

Abstract
Antibiotic resistance of bacteria is a growing problem and presents a challenge for prompt treatment in patients with sepsis. Currently used methods rely on culturing or amplification; however, these steps are either time consuming or suffer from interference issues. A microfluidic device was made from black polypropylene, with a monolithic column modified with a capture oligonucleotide for sequence selective solid-phase extraction of a complementary target from a lysate sample. Porous properties of the monolith allow flow and hybridization of a target complementary to the probe immobilized on the column surface. Good flow through properties enable extraction of a 100 μL sample and elution of target DNA in 12 minutes total time. Using a fluorescently labeled target oligonucleotide related to Verona Integron-Mediated Metallo-β-lactamase it was possible to extract and detect a 1 pM sample with 83% recovery. Temperature-mediated elution by heating above the duplex melting point provides a clean extract without any agents that interfere with base pairing, allowing various labeling methods or further downstream processing of the eluent. Further integration of this extraction module with a system for isolation and lysis of bacteria from blood, as well as combining with single-molecule detection should allow rapid determination of antibiotic resistance.
Keywords: hybridization, microfluidics, monolith, sepsis
1. Introduction
Antibiotic resistance of bacteria to commonly used antibiotics calls for rapid methods of identification of the source of infection, often in critical sepsis patients. As antibiotic resistance is a growing problem, it makes determination of proper treatment even more important than before. Commonly used methods rely on culturing bacteria to enable their detection; however, this procedure is time consuming (1 or more days) and limiting for appropriately timed treatment [1]. Improved analysis methods for specific DNA sequences related to antibiotic-resistant bacteria could allow their rapid identification, without the need for amplification.
Extraction and purification of DNA from a biological matrix presents a critical process for diagnostics [2]. For methods based on amplification [3], sequencing [4] or hybridization [5] it is necessary to isolate and concentrate DNA from a complex sample containing various classes of compounds. Traditional methods involving solvent precipitation are replaced by solid-phase extraction on silica or organic polymers [6, 7] or use of magnetic particles [8], which offer automation and miniaturization for applications where sample is available in limited quantity [6]. However, due to lack of selectivity, DNA extraction efficiency on silica or similar materials suffers from competing molecules like proteins that must be removed from the sample, increasing process complexity [9].
Oligonucleotides immobilized on substrates have extensive implementation in molecular biology applications, including DNA arrays widely used in genomics, food characterization and disease diagnosis, enabling screening for a large number of sequences on a surface grafted with complementary DNA [10]. Hybridization between an immobilized oligonucleotide and its single-stranded complementary sequence in a sample allows sequence-specific retention of DNA on a solid support. For example, a column packed with particles modified with oligonucleotides was used for sequence-specific DNA analysis, using elution by denaturing with sodium hydroxide and detection by UV absorption [11]. Satterfield et al. [12] prepared a porous monolith in a fused silica capillary with the surface modified with a poly(dT) 30 mer to capture mRNA from eukaryotic cells. Polyacrylamide hydrogel plugs modified with immobilized oligonucleotides were used in a polycarbonate microfluidic device for electrophoresis of a fluorescently tagged complementary sequence [13]. This work demonstrated selectivity of the hybridization of multiple targets on porous media that withstood low pressures (<3 psi or 200 mbar). Root et al. [14] demonstrated a polyacrylamide gel with immobilized oligonucleotides for capture of target mRNA from serum using voltage to pass the sample through the polymer matrix; the extraction procedure was followed by off-chip PCR amplification for detection of low target copy numbers.
Microfluidic systems are becoming widely used in sample preparation for DNA extraction and purification, allowing miniaturization and automation of complex workflows, and processing of small quantities of sample [15,16]. Porous polymers in microfluidics have been used for sample preparation and extraction of DNA, enabling rapid purification for amplification by polymerase chain reaction [17–19]. Silica beads incorporated in a porous polymer monolith were also used for non-sequence-specific extraction of DNA and RNA [20,21].
In this work, we describe a platform for selective DNA extraction utilizing hybridization, with selectivity governed by DNA base complementarity. A sample containing target DNA is flowed through a porous polymer monolith modified with a capture oligonucleotide designed to be complementary to the target. After a rinse step, captured target DNA is released by heating the monolith above melting point of the DNA hybrid and detected by laser induced fluorescence during flow. Preparing the monolith in a microfluidic channel enables integration of such a module into a platform for rapid diagnosis of bacterial infection as well as possible antibiotic resistance.
