4.6 mg per deciliter (1.5 mmol per liter); and magnesium, 1.1 mg per deciliter (0.45 mmol per liter). Urinalysis and renal ultrasonography were normal. The 24-hour creatinine clearance was 102 ml per minute. He was prescribed a potassium-rich diet, oral potassium chloride (at a dose of 200 mmol per day), and magnesium oxide (400 to 800 mg as tolerated).
A 24-hour urine collection showed a potassium excretion consistent with his prescribed regimen and the absence of detectable diuretics on multiple clinic visits, but the plasma potassium level ranged from 2.7 to 2.9 mmol per liter. Gitelman's syndrome was diagnosed. The addition of amiloride (5 mg per day) had a marginal effect on increasing the plasma potassium level. Spironolactone (25 mg per day) was added to his regimen with only a small effect on the plasma potassium level, which ranged from 3.1 to 3.3 mmol per liter, but was discontinued because of unacceptable side effects. The patient has been followed for more than 25 years with normal renal function and plasma potassium values of 2.9 to 3.2 mmol per liter.
This case illustrates that the phenotype of Gitelman's syndrome can be relatively mild. The diagnosis of Gitelman's syndrome is usually made in the third decade of life and typically later than the diagnosis of Bartter's syndrome. Muscle cramps and weakness associated with a variable degree of impairment in daily activities are the most common symptoms. However, debilitating muscle weakness and cramps, symptoms related to hypotension, paresthesia, and frank paralysis are known to occur.39,40
In contrast to the hypokalemia and hypomagnesemia that can be seen with diuretic use, in which case correction of the hypomagnesemia may allow for correction of the hypokalemia, both the hypokalemia and hypomagnesemia were resistant to correction by oral supplementation. The effect of increasing potassium intake in this patient is in clear distinction to that of the fourth patient, in whom the origin of the hypokalemia was extrarenal.
In Case 4, a 58-year-old woman was admitted for treatment of hypokalemia. She had seen her physician earlier and received a message to seek medical attention because her plasma potassium level was 2.0 mmol per liter. The patient had no history of abnormal blood electrolytes until approximately 3 years earlier, when she was noted to have intermittent hypokalemia. In the emergency department, the patient reported fatigue but otherwise was in no acute distress. She reported no use of laxatives, diuretics, or nonprescription medicine except for occasional ibuprofen for back pain, but she had not taken it during the past week.
Renal and electrolyte laboratory tests in the emergency department were as follows: sodium, 142 mmol per liter; potassium, 2.1 mmol per liter; chloride, 104 mmol per liter; bicarbonate, 29 mmol per liter; blood urea nitrogen, 15 mg per deciliter (5.4 mmol per liter); creatinine, 1.5 mg per deciliter (133 μmol per liter); glucose, 77 mg per deciliter (4.3 mmol per liter); calcium, 8.5 mg per deciliter (2.1 mmol per liter); and magnesium, less than 0.4 mg per deciliter (<0.16 mmol per liter). Repeat electrolyte measurements confirmed the presence of hypokalemia (potassium, 2.1 mmol per liter) and hypomagnesemia (magnesium, <0.4 mg per deciliter).
Potassium chloride was administered (60 mmol orally and 40 mmol intravenously), along with 16 mmol of magnesium sulfate. On repeat measurement, the potassium level was 2.5 mmol per liter, and magnesium was 3 mg per deciliter.
The nephrology department was consulted for the possibility of Bartter's or Gitelman's syndrome. The physical examination was unremarkable. Renal ultrasonography showed normal-sized kidneys and no obstruction. Determination of the rate of urine potassium excretion and continued potassium replacement was recommended.
The urinary potassium excretion over a 24-hour period was approximately 12 mmol. She received 60 mmol of potassium chloride orally and 50 mmol of potassium chloride intravenously. Over the subsequent 24 hours, she received an additional 30 mmol of potassium chloride intravenously and 160 mmol orally. The urine potassium excretion increased to approximately 42 mmol during a 24-hour period, the plasma potassium level increased to 3.7 mmol per liter, and the plasma magnesium level was 2.7 mg per deciliter. She subsequently admitted that she had been using laxatives as a weight-control measure for several months. She was discharged with normal plasma potassium and magnesium levels, and her renal function had returned to its baseline value.
This case illustrates the challenges confronting the physician in the diagnosis of hypokalemia. The use of laxatives and diuretics and surreptitious vomiting are frequent causes of unexplained hypokalemia, and the patient may be reluctant to disclose this information. The concurrent hypomagnesemia also suggests the possibility of the use of diuretics or laxatives as an explanation for the hypokalemia. Under normal conditions, renal adaptation to a potassium-poor diet will result in urinary potassium excretion of less than 20 mmol per 24 hours, and the collection on the first hospital day provides strong evidence that the source of the hypokalemia is not of renal origin. Chloride excretion among patients consuming a typical Western diet will usually reflect sodium chloride intake. In contrast, patients with surreptitious vomiting often have low urinary chloride excretion (typically, <20 mmol per 24 hours and frequently <10 mmol per 24 hours), a finding rarely seen under normal conditions unless the intake of sodium chloride is drastically reduced.41
If prompt and substantial potassium replacement occurs before the measurement of urinary potassium excretion, the rate of potassium excretion may exceed 20 mmol per day and could lead to the misinterpretation that the hypokalemia is of renal origin. The correction of the hypokalemia in the present case with moderate potassium administration makes Gitelman's or Bartter's syndrome unlikely.
