

Abstract
The regulation effect of surface charges on the transport of electrons in nanomaterials and ions in nanofluidic devices has been widely used to develop highly sensitive and label-free electrical biosensors. The intrinsic limitation to the clinical application of surface charge-effect nano-electrical biosensors is that they usually do not function in physiological conditions normally with high ionic concentrations (~160 mM), in which the surface charges are screened within a short distance (<1 nm at 160 mM). In this work, we developed a general strategy that enables surface charge-effect electrical biomolecular detection in physiological conditions with integrated mechanism for enhancement of limit of detection (LOD) by in-situ preconcentration of target molecules during incubation and creating a transient low ionic concentration environment during the signal readout step using reconfigurable ion concentration polarization (ICP). We demonstrated the effectiveness of this strategy in a facile nanofluidic biosensor named nanofluidic crystal (NFC), which can be prepared within hours and without expensive equipment. Our results indicate that the ion depletion effect of ICP could lower the ionic concentration by at least 200 folds and provide a stable ionic environment for over 15 s, enabling electrical detection of proteins and DNAs in serum and urine with LODs of 1–10 nM. We further reconfigured the device to preconcentrate target biomolecules before detection using the enrichment effect of ICP, obtaining LODs of 10–100 pM for proteins and DNAs in physiological conditions. By overcoming the inherent constraint on buffer conditions and the issues regarding fabrication, we believe this work represents a significant progress towards the practical application of surface charge-effect nano-electrical biosensors in point-of-care diagnostics and clinical medicine.
TOC image
Electrical biosensing in high ionic conentrations is enabled by the depletion effect of ion concentration polarization and further enhanced by its enrichment effect.
Introduction
Because of the paramount role biomolecular detection plays in medicine and life sciences, there have been extensive efforts devoted to developing sensitive and specific biosensing techniques. Amongst others, nanostructure-based electrical biosensors have been intriguing due to their demonstrated ability in ultra-sensitive and label-free biomolecular detection.1, 2 Nanofluidic biosensors, which exploit the unique ion transport behaviors in nanometer-sized confined space, attract much attention thanks to the direct signal transduction from bio-recognition to electrical readout.3–8 Similar to nanomaterial-based field-effect transistors (FETs),9–13 the binding of biomolecules to affinity probes grafted on the surfaces of nanofluidic channels changes their surface charge densities, which in turn regulate the densities of electric current carriers (ions) in the channels. The current-voltage characteristics of nanofluidic channels thus sensitively report the levels of specific biomolecules in the samples. Duan et al. realized detection of trypsin down to 0.2 nM in 52 nm deep nanochannels in DI water.6 Liu et al. achieved an LOD of 0.3 fM for troponin T protein in 1.6 mM phosphate buffered saline (0.01×PBS) by using conical nanochannels with a tip opening size of 70 nm.14 Despite the promising performances of nanofluidic biosensors and nanomaterial-based FETs, they normally do not function in high ionic strengths or physiological conditions (~160 mM), in which the surface charges are screened by buffer ions within a short distance (thickness of electrical double layer (EDL) <1 nm at 160 mM).15–17 To enable biomolecular detection in physiological conditions, a few research groups proposed the use of size-reduced affinity probes to confine biomolecules in the EDL,18 and engineering of the electrolyte dielectric constant to increase the thickness of the EDL in nanomaterial-based FET biosensors.19, 20 Besides the issues regarding the general applicability and complexity of these methods for nanomaterial-based FETs, they do not apply to nanofluidic biosensors. The Dukhin length characterizes the critical dimension of a nanochannel below which the conductance of additional ions induced by the surface charges dominates over the bulk conductance of the nanochannel at a certain ionic concentration, which is defined as |σ/zFc| (σ: surface charge density, z: valence of ions, F: Faraday constant, c: bulk ionic concentration).21 Accordingly, in order for nanofluidic biosensors to work in physiological conditions (~160 mM), the critical dimensions of the nanochannels (typically with surface charge densities of 10 mC/m2 for silica, ref. 22) cannot exceed the Dukhin length of 0.63 nm, which is impractical for detecting biomolecules that are usually >1 nm. Alternatively, in some works high ionic concentration buffers were used for the incubation step, but extensive washing steps with low ionic concentration buffers were indispensible in reading out the signals, which may compromise the reproducibility, reliability, and sensitivity of the biosensors.6, 14 On top of the intrinsic limitation on the buffer conditions, issues regarding the fabrication of nanofluidic devices, the critical dimensions of which typically fall below 100 nm, should be addressed to meet the needs of clinical and point-of-care applications.23 Although pushing the limits of standard silicon micro/nanofabrication techniques and the polymer track etch technique could meet the needs of laboratory research, concerns regarding the mass manufacturing and costs of the devices in practical applications were raised. Moreover, surface functionalization of nanochannels for target-specific binding can be technically challenging, time-consuming and hard to characterize.6, 24, 25
In this paper, we developed a universal strategy for nanofluidic biosensors and nanomaterial-based FETs that enables the detection of biomolecules in physiological conditions with in-situ mechanism of LOD enhancement by using reconfigurable ion concentration polarization (ICP). ICP is a fundamental electrochemical transport phenomenon that occurs near an ion-selective membrane when a DC voltage is applied.26, 27 In principle, the selective transport of ions through the membrane leads to the generation of an ion depletion zone, which could result in near-complete ion removal at sufficiently high electric fields.28–31 This phenomenon has been studied for a long time theoretically26, 32 and experimentally30 in the context of electrodes and membranes. More recently, nanochannels in microfluidics systems were shown to exhibit similar behaviors,28 which led to the development of several interesting micro-nanofluidic devices. Negatively charged target biomolecules are repelled from the depletion zone by the electrostatic force, and in the meantime, moved towards the depletion zone by an electroosmotic flow (EOF) in the presence of an additional tangential electric field, which leads to the continuous accumulation (preconcentration) of target biomolecules in the vicinity of the depletion zone by as high as several million folds.33 ICP has been widely used in the context of desalination of seawater,34, 35 preconcentration of biosamples,36 and recently as pH regulators,37 and biosensors.38, 39 In this work, we explore the role of ICP as an ionic environment modulator for electrical biosensing. The locations of the depletion and preconcentration zones in an ICP device are controllable by tuning the interplay between the electrophoresis and EOF, which can be achieved by changing the geometry/fluid path of the device.31, 40, 41 By controlling the locations of the depletion and preconcentration zones through the reconfiguration of the device, we preconcentrated the target biomolecules during incubation and transiently created a low ionic concentration environment during the signal readout for the biosensors, thereby enabling highly sensitive biomolecular detection in high ionic concentration buffers and physiological samples. We demonstrated the effectiveness of this strategy in a facile nanofluidic biosensor named nanofluidic crystal (NFC).22, 42–47 Affinity probe-functionalized nanoparticles are self-assembled into a close-packed array in a microchannel, forming a highly ordered nanochannel network from the interstices of nanoparticles. Using this method, we are able to obtain nanofluidic channels with equivalent sizes of tens of nanometers by simply using nanoparticles with diameters of 100–500 nm.22 Moreover, the variety of nanoparticles commercially available enables us to graft specific affinity probes on nanoparticles easily and flexibly. By coupling NFC with reconfigurable ICP, we realized nanofluidic detection of proteins and nucleic acids in physiological samples with LODs of 10–100 pM. We believe this work represents a significant progress towards the practical application of nanofluidic biosensors and other surface charge-effect electrical biosensors in point-of-care diagnostics and clinical medicine.