2. Materials and Methods
2.1 Chemicals
Tris hydrochloride, Tris base, sulfuric acid, sodium hydroxide, sodium dodecylsulfate, sodium bicarbonate, ethylenediaminetetraacetic acid (EDTA), sodium chloride, magnesium chloride, sodium periodate, glycidyl methacrylate (GMA), poly(ethyleneglycol) diacrylate (PEGDA, MW 575), 1-propanol, 1,4-butanediol, benzoin methyl ether (BME), sodium cyanoborohydride, 2-propanol and ethanol were obtained from Sigma-Aldrich (St. Louis, MO), Merck (Darmstadt, Germany) or Fisher Scientific (Pittsburgh, PA). All chemicals were analytical grade purity or higher.
2.2 Sequence design methods
For extracting and detecting genes associated with antibiotic resistance, Verona Integron-Mediated Metallo-β-lactamase (VIM) gene was selected. The capture sequence was designed by querying GenBank (National Center for Biotechnology Information, NIH) for coding sequences for the VIM gene. At least 50 sequences were downloaded and analyzed using the MUSCLE algorithm for multiple sequence alignment in MEGA 5.1 (Molecular Evolutionary Genetics Analysis). A consensus sequence for the gene was constructed and used for capture sequence design using PrimerQuest (Integrated DNA Technologies, Coralville, IA). The putative capture sequence was evaluated for optimum theoretical sensitivity and specificity using BLAST (National Center for Biotechnology Information, NIH). A list of the bacterial genomes evaluated for sequence specificity in this study is provided in the Supplementary Information. The oligonucleotides used in these experiments (see Table 1) were obtained from Eurofins Scientific (Louisville, KY).
Table 1.
DNA oligonucleotide sequences. The modified base in the mismatched target is underlined. Sequences are given 5′ to 3′.
| name | sequence |
|---|---|
| NH2-VIM | [AminoC6][Sp-C18]TCGGRGAGGTCCGRCTTTACCAGA |
| Fl-VIM | [Fl]TCTGGTAAAGYCGGACCTCYCCGA |
| Fl-VIM-M1 | [Fl]TCTGGTAAAGYCAGACCTCYCCGA |
| Fl-KPC | [Fl]CAAGACAGCAGAACTAGACGGCGATA |
2.3 Device and monolith fabrication
Polypropylene microfluidic devices were prepared by a hot embossing method to transfer the channel features using custom-made CNC machined aluminum masters and black polypropylene sheets used in a commercial sepsis diagnostic system (Great Basin Corp., Salt Lake City, UT). Next, holes were drilled for channel inlets and outlets, followed by thermal sealing of the channels with a transparent polypropylene foil (Great Basin Corp.).
After cleaning of the channels with 2-propanol, the device was filled with a polymerization mixture containing GMA (24% m/m), PEGDA (16% m/m), 1-propanol (40% m/m) and 1,4-butanediol (20% m/m) with BME (3% m/m, relative to monomers) as a photoinitiator. The device was masked using black tape leaving a 5 mm gap close to the device outlet and exposed to UV light using a SunRay 600 UV lamp (Uvitron international, West Springfield, MA). After the polymerization step, the device was rinsed with 2-propanol for 15 minutes and water for 10 minutes. Scanning electron microscopy (SEM) images of monoliths and devices were taken using a Phillips XL30 environmental scanning electron microscope (Hillsboro, OR).
2.4 Modification of monoliths with capture probe
Amine-terminated capture probes were attached to epoxy groups on the monolith via the Schiff base method [22]. Briefly, the monolith was treated with 0.5 M sulfuric acid for 24 hours at 50 °C, followed by oxidation with 0.1 M sodium periodate. Amine-terminated oligonucleotide (20 μM) in 0.1 M sodium bicarbonate pH 9 containing 5 mg/mL sodium cyanoborohydride was passed through the monolith at 5 μL/min for 1 hour and then left to react overnight at room temperature. Next, the monolith was rinsed with 20 mM Tris-HCl pH 8 containing 5 mg/mL sodium cyanoborohydride at 5 μL/min for 1 hour and then rinsed and stored at 4 °C in 20 mM Tris-HCl pH 8.