The mechanisms for potassium secretion and reabsorption in the collecting duct are shown in Figure 2. Apical cellular sodium entry through the amiloride-sensitive epithelial sodium channel (ENaC) promotes active basolateral cellular potassium uptake in exchange for sodium extrusion by the sodium–potassium pump. Apical sodium entry through ENaC depolarizes the apical membrane, which stimulates potassium secretion through apical potassium channels. Functional cotransport of potassium chloride also effects potassium secretion and is particularly important when the luminal chloride level is substantially reduced, as in the administration of a non-reabsorbable anion or during chloride-dependent metabolic alkalosis.2
Figure 2. Model of the Major Cell Types of the Cortical Collecting Duct.
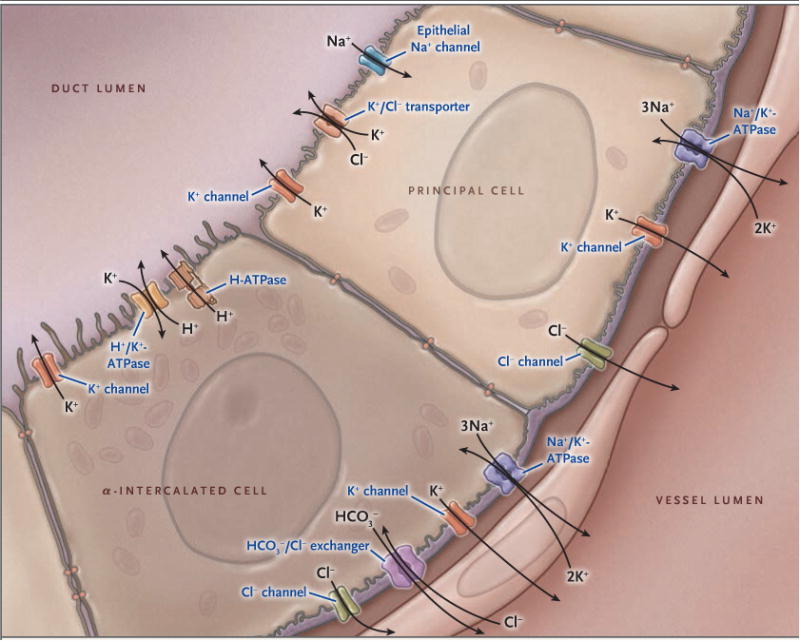
Shown are important potassium ion (K+) transport proteins of the principal cell and the α-intercalated cell, illustrating the mechanism of active potassium secretion and active potassium reabsorption. In principal cells, potassium is actively pumped into the cell from the peritubular fluid by basolateral sodium-potassium adenosine triphosphatase (Na+/K+-ATPase, also called sodium-potassium pump) and is secreted at the apical membrane by potassium channels and by functional potassium chloride (K+/Cl−) cotransporters. (The sodium-potassium pump moves out three sodium ions [3NA+] and moves in two potassium ions [2K+], thus removing one positive charge.) In the α-intercalated cell, potassium is actively absorbed from the lumen and can exit the cell apically during potassium-replete states or basolaterally during conditions of potassium deficiency. The collecting duct is part of the aldosterone-sensitive distal nephron, which also includes the distal convoluted tubule and connecting segment. These segments also have the capacity for substantial net potassium secretion.
Active potassium reabsorption is driven by an apical membrane proton–potassium pump (Fig. 2). The activity of this pump is pH-sensitive and activated by acidosis,42-44 potassium restriction,45 and mineralocorticoids.34 The mineralocorticoid effect may explain the lack of substantial renal potassium loss with chronic mineralocorticoid stimulation.34-36,46 Thus, mineralocorticoids can enhance potassium reabsorption or secretion depending on the potassium balance.
The Circadian Clock in Cellular Physiology
In vertebrates, a central clock in the suprachiasmatic nucleus of the brain and peripheral clocks that are present in virtually all cells regulate circadian rhythms.47 Although ablation of the suprachiasmatic nucleus disrupts many circadian rhythms, particularly those related to activity, the circadian rhythm of potassium excretion is preserved, presumably due to continued activity of renal-cell clocks. Indeed, this rhythm persists after adrenalectomy and requires no environmental stimuli.7,48,49
Among the many physiologic functions in humans that show circadian rhythms, few are more consistent and stable than the circadian rhythm of urinary potassium excretion.7,50 Increasing potassium intake magnifies the amplitude of this rhythm, but the intrinsic circadian periodicity is retained (Fig. 3).23 For example, after transatlantic air travel, the circadian rhythm of renal potassium excretion adjusts slowly over several days and finally resynchronizes to the local day–night cycle.7,51
Figure 3. Circadian Rhythm of Urinary Potassium Excretion in Humans during Two Levels of Potassium Intake.