The principle of the device is illustrated in Fig. 1. The electrical resistance of an NFC biosensor is only sensitive to biomolecular binding in low ionic concentrations, which disables its application in physiological conditions (Fig. 1(a)). To solve this problem, a micro-nanofluidic device is designed to control the ionic environment of the NFC. The device consists of four orthogonal microchannels connected by three nanofilter arrays. Functionalized nanoparticles are assembled into the NFC at the central inter-connecting region through channel A, with a micropatterned electrode pair at the two sides for electrical resistance measurement. The ion-selective membrane (Nafion) for generating the ICP phenomenon is patterned in channel B. The protocol of biomolecular detection in the chip includes five steps: sample loading, preconcentration (optional), incubation, depletion, and measurement. After loading physiological samples into the chip through channel A, a preconcentration step is performed to concentrate target biomolecules in the NFC region. Benefitting from the high ionic concentrations of physiological samples, the size of the ion depletion zone is minimal,48, 49 allowing most parts of the NFC to be immersed in the preconcentration plug. Following the incubation step, a large ion depletion zone is induced by ICP to create the low ionic concentration environment required for nanofluidic biosensing. During the depletion, EOF is mechanically inhibited by blocking the outlet of channel B, forcing the depletion zone to continuously grow as more ions are transported through the ion-selective membrane.48 At last, upon turning off the voltage for depletion, the electrical resistance of the NFC is measured by the micropatterned electrode pair, before ions diffuse back to the central region. The actual images of the device are shown in Figs. 1(c–e). The microchannels are 10 μm deep, 100 μm wide and 5 mm long, connected by three nanofilter arrays with a cutoff size of 400 nm (Fig. 1(c)). Due to the additional pressure required for the resin to pass the geometrical expansion, the Nafion resin self-stopped after reaching the nanofilter array, which formed the Nafion membrane after solvent evaporation (Fig. 1(d)).51 Nanoparticles of 110/200/510 nm were assembled into the NFC in the central region due to the filtration effect of the nanofilter arrays (Fig. 1(e)). It is worth mentioning that, although in this work we required electron beam lithography to fabricate the nanofilters that may add to the cost of the device, we believe that the device can be made disposable via injection molding, using low cost plastic materials in the future, in which assembly of nanoparticles will be controlled by the capillary force instead of nanofilters.51 Finally, the device is ready for experiments after the assembled nanoparticles were dried and stabilized. The photos of the whole chip and testing system were shown in the Supplementary Information.
Figure 1.
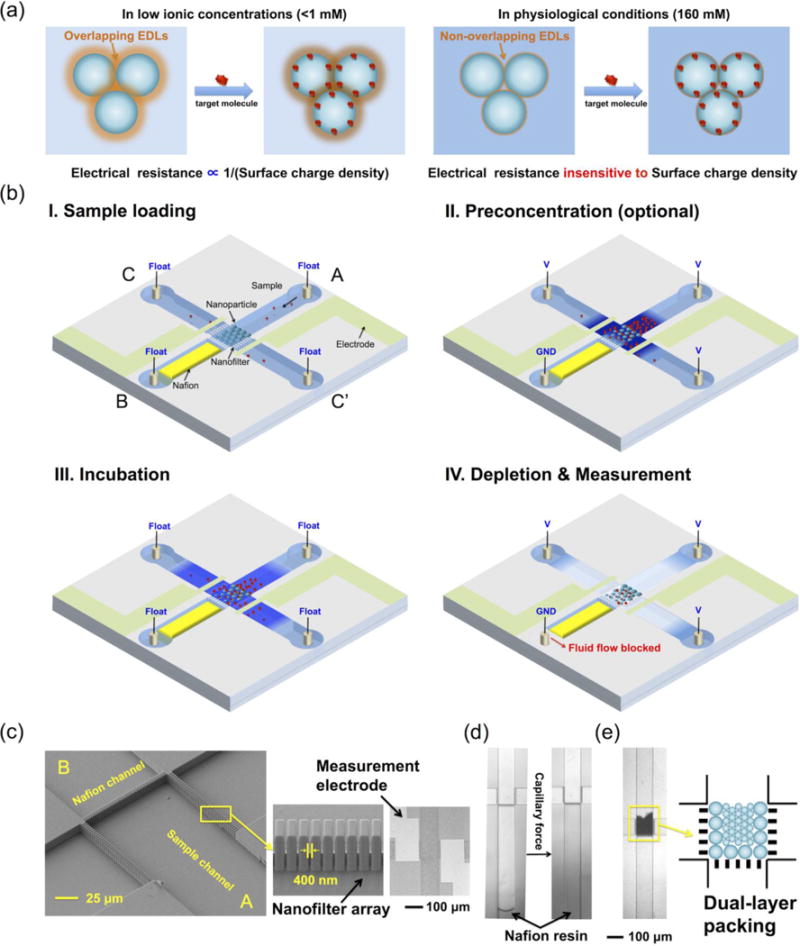
Principle and design of the nanofluidic biosensing platform. (a) Electrostatic effect-based nanofluidic biosensing does not work in normal physiological conditions due to the suppression of the Debye length at high ionic concentrations. (b) Principle of ion concentration polarization coupled nanofluidic crystal for biosensing. (c) SEM image of the central sensing region on the silicon substrate, a magnified view of the nanofilter array, and bright-field microscope photo of the chip after anodic bonding, which shows the micropatterned Ag/AgCl electrode pair on the glass substrate. (d) Filling of nafion resin by capillary force. (e) Assembled nanofluidic crystal in the central region. Dual-layer packing allows the packing of nanoparticles smaller than the nanofilter cutoff size (400 nm), as described in Materials and Methods.
