

Abstract
Introduction
The objective of this study was to evaluate the relationship between self-reported exercise levels and Alzheimer's disease biomarkers, in a cohort of autosomal dominant Alzheimer's disease (ADAD) mutation carriers.
Methods
In 139 pre-symptomatic mutation carriers from the Dominantly Inherited Alzheimer Network, the relationship between self-reported exercise levels and brain amyloid load, CSF Aβ42 and tau levels was evaluated using linear regression.
Results
No differences in brain amyloid load, CSF Aβ42 or tau were observed between low and high exercise groups. Nevertheless, when examining only those already accumulating AD pathology (i.e. amyloid positive), low exercisers had higher mean levels of brain amyloid than high exercisers. Furthermore, the interaction between exercise*estimated years from expected symptom onset was a significant predictor of brain amyloid levels.
Discussion
Our findings indicate a relationship exists between self-reported exercise levels and brain amyloid in ADAD mutation carriers.
Keywords: Physical activity, beta-amyloid, genetics, tau, Alzheimer's disease, dementia
Introduction
Deposition of amyloid plaque within the brain contributes to the neuronal and synaptic loss consistent with Alzheimer's disease (AD), while hyperphosphorylation of tau, believed to occur downstream of amyloid plaque formation, is associated with AD symptom severity [1]. Autosomal dominant AD (ADAD) caused by a mutation in one of three genes: amyloid precursor protein (APP), presenilin 1 (PSEN1), or presenilin 2 (PSEN2), is a rare form of AD resulting in alteration of beta-amyloid (Aβ) processing, leading to AD with full penetrance and at an early age (typically <50 years). In both sporadic late-onset AD (LOAD) and ADAD, accumulation of amyloid manifests up to two decades prior to the presentation of clinical symptoms [2, 3], thus, providing a window of opportunity for intervention. However, in both LOAD and ADAD there are currently no available pharmaceutical treatments known to alter the trajectory of brain amyloid accumulation, nor significantly alter the course of cognitive decline.
Numerous observational studies indicate that high levels of physical activity are associated with reduced risk of clinical LOAD [4-7], as well as risk of LOAD mortality [8]. It is likely this association is governed by underlying mechanisms, including an effect of physical activity on Aβ and/or tau. Indeed, animal studies have demonstrated both soluble and insoluble Aβ levels are lowered by exercise in AD transgenic mice [9-15]. Within the small number of human studies, greater levels of self-reported physical activity have been associated with lower levels of brain amyloid, as measured through amyloid-binding tracers coupled with positron emission tomography (PET) [16-18]. Furthermore, Liang and colleagues [16] reported higher physical activity levels in older adults were associated with greater levels of cerebrospinal fluid (CSF) Aβ42 (an indicator of lower brain amyloid) and lower levels of CSF tau (a marker of neuronal injury). The evidence that exercise is associated with lower brain amyloid levels, and less neuropathology reflected by CSF Aβ and tau measurements, provides important insight into the possible use of exercise as a therapeutic modality in AD. These associations; however, are yet to be examined in individuals with mutations causing ADAD; a gap in knowledge that the current study seeks to address.
The ADAD mutation carriers in the Dominantly Inherited Alzheimer Network (DIAN) study [2] provide an excellent model to determine whether exercise is associated with amyloid load, as these mutation carriers are destined to accumulate cerebral amyloid at an early age. In the current study, we explored relationships between self-reported exercise habits, AD mutation carrier status, and AD biomarkers, hypothesising the following among pre-symptomatic ADAD mutation carriers: 1) exercise habits are associated with biomarker evidence of Aβ and tau (as measured by brain amyloid, CSF Aβ42, and CSF tau), 2) among individuals with evidence of brain amyloid, those with higher amounts of exercise would demonstrate less evidence of AD biomarkers, and 3) exercise level modifies the relationship between expected age of AD symptom onset and AD biomarkers. We also investigated the association between exercise and AD neuroimaging and CSF biomarkers, as described above, in mutation non-carriers included in the DIAN observational study, to evaluate this relationship in those with similar demographic characteristics, but without a dominant gene mutation.
Methods
Participants
Participants at risk for carrying an ADAD mutation were enrolled in the DIAN study. To be eligible for the DIAN study, participants were recruited only if they were a member of a family pedigree with known ADAD mutations, with 197 families (from USA, UK and Australia, Japan, Germany and Argentina) comprising the DIAN cohort. Information regarding participant enrolment and procedures has previously been described in detail [2]. In the current cross-sectional analysis, we used baseline data from mutation non-carriers and mutation carriers with no cognitive impairment. From DIAN data freeze-10, a total of 435 (mutation non-carriers = 172, mutation carriers = 263) participants had baseline data. Individuals with missing exercise and PET data, and/or a Clinical Dementia Rating Global score of greater than 0 were excluded from the analysis (See Figure 1 for full description of participant numbers). Participants without available CSF data, but with available PET data, were included in the brain amyloid analyses only. All participants underwent a comprehensive clinical assessment regarding self and family medical history, medication use, and a physical/medical examination.
Figure 1.