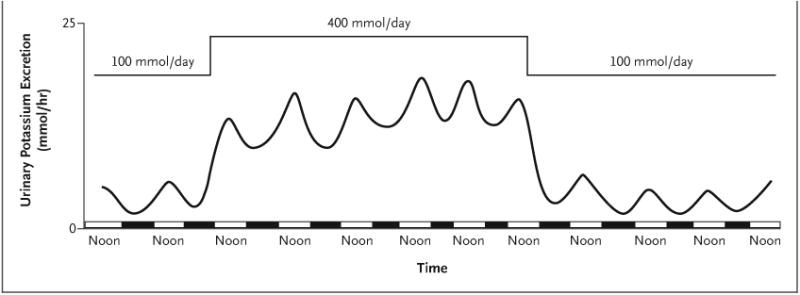
Shown is the approximate hourly rate of urinary potassium excretion (based on urine collections every 6 hours) in multiple patients receiving four identical meals every 6 hours, with a normal amount of potassium (100 mmol per day) for the first 2 days, a high-potassium diet (400 mmol per day) for the next 6 days, and a normal amount of potassium for the next 4 days. Rapid renal potassium adaptation occurs in response to either an increase or a decrease in potassium intake. The hourly rate of potassium excretion over a 24-hour period varies from noon, when the largest rate of potassium excretion typically occurs (midpoint of the white bar on the x axis), to midnight, when it is typically the least (midpoint of the black bar). This circadian variation is approximately 40% in persons consuming a high-potassium diet and by approximately 300% in persons consuming a normal level of potassium. This circadian rhythm occurs despite evenly spaced meals every 6 hours during a 24-hour period. Data are adapted from Rabelink et al.23
The magnitude of the daily change in the clock-driven rate of renal potassium excretion can be substantial. For example, during human consumption of a high-potassium diet (400 mmol per day), potassium excretion can increase by a factor of approximately 1.6 from nadir to maximum within a 24-hour period, even though similar meals may be evenly spaced throughout the day. Greater variations — by a factor of 2 to 4 — may be present during normal potassium intake.23
This circadian rhythm of potassium excretion can serve to minimize the change in the potassium content of extracellular fluid. For example, in one study, intravenous administration of potassium at noon (when clock-driven potassium excretion was near its maximum and food intake typically occurs) resulted in a smaller increase in the plasma potassium level than when the same amount of potassium was administered at midnight (when clock-driven potassium excretion was minimal).11 Potassium homeostasis, therefore, is not merely due to input-mediated systems but is also modulated by the central and peripheral circadian clocks.
The molecular mechanism that produces these rhythmic oscillations remained unknown until seminal work in the fruit fly, Drosophila melanogaster, showed that mutations in a single gene could affect circadian activity.52 Subsequent work by many investigators over nearly three decades established that core circadian-clock genes encode transcription factors.47,53-55 These proteins control a transcription-translation feedback loop that is the core component of the circadian clock (Fig. 4).53,56 Control of the circadian clock is robust and redundant so that the disruption of single genes may have little effect on clock activity.57,58
Figure 4. Molecular Mechanism of the Mammalian Circadian Clock.
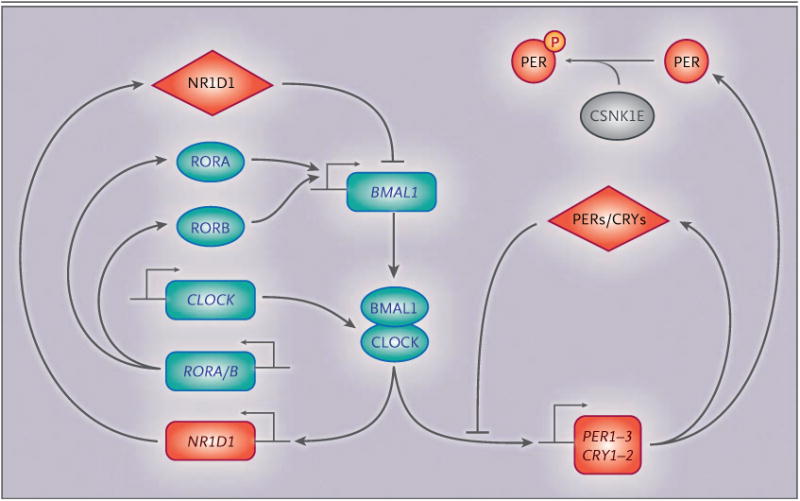
The core cycle is shown on the right, with an increase in the amount of period circadian clock proteins 1 through 3 (PER1–3) and cryptochrome proteins 1 and 2 (CRY1–2) throughout the day, which acts to suppress their transcription. The red diamonds represent repressor proteins, and the blue ovals, activator proteins. Both activators and repressors are transcription factors or transcription regulators. The rectangles represent genes that encode the respective proteins. CSNK1E encodes casein kinase 1 epsilon, a kinase that is known to alter the period of the circadian oscillator through the phosphorylation (P) of core clock proteins, as shown. CSNK1E (also called CK1ε) is shown in gray because it is neither a transcriptional activator nor a repressor. BMAL1 (also called ARNTL) denotes aryl hydrocarbon receptor nuclear translocator-like protein 1, CLOCK clock circadian regulator, NR1D1 (also called REV-ERBα) nuclear receptor subfamily 1 group D member 1, and ROR retinoic acid receptor–related orphan receptor.