Materials and methods
Reagents and Chemicals
Streptavidin-coated polystyrene (PS) nanoparticles with diameters of 110 nm, 200 nm, 510 nm were purchased from Bangs Laboratories (Catalog Code: CP01N, Lot Numbers: 12320, 12612, 12683, respectively). According to the Certificate of Analysis, nanoparticles of 110 nm, 200 nm, 510 nm have biotin-FITC binding capacities of 3.6 μg, 4.2 μg, 3.4 μg/mg nanoparticles, respectively. Although the total surface area per mg nanoparticles increases as the diameter decreases, the biotin-FITC binding capacity per mg nanoparticles remains almost constant at different diameters (3.73 μg/mg nanoparticles on average), owing to the proprietary manufacturing processes of the vendor. Nafion resin (20 wt % solution in lower aliphatic alcohol/H2O mix) was purchased from Sigma-Aldrich (Catalog Number: 527122-25ML). Alexa Fluor 488 (carboxylic acid succinimidyl ester, A20000, Invitrogen) was used as tracers in the characterization of ICP. Ag/AgCl wire electrodes (A-M Systems, Sequim, WA) were used to apply the voltages that induce the ICP phenomenon (Catalog Number: 531500). Phosphate Buffered Saline (PBS, pH=7.4) was purchased from Invitrogen (Catalog Number: 10010-031). R-Phycoerythrin (R-PE) and Alexa Fluor 488 conjugated Albumin from Bovine Serum (BSA) were purchased from Invitrogen (Catalog Numbers: P801 and A13100, respectively). Positively charged biotinylated peptide (biotin-GRGGRG) was synthesized by the Biopolymers Laboratory at Koch Institute of MIT. Recombinant HIV-1 (CN54) gp120 was purchased from ACRO Biosystems (Catalog Number: GP4-V15227-100ug). Monoclonal mouse HIV-1 gp120 antibody was purchased from Sino Biological (Catalog Number: 11233-MM01-50). HIV-1 gp120 antibody was biotinylated using EZ-Link™ NHS-PEG4-Biotin Linker from Thermo Scientific (Catalog Number: 21329). Recombinant ESAT6 protein was purchased from Abcam (Catalog Number: ab124574). Mycobacterium tuberculosis specific DNA probe (5′-biotin-GT AGG CGA ACC CTG CCC AGG TC-3′), unlabeled DNA target (5′-ACC AGC ACC TAA CCG GCT GTG GGT AGC AGA CCT CAC CTA TGT GTC GAC CTG GGC AGG GTT CGC CTA C-3′), fluorescence-labeled DNA target (5′-ACC AGC ACC TAA CCG GCT GTG GGT AGC AGA CCT CAC CTA TGT GTC GAC CTG GGC AGG GTT CGC CTA C-(Alexa Fluor 488)-3′), non-target DNA (5′-AG CTA GCT AGC TAG CTA GCT AGC TAG CTA GCT AGC TAG CTA GCT AGC TAG CTA GCT AGC TAG CT-3′) were synthesized by Integrated DNA Technologies. Human thrombin protein was purchased from R&D Systems (Catalog Number: 2196-SE-200). Thrombin binding aptamer (5′-biotin-GGT TGG TGT GGT TGG-3′) was synthesized by Integrated DNA Technologies. Pooled human serum and pooled human urine were purchased from Lee Biosolutions (Catalog Numbers: 991-24-P and 991-03-P-1, respectively).
Chip Fabrication
The chip consists of fluidic channels on a silicon substrate and an Ag electrode pair on a glass substrate, which were anodically bonded after separate processes on each substrate. Briefly, the fabrication process is as follows: A SiO2 layer of 2500 Å thick was thermally grown on the silicon substrate. Nanofilter arrays were first defined by Electron Beam Lithography (EBL) and transferred to the SiO2 layer by Reactive Ion Etching (RIE). Microchannels and contact holes were then patterned by photolithography, followed by a RIE process to etch off 2500 Å SiO2. After stripping the photoresist, the micro-nano patterns, including microchannels, nanofilters and contact holes were obtained by etching the silicon substrate to 10 μm deep in a Deep RIE (DRIE) machine with the patterned SiO2 layer as the etching mask. Afterwards, the etched structures were protected by 100 nm thick low pressure chemical vapor deposition (LPCVD) SiNx, then the contact holes were opened to function as inlets/outlets by etching through the silicon substrate from the backside of the wafer in a 80°C KOH bath for 4 hours. After removing the protecting layer, the whole wafer was covered by a thermally oxidized SiO2 layer of 2000 Å for electrical insulation. Electrodes were patterned on a glass substrate by photolithography, after which an Ag layer of 2000 Å thick was sputtered onto the glass substrate. Next, a lift-off process was performed to form the Ag electrode pair. The silicon and glass substrates were anodically bonded. Finally, the bonded wafer was diced into 1.5 cm×1.5 cm chips.