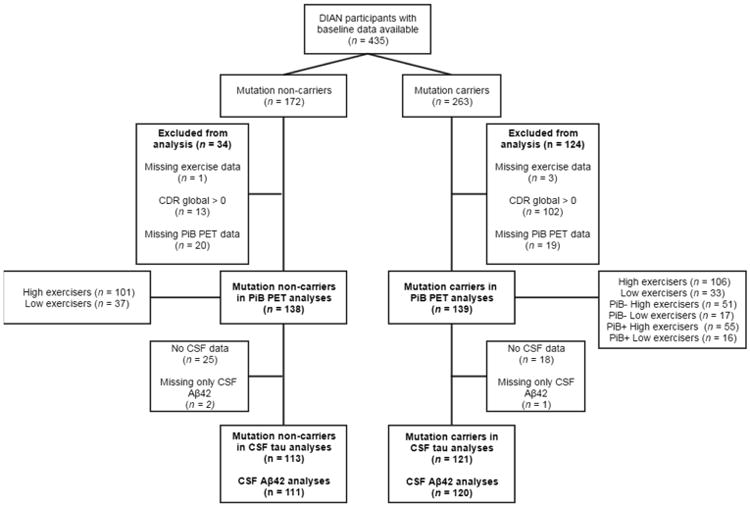
Flow diagram indicating number of participants with data available for inclusion in this study. PiB -, PiB negative with an SUVR of less than 1·3; PiB +, PiB positive with an SUVR of 1·3 or higher. Low exercisers reported less than 150 minutes per week of exercise, high exercisers reported 150 or more minutes of exercise per week. Abbreviations: DIAN, Dominantly Inherited Alzheimer Network study; CDR, Clinical Dementia Rating; PiB PET, Pittsburgh Compound B positron emission tomography; CSF, cerebrospinal fluid; Aβ, beta-amyloid.
Neuroimaging
Images obtained through PET with the use of Pittsburgh Compound B (PiB; an Aβ binding ligand), were co-registered with individual magnetic resonance images for the identification of regions of interest. All studies were collected contemporaneously to baseline determination of self-reported exercise. For each region of interest (FreeSurfer defined, MA, USA), a standardised uptake value ratio (SUVR) was calculated with the cerebellar cortex used as the reference region [19]. The SUVR of the prefrontal cortex, temporal lobe, gyrus rectus and precuneus were averaged to calculate a total cortex SUVR. An SUVR of 1·3 was used to stratify the cohort based on amyloid positivity (PiB- < 1·3, PiB+ ≥ 1·3) [20].
CSF collection and biochemical analyses
Fasting CSF was collected in the morning via lumbar puncture. Samples were snap frozen and shipped on dry ice to the DIAN Biomarker Core laboratory. Levels of Aβ42 and total tau were measured by immunoassay (INNO-BIA AlzBio3, Innogenetics, Ghent, Belgium). All values included in the analysis met quality control standards, which included; a coefficient of variation of 25% or less (typical % CVs were <10%), kit “controls” within the expected range, and measurement consistency between plates of a common sample included in each run.
Genotyping
For the identification of ADAD genetic mutations in the APP, PSEN1, or PSEN2 genes, genotyping was performed on extracted DNA from blood samples. Genotyping was performed at The Genome Technology Access Centre at Washington University using the Infinium HumanExomeCore V1.0 Beadchip (Illumina, Inc., USA). Genotype data were cleaned by applying a minimum call rate for single nucleotide polymorphisms (SNPs) and individuals, set at 98%.
Exercise level evaluation
Participants reported, via questionnaire, their average time spent partaking in 10 various leisure-time exercise activities over the past 12 months in a measurement of ‘minutes per week’. This exercise questionnaire has not been previously validated; thus using data available from DIAN participants reporting exercise at baseline and at a 1 year follow-up (n = 107), we assessed the consistency of exercise reports in this cohort, and report a significant correlation with a moderate effect size (r = 0·53, p < 0·0001). Participation in activities such as walking, running, cycling, swimming, tennis, aerobics and weight training was recorded. In the questionnaire instructions, participants were encouraged to have their responses corroborated by their collateral source (e.g. family member or friend). Outliers were minimised by truncation of individual item responses to a maximum of 600 minutes per week (an adaptation of similar guidelines regarding maximum reports of daily activities to those recommended for the International Physical Activity Questionnaire [21]); this truncation did not alter categorisation into the exercise groups (described below). A continuous score was calculated from all items by the addition of minutes per week spent exercising in each activity. This continuous score was stratified based on current recommendations from the World Health Organisation and the American College of Sports Medicine of a minimum of 150 minutes per week of exercise [22, 23]. Individuals reporting less than 150 minutes of exercise per week were categorised into a ‘low exercise’ group (mutation non-carriers: n = 37, mutation carriers: n = 33), and those participating in more than or equal to 150 minutes of activity per week were categorised into a ‘high exercise’ group (mutation non-carriers: n = 101, mutation carriers: n = 106).
Statistical analysis
Expected years from symptom onset (EYO) was calculated using previously published mutation data, if available; if data for a specific mutation was not available then parental age of symptom onset was used. The specified age of symptom onset from previously published data (or if unavailable, parental age of onset) was taken from the participant's age at the time of assessment to calculate EYO (e.g. a participant aged 30 years at assessment, minus previously published age of symptom onset, 37 years: EYO = -7, i.e. 7 years to expected symptom onset). Due to the high collinearity between EYO and age (r = 0.79, p < 0.0001), the age variable was residualized from EYO for use as a covariate in the linear models. Descriptive data was calculated in the form of mean (standard deviation) and percentage (n) for important clinical and demographic data. Independent sample t-tests were used to evaluate differences in continuous variables, and Chi-square to calculate differences in categorical variables, between the low exercise and high exercise groups for both mutation non-carriers and mutation carriers.
Following stratification of the study cohort into mutation non-carriers and mutation carriers, a series of linear models was used to examine differences in brain amyloid burden and CSF biomarker levels between the low exercise and high exercise groups. These analyses were conducted in the mutation non-carriers to examine whether associations observed in the mutation carriers were unique to this genetic status, or were common to all individuals of a similar age group regardless of genetic status. Brain amyloid burden, CSF Aβ and CSF tau were entered individually as dependent variables, with exercise group entered as a dichotomous independent variable. Furthermore, age (residualized from EYO), family mutation (i.e. APP, PSEN1 or PSEN2) and EYO were entered as covariates in all models, with the inclusion of an exercise group*EYO interaction. The mutation carrier group was further stratified into those who were PiB- versus those who were PiB+, and the models were re-run. The number of participants in each group, and stratified groupings, is described in detail in Figure 1.