2.5 Lysis of bacteria
E. coli strain BL21 Star (DE3) from Thermo Fisher (Waltham, MA) was grown for 18 hours in 250 mL of Luria-Bertani broth (2.5 g tryptone, 1.25 g yeast extract and 2.5 g sodium chloride) in a shaker flask at 37 °C. Growth medium (5 mL) was taken and centrifuged at 3000 rpm for 2 minutes and decanted. Phosphate-buffered saline (3 mL) was added, the solution was centrifuged and this cleaning process was repeated twice. Then, 350 μL of this solution was mixed with 350 μL of 1% SDS, 0.2 M NaOH, 10 mM EDTA in 50 mM Tris-HCl pH 8 and incubated for 10 minutes with vortexing every 2 minutes. A resuspended Dynabeads MyOne SILANE suspension (50 μL, Thermo Fisher) was added followed by 400 μL of 2-propanol and was then vortexed and incubated for 5 minutes. Dynabeads were collected by a magnet and the supernatant was removed. A solution of 5 mM NaCl, 10 mM Tris-HCl pH 8, 1 mM EDTA and 40% 2-propanol (950 μL) was added for rinsing. After vortexing and incubating for 2 minutes the Dynabeads were collected by a magnet and the supernatant removed. A similar rinse step was performed with 950 μL of 50 mM NaCl, 10 mM Tris-HCl (pH 8), 1 mM EDTA and 70% ethanol. Finally, 100 μL of 10 mM Tris-HCl pH 8 was added, mixed and incubated for 2 minutes, the Dynabeads were collected by a magnet, and the supernatant collected.
2.6 Device operation and instrumental setup
Monolith-modified devices were connected to a Fluigent (Lowell, MA) pressure driven pumping and valving system for introduction of buffer through one arm of the device and sample through the other one (see Figure 1A). First, the device was filled with hybridization buffer (20 mM Tris-HCl pH 8 with 500 mM NaCl and 50 mM MgCl2). Next, 100 μL of sample was passed through the monolith at 20 μL/min, typically requiring ~5 bars of pressure. Then, the monolith was rinsed with 20 μL of buffer and flow was stopped. Heating of the monolith section of the device was performed by using a thermoelectric module (TE-23-1.0-1.3P, TE Technology, Traverse City, MI) operated by a controller (TC-48-20, TE Technology) and equipped with control thermistors (MP-2444, TE Technology). Thermistors were placed on the edge of the thermoelectric module and on the microfluidic device on the opposite side from the heater, to measure heater and device temperature, respectively. When the desired temperature on the monolith was established, 500 mbar was applied and the eluted nucleic acid band was detected using a confocal laser-induced fluorescence setup probing a point 2 mm beyond the monolith as described in a prior publication [23].
Figure 1.

Device characterization. (A) Photograph of a black polypropylene microdevice containing a porous polymer. (B–C) Scanning electron micrographs of the porous polymer.
3. Results and Discussion
3.1 Monolith preparation in microdevices
Porous polymers are widely used in separation science as solid supports that increase surface area and provide various chromatographic interactions. These materials are easily prepared in-situ by photoinitiated polymerization, and their surface and porous properties can be tuned by monomers or porogens in the polymerization mixture. Batch-to-batch variability with free-radical polymerization is less of an issue for sample preparation than for high-performance separations [24]. Thus we prepared a porous polymer modified with a capture probe for selective nucleic acid capture. Capture probes are often immobilized on flat substrates or particles [10]; however, these are limited in surface area for interaction, which often leads to long incubation periods (many hours) with sample. On the other hand, tortuous sample flow through the monolith [25] increases the likelihood of nucleic acid hybridization with the immobilized probe.
GMA-based monoliths were chosen for experiments, as they are a commonly used support for immobilization of molecules with primary amines by the Schiff base method [22]. In initial studies, ethylene dimethacrylate was used as a crosslinker; however, this monolith formulation suffered from nonspecific adsorption of fluorescent molecules when bacterial lysate samples were introduced. Monoliths prepared with PEGDA as an alternate, more hydrophilic crosslinker [26] showed much less nonspecific adsorption and were therefore used for the reported experiments. The ratio of the porogens, 1-propanol and 1,4-butanediol, was studied regarding monolith morphology (evaluated by SEM) and flow-through properties. The optimal ratio was found to be 2:1 1-propanol: 1,4-butanediol. SEM images of representative monoliths in microchips are shown in Figures 1B–C.
The microfluidic device material was chosen to be black polypropylene due to its inertness that is useful in clinical diagnostics [27]. On the other hand, polypropylene's low reactivity makes anchoring of the monolith more challenging, since attachment of a porous polymer to the wall through covalent bonds is complicated by polypropylene's lack of reactive sites. However, using sufficiently concentrated type I photoinitiators like BME, it is possible to link the monolith to the substrate through hydrogen abstraction, allowing formation of covalent bonds to the polymer network in one step [28]. Monoliths prepared with concentrations of 1%, 2% and 3% BME in the polymerization mixture and exposed for 10, 12 and 15 minutes were evaluated by observing monolith stability in the microfluidic channel under 6 bars applied pressure at 70 °C. The best results for anchoring of the monolith (shown in Figure 1C) were found with 3% BME and 12 minutes exposure.