Device Preparation
(1) Nafion patterning: A 2 μL droplet of nafion resin was loaded into the contact hole connecting to the nafion channel. The nafion resin propagated along the channel automatically due to capillary force, and self-stopped at the nanofilter-microchannel neck, because an additional pressure was needed for the nafion resin to pass the geometrical expansion.51 The resin was cured by heating the chip at 95°C on the hotplate for 10 min. (2) Reservoir fabrication: A PDMS slab with four punched holes was bonded to the backside of the silicon substrate by plasma bonding, with the punched holes aligned to the contact holes of the chip. 10 μL pipet tips were inserted into the punched holes as reservoirs. In the depletion step of the experiment, a pipet tip sealed by PDMS was used instead for the nafion channel, with an Ag/AgCl wire electrode inserted. (3) AgCl formation on the micropatterned Ag electrode pair: The micropatterned Ag electrode pair for electrical resistance measurement needs to be oxidized to form Ag/AgCl electrodes. The oxidation was realized by filling the chip with 100 mM KCl solution and applying a voltage of 0.3 V for 3 h.50 After the electrochemical oxidation, the chip was rinsed with DI water and dried at room temperature. (4) Nanoparticle functionalization: Streptavidin-coated nanoparticles were rinsed in PBS for three times, followed by incubation with excess biotinylated biomolecule probes (antibody, ssDNA, aptamer) for 30 min. The incubation was performed on a vortex mixer to accelerate the reaction. After incubation, functionalized nanoparticles were rinsed in PBS for three times to remove unbound biomolecule probes, and stored at 4°C for use within 3 days. (5) Nanoparticle assembly: 10 μL of 0.01 mg/ml functionalized nanoparticles were loaded into the reservoir of the sample-loading channel. A pressure was manually applied with a syringe to introduce the nanoparticles into the channel. For 510 nm nanoparticles, which were larger than the nanofilter size (400 nm), they were unable to pass the nanofilter and consequently assembled in the central sensing region. The dual-layer packing method was used to assemble 110 nm and 200 nm nanoparticles in the chip: first, thin layers of close-packed 510 nm nanoparticles were formed adjacent to the nanofilter arrays, which served as secondary nanofilters with a cutoff size of <100 nm; subsequently, 110 nm or 200 nm nanoparticles were introduced into the chip and assembled in the central region. Finally, the chip was dried at room temperature to stabilize the assembled NFC.
Apparatus
DC voltages of 30–90 V for ICP were applied by a DC power supply (Stanford Research Systems, Sunnyvale, CA). Fluorescence images were acquired using an inverted fluorescence microscope (Olympus, IX71) and a CCD camera (Sensicam qe, Cook Corp.), with an exposure time of 100 ms. A mechanical shutter was used to reduce the photo-bleaching effect, which was synchronized with the CCD camera by the open source software Micro-manager. The electrical current v.s. time (I-t) plot during the back diffusion of ions (Fig. 2(i)) was measured by VersaSTAT 3 (Princeton Applied Research) via the micropatterned Ag/AgCl electrode pair, with the voltage set to 100 mV. The Nyquist Plot of the NFC (Fig. 4(c)) was measured by the HP4294A Precision Impedance Analyzer (Frequency range: 40 Hz to 110 MHz) via the micropatterned Ag/AgCl electrode pair, with the peak-to-peak voltage set to 500 mV and frequency range set to 40 Hz to 1 MHz. Electrical resistance of the NFC was also measured by HP4294A via the micropatterned Ag/AgCl electrode pair, with the peak-to-peak voltage set to 500 mV and frequency set to 40 Hz.
Figure 2.

Characterization of the ion depletion effect. (a) Configuration of the device. (b) The electroosmotic flow was mechanically blocked, forcing the ion depletion zone to continuously grow. (c) Fluorescence images the of the ion depletion process using 50 μM Alexa Fluor 488 in 1×PBS. (d) Fluorescence profile along the channel at different times shown in (c). (e) Fluorescence intensity and depletion factor in the vicinity of the NFC over time. 400 μM Alexa Fluor 488 in 1×PBS was used for the measurement. (f) Electrical resistance of a 510 nm NFC after different time lengths of depletion at 90 V. (g) COMSOL simulation of the back diffusion process of ions over time, when the depletion zone is 1000 μm long. (h) Simulated concentration in the vicinity of the central zone during the back diffusion of ions, at different initial depletion lengths and flow rates. The fluid flow is mainly caused by the uneven fluid levels in different reservoirs. (i) Electrical current through a 200 nm NFC during the back diffusion of ions. 100 mV was applied between the micropatterned Ag/AgCl electrode pair.
Figure 4.
Detection of a biotinylated positively charged peptide (GRGGRG). (a) Schematics of the biotinylated peptide detection. (b) Equivalent electrical circuit diagram of the device. (c) Complex impedance plot of the chip at different concentrations of peptide (10−3xPBS, without depletion). (d–f) Detection of the peptide in low ionic strength buffers without depletion. (g–i) Detection of the peptide in high ionic strength buffers with depletion. Samples were injected at a flow rate of 0.5 μL/min for 15 min, unless otherwise specified.
Data Processing
All fluorescence images were analyzed by the NIH ImageJ software. The fluorescence intensity profiles in the characterization of ICP (Fig. 2 and Fig. 3) were measured along the centerline of the channel. The electrical resistance was determined by the mean of at least three measurements, and the corresponding standard deviation was taken as the uncertainty of measurement. In each set of biomolecular detection data (corresponding to single dotted curves in Fig. 4 and Fig. 5), the electrical resistance measured in buffer (spiked target biomolecule concentration=0) was taken as R0. The ratio of the electrical resistance measured in samples (R′) to R0, R′/R0, was used to quantify the concentrations of target biomolecules. The error bars in Fig. 4 and Fig. 5 were determined by the propagation of uncertainty formula. The LOD is defined as the minimum concentration of target biomolecules that generates a relative change of electrical resistance (|R′ − R0|/R0) larger than 5%. This definition is based on the fact that the coefficient of variation of R0 between measurements is less than 4% (detailed data not shown). In fluorescence-based assays (Fig. 5), the average fluorescence intensity over the whole NFC area was taken as the fluorescence intensity in each single measurement, and the mean of at least three measurements was used as the final fluorescence intensity under each condition, with the corresponding standard deviation as the value of the error bar.
Figure 3.
Characterization of sample preconcentration and incubation. (a) Configuration of the device in the preconcentration step. (b) Fluid flow through the nafion channel suppresses the expansion of the depletion zone and pins the preconcentration zone near the NFC. (c) Fluorescence images of the preconcentration process. (d) Preconcentration factor of AF 488 over time at different voltages. (e) Distance of the preconcentration plug peaks from the NFC over time at different voltages. (f) Preconcentration factor of different species over time at 30 V. (g) Distance of the preconcentration plug peaks of different species from the NFC over time. (h) Configuration of the device in the incubation step. (i) Fluorescence images of the incubation process. (j) Preconcentration factor of AF 488 over time during incubation.