Mutations in critical clock proteins in vertebrates can also influence the circadian behavior in animals. In the golden hamster, the tau mutation in the gene encoding casein kinase 1 epsilon causes profound disturbances in activity and sleep–wake patterns when the animals are housed in total darkness. This mutation results in a defective casein kinase 1 epsilon, a kinase that is known to alter the period of the circadian oscillator.59,60 Thus, the circadian clock has post-translational mechanisms that affect the circadian cycle (Fig. 4).47
The molecular components that regulate the core elements of the circadian clock are remarkably similar in widely differing organisms, from drosophila to humans, both in terms of structure and function (Table S1 in the Supplementary Appendix, available with the full text of this article at NEJM.org).55,58,60,61 Such similarity across highly diverse species indicates that the circadian clock serves a critical physiologic role.62,63 In mammals, as in drosophila, BMAL1 (brain and muscle Arnt-like protein 1) and its cognate binding partner CLOCK (circadian locomotor output cycles kaput) bind to specific DNA sequences, referred to as E-box response elements, that are critical for circadian clock–mediated gene transcription. Several studies have suggested that nearly 50% of tissue-specific gene expression has a circadian variation.64-67 The circadian clock also modulates the activity of genes that regulate potassium homeostasis.64,68-71 In particular, mice that are deficient in the gene encoding CLOCK have substantial disruption in normal circadian rhythms for renal potassium and sodium excretion and the plasma aldosterone level.71 Since the CLOCK knockout was global, cell-specific disruption of the protein will be necessary to determine the cell types that sustain the circadian pattern of potassium excretion, sodium excretion, and plasma aldosterone. Other core clock proteins also affect these rhythmic oscillations. For example, mice that are deficient in the genes encoding two cryptochrome proteins, CRY1 and CRY2, have salt-sensitive hypertension owing to excessive aldosterone synthesis, which supports the role of the circadian clock in steroidogenesis.72
Clock Synchronization and Hormone Signaling
The timing signals from the central clock to the peripheral clocks remain uncertain, but adrenal corticosteroids and agents from other loci have been proposed or identified.10,73 Circadian rhythm can also influence the action of both aldosterone and cortisol on renal function. Mills and co-workers found that aldosterone consistently enhanced sodium retention and that cortisol consistently enhanced potassium excretion regardless of the time of day. However, aldosterone increased potassium excretion modestly in the afternoon and had no substantial effect in the morning or at night.10 Thus, in these studies, a kaliuretic effect of aldosterone was apparently dependent on the phase of the circadian cycle.
Although the action of cortisol in promoting potassium excretion would suggest a direct (nonclock) hormonal effect, studies by Moore-Ede and colleagues indicate that cortisol serves as a clock synchronizer.73 Specifically, the squirrel monkey has a prominent circadian pattern of urinary potassium excretion that is unaffected by adrenalectomy. Adrenalectomized animals receiving a single morning infusion of cortisol had a significant increase in potassium excretion that was similar to that in intact animals, but when the cortisol was administered 8 hours later, a gradual shift in the time of peak potassium excretion occurred over 72 hours. These early observations are consistent with molecular observations that glucocorticoids can act as a zeitgeber (“time giver,” or synchronizing signal).74
In one study, cortisol was shown to act in the synchronization of some peripheral clocks, including those in the kidney, with the suprachiasmatic nucleus central clock.75 Dexamethasone transiently changed the phase of circadian gene expression in liver, kidney, and heart.76 Cortisol acts as a strong synchronizing signal for most peripheral circadian oscillators, including the kidney.74 These studies provide a molecular basis for the observations by Moore-Ede et al.12,73 and suggest that glucocorticoids serve as an important signal that coordinates the central and specific peripheral clocks.
Aldosterone also affects certain circadian clocks and, in particular, acutely induces the expression of period circadian clock 1 (PER1) in the kidney. PER1 stimulates the expression of the alpha subunit of ENaC (αENaC), a finding that is consistent with the effect of aldosterone in enhancing sodium retention and, consequently, sodium balance.77 Underscoring this effect, PER1-null mice have a substantially lower systemic blood pressure than wild-type controls.78 The molecular mechanisms that are responsible for circadian rhythms have been studied in mice that are deficient in core clock genes. These studies have identified a substantial number of renally expressed potassium channels and transporters as potential candidate genes that contribute to the circadian variation of potassium excretion.69
The effects of cortisol (or its analogues) and aldosterone on circadian-clock genes can be blocked by glucocorticoid and mineralocorticoid-receptor antagonists, respectively, indicating that they act through their respective nuclear hormone receptors. Other members of the nuclear hormone receptor superfamily also appear to be connected to the circadian clock.79,80 For example, the clock-controlled gene ATP12A (also called HKα2) is also under control of the progesterone receptor.81 Intriguingly, one regulator of the circadian clock is adenosine monophosphate kinase,66 and activation of this kinase produces substantial hypokalemia that is largely due to redistribution.82 Whether this effect involves the circadian clock deserves further investigation.15
Finally, potassium depletion produces striking pathological changes in the kidney, including interstitial fibrosis.83,84 The tau mutation in the golden hamster reduces life span and produces profound cardiorenal disease associated with scarring and fibrosis in heterozygotes, but not homozygotes, when maintained on a 24-hour light–dark cycle.85 Surprisingly, when the animals were subjected to their endogenous 22-hour light–dark cycle, longevity was restored without cardiorenal disease. Future studies should examine how clock mutations may contribute to chronic cardiac or renal disease.
Conclusions
Circadian clocks are involved in many fundamental cellular processes and exert important control over physiologic functions. A striking degree of conservation of the core elements of the circadian clock exists from bread mold to fruit fly and from mice to humans.
In humans, there are marked, transient, meal-related increases in renal potassium excretion that depend on rapid changes in active potassium secretion and reabsorption in the distal nephron. These reactive responses are superimposed on a predictive enhancement of these transport mechanisms that occurs at the time of day when meal intake conventionally occurs. This predictive component of potassium homeostasis involves circadian rhythms generated by tubule-cell circadian clocks, which are synchronized with the central circadian clock in the brain. Much remains to be learned about both reactive and predictive mechanisms of potassium homeostasis and their integration.