3.2 DNA extraction
Hybridization of an oligonucleotide target to complementary immobilized probes is an essential part of DNA based biosensors [29]. Studies with microarray technology widely used in gene expression, sequencing, and drug discovery reveal the importance of employing a spacer between the surface and the immobilized sequence itself, allowing hybridization of the target to happen further away from the solid phase [30,31]. Hence, the amine-terminated capture probe was designed with an 18 atom long polyethylene glycol spacer.
For evaluation of hybridization conditions for solid-phase extraction of the target sequence, a fluorescently labeled oligonucleotide is flowed through the porous polymer modified with an immobilized complementary probe. The target sequence then hybridizes to the surface, while other molecules can be washed away. Multiple hybridization buffers were tested, including saline sodium citrate, phosphate buffered saline and Tris-HCl [32]. Buffer type influenced hybridization less than ionic strength, which is a significant experimental parameter for hybridization, and is crucial for fast and stable duplex formation [33,34]. Several buffer additives have been tested for their effect on hybridization; for instance, Mg2+ improved the discrimination of complementary and point mismatched targets [34].
The composition of the hybridization buffer was studied for its effect on the amount of DNA captured on and eluted from the monolith. Various concentrations of Na+ and Mg2+ (in a 10:1 ratio) in 20 mM Tris-HCl buffer pH 8 were tested with the results shown in Figure 2. We found that buffers with higher ionic strength (200 mM NaCl and 20 mM MgCl2) significantly increased the amount of DNA eluted. At still higher ionic strengths only a small effect on the eluted peak area was observed, and the best results were found with 500 mM NaCl and 50 mM MgCl2. An oligonucleotide with a single-base mismatch was also compared to the complementary target. Capture experiments with 100 pM mismatched sequence provided signal near the limit of detection, so 1 nM mismatched sequence was used to compare capture selectivity (Figure 2). Moreover, loading a 10 nM sample containing an oligonucleotide (KPC) with a fully mismatched sequence did not provide any signal during elution.
Figure 2.
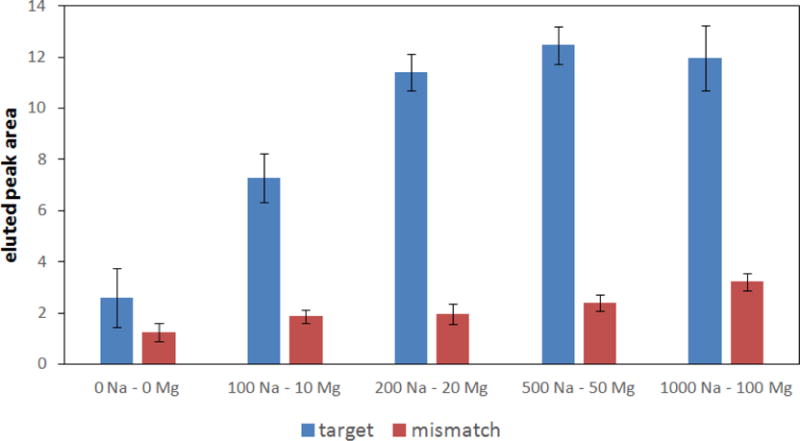
Dependence of peak area (see Figure 3 for an elution trace) on concentration (mM) of NaCl and MgCl2 in the hybridization buffer for complementary target (100 pM) and a single-base-mismatched sequence (1 nM).
In order to release bound target from the column, denaturation of the hybrid has to be performed; using high pH [11,12] or chaotropic agents like urea [35] or guanidinium [36] can disrupt hydrogen bonding between bases. These elution procedures could, however, limit some DNA labeling strategies for fluorescence detection, so elution by heating the column above the melting point of the duplex was selected. For the 24 base long oligonucleotide related to the VIM carbapenemase gene used in these experiments, the theoretical melting point was 62.5 °C (Oligo Calc: Oligonucleotide Properties Calculator, http://biotools.nubic.northwestern.edu/OligoCalc.html). Using the thermoelectric module placed on the monolith section of the device, temperatures ranging from 50 to 80 °C were tested for releasing of the extracted target. Elution at 50–65 °C resulted in a broad peak due to slow release of DNA hybridized to the monolith. Elution at 70 °C showed DNA eluting with increased concentration in a narrow peak, and temperatures over 70 °C showed similar results; therefore, elution at 70 °C was selected. The use of higher temperature degrades the anchoring of the monolith to the channel; we found that devices sustained 7–10 elutions on average before a change in monolith flow-through properties was observed, degrading performance. Devices were used repeatedly in this study for optimization purposes; however, for real sample analysis the microfluidic device is intended to be a disposable single-use system. Fluorescence signal after the monolith during an extraction and elution experiment under optimized conditions with 100 pM Fl-VIM is shown in Figure 3.