All statistical analyses were conducted using the Statistical Package for the Social Sciences (IBM SPSS Statistics for Windows, Version 22·0. Armonk, NY: IBM Corp). A p-value of 0·05 or smaller determined a significant result. False discovery rate (FDR; R environment, version 3.3.2) was used for group corrections for multiple comparisons [24]. Data were visually inspected for outliers and all data-points were within 3.29SD of the mean, a cut-off described by Tabachnick and Fidell [25]. Values for individual participants are not displayed on graphs (i.e. as a scatter plot) to protect the confidentiality of the mutation status of participants (e.g. based on EYO alone, a participant could potentially deduce their mutation status).
Results
Descriptive statistics
Descriptive statistics relating to demographics and relevant medical history are detailed in Table 1. No differences were observed between the ‘low’ and ‘high’ exercise groups within the mutation non-carriers. The mutation carrier ‘low exercise’ group (38·6 ± 7·9 years) was significantly older than the mutation carrier ‘high exercise’ group (33·7 ± 9·3 years; t = 2·68, p = 0·008). Depressive symptoms (as measured by the geriatric depression scale; GDS) were significantly higher among the mutation carrier ‘low exercise’ group (2·2 points ± 2·2 points) compared to the mutation carrier ‘high exercise’ group (1·4 points ± 1·8 points; t = 2·04, p = 0·04). To evaluate the effect of depressive symptoms on the relationship between exercise and AD neuroimaging and CSF biomarkers, GDS was entered as a covariate into the models reported in Table 2; however, the inclusion of GDS did not alter the findings (data not reported), and thus this variable was not included in the final analysis.
Table 1. Demographic and clinical cohort characteristics stratified by non- carriers and carriers of autosomal dominant Alzheimer's disease mutations, split by exercise level.
| NC Low exercise (n = 37) | NC High Exercise (n = 101) | NC low exercise Vs. NC high exercise* p value | MC Low exercise (n = 33) | MC High exercise (n = 106) | MC low exercise Vs. MC high exercise* p value | |
|---|---|---|---|---|---|---|
| Age, y | 39·2 ± 11·6 | 390 ± 11·3 | 0·94 | 38·6 ± 7·9 | 33·7 ± 9·3 | 0008 |
| EYO | N/A | N/A | N/A | -10·2 ± 9·4 | -13·6 ± 9·2 | 007 |
| Years of Education | 150 ± 3·2 | 14·9 ± 2·5 | 0·89 | 14·9 ± 2·8 | 14·7 ± 2·8 | 0·66 |
| Gender, Female % (n) | 65 (24) | 56 (57) | 0·37 | 64 (21) | 56 (59) | 0·42 |
| APOE ε4 allele carriers, % (n) | 21.6 (8) | 30.7 (31) | 0.29 | 36.4 (12) | 25.7 (27) | 0.24 |
| PiB SUVR | 1 03 ± 008 | 105 ± 007 | 0·21 | 1·77 ± 103 | 1·59 ± 0·64 | 0·23 |
| PiB+**, % (n) | 0 (0) | 0 (0) | N/A | 49 (16) | 52 (55) | 0·73 |
| GDS | 1·4 ± 1·7 | 1·3 ± 1 ·6 | 0·66 | 2·2 ± 2·2 | 1·4 ± 1·8 | 004 |
| BMI, kg/m2 | 29·3 ± 8·7 | 29·4 ± 9·8 | 0·96 | 29·8 ± 9·6 | 27·2 ± 6·2 | 007 |
| High cholesterol, % (n) | 11 (4) | 17 (17) | 0·54 | 21 (7) | 12 (13) | 0·24 |
| Hypertension, % (n) | 13 (5) | 18 (18) | 0·82 | 12 (4) | 5 (5) | 0·13 |
| Exercise duration/week, minutes | 68 ± 43 | 427 ± 218 | <0.001 | 64 ± 51 | 522 ± 343 | <0.001 |
| Mutation carriers, n, APP/PSEN1/PSEN2 | N/A | N/A | N/A | 17 / 7 / 9 | 85 / 9 / 12 |
Unless otherwise described, data are presented as mean ± standard deviation of the mean
From independent samples t-test for continuous variables and chi-square for categorical variables.
Those with PiB SUVR 1·3 and greater were categorised as PiB positive (PiB+).
Abbreviations: APOE, Apolipoprotein E; APP, amyloid precursor protein; BMI, body mass index; EYO, estimated years from expected symptom onset; GDS, Geriatric Depression Scale; kg/m2, kilograms per metre squared; MC, Mutation Carriers; NC, Mutation Non-Carriers; y, years; PiB SUVR, Pittsburgh Compound B standardised uptake value ratio; PSEN1, Presenilin 1; PSEN2, Presenilin 2.
Table 2. Mutation non-carriers.