Figure 5.
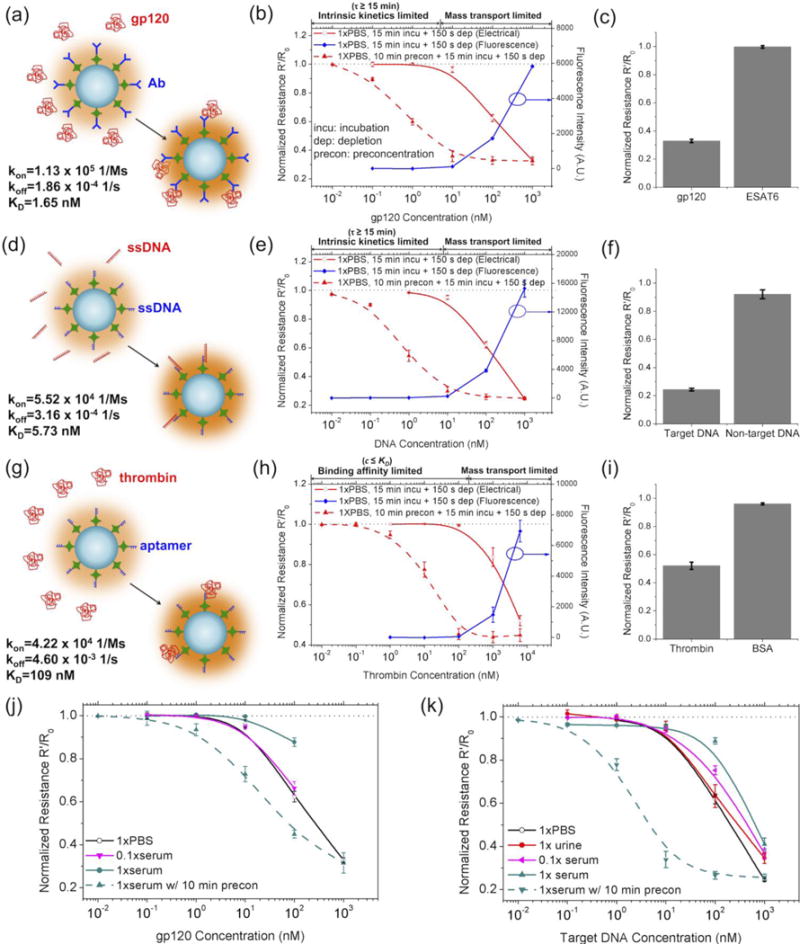
Detection of biomolecules in normal physiological conditions by coupled NFC-ICP. (a–c) Detection of HIV antigen gp120 using an antibody-functionalized 200 nm NFC in 1×PBS. MTB ESAT6 antigen was used as a positive control. The fluorescence-based assays were conducted following exactly the same protocol as the electrical resistance-based assay, except that the target biomolecules were labeled by AF 488 for fluorescence imaging. The processing of the fluorescence images was described in Materials and Methods. (d–f) Detection of MTB DNA fragment using a complementary-DNA functionalized NFC in 1×PBS. Non-target DNA was used as a positive control. (g–i) Detection of human thrombin using an aptamer-functionalized 200 nm NFC in 1×PBS. BSA was used as a positive control. (j) Detection of gp120 in human serum. (k) Detection of MTB DNA in human serum and urine. In all the experiments, 10 μL sample was first loaded into reservoir A, then wetted the channels by manual injection with a syringe. After that, the other three reservoirs were loaded with 10 μL 1×PBS. The sample was not continuously injected during the incubation.
Results and discussion
Characterization of the depletion step
First and foremost, we studied the key problem: can the ion depletion effect of ICP create an ionic environment sufficiently low and stable for nanofluidic biosensing? The equivalent nanochannel sizes of NFCs with nanoparticle diameters of 110/200/510 nm are 26.4/48.0/122.4 nm (24%, as derived in ref. 22). According to the definition of the Dukhin length, ionic concentrations lower than 4.5/2.1/0.82 mM should be achieved to bring 110/200/510 nm NFCs into the surface charge-governed regime (assuming a surface charge density of 10 mC/m2), which correspond to at least 35.6/76.2/195 folds of ion depletion from the physiological ionic concentration (160 mM), respectively. Figs. 2(a–b) show the configuration of the device during the ion depletion step. While other reservoirs were electrically biased and open to air, the reservoir of the nafion channel was grounded and sealed by PDMS to inhibit EOF. 50 μM Alexa Fluor 488 (AF 488) dye in 1×PBS (160 mM) was used to characterize the ion depletion effect. The ion depletion zone kept propagating even at high ionic concentrations, as there was minimal EOF to supplement ions that were transported through nafion (Figs. 2(c–d)). Fig. 2(e) shows the temporal evolution of the fluorescence intensity in the vicinity of the NFC. A higher concentration of AF 488 (400 μM) in 1×PBS was used in order to better quantify the low concentration zone. Upon the application of the DC voltage, the fluorescence intensity dramatically dropped to the baseline level within 15 s. The depletion factor of AF 488 was estimated to be ~200 at 90 V, which may have been underestimated due to the background intensity from the adsorption of the dye molecules on the channel wall. Since dye is not a quantitative reporter for ion concentration within the depletion zone, we further measured the electrical resistance of 510 nm NFC immediately after different time lengths of depletion and observed an ion depletion factor of ~800 after 150 s of depletion, based on the comparison with electrical resistances of the NFC in known concentrations of PBS (Fig. 2(f)).