Figure 3.
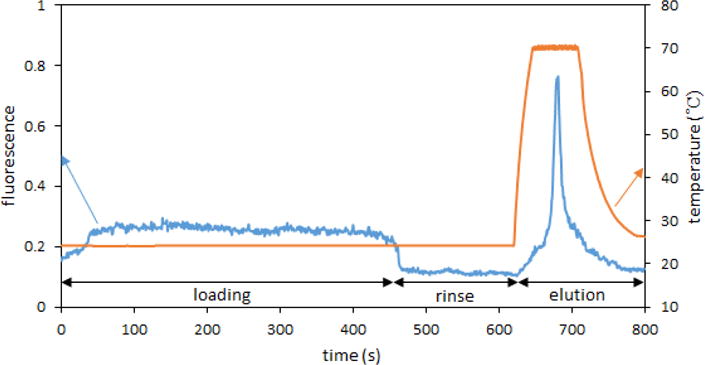
Fluorescence signal recorded after the monolith during loading of 100 pM Fl-VIM, rinsing, and elution at 70 °C.
Next, the effect of sample flow rate through the column (from 10 – 20 μL/min) was studied. Figure 4 shows a comparison of fluorescence recorded during the elution step for different loading flow rates; a slight increase in the eluted peak area was observed for 20 μL/min sample loading, possibly due to improved mass transfer with increased convection from the higher flow [37]. Employing a faster flow rate is advantageous for shortening the assay time, allowing extraction and elution to be finished within 12 minutes, which is beneficial in speeding up the determination of antibiotic resistance.
Figure 4.
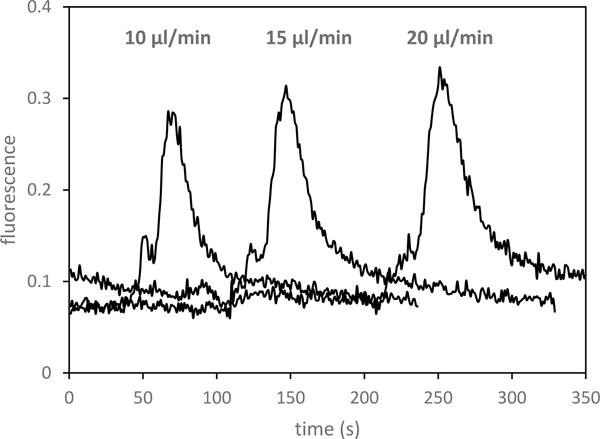
Dependence of eluted peak on flow rate during loading of 100 pM Fl-VIM.
Elutions recorded for extractions with different loaded volumes of sample are shown in Figure 5A, demonstrating an increase in the amount of captured DNA eluted with a larger sample volume passed through the monolith. Similarly, elutions after loading a range of concentrations of target oligonucleotide are shown in Figure 5B, demonstrating the ability to extract samples with sub-nanomolar concentrations and detect target DNA present as low as 1 pM in a 100 μL volume. The binding capacity, determined by passing 50 μL of a 5 nM sample through the column, was measured to be ~10 fmol. This observed binding capacity is sufficient for extraction from samples containing billions of copies of the targeted antibiotic resistance gene. Repeatability of the peak area for elutions with a 100 pM sample was 7.2% RSD (n=4) on a single device and 13% RSD for 4 separate devices. Passing 100 μL of sample through the monolith also allows enrichment of the target DNA, and the enrichment factor for a 1 pM loaded sample was 160×. Recoveries for the extractions in Figure 5B are shown in Table 2. The lower percent recovery for higher target concentrations could be caused by partial saturation of the monolith if sample flows through preferential paths during loading [38]. Improved recovery could be achieved by using a longer monolithic column, but that would increase backpressure and elution volume, which would not work well for our intended analysis. The 100 μL of 1 pM sample corresponded to 60 million target copies loaded; much lower copy numbers could be detected by integrating our extraction module with a single-molecule counting setup [39].
Figure 5.