Supplementary Material
Footnotes
Dr. Wingo reports receiving consulting fees from ZS Pharma. No other potential conflict of interest relevant to this article was reported.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Weiner ID, Linus S, Wingo CS. Disorders of potassium metabolism. In: Free-hally J, Johnson RJ, Floege J, editors. Comprehensive clinical nephrology. 5th. St. Louis: Saunders; 2014. p. 118. [Google Scholar]
- 2.Malnic G, Giebisch G, Muto S, Wang W, Bailey MA, Satlin LM. Regulation of K+ excretion. In: Alpern RJ, Caplan MJ, Moe OW, editors. Seldin and Giebisch's the kidney: physiology and pathophysiology. 5th. London: Academic Press; 2013. pp. 1659–716. [Google Scholar]
- 3.Mount DB, Zandi-Nejad K. Disorders of potassium balance. In: Taal MW, Chertow GM, Marsden PA, Skorecki KL, Yu ASL, Brenner BM, editors. The kidney. 9th. Philadelphia: Elsevier; 2012. pp. 640–88. [Google Scholar]
- 4.Goyal A, Spertus JA, Gosch K, et al. Serum potassium levels and mortality in acute myocardial infarction. JAMA. 2012;307:157–64. doi: 10.1001/jama.2011.1967. [DOI] [PubMed] [Google Scholar]
- 5.Torlén K, Kalantar-Zadeh K, Molnar MZ, Vashistha T, Mehrotra R. Serum potassium and cause-specific mortality in a large peritoneal dialysis cohort. Clin J Am Soc Nephrol. 2012;7:1272–84. doi: 10.2215/CJN.00960112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smyth A, Dunkler D, Gao P, et al. The relationship between estimated sodium and potassium excretion and subsequent renal outcomes. Kidney Int. 2014;86:1205–12. doi: 10.1038/ki.2014.214. [DOI] [PubMed] [Google Scholar]
- 7.Moore-Ede MC. Physiology of the circadian timing system: predictive versus reactive homeostasis. Am J Physiol. 1986;250:R737–R752. doi: 10.1152/ajpregu.1986.250.5.R737. [DOI] [PubMed] [Google Scholar]
- 8.Hermida RC, Ayala DE, Mojón A, Fernández JR. Bedtime dosing of anti-hypertensive medications reduces cardiovascular risk in CKD. J Am Soc Nephrol. 2011;22:2313–21. doi: 10.1681/ASN.2011040361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hermida RC, Ayala DE, Smolensky MH, et al. Chronotherapy improves blood pressure control and reduces vascular risk in CKD. Nat Rev Nephrol. 2013;9:358–68. doi: 10.1038/nrneph.2013.79. [DOI] [PubMed] [Google Scholar]
- 10.Mills JN, Thomas S, Williamson KS. The effects of intravenous aldosterone and hydrocortisone on the urinary electrolytes of the recumbent human subject. J Physiol. 1961;156:415–23. doi: 10.1113/jphysiol.1961.sp006684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moore-Ede MC, Meguid MM, Fitzpatrick GF, Boyden CM, Ball MR. Circadian variation in response to potassium infusion. Clin Pharmacol Ther. 1978;23:218–27. doi: 10.1002/cpt1978232218. [DOI] [PubMed] [Google Scholar]
- 12.Moore Ede MC, Brennan MF, Ball MR. Circadian variation of intercompartmental potassium fluxes in man. J Appl Physiol. 1975;38:163–70. doi: 10.1152/jappl.1975.38.1.163. [DOI] [PubMed] [Google Scholar]
- 13.Moore-Ede MC, Herd JA. Renal electrolyte circadian rhythms: independence from feeding and activity patterns. Am J Physiol. 1977;232:F128–F135. doi: 10.1152/ajprenal.1977.232.2.F128. [DOI] [PubMed] [Google Scholar]
- 14.Crambert GH. H-K-ATPase type 2: relevance for renal physiology and beyond. Am J Physiol Renal Physiol. 2014;306:F693–F700. doi: 10.1152/ajprenal.00605.2013. [DOI] [PubMed] [Google Scholar]
- 15.Greenlee M, Wingo CS, McDonough AA, Youn JH, Kone BC. Narrative review: evolving concepts in potassium homeostasis and hypokalemia. Ann Intern Med. 2009;150:619–25. doi: 10.7326/0003-4819-150-9-200905050-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oh YT, Kim J, Youn JH. Role of pituitary in K+ homeostasis: impaired renal responses to altered K+ intake in hypophy-sectomized rats. Am J Physiol Regul Integr Comp Physiol. 2013;304:R1166–R1174. doi: 10.1152/ajpregu.00495.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rabinowitz L, Aizman RI. The central nervous system in potassium homeostasis. Front Neuroendocrinol. 1993;14:1–26. doi: 10.1006/frne.1993.1001. [DOI] [PubMed] [Google Scholar]
- 18.Oh KS, Oh YT, Kim SW, Kita T, Kang I, Youn JH. Gut sensing of dietary K intake increases renal K excretion. Am J Physiol Regul Integr Comp Physiol. 2011;301:R421–R429. doi: 10.1152/ajpregu.00095.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aschoff J. Circadian rhythms in man. Science. 1965;148:1427–32. doi: 10.1126/science.148.3676.1427. [DOI] [PubMed] [Google Scholar]
- 20.Calò L, Borsatti A, Favaro S, Rabinowitz L. Kaliuresis in normal subjects following oral potassium citrate intake without increased plasma potassium concentration. Nephron. 1995;69:253–8. doi: 10.1159/000188466. [DOI] [PubMed] [Google Scholar]