We next studied how long the low ionic concentration environment could be maintained after turning off the voltage. We first simulated the back diffusion of ions using COMSOL. The initial ionic concentration profile was simplified in the model such that the depletion zone had a uniform ionic concentration of 0.16 mM (1000-fold depletion), which restored to the bulk value (160 mM) at the edge of the depletion zone, approximately demarcated by the dye depletion (see Supplementary Information for details).31 We simulated the ionic concentration in the vicinity of the central inter-connecting zone at different initial depletion lengths. The simulation indicates that the low ionic concentration in the central zone (100 μm×100 μm) could be stably maintained for 7 s, 20 s, and >60 s when the depletion zone is 500 μm, 1000 μm, and 1500 μm long (Figs. 2(g–h)), until trace amount of ions diffuse into this zone. We also noticed that fluid flow between channels could significantly accelerate the diffusion and shorten the time window for nanofluidic biosensing (Fig. 2(h)). In the presence of residual flows of 40 μm/s (created by slight differences in reservoir liquid level or capillarity), the stable time window decreases from 20 s to <15 s when the depletion length is 1000 μm. Therefore, it is important to minimize residual flows. We experimentally monitored the back diffusion of ions by measuring the electrical current through a 200 nm NFC immediately after 150 s of depletion at 90 V, with a voltage of 100 mV applied between the micropatterned Ag/AgCl electrode pair. As shown in Fig. 2(i), the current was relatively stable for at least 15 s, which may have benefitted from the surface charge-governed property of the NFC. In other words, the electrical resistance of the 200 nm NFC would change only after diffusion raised the ionic concentration to a point (2.1 mM) where the NFC was no longer in the surface charge-governed regime. Therefore, we were able to create a time window of about 15 s for nanofluidic biosensing (sufficient for electrical impedance readout) by 150 s of depletion at 90 V in 1×PBS. In summary, our simulation and experimental results suggest that nanofluidic biosensing is feasible in the low ionic concentration environment created by the ion depletion effect.
Characterization of the preconcentration step
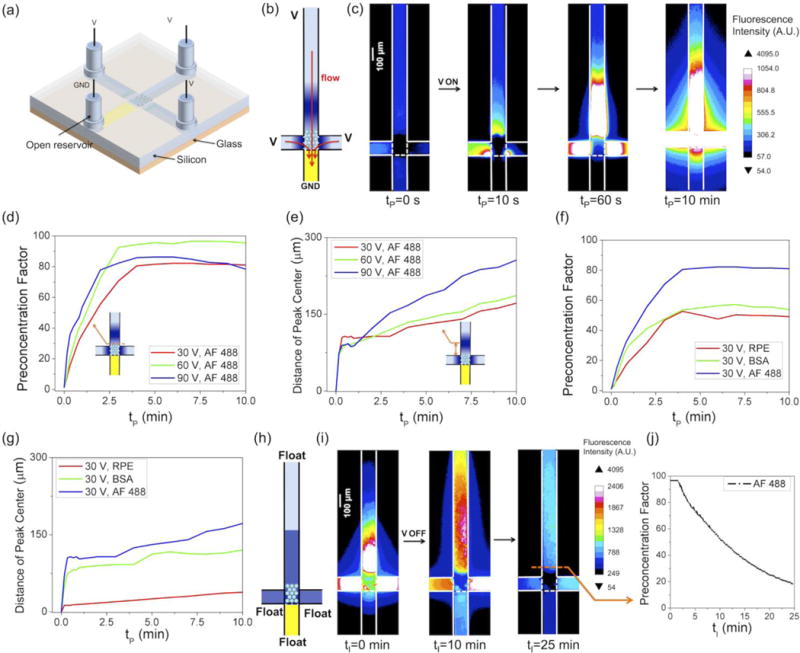
In the preconcentration step, the configuration of the device is the same as that in the depletion step, except that the reservoir of the nafion channel is not sealed (Fig. 3(a)). In this configuration, EOF is induced concurrently with ICP, which suppresses the propagation of the ion depletion zone and causes the preconcentration of negatively charged molecules in the central NFC region (Fig. 3(b)). 100 nM AF 488 dye in 1×PBS was used to characterize the concentration effect. As shown in Fig. 3(c), AF 488 molecules were continuously concentrated in the central region. Increase of fluorescence intensity was also observed in the NFC, suggesting that the depletion zone was localized to the close vicinity of the nafion membrane due to the high bulk ionic concentration.48 The preconcentration factor of AF 488 at the boundary of the NFC increased over time until saturation was reached (Fig. 3(d)). Higher voltages resulted in higher preconcentration factors initially, but also caused the peak of the concentration plug to shift towards the reservoir faster (Fig. 3(e)), which in turn reduced the preconcentration factor near the NFC. Therefore, the highest preconcentration factor for AF 488 (~95) was achieved at 60 V rather than at 30 V and 90 V (Fig. 3(d)). The preconcentration factor and migration speed of the concentration plug were also dependent on the electrical mobility and other properties of the concentrated molecules. At the same voltage, the preconcentration factor of AF 488 was higher than that of BSA (66.5 kDa) and R-PE (240 kDa) (Fig. 3(f)). Meanwhile, the concentration plug of AF 488 migrated fastest and that of R-PE slowest (Fig. 3(g)). It is also worth noting that the preconcentration factors measured in this work (<100) are lower than many other nafion-based electrokinetic concentrators (typically 102–104 in 10 min),48, 53–55 which is attributed to the leakage of biomolecules through the depletion zone. Due to the small thickness of nafion film (<1 μm) and high ionic concentration, the blocking effect by the depletion zone was weak for proteins, which led to the leakage of the concentrated molecules to the nafion channel. After 10 minutes of preconcentration, the NFC was incubated in the preconcentration plug with all reservoirs electrically floated (Fig. 3(h)). As diffusion proceeded, the preconcentration factor gradually decreased from 95 to ~55 after 10 min, and to ~20 after 25 min, which could still effectively improve binding efficiency of the target molecules (Figs. 3(i–j)).
Basic properties of NFC-ICP coupled biomolecular detection
We next coupled the NFC with the depletion and concentration effects of ICP for nanofluidic biosensing. We will only consider negatively charged nanoparticles in the following analyses. For an NFC with a nanoparticle diameter of D packed in a volume of W×L×H (width×length×height), its electrical resistance in the surface charge-governed regime is,22
| (1) |
where σ is the surface charge density of the nanoparticles (<0) and μ+ is the electrical mobility of cations (see Supplementary Information for details). The surface charge density of the nanoparticles before and after target binding is,
| (2) |
| (3) |
where F is the Faraday constant, zprobe, nprobe, zSA, nSA, ztgt, ntgt are the valence and surface density of the grafted affinity probe, surface-coated streptavidin and captured target biomolecule on the nanoparticles, respectively. Let Vsample and ctgt be the volume and target concentration of a sample, ηcapture be the capture efficiency of the target by the NFC, α be the occupation rate of the affinity probes onto the streptavidin binding sites (nprobe=αnSA), then the average surface density of the captured target biomolecules on the nanoparticles is,
| (4) |
The ratio of the electrical resistance after and before target binding is,
| (5) |
In this work, we used streptavidin-coated polystyrene (PS) nanoparticles with diameters of 110/200/510 nm for the biosensing experiments. Because that the affinity probes with large molecular weights (MWs) can hide other biotin binding sites on streptavidin, α is typically less than 100%, and can be reasonably assumed to be proportional to 1/MW of the affinity probe (regardless of the nanoparticle diamter). According to the Certificates of Analysis, nSA/D is roughly constant for nanoparticles of the three diameters (0.76 mol/m3). Therefore, for a specific probe-target pair, the key to achieving a low LOD is enhancing ηcapture, which is affected by sample flow rate, target concentration, nanoparticle diameter, binding kinetics and affinity of the probe and target, and incubation time.