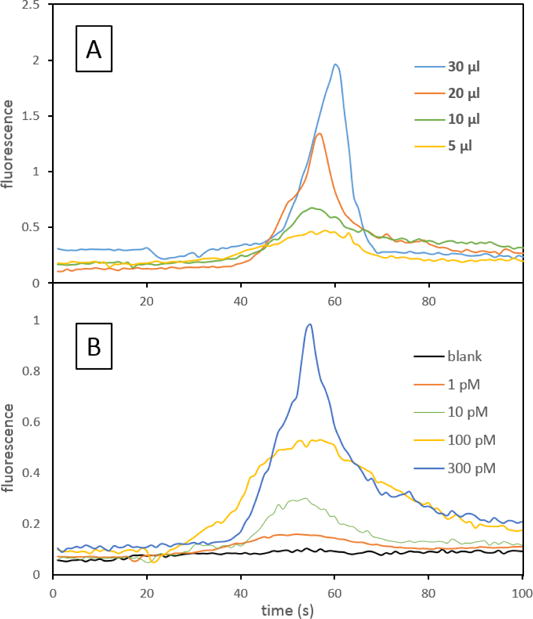
Dependence of eluted peak on loaded volume or concentration. (A) Loading different volumes of 1 nM Fl-VIM. (B) Loading 100 μL of different concentrations of Fl-VIM.
Table 2.
Peak areas and moles of sample loaded and eluted for different loaded Fl-VIM concentrations with corresponding percent recovery values.
| sample | peak area | DNA loaded (amol) | DNA eluted (amol) | recovery (%) |
|---|---|---|---|---|
| 1 pM | 2.9 | 100 | 83 | 83 |
| 10 pM | 7.7 | 1000 | 430 | 43 |
| 100 pM | 16.3 | 10,000 | 1,100 | 11 |
| 300 pM | 22.9 | 30,000 | 1,500 | 5 |
3.3 DNA extraction from bacterial lysate
Nucleic acid based detection of antibiotic resistant bacterial pathogens in blood involves separation and lysis of bacteria, and this lysate will be flowed through the monolithic column with immobilized probes for selective extraction of targeted genes. In order to test the suitability of this approach, a sample of processed bacterial lysate mixed with hybridization buffer was spiked with Fl-VIM and extracted using the monolithic column. Figure 6 shows fluorescence signal after the monolith for extraction of unspiked and spiked lysate samples, together with extraction from the hybridization buffer. The loading part of the trace shows low fluorescence signal coming from the lysate sample, and elution from lysate without added Fl-VIM showed a very small peak. We found that using PEGDA instead of ethylene dimethacrylate as the crosslinker significantly decreased the amount of adsorbed fluorescent material on the monolith; however, it was not possible to eliminate it completely either with extensive (up to 100 μL) rinsing or rinsing at 40 °C. Although the source of the minor co-eluting fluorescent compound is unknown, we believe it could be related to the broth medium or the high concentration of bacteria in the lysis input, which exceeded the expected number of bacteria present in a typical infected blood sample. Thus, this small peak should decrease to undetectable levels in blood samples with lower bacterial counts. Despite this minor contaminant, it was possible to selectively extract target DNA from a sample containing bacterial lysate. Extracting 100 pM Fl-VIM from 100 μL of lysate resulted in a recovery of 9,3%, which is comparable to the recovery obtained in extracting the same target concentration from buffer (10.8%).
Figure 6.
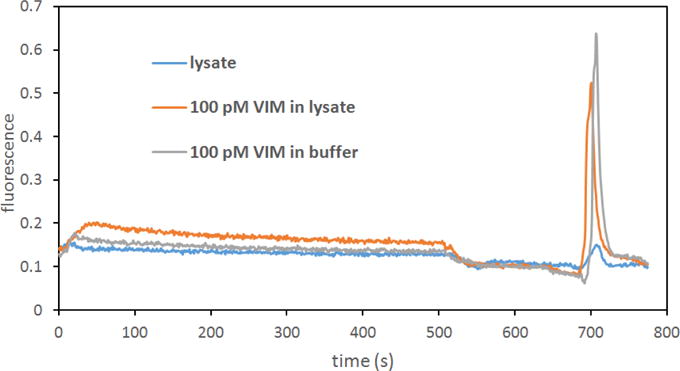
Extraction of 100 μL of bacterial lysate, lysate spiked with 100 pM Fl-VIM and 100 pM VIM in hybridization buffer.
4. Conclusions
We prepared microfluidic devices from black polypropylene, having monolithic columns modified with a capture oligonucleotide for sequence-selective solid-phase extraction of a complementary target from a bacterial lysate sample. Employing a porous polymer monolith enables interaction with the target DNA on a large surface area modified with the probe. Good flow through properties of the monolith allow flow rates of 20 μL/min at 5 bars applied pressure, making it possible to pass 100 μL of a bacterial lysate sample through the monolith and elute the target DNA within 12 minutes. Temperature-mediated elution by heating the column above the duplex melting point provides a clean extract without chemical agents that could interfere with base pairing, potentially allowing a wide variety of labeling methods or further processing of the eluate. The extraction procedure was optimized regarding hybridization buffer composition, flow rate of the sample and elution temperature. Under optimized conditions it was possible to extract a 1 pM target oligonucleotide from a 100 μL solution with 83% recovery. A 100 pM target oligonucleotide was also extracted from bacterial lysate with 9% recovery.