- 21.Youn JH. Gut sensing of potassium intake and its role in potassium homeostasis. Semin Nephrol. 2013;33:248–56. doi: 10.1016/j.semnephrol.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Youn JH, McDonough AA. Recent advances in understanding integrative control of potassium homeostasis. Annu Rev Physiol. 2009;71:381–401. doi: 10.1146/annurev.physiol.010908.163241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rabelink TJ, Koomans HA, Hené RJ, Dorhout Mees EJ. Early and late adjustment to potassium loading in humans. Kidney Int. 1990;38:942–7. doi: 10.1038/ki.1990.295. [DOI] [PubMed] [Google Scholar]
- 24.Cheng CJ, Kuo E, Huang CL. Extracellular potassium homeostasis: insights from hypokalemic periodic paralysis. Semin Nephrol. 2013;33:237–47. doi: 10.1016/j.semnephrol.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cope TE, Samaraweera AP, Burn DJ. Thyrotoxic periodic paralysis: correct hypokalemia with caution. J Emerg Med. 2013;45:338–40. doi: 10.1016/j.jemermed.2012.11.107. [DOI] [PubMed] [Google Scholar]
- 26.Fontaine B, Lapie P, Plassart E, et al. Periodic paralysis and voltage-gated ion channels. Kidney Int. 1996;49:9–18. doi: 10.1038/ki.1996.2. [DOI] [PubMed] [Google Scholar]
- 27.Ptácek LJ. Channelopathies: ion channel disorders of muscle as a paradigm for paroxysmal disorders of the nervous system. Neuromuscul Disord. 1997;7:250–5. doi: 10.1016/s0960-8966(97)00046-1. [DOI] [PubMed] [Google Scholar]
- 28.Gordon DL, Agrawal L, Swade TF, Lawrence AM. Thyrotoxic hypokalemic periodic paralysis: six cases in non-Asian patients. Endocr Pract. 1998;4:142–5. doi: 10.4158/EP.4.3.142. [DOI] [PubMed] [Google Scholar]
- 29.Lin SH, Huang CL. Mechanism of thyrotoxic periodic paralysis. J Am Soc Nephrol. 2012;23:985–8. doi: 10.1681/ASN.2012010046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumar V, Armstrong L, Seshadri MS, Finny P. Hypokalaemic periodic paralysis in rural northern India — most have secondary causes. Trop Doct. 2014;44:33–5. doi: 10.1177/0049475513512643. [DOI] [PubMed] [Google Scholar]
- 31.Yılmaz H, Kaya M, Özbek M, ÜUreten K, Safa Yıldırım İ. Hypokalemic periodic paralysis in Sjogren's syndrome secondary to distal renal tubular acidosis. Rheumatol Int. 2013;33:1879–82. doi: 10.1007/s00296-011-2322-z. [DOI] [PubMed] [Google Scholar]
- 32.Agarwal A, Wingo CS. Treatment of hypokalemia. N Engl J Med. 1999;340:154–5. doi: 10.1056/nejm199901143400220. [DOI] [PubMed] [Google Scholar]
- 33.van den Wildenberg MJ, Hoorn EJ, Mohebbi N, et al. Distal renal tubular acidosis with multiorgan autoimmunity: a case report. Am J Kidney Dis. 2015;65:607–10. doi: 10.1053/j.ajkd.2014.09.026. [DOI] [PubMed] [Google Scholar]
- 34.Greenlee MM, Lynch IJ, Gumz ML, Cain BD, Wingo CS. Mineralocorticoids stimulate the activity and expression of renal H+,K+-ATPases. J Am Soc Nephrol. 2011;22:49–58. doi: 10.1681/ASN.2010030311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pan YJ, Young DB. Experimental aldosterone hypertension in the dog. Hypertension. 1982;4:279–87. doi: 10.1161/01.hyp.4.2.279. [DOI] [PubMed] [Google Scholar]
- 36.Grekin RJ, Terris JM, Bohr DF. Electrolyte and hormonal effects of deoxycorticosterone acetate in young pigs. Hypertension. 1980;2:326–32. doi: 10.1161/01.hyp.2.3.326. [DOI] [PubMed] [Google Scholar]
- 37.Schwartz WB. Potassium and the kidney. N Engl J Med. 1955;253:601–8. doi: 10.1056/NEJM195510062531405. [DOI] [PubMed] [Google Scholar]
- 38.El Moghrabi S, Houillier P, Picard N, et al. Tissue kallikrein permits early renal adaptation to potassium load. Proc Natl Acad Sci U S A. 2010;107:13526–31. doi: 10.1073/pnas.0913070107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Graziani G, Fedeli C, Moroni L, Cosmai L, Badalamenti S, Ponticelli C. Gitelman syndrome: pathophysiological and clinical aspects. QJM. 2010;103:741–8. doi: 10.1093/qjmed/hcq123. [DOI] [PubMed] [Google Scholar]
- 40.Bettinelli A, Ciarmatori S, Cesareo L, et al. Phenotypic variability in Bartter syndrome type I. Pediatr Nephrol. 2000;14:940–5. doi: 10.1007/pl00013418. [DOI] [PubMed] [Google Scholar]
- 41.Asmar A, Mohandas R, Wingo CS. A physiologic-based approach to the treatment of a patient with hypokalemia. Am J Kidney Dis. 2012;60:492–7. doi: 10.1053/j.ajkd.2012.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Silver RB, Mennitt PA, Satlin LM. Stimulation of apical H-K-ATPase in intercalated cells of cortical collecting duct with chronic metabolic acidosis. Am J Physiol. 1996;270:F539–F547. doi: 10.1152/ajprenal.1996.270.3.F539. [DOI] [PubMed] [Google Scholar]
- 43.Silver RB, Soleimani MH. H+-K+-ATPases: regulation and role in pathophysiological states. Am J Physiol. 1999;276:F799–F811. doi: 10.1152/ajprenal.1999.276.6.F799. [DOI] [PubMed] [Google Scholar]