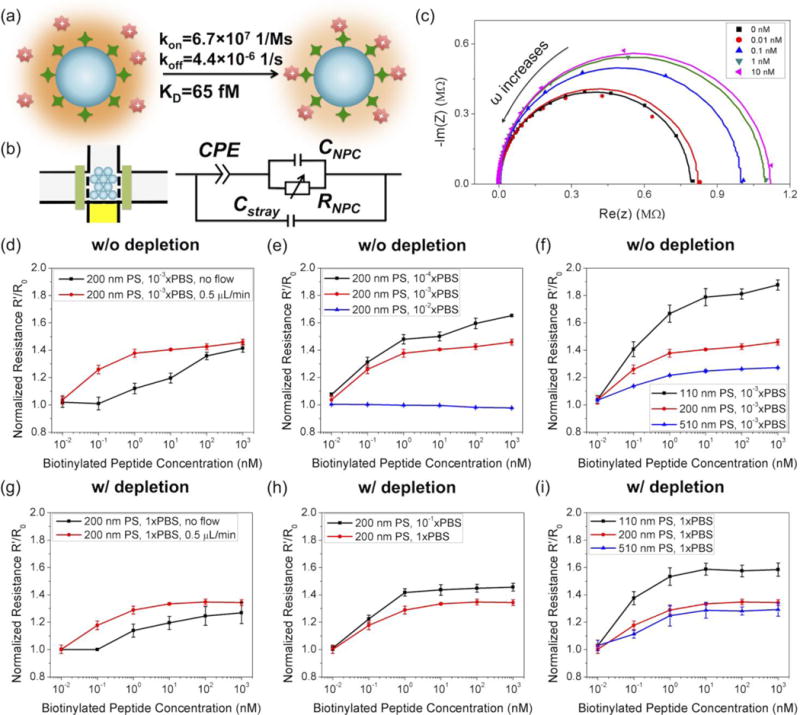
We first studied NFC-ICP coupled biosensing using the high association rate and high affinity streptavidin-biotin model (Fig. 4(a)), which enabled us to exclude kinetics-related and affinity-related issues and focus solely on the parameters of the NFC-ICP platform. Fig. 4(b) shows the electrical model of the system, where CPE is the constant phase element modeling the EDLs on the electrodes, RNFC and CNFC are the resistance and capacitance of the NFC, and Cstray is the stray capacitance of the chip.56 At low frequencies the resistance of the NFC dominates the impedance, while at high frequencies the stray capacitance dominates. We used streptavidin as the probe and a biotinylated peptide (biotin-GRGGRG) as the target molecule, which has one positive charge in 1×PBS (pH=7.4). The impedance of the system was measured by the HP4294A Precision Impedance Analyzer, with a frequency range of 40 Hz to 1 MHz and a peak-to-peak voltage of 500 mV. Fig. 4(c) shows the complex impedance plot of a 200 nm NFC in 10−3×PBS after loading different concentrations of the biotinylated peptide at a flow rate of 0.5 μL/min for 15 min. The experimental data were fitted with the electrical model using the EIS Spectrum Analyser (http://www.abc.chemistry.bsu.by/vi/analyser/). The resistance of the NFC corresponds to the high intercept point of the semicircle on the real axis. As shown in Fig. 4(c), the resistance of the NFC increased at higher concentrations of peptide, because binding of the positively charged peptide decreased the surface charge density of the streptavidin-coated nanoparticles. Additionally, at low frequencies we did not observe the typical linear line representing the Warburg impedance,56, 57 probably because frequencies lower than 40 Hz is needed to obtain the full impedance spectrum.58
We compared the peptide detection results in the NFC without depletion (Figs. 4(d–f)) and with depletion coupled (Fig. 4(g–i)). In a low ionic concentration (10−3×PBS) without the need of depletion, we found that continuous injection of the sample (0.5 μL/min) considerably increased the capture efficiency ηcapture, thereby lowering the LOD from 0.1–1 nM to approximately 0.01 nM (Fig. 4(d)), which agrees with the observation of Schoch et al.57 Similar effect was also seen in the case of depletion-coupled detection in 1×PBS (Fig. 4(g)), with a 10-fold lower LOD achieved in the presence of a flow rate of 0.5 μL/min. In a 200 nm NFC, the change of resistance decreased as we increased the buffer concentration from 10−4×PBS to 10−3×PBS, until no change of resistance was detected in 10−2×PBS (Fig. 4(e)). This is because that, the dominating effect of surface charge on the resistance of the NFC decreased as bulk conductance increased at higher buffer concentrations, until vanishing at the previously calculated upper limit (2.1 mM ~ 10−2×PBS). In contrast, we were still able to perform peptide detection in 10−1×PBS and 1×PBS by coupling depletion, with slightly stronger signal achieved in 10−1×PBS (Fig. 4(h)). Finally, in 10−3×PBS without depletion (Fig. 4(f)) and 1×PBS with depletion (Fig. 4(i)), the changes of resistance both increased as the diameter of the nanoparticle decreased, which was due to the higher capture efficiency ηcapture and stronger EDL overlapping in smaller nanometer-sized interstices.