Integrating monolithic column sequence-specific capture of targeted genes with a device with modules for separation and lysis of bacteria from blood [40], followed by single-molecule fluorescence detection on an optofluidic platform [39], should allow rapid determination of antibiotic resistance in patients. Such a device, currently under development in an instrument that includes a pumping mechanism, is expected to allow detection of genes related to antibiotic resistance in approximately one hour, compared to commonly used methods that currently take 24–72 hours.
Supplementary Material
Highlights.
A polypropylene microfluidic device with a porous polymer monolith modified with a covalently linked capture oligonucleotide was prepared.
Solid-phase extraction conditions for hybridization of target DNA were optimized.
The device allows fast extraction of 1 pM DNA from a 100 μL sample with recovery up to 83%.
Acknowledgments
This work was supported by the U.S. National Institutes of Health (R01 AI116989). We are grateful to Dr. William Pitt and Ryan Wood for providing a sample of bacterial lysate.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Daikos GL, Petrikkos P, Psichogiou M, Kosmidis C, Vryonis E, Skoutelis A, Georgousi K, Tzouvelekis LS, Tassios PT, Bamia C, Petrikkos G. Antimicrob Agents Chemother. 2009;53:1868–73. doi: 10.1128/AAC.00782-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barken KB, Haagensen JAJ, Tolker-Nielsen T. Clin Chim Acta. 2007;384:1–11. doi: 10.1016/j.cca.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 3.Yanga S, Rothman DE. Lancet Infect Dis. 2004;4:337–348. doi: 10.1016/S1473-3099(04)01044-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petti CA. Clin Infect Dis. 2007;44:1108–1114. doi: 10.1086/512818. [DOI] [PubMed] [Google Scholar]
- 5.Fredricks DN, Schubert MM, Myerson D. Clin Infect Dis. 2005;41:e1–4. doi: 10.1086/430824. [DOI] [PubMed] [Google Scholar]
- 6.Kashkary L, Kemp C, Shaw KJ, Greenway GM, Haswell SJ. Anal Chim Acta. 2012;750:127–131. doi: 10.1016/j.aca.2012.05.019. [DOI] [PubMed] [Google Scholar]
- 7.Percin I, Saglar E, Yavuz H, Aksoz E, Denizli A. Int J Biol Macromolec. 2011;48:577–582. doi: 10.1016/j.ijbiomac.2011.01.022. [DOI] [PubMed] [Google Scholar]
- 8.Rittich B, Spanova A. J Sep Sci. 2013;36:2472–2485. doi: 10.1002/jssc.201300331. [DOI] [PubMed] [Google Scholar]
- 9.Wen J, Guillo C, Ferrance JP, Landers JP. J Chromatogr A. 2007;1171:29–36. doi: 10.1016/j.chroma.2007.09.057. [DOI] [PubMed] [Google Scholar]
- 10.Wang L, Li PCH. Anal Chim Acta. 2011;687:12–27. doi: 10.1016/j.aca.2010.11.056. [DOI] [PubMed] [Google Scholar]
- 11.Lee GY, Chen CH, Wang TH, Lee WC. Anal Biochem. 2003;312:235–241. doi: 10.1016/s0003-2697(02)00507-9. [DOI] [PubMed] [Google Scholar]
- 12.Satterfield BC, Stern S, Caplan MR, Hukari KW, West JAA. Anal Chem. 2007;79:6230–6235. doi: 10.1021/ac0709201. [DOI] [PubMed] [Google Scholar]
- 13.Olsen KG, Ross DJ, Tarlov MJ. Anal Chem. 2002;74:1436–1441. doi: 10.1021/ac0156969. [DOI] [PubMed] [Google Scholar]
- 14.Root BE, Agarwal AK, Kelso DM, Barron AE. Anal Chem. 2011;83:982–988. doi: 10.1021/ac102736g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cui F, Rhee M, Singh A, Tripathi A. Annu Rev Biomed Eng. 2015;7:267–286. doi: 10.1146/annurev-bioeng-071114-040538. [DOI] [PubMed] [Google Scholar]