Results from linear models, examining the differences in Pittsburgh Compound B positron emission tomography (PiB PET) measured brain amyloid burden and CSF biomarkers between the low and high exercise groups (models also included family mutation, EYO, age, and an EYO*exercise interaction).
| Exercise* | Family Mutation** | EYO** | Age*** | EYO* Exercise | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dependent variable | Low exercise Mean (SE) Ɣ | High exercise Mean (SE) Ɣ | F | p | ηP2 | F | p | ηp2 | F | p | ηp2 | F | p | ηp2 | F | p | ηp2 |
| PiB SUVR (n = 138) | 1.03 (0.01) | 1.04 (0.01) | 0.33 | 0.60 | 0.003 | 1.53 | 0.22 | 0.023 | 2.64 | 0.11 | 0.020 | 1.71 | 0.19 | 0.013 | 0.43 | 0.51 | 0.003 |
| CSF Aβ42 (ng/L) (n = 111) | 387.2 (29.1) | 439.6 (18.0) | 1.48 | 0.23 | 0.014 | 1.13 | 0.33 | 0.021 | 0.65 | 0.42 | 0.006 | 2.74 | 0.10 | 0.026 | 0.01 | 0.97 | 0.001 |
| CSF tau (ng/L) (n = 113) | 53.0 (4.8) | 60.4 (3.0) | 2.26 | 0.14 | 0.021 | 1.40 | 0.25 | 0.026 | 2.78 | 0.10 | 0.026 | 0.04 | 0.84 | 0.001 | 0.64 | 0.43 | 0.006 |
Low exercisers reported less than 150 minutes per week of exercise, high exercisers reported 150 or more minutes of exercise per week.
For consistency with the mutation carrier models (Tables 3 and 4), EYO and family mutation were entered into the mutation non-carrier models.
The age variable was residualized from EYO.
Adjusted marginal means (standard error). Abbreviations: CSF, cerebrospinal fluid; EYO, estimated years from expected symptom onset; ng/L, nanograms per litre; PiB SUVR, Pittsburgh Compound B standardised uptake value ratio; SE, standard error.
The impact of exercise on Aβ and tau
Within the mutation non-carriers (Table 2), no differences in PiB SUVR (F = 0·33, p = 0·60), CSF Aβ42 (F = 1.48, p = 0·23) and CSF tau (F = 2·26, p = 0·14) were evident between the ‘low exercise’ and ‘high exercise’ groups. On examination of all mutation carriers (Table 3), there were also no differences in PiB SUVR (F = 2.68, p = 0.10), CSF Aβ42 (F = 0.01, p = 0.95) and CSF tau levels (F = 0.25, p = 0.62) between the ‘low exercise’ and ‘high exercise’ groups. In order to examine only those individuals in whom significant brain amyloid load was already present, we stratified the mutation carriers based on PiB positivity (cut-off: 1·3), and re-ran the linear models. In the PiB+ group, lower brain amyloid burden was observed in the MC ‘high exercisers’ (SUVR: 2·16 ± 0·15) compared with the mutation carrier ‘low exercisers’ (SUVR: 2·36 ± 0·19; F = 8·20, p = 0·006, FDR-adjusted p = 0.018).
Table 3. Mutation carriers.
Results from linear models, examining the differences in Pittsburgh Compound B positron emission tomography (PiB PET) measured brain amyloid burden and CSF biomarkers between the low and high exercise groups (models also included family mutation, EYO, age, and an EYO*exercise interaction).
| Exercise* | Family Mutation | EYO | Age** | EYO*Exercise | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dependent variable | Low exerciseMean (SE) Ɨ | High exerciseMean (SE) Ɨ | F | p | ηp2 | F | p | ηp2 | F | p | ηp2 | F | p | ηp2 | F | p | ηp2 |
| PiB SUVR (n = 139) | 1.59 (0.12) | 1.58 (0.09) | 2.68 | 0.10 | 0.020 | 2.42 | 0.09 | 0.035 | 41.30 | <0.001 | 0.238 | 1.86 | 0.17 | 0.014 | 3.32 | 0.07 | 0.025 |
| CSF Aβ42 (ng/L) (n = 120) | 337.5 (30.7) | 349.1 (22.9) | 0.01 | 0.95 | 0.001 | 1.54 | 0.22 | 0.026 | 21.05 | <0.001 | 0.157 | 0.01 | 0.99 | 0.001 | 0.04 | 0.85 | 0.001 |
| CSF tau (ng/L) (n = 121) | 81.0 (9.4) | 82.1 (6.9) | 0.25 | 0.62 | 0.002 | 0.85 | 0.43 | 0.015 | 13.4 | <0.001 | 0.105 | 0.14 | 0.71 | 0.001 | 0.25 | 0.61 | 0.002 |
Low exercisers reported less than 150 minutes per week of exercise, high exercisers reported 150 or more minutes of exercise per week.
The age variable was residualized from EYO.
Adjusted marginal means (standard error).
Abbreviations: CSF, cerebrospinal fluid; EYO, estimated years from expected symptom onset; ng/L, nanograms per litre; PiB SUVR, Pittsburgh Compound B standardised uptake value ratio; SE, standard error.
Previous studies have reported an effect of the APOE ε4 allele on the relationship between exercise and brain amyloid in LOAD; thus, we re-ran the linear models including APOE ε4 carriage as a covariate and an exercise*APOE ε4 interaction. Neither the APOE ε4 carriage variable, nor the interaction term was a significant predictor of brain amyloid in the mutation non-carriers, mutation carrier PiB+ and mutation carrier PiB- groups (data not reported).
Effect of exercise*EYO interaction on PiB SUVR, CSF Aβ42 and CSF tau
On examination of the mutation carrier group as a whole, there was no significant effect of the exercise group*EYO interaction on PiB SUVR (F = 3·32, p = 0·07), CSF Aβ42 (F = 0·04, p = 0·85) and CSF tau levels (F = 0·25, p = 0·61; Table 3). After stratification of the mutation carrier group by PiB positivity, an interaction was observed between exercise group*EYO on PiB SUVR in the PiB+ group (F = 7·04, p = 0·01, FDR-adjusted p = 0.03; Table 4, Figure 2), indicative of a stronger association between brain amyloid and EYO in the low exercisers, compared to the high exercisers. No effect of the exercise*EYO interaction was noted on levels of CSF Aβ42 and CSF tau in either the PiB- or PiB+ groups.