- 44.Wingo CS, Smolka AJ. Function and structure of H-K-ATPase in the kidney. Am J Physiol. 1995;269:F1–F16. doi: 10.1152/ajprenal.1995.269.1.F1. [DOI] [PubMed] [Google Scholar]
- 45.Gumz ML, Lynch IJ, Greenlee MM, Cain BD, Wingo CS. The renal H+-K+-ATPases: physiology, regulation, and structure. Am J Physiol Renal Physiol. 2010;298:F12–F21. doi: 10.1152/ajprenal.90723.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hulter HN, Sigala JF, Sebastian A. K+ deprivation potentiates the renal alkalosis-producing effect of mineralocorticoid. Am J Physiol. 1978;235:F298–F309. doi: 10.1152/ajprenal.1978.235.4.F298. [DOI] [PubMed] [Google Scholar]
- 47.Dibner C, Schibler U, Albrecht U. The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu Rev Physiol. 2010;72:517–49. doi: 10.1146/annurev-physiol-021909-135821. [DOI] [PubMed] [Google Scholar]
- 48.Ernsberger P, Azar S, Tarbell M. Effects of preoptic-suprachiasmatic lesions on renal excretion of electrolytes. Life Sci. 1981;28:1387–90. doi: 10.1016/0024-3205(81)90413-6. [DOI] [PubMed] [Google Scholar]
- 49.Stoynev AG, Ikonomov OC, Usunoff KG. Feeding pattern and light-dark variations in water intake and renal excretion after suprachiasmatic nuclei lesions in rats. Physiol Behav. 1982;29:35–40. doi: 10.1016/0031-9384(82)90362-6. [DOI] [PubMed] [Google Scholar]
- 50.Mills JN. Human circadian rhythms. Physiol Rev. 1966;46:128–71. doi: 10.1152/physrev.1966.46.1.128. [DOI] [PubMed] [Google Scholar]
- 51.Elliott AL, Mills JN, Minors DS, Waterhouse JM. The effect of real and simulated time-zone shifts upon the circadian rhythms of body temperature, plasma 11-hydroxycorticosteroids, and renal excretion in human subjects. J Physiol. 1972;221:227–57. doi: 10.1113/jphysiol.1972.sp009750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Konopka RJ, Benzer S. Clock mutants of Drosophila melanogaster. Proc Natl Acad Sci U S A. 1971;68:2112–6. doi: 10.1073/pnas.68.9.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.King DP, Zhao Y, Sangoram AM, et al. Positional cloning of the mouse circadian clock gene. Cell. 1997;89:641–53. doi: 10.1016/s0092-8674(00)80245-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Darlington TK, Wager-Smith K, Ceriani MF, et al. Closing the circadian loop: CLOCK-induced transcription of its own inhibitors per and tim. Science. 1998;280:1599–603. doi: 10.1126/science.280.5369.1599. [DOI] [PubMed] [Google Scholar]
- 55.Shearman LP, Sriram S, Weaver DR, et al. Interacting molecular loops in the mammalian circadian clock. Science. 2000;288:1013–9. doi: 10.1126/science.288.5468.1013. [DOI] [PubMed] [Google Scholar]
- 56.Vitaterna MH, King DP, Chang AM, et al. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science. 1994;264:719–25. doi: 10.1126/science.8171325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Debruyne JP, Noton E, Lambert CM, Maywood ES, Weaver DR, Reppert SM. A clock shock: mouse CLOCK is not required for circadian oscillator function. Neuron. 2006;50:465–77. doi: 10.1016/j.neuron.2006.03.041. [DOI] [PubMed] [Google Scholar]
- 58.DeBruyne JP, Weaver DR, Reppert SM. CLOCK and NPAS2 have overlapping roles in the suprachiasmatic circadian clock. Nat Neurosci. 2007;10:543–5. doi: 10.1038/nn1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lowrey PL, Shimomura K, Antoch MP, et al. Positional syntenic cloning and functional characterization of the mammalian circadian mutation tau. Science. 2000;288:483–92. doi: 10.1126/science.288.5465.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee HM, Chen R, Kim H, Etchegaray JP, Weaver DR, Lee C. The period of the circadian oscillator is primarily determined by the balance between casein kinase 1 and protein phosphatase 1. Proc Natl Acad Sci U S A. 2011;108:16451–6. doi: 10.1073/pnas.1107178108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang EE, Kay SA. Clocks not winding down: unravelling circadian networks. Nat Rev Mol Cell Biol. 2010;11:764–76. doi: 10.1038/nrm2995. [DOI] [PubMed] [Google Scholar]
- 62.Miller BH, McDearmon EL, Panda S, et al. Circadian and CLOCK-controlled regulation of the mouse transcriptome and cell proliferation. Proc Natl Acad Sci U S A. 2007;104:3342–7. doi: 10.1073/pnas.0611724104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Su AI, Wiltshire T, Batalov S, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A. 2004;101:6062–7. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pizarro A, Hayer K, Lahens NF, Hogenesch JB. CircaDB: a database of mammalian circadian gene expression profiles. Nucleic Acids Res. 2013;41:D1009–D1013. doi: 10.1093/nar/gks1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klevecz RR, Li CM. Evolution of the clock from yeast to man by period-doubling folds in the cellular oscillator. Cold Spring Harb Symp Quant Biol. 2007;72:421–9. doi: 10.1101/sqb.2007.72.