- 16.Nge PN, Rogers CI, Woolley AT. Chem Rev. 2013;113:2550–2583. doi: 10.1021/cr300337x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kendall EL, Wienhold E, DeVoe DL. Biomicrofluidics. 2014;8:044109. doi: 10.1063/1.4891100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaw KJ, Joyce DA, Docker PT, Dyer CE, Greenway GM, Greenman J, Haswell SJ. Lab Chip. 2011;11:443–448. doi: 10.1039/c0lc00346h. [DOI] [PubMed] [Google Scholar]
- 19.Kashkary L, Kemp C, Shaw KJ, Greenway GM, Haswell SJ. Anal Chim Acta. 2012;750:127–131. doi: 10.1016/j.aca.2012.05.019. [DOI] [PubMed] [Google Scholar]
- 20.Mahalanabis M, Al-Muayad H, Kulinski MD, Altman D, Klapperich CM. Lab Chip. 2009;7:2811–2817. doi: 10.1039/b905065p. [DOI] [PubMed] [Google Scholar]
- 21.Bhattacharyya A, Klapperich CA. Sens Actuators. 2008;B 129:693. [Google Scholar]
- 22.Mallik R, Jiang T, Hage DS. Anal Chem. 2004;76:7013–7022. doi: 10.1021/ac049001q. [DOI] [PubMed] [Google Scholar]
- 23.Sonker M, Yang R, Sahore V, Kumar S, Woolley AT. Anal Meth. 2016;8:7739–7746. doi: 10.1039/C6AY01803C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knob R, Sahore V, Sonker M, Woolley AT. Biomicrofluidics. 2016;10:032901. doi: 10.1063/1.4948507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aggarwal P, Asthana V, Lawson JS, Tolley HD, Wheeler DR, Mazzeo BA, Lee ML. J Chromatogr A. 2014;1334:20–29. doi: 10.1016/j.chroma.2014.01.056. [DOI] [PubMed] [Google Scholar]
- 26.Li Y, Lee ML. J Sep Sci. 2009;32:3369–3378. doi: 10.1002/jssc.200900478. [DOI] [PubMed] [Google Scholar]
- 27.Davies J, Nunnerley CS, Paul AJ. Colloids Surf, B. 1996;6:181–190. [Google Scholar]
- 28.Ladner Y, Bruchet A, Cretier G, Dugas V, Randon J, Faure K. Lab Chip. 2012;12:1680–1685. doi: 10.1039/c2lc21211k. [DOI] [PubMed] [Google Scholar]
- 29.Levicky R, Horgan A. Trends Biotechnol. 2005;23:143–149. doi: 10.1016/j.tibtech.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 30.Zammatteo N, Jeanmart L, Hamels S, Courtois S, Louette P, Hevesi L, Remacle J. Anal Biochem. 2000;280:143–150. doi: 10.1006/abio.2000.4515. [DOI] [PubMed] [Google Scholar]
- 31.Shchepinov MS, Case-Green SC, Southern EM. Nucleic Acids Res. 1997;25:1155–1561. doi: 10.1093/nar/25.6.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Owczarzy R, Moreira BG, You Y, Behlke MA, Walder JA. Biochemistry. 2008;47:5336–5353. doi: 10.1021/bi702363u. [DOI] [PubMed] [Google Scholar]
- 33.Tan ZJ, Chen SJ. Biophys J. 2006;90:1175–1190. doi: 10.1529/biophysj.105.070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Springer T, Sipova H, Vaisocherova H, Stepanek J, Homola J. Nucleic Acids Res. 2010;38:7343–7351. doi: 10.1093/nar/gkq577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao Y, Wolf LK, Georgiadis RM. Nucleic Acids Res. 2006;34:3370–3377. doi: 10.1093/nar/gkl422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hagan MF, Chakraborty AK. J Chem Phys. 2004;120:4958–4968. doi: 10.1063/1.1645786. [DOI] [PubMed] [Google Scholar]
- 37.Svec F, Frechet JMJ. Anal Chem. 1992;64:820–822. doi: 10.1021/ac00035a008. [DOI] [PubMed] [Google Scholar]
- 38.Koku H, Maier RS, Czymmek KJ, Schure MR, Lenhoff AM. J Chromatogr A. 2011;1218:3466–3475. doi: 10.1016/j.chroma.2011.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ozcelik D, Parks JW, Wall TA, Stott MA, Cai H, Parks JW, Hawkins AR, Schmidt H. Proc Natl Acad Sci USA. 2015;112:12933–12937. doi: 10.1073/pnas.1511921112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alizadeh M, Wood RL, Buchanan CM, Bledsoe CG, Wood ME, McClellan DS, Blanco R, Ravsten TV, Husseini GA, Hickey CL, Robison RA, Pitt WG. Colloids Surf B. 2017;154:365–372. doi: 10.1016/j.colsurfb.2017.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.