Table 4. Mutation carriers stratified by brain amyloid load.
Results from linear models, examining the differences in Pittsburgh Compound B positron emission tomography (PiB PET) measured brain amyloid burden and CSF biomarkers between the low and high exercise groups, following stratification of the mutations carriers by PiB PET SUVR status (models also included family mutation, EYO, age, and an EYO*exercise interaction).
| Exercise* | Family Mutation | EYO | Age** | EYO*Exercise | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Low exercise Mean (SE) Ɨ | High exercise Mean (SE) Ɨ | F | p | ηp2 | F | p | ηp2 | F | p | ηp2 | F | p | ηp2 | F | p | ηp2 | ||
| PiB SUVR | MC PiB − (n = 68) | 1.09 (0.03) | 1.11 (0.02) | 1.16 | 0.28 | 0.019 | 0.30 | 0.74 | 0.010 | 2.27 | 0.14 | 0.036 | 0.97 | 0.33 | 0.016 | 0.48 | 0.49 | 0.008 |
| MC PiB + (n = 71) | 2.36 (0.19) | 2.16 (0.15) | 8.20 | 0.006a | 0.114 | 0.51 | 0.60 | 0.016 | 16.22 | <0.001 | 0.202 | 1.92 | 0.17 | 0.029 | 7.04 | 0.01a | 0.099 | |
| CSF Aβ42 (ng/L) | MC PiB − (n = 57) | 387.0 (47.3) | 427.8 (31.3) | 0.15 | 0.70 | 0.003 | 3.02 | 0.06 | 0.108 | 1.27 | 0.26 | 0.025 | 0.47 | 0.50 | 0.009 | 0.00 | 0.99 | 0.001 |
| MC PiB + (n = 63) | 269.0 (34.9) | 290.3 (30.4) | 0.34 | 0.56 | 0.006 | 0.06 | 0.94 | 0.002 | 7.36 | 0.01 | 0.116 | 0.38 | 0.54 | 0.007 | 0.02 | 0.88 | 0.001 | |
| CSF tau (ng/L) | MC PiB − (n = 57) | 57.5 (8.6) | 64.1 (5.7) | 0.01 | 0.98 | 0.001 | 0.30 | 0.74 | 0.012 | 1.03 | 0.31 | 0.020 | 0.15 | 0.70 | 0.003 | 0.16 | 0.69 | 0.003 |
| MC PiB + (n = 64) | 109.9 (16.1) | 94.3 (13.4) | 0.51 | 0.48 | 0.009 | 1.16 | 0.32 | 0.039 | 2.22 | 0.14 | 0.037 | 1.16 | 0.29 | 0.020 | 3.16 | 0.08 | 0.052 | |
Low exercisers reported less than 150 minutes per week of exercise, high exercisers reported 150 or more minutes of exercise per week.
The age variable was residualized from EYO.
Adjusted marginal means (standard error).
p-value remained significant (p < 0.05) after false discovery rate correction.
Abbreviations: CSF, cerebrospinal fluid; EYO, estimated years from expected symptom onset; MC, mutation carriers; ng/L, nanograms per litre; PiB SUVR, Pittsburgh Compound B standardised uptake value ratio; PiB -, PiB negative with an SUVR of less than 1·3; PiB +, PiB positive with an SUVR of 1·3 or higher SE, standard error.
Figure 2. The association between brain amyloid and EYO is more marked in low exercisers, compared with high exercisers.
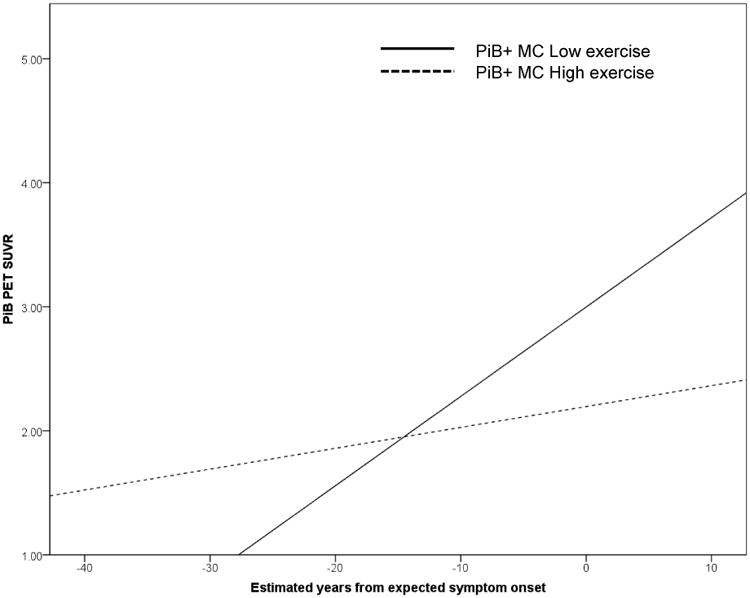
PiB positive (PiB+, i.e. those with an SUVR ≥ 1·3) mutation carriers (MC) reporting less than 150 minutes of exercise per week (low exercise) have a more marked association between estimated years from expected symptom onset and Pittsburgh Compound B positron emission tomography (PiB PET) standardised uptake value ratio (SUVR), compared with PiB+ mutation carriers reporting more than 150 minutes of exercise per week (high exercise).
Discussion
Previous studies of cognitively healthy older adults have established a link between higher physical activity levels and lower levels of AD biomarkers (through PET imaging and CSF biomarker analysis). This study reports, for the first time, an association between higher exercise levels and lower brain amyloid in individuals who have already accumulated high levels of brain amyloid and are carriers of mutations in APP, PSEN1 or PSEN2 genes, which are known to cause AD with full penetrance. More specifically, we observed mutation carriers reporting less than 150 minutes of exercise per week had a higher mean level of brain amyloid, compared with those reporting 150 or more minutes of exercise per week. Furthermore, we report a significant interaction of exercise group*EYO on amyloid load, whereby in PiB+ mutation carrier low exercisers the relationship between brain amyloid and EYO was more marked, compared with high exercisers (Figure 2).