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bass J, Takahashi JS. Circadian integration of metabolism and energetics. Science. 2010;330:1349–54. doi: 10.1126/science.1195027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang R, Lahens NF, Ballance HI, Hughes ME, Hogenesch JB. A circadian gene expression atlas in mammals: implications for biology and medicine. Proc Natl Acad Sci U S A. 2014;111:16219–24. doi: 10.1073/pnas.1408886111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Salhi A, Centeno G, Firsov D, Crambert G. Circadian expression of H,K-ATPase type 2 contributes to the stability of plasma K levels. FASEB J. 2012;26:2859–67. doi: 10.1096/fj.11-199711. [DOI] [PubMed] [Google Scholar]
- 69.Zuber AM, Centeno G, Pradervand S, et al. Molecular clock is involved in predictive circadian adjustment of renal function. Proc Natl Acad Sci U S A. 2009;106:16523–8. doi: 10.1073/pnas.0904890106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gumz ML, Rabinowitz L. Role of circadian rhythms in potassium homeostasis. Semin Nephrol. 2013;33:229–36. doi: 10.1016/j.semnephrol.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nikolaeva S, Pradervand S, Centeno G, et al. The circadian clock modulates renal sodium handling. J Am Soc Nephrol. 2012;23:1019–26. doi: 10.1681/ASN.2011080842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Doi M, Takahashi Y, Komatsu R, et al. Salt-sensitive hypertension in circadian clock-deficient Cry-null mice involves dysregulated adrenal Hsd3b6. Nat Med. 2010;16:67–74. doi: 10.1038/nm.2061. [DOI] [PubMed] [Google Scholar]
- 73.Moore-Ede MC, Schmelzer WS, Kass DA, Herd JA. Cortisol-mediated synchrinization of circadian rhythm in urinary potassium excretion. Am J Physiol. 1977;233:R230–R238. doi: 10.1152/ajpregu.1977.233.5.R230. [DOI] [PubMed] [Google Scholar]
- 74.Pezük P, Mohawk JA, Wang LA, Menaker M. Glucocorticoids as entraining signals for peripheral circadian oscillators. Endocrinology. 2012;153:4775–83. doi: 10.1210/en.2012-1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Le Minh N, Damiola F, Tronche F, Schütz G, Schibler U. Glucocorticoid hormones inhibit food-induced phase-shifting of peripheral circadian oscillators. EMBO J. 2001;20:7128–36. doi: 10.1093/emboj/20.24.7128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Balsalobre A, Brown SA, Marcacci L, et al. Resetting of circadian time in peripheral tissues by glucocorticoid signaling. Science. 2000;289:2344–7. doi: 10.1126/science.289.5488.2344. [DOI] [PubMed] [Google Scholar]
- 77.Gumz ML, Stow LR, Lynch IJ, et al. The circadian clock protein Period 1 regulates expression of the renal epithelial sodium channel in mice. J Clin Invest. 2009;119:2423–34. doi: 10.1172/JCI36908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stow LR, Richards J, Cheng KY, et al. The circadian protein period 1 contributes to blood pressure control and coordinately regulates renal sodium transport genes. Hypertension. 2012;59:1151–6. doi: 10.1161/HYPERTENSIONAHA.112.190892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bass J, Takahashi JS. Circadian rhythms: redox redux. Nature. 2011;469:476–8. doi: 10.1038/469476a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Marcheva B, Ramsey KM, Buhr ED, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. 2010;466:627–31. doi: 10.1038/nature09253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Elabida B, Edwards A, Salhi A, et al. Chronic potassium depletion increases adrenal progesterone production that is necessary for efficient renal retention of potassium. Kidney Int. 2011;80:256–62. doi: 10.1038/ki.2011.15. [DOI] [PubMed] [Google Scholar]
- 82.Zheng D, Perianayagam A, Lee DH, et al. AMPK activation with AICAR provokes an acute fall in plasma [K+] Am J Physiol Cell Physiol. 2008;294:C126–C135. doi: 10.1152/ajpcell.00464.2007. [DOI] [PubMed] [Google Scholar]
- 83.Sinha AD, Agarwal R. Chronic renal disease progression: treatment strategies and potassium intake. Semin Nephrol. 2013;33:290–9. doi: 10.1016/j.semnephrol.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 84.Walsh SB, Unwin E, Vargas-Poussou R, Houillier P, Unwin R. Does hypokalaemia cause nephropathy? An observational study of renal function in patients with Bartter or Gitelman syndrome QJM. 2011;104:939–44. doi: 10.1093/qjmed/hcr095. [DOI] [PubMed] [Google Scholar]
- 85.Martino TA, Oudit GY, Herzenberg AM, et al. Circadian rhythm disorganization produces profound cardiovascular and renal disease in hamsters. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1675–R1683. doi: 10.1152/ajpregu.00829.2007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.