High levels of physical activity have been previously associated with lower brain amyloid levels in cognitively healthy older adults at increased risk of late onset Alzheimer's disease, due to carriage of a major genetic risk factor (APOE ε4 carriage) [17, 18]. Consequently, we hypothesised that exercise may also positively influence individuals carrying mutations for ADAD through a reduction in brain Aβ and/or slowed Aβ accumulation. Within the current study, in a cohort of individuals with ADAD genetic mutations who were also dichotomized as PiB positive (i.e. in whom significant levels of aggregated Aβ are already present in the brain), we observed significantly lower levels of cortical amyloid consistent with high levels of exercise. We also observed a significant effect of the interaction term exercise group*EYO on brain amyloid. Our results indicate that in PiB+ low exercisers, the expected strong association between brain amyloid load and EYO exists [2]. Conversely, and importantly, we observed no association between brain amyloid and EYO in the PiB+ high exercisers, which is not the expected course of the disease for individuals with ADAD mutations. Our findings may reflect the notion that low exercisers are more likely to follow the usual disease course of ADAD (i.e. increasing amyloid accumulation with increasing EYO), than high exercisers. Although our findings are novel and promising, the cross-sectional study design does not allow causal inferences. In order to further examine the relationship between exercise and the trajectory of amyloid accumulation, and subsequent symptom onset in individuals with ADAD mutations, longitudinal analyses of the impact of exercise habits on disease course are vital.
The reported association between exercise and brain amyloid levels was limited to participants who were mutation carriers and PiB positive (i.e. in those with significant pathology present, at levels comparable to a positive PiB scan in sporadic AD). It is possible, however, that the low variability in PiB SUVR levels in those deemed PiB negative may account for the lack of findings in this group. Bateman et al. [2] showed Aβ deposition in ADAD mutation carriers begins approximately 20 years before the onset of clinical symptoms. Should exercise be effective in delaying Aβ accumulation, it would be reasonable to assume that this mechanism of action would also be vital in the early stages of neuropathological changes (i.e. those who are PiB-). Thus the lack of association in this study between exercise and brain Aβ in the PiB- group is unexpected. To further understand this relationship, a longitudinal study evaluating exercise levels and brain Aβ over long periods (i.e. from -15 EYO until 0 EYO) is necessary.
In contrast to the observed association between exercise and brain amyloid levels (quantified by PiB PET), we did not observe an association between exercise and CSF levels of Aβ42, or tau, in the mutation carriers. It is possible that exercise plays a role in reducing the deposition of soluble Aβ into cerebral amyloid plaques, rather than modulating the production of soluble Aβ (levels of which are quantified by the CSF assays). Recent studies have reported a close association between changes in CSF Aβ42 and brain amyloid in the earliest stages of AD pathology.[26] Nevertheless, CSF Aβ42 levels and brain amyloid levels appear to diverge once significant plaque load is present; which may explain the lack of association between CSF Aβ42 and exercise levels in the PiB+ group. The use of amyloid brain imaging may provide a more robust measurement for the evaluation of the relationship between exercise and aggregation of Aβ into amyloid plaques. However, due to the vast literature supporting the use of CSF Aβ42 and tau as biomarkers of AD [27], these measurements should be considered in future longitudinal studies of exercise and AD biomarkers.
We report an association between exercise levels and brain amyloid in those with genetic mutations known to cause increased Aβ; nevertheless, whether exercise is associated with reduced Aβ deposition, or enhanced Aβ clearance remains to be established. Aβ is produced from the amyloid precursor protein (APP), which is cleaved via one of two competing pathways: the non-amyloidogenic pathway and the amyloidogenic pathway [28]. Evidence from animal studies indicates exercise may contribute to both the alteration of APP processing towards the non-amyloidogenic pathway resulting in reduced Aβ production, and to improvement of Aβ clearance in the brain. Indeed, decreased levels of APP cleavage fragments (αCTFs and βCTFs), but not levels of APP itself, have been observed in exercising AD transgenic mice, suggesting increased non-amyloidogenic processing [9, 11]. Furthermore, exercise-induced increases in activity of neprilysin and insulin degrading enzyme, both known Aβ proteases, indicates a positive effect of exercise on Aβ degradation [29, 30]. It is possible that through the conduct of both longitudinal studies and well-designed exercise intervention trials, we may have the opportunity to establish whether exercise contributes to decreased Aβ deposition or enhanced clearance (or possibly both), in this unique cohort of individuals carrying ADAD genetic mutations.
To our knowledge, this preliminary study is the first report of an association between exercise level and brain amyloid in a cohort of pre-symptomatic ADAD mutation carriers. Nevertheless, this study is not without limitations. As stated earlier, this is a cross-sectional analysis, and thus the direction of the reported associations cannot be inferred. Although it is possible that low exercise might be an early symptom of amyloid accumulation, the association between exercise and amyloid remained stable following adjustment for EYO (i.e. years from expected symptom onset). A more likely hypothesis is that higher exercise alters amyloid accumulation, which is supported by previous animal work; however, this hypothesis requires further investigation using longitudinal and intervention trial designs. Furthermore, we utilised an exercise questionnaire specifically designed for this study (i.e. not previously validated) and also acknowledge that the reported exercise levels are higher than that of the wider community. Nevertheless, it is likely that our cohort is highly motivated to participate in exercise, given the increasing literature linking a healthy lifestyle (including exercise) to reduced biomarkers of Alzheimer's disease, and enhanced overall cognitive health. Coupled with corroboration of reports by a collateral source and truncation of exceptionally high reports, we believe the reports of exercise are a relatively true representation in this unique genetic group. It is important to note that we only quantified the duration of exercise undertaken by participants. Future studies should evaluate intensity, frequency, duration and type of exercise and physical activities, in an attempt to identify the optimum exercise/physical activity regimen, in terms of modulating Alzheimer's disease biomarkers. We attempted to control for factors which may confound the relationship between exercise and AD pathology, including EYO, age and APOE ε4 allele carriage. Nonetheless, we were limited by small sample size which thus requires limited inclusion of additional variables of interest in the model. Importantly, exercise may be a proxy for other healthy lifestyle decisions and behaviours, including dietary habits, midlife obesity, body mass index, and tobacco abuse, among other potential modifiable risk factors shown to modify AD risk and pathology in non-ADAD cohorts.
To our knowledge, this study is the first to demonstrate an association between high levels of exercise and lower brain amyloid as a function of expected years from symptoms onset in those known to have ADAD genetic mutations and in whom high levels of brain amyloid are already present. These findings support previous work conducted in cognitively healthy older adults; however, the relationship between brain amyloid and exercise in ADAD mutation carriers requires further confirmation. Future research should include longitudinal studies of exercise and brain amyloid levels to inform intervention trials with amyloid as the primary outcome measure.
Research in context.
Systematic review
The authors reviewed previous literature via usual methods (e.g., PubMed). Previous cross-sectional observational studies have demonstrated an association between higher physical activity levels and lower cerebral amyloid load. This relationship has also been reported to be more prominent in carriers of the apolipoprotein E ε4 allele, the greatest known genetic risk factor for late-onset sporadic Alzheimer's disease.
Interpretation
This study utilised data from mutation carriers of autosomal dominant Alzheimer's disease genes; which are known to cause Alzheimer's disease with full penetrance at an early age (usually less than 50 years). By studying this unique cohort of individuals, we are able to evaluate the relationship between self-reported exercise levels and markers of Alzheimer's disease pathology, in those we know will develop the condition at a young age. We report a relationship between self-reported exercise levels and cerebral amyloid load in mutation carriers already accumulating Alzheimer's disease pathology, as a function of their estimated years from expected symptom onset.
Future directions
Although numerous studies, including the current study, have demonstrated a cross-sectional relationship between physical activity levels and cerebral amyloid load, this evidence requires further validation in longitudinal and intervention studies.
Acknowledgments
Data collection and sharing for this project was supported by The Dominantly Inherited Alzheimer Network (DIAN; UF1 AG032438; to RJB and JCM), funded by the National Institute on Aging, the German Center for Neurodegenerative Diseases (DZNE), the Medical Research Council (MRC; to NCF and MNR) Dementias Platform UK (MR/L023784/1 and MR/009076/1), and National Institute for Health Research Queen Square Dementia Biomedical Research Unit. BB receives research support from Alzheimer's Australia Dementia Research Foundation, NHMRC National Institute of Dementia and the Brain Foundation. JCM and RJB receive research support from National Institute of Health. RJB receives research support from the Alzheimer's Association, Foundation for Biomedical Research and Innovation, BrightFocus Foundation, Cure Alzheimer's Fund, Glenn Foundation for Medical Research, Metropolitan Life Foundation, and Ruth K Broadman Biomedical Research Foundation. This manuscript has been reviewed by DIAN Study investigators for scientific content and consistency of data interpretation with previous DIAN Study publications. The DIAN Expanded Registry welcomes contact from any families or treating clinicians interested in research about autosomal dominant familial Alzheimer's disease.
Footnotes
Declarations of Interest: RJB reports grants from Eli Lilly, Roche, Pharma Consortium (Abbvie, AstraZeneca, Biogen, Eisai, Eli Lilly and Co., Hoffman La-Roche Inc., Janssen, Pfizer, Sanofi-Aventi), and Tau SILK/PET Consortium (Biogen/Abbvie/Lilly), non-financial support from Avid Radiopharmaceuticals, personal fees and other from Washington University, personal fees and non-financial support from Roche, IMI, FORUM, and Pfizer, and personal fees from Merck, Johnson and Johnson, outside the submitted work. JCM is currently participating in clinical trials of antidementia drugs from Eli Lilly and Company, Biogen, and Janssen. JCM serves as a consultant for Lilly USA and receives research support from Eli Lilly/Avid Radiopharmaceuticals. TB has receives grant funding from Avid Radiopharmaceuticals/Eli Lilly and participates in clinical trials sponsored by Eli Lilly, Avid Radiopharmaceuticals, Roche, and Pfizer.
Contributors: BB completed the literature search and prepared the figures. BB and RNM designed this sub-study, RJB and JCM designed larger DIAN study. BB, HRS, SG, KT, TS, CX, AMF, TB, RC, CLM, MW, NC, MR, NRG, SS, JV, CL, VB, JN, PS and RNM, with the Dominantly Inherited Alzheimer Network (DIAN) collected the data. BB and CX conducted the analysis. BB, RNM, JP, SRS and KE interpreted the data and wrote the report. All co-authors critically reviewed the report.
For all other authors; Conflicts of interest: none.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ittner LM, Gotz J. Amyloid-beta and tau--a toxic pas de deux in Alzheimer's disease. Nat Rev Neurosci. 2011;12(2):65–72. doi: 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- 2.Bateman RJ, et al. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer's Disease. N Engl J Med. 2012 doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Villemagne VL, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357–67. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- 4.Buchman AS, et al. Total daily physical activity and the risk of AD and cognitive decline in older adults. Neurology. 2012;78(17):1323–9. doi: 10.1212/WNL.0b013e3182535d35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rovio S, et al. Leisure-time physical activity at midlife and the risk of dementia and Alzheimer's disease. Lancet Neurol. 2005;4(11):705–11. doi: 10.1016/S1474-4422(05)70198-8. [DOI] [PubMed] [Google Scholar]
- 6.Abbott RD, et al. Walking and dementia in physically capable elderly men. JAMA. 2004;292(12):1447–53. doi: 10.1001/jama.292.12.1447. [DOI] [PubMed] [Google Scholar]
- 7.Scarmeas N, et al. Physical activity, diet, and risk of Alzheimer disease. JAMA. 2009;302(6):627–37. doi: 10.1001/jama.2009.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scarmeas N, et al. Physical activity and Alzheimer disease course. Am J Geriatr Psychiatry. 2011;19(5):471–81. doi: 10.1097/JGP.0b013e3181eb00a9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adlard PA, et al. Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer's disease. J Neurosci. 2005;25(17):4217–21. doi: 10.1523/JNEUROSCI.0496-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Um HS, et al. Exercise training acts as a therapeutic strategy for reduction of the pathogenic phenotypes for Alzheimer's disease in an NSE/APPsw-transgenic model. Int J Mol Med. 2008;22(4):529–39. [PubMed] [Google Scholar]
- 11.Liu HL, et al. Long-term treadmill exercise inhibits the progression of Alzheimer's diseaselike neuropathology in the hippocampus of APP/PS1 transgenic mice. Behav Brain Res. 2013;256:261–72. doi: 10.1016/j.bbr.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 12.Yuede CM, et al. Effects of voluntary and forced exercise on plaque deposition, hippocampal volume, and behavior in the Tg2576 mouse model of Alzheimer's disease. Neurobiol Dis. 2009;35(3):426–32. doi: 10.1016/j.nbd.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nichol KE, et al. Exercise alters the immune profile in Tg2576 Alzheimer mice toward a response coincident with improved cognitive performance and decreased amyloid. J Neuroinflammation. 2008;5:13. doi: 10.1186/1742-2094-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao G, et al. Treadmill exercise enhances synaptic plasticity, but does not alter beta-amyloid deposition in hippocampi of aged APP/PS1 transgenic mice. Neuroscience. 2015;298:357–66. doi: 10.1016/j.neuroscience.2015.04.038. [DOI] [PubMed] [Google Scholar]
- 15.Moore KM, et al. A spectrum of exercise training reduces soluble Abeta in a dose-dependent manner in a mouse model of Alzheimer's disease. Neurobiol Dis. 2016;85:218–24. doi: 10.1016/j.nbd.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 16.Liang KY, et al. Exercise and Alzheimer's disease biomarkers in cognitively normal older adults. Ann Neurol. 2010;68(3):311–8. doi: 10.1002/ana.22096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Head D, et al. Exercise Engagement as a Moderator of the Effects of APOE Genotype on Amyloid Deposition. Arch Neurol. 2012 doi: 10.1001/archneurol.2011.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown BM, et al. Physical activity and amyloid-beta plasma and brain levels: results from the Australian Imaging, Biomarkers and Lifestyle Study of Ageing. Mol Psychiatry. 2013;18(8):875–81. doi: 10.1038/mp.2012.107. [DOI] [PubMed] [Google Scholar]
- 19.Benzinger TL, et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer's disease. Proc Natl Acad Sci U S A. 2013;110(47):E4502–9. doi: 10.1073/pnas.1317918110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su Y, et al. Partial volume correction in quantitative amyloid imaging. Neuroimage. 2015;107:55–64. doi: 10.1016/j.neuroimage.2014.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Craig CL, et al. International physical activity questionnaire: 12-country reliability and validity. Med Sci Sports Exerc. 2003;35(8):1381–95. doi: 10.1249/01.MSS.0000078924.61453.FB. [DOI] [PubMed] [Google Scholar]
- 22.World Health Organisation. Global Recommendations on Physical Activity for Health. 2010 [PubMed] [Google Scholar]
- 23.Garber CE, et al. American College of Sports Medicine position stand. Quantity and quality of exercise for developing and maintaining cardiorespiratory, musculoskeletal, and neuromotor fitness in apparently healthy adults: guidance for prescribing exercise. Med Sci Sports Exerc. 2011;43(7):1334–59. doi: 10.1249/MSS.0b013e318213fefb. [DOI] [PubMed] [Google Scholar]
- 24.Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. The Annals of Statistics. 2001;29(4):1165–1188. [Google Scholar]
- 25.Tabachnick BG, Fidell LS. Using multivariate statistics. 5th. Needham Heights, MA, USA: Pearson Education; 2007. [Google Scholar]
- 26.Vlassenko AG, et al. Imaging and cerebrospinal fluid biomarkers in early preclinical alzheimer disease. Ann Neurol. 2016;80(3):379–87. doi: 10.1002/ana.24719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fagan AM, et al. Longitudinal change in CSF biomarkers in autosomal-dominant Alzheimer's disease. Sci Transl Med. 2014;6(226):226ra30. doi: 10.1126/scitranslmed.3007901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verdile G, et al. The role of beta amyloid in Alzheimer's disease: still a cause of everything or the only one who got caught? Pharmacol Res. 2004;50(4):397–409. doi: 10.1016/j.phrs.2003.12.028. [DOI] [PubMed] [Google Scholar]
- 29.Lazarov O, et al. Environmental enrichment reduces Abeta levels and amyloid deposition in transgenic mice. Cell. 2005;120(5):701–13. doi: 10.1016/j.cell.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 30.Tabuchi M, et al. Sleep interacts with abeta to modulate intrinsic neuronal excitability. Curr Biol. 2015;25(6):702–12. doi: 10.1016/j.cub.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]