Biomolecular detection in physiological samples
We next showcased the capability of NFC-ICP as a platform technique for detecting various biomolecules in physiological samples via various types of interactions, including antigen-antibody binding, DNA hybridization, and protein-aptamer binding. We first demonstrated the detection of the HIV gp120 antigen using a 200 nm NFC functionalized with a monoclonal anti-gp120 antibody (Fig. 5(a)). Before testing in our platform, we first measured the association rate kon, dissociation rate koff, and dissociation constant KD of the target-probe binding using Octet RED 96 (Fig. 5(a)) (see Supplementary Information for details). The binding of gp120 to the immobilized antibody on the nanoparticles, which were both negatively charged in physiological conditions (pH=7.4), increased the surface charge density of the nanoparticles and thus decreased the electrical resistance of the NFC. Following 15 min incubation and 150 s ion depletion at 90 V, we achieved an LOD of ~10 nM for gp120 in 1×PBS using the NFC-ICP platform, which is comparable to that obtained by fluorescence microcopy but in a label-free manner (Fig. 5(b)). By adding 10 min preconcentration at 60 V before the incubation, we were able to lower the LOD to less than 0.1 nM, which is an improvement of more than two orders of magnitude (Fig. 5(b)). The specificity of the device was confirmed by using mycobacterium tuberculosis (MTB) antigen ESAT6 as a control, which barely induced any changes of resistance even at 1 μM (Fig. 5(c)). Following the same protocol, we also studied the detection of the MTB pathogen fragment DNA (ACC AGC ACC TAA CCG GCT GTG GGT AGC AGA CCT CAC CTA TGT GTC GAC CTG GGC AGG GTT CGC CTA C) in 1×PBS using a 200 nm NFC functionalized with a complementary DNA probe (biotin-GT AGG CGA ACC CTG CCC AGG TC)59, achieving LODs of 10 nM without preconcentration and 0.01 nM with preconcentration(Figs. 5(d–f)). Similarly, we detected human thrombin protein in 1×PBS using a thrombin-binding aptamer (biotin-GGT TGG TGT GGT TGG),60 achieving LODs of >100 nM without preconcentration and 0.1 nM with preconcentration (Figs. 5(g–i)).
The main factors limiting the LOD of NFC-ICP are the binding reaction limitation (i.e. either too low kon or too high koff), and the mass transport limitation. The characteristic time for the binding between target molecules in the solution and immobilized probes on the nanoparticles to reach equilibrium,61 disregarding the mass transport limitation, is
| (6) |
In other words, for certain incubation time tI, the device is intrinsic reaction-limited when the target molecule concentration is lower than
| (7) |
Accordingly, for incubation time of 15 min, the detection was limited by the intrinsic kinetics at gp120 concentrations lower than 5.16 nM and MTB DNA concentrations lower than 8.2 nM, which generally agree with the LOD of 10 nM achieved in our devices for the two biomolecules without preconcentration. In the detection of human thrombin (LOD>100 nM in this work), however, the poor binding affinity between the aptamer and the thrombin (KD=109 nM) is the major limiting factor. Lastly, even beyond the binding kinetics and affinity limited regimes, changes of the resistance did not immediately reach saturation, indicating that the diffusion-limited transport of target molecules to the surface of nanoparticles was also an affecting factor.
Finally, we simulated real physiological samples by spiking known concentrations of gp120 and MTB DNA into human serum and urine. At the same concentrations of gp120, slightly smaller change of resistance was observed in 0.1×serum and much smaller in 1×serum than in 1×PBS (Fig. 5(j)). Similarly, at the same concentrations of MTB DNA, the relative change of resistance (ΔR/R) was greatest in 1×PBS, and decreased in the order of 1×urine, 0.1×serum, and 1×serum (Fig. 5(k)). Reduction in the signal was mainly due to the interference from the condense background biomolecules in real physiological fluids, which hindered the target-probe recognition by non-specifically binding to the surface of the nanoparticles and hiding the binding sites, and retarded mass transport by increased viscosity.62, 63 As a result, the detection sensitivity was most significantly weakened in 1×serum, which has a total background protein concentration of ~ 50 mg/ml.64 We were also able to perform the preconcentration of gp120 and MTB DNA in 1×serum and lowered the LODs by 10~102 folds (Figs. 5(j–k)). Compared to that of MTB DNA, the improvement of LOD from preconcentration is less for gp120. We speculate that the co-preconcentration of background proteins further suppressed the recognition between gp120 and anti-gp120 antibody, while this suppression effect is relatively weak for DNA due to the lower level of background DNAs in serum. Besides, the high-abundance background molecules may have also reduced the electric field gradient in the ion depletion zone, thus resulting in slightly lower preconcentration effect compared to that of 1×PBS.
Conclusion
We investigated the feasibility of electrical biomolecular detection in normal physiological conditions and enhancement of detection limit thereof by coupling NFC with the depletion and enrichment effects of ICP, achieving detection of proteins and DNAs in serum and urine with LODs of 10–100 pM. We believe this work is significant in that it broke the low ionic concentration constraint on nanofluidic biosensing and eliminated one of the major obstacles towards the field application of nanofluidic biosensors, which can be potentially applied to nanomaterial-based FETs and other surface charge-effect biosensors. As predicted by our model, we will work towards achieving lower LODs by improving the capture efficiency and the mass transport of target molecules. It’s also worthy of mentioning that according to Eq. (5), the relative change of resistance increase as the volume of the nanofluidic crystal decreases, which gives us a hint that reducing the number of nanoparticles (ultimately to one single nanoparticle) in the sensors would be another strategy to further improve the LOD in the future.
Supplementary Material
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (Grant No. 81471750 and 91323304), the Major State Basic Research Development Program (973 Program) (Grant No. 2015CB352100) and the MIT-Greater China MISTI program. J. Han and W. Ouyang were also supported by NIH (U19AI109755).
References
- 1.Luo X, Davis JJ. Chem Soc Rev. 2013;42:5944–5962. doi: 10.1039/c3cs60077g. [DOI] [PubMed] [Google Scholar]
- 2.Roy S, Gao Z. Nano Today. 2009;4:318–334. [Google Scholar]
- 3.Vlassiouk I, Kozel TR, Siwy ZS. J Am Chem Soc. 2009;131:8211–8220. doi: 10.1021/ja901120f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ali M, Yameen B, Neumann R, Ensinger W, Knoll W, Azzaroni O. J Am Chem Soc. 2008;130:16351–16357. doi: 10.1021/ja8071258. [DOI] [PubMed] [Google Scholar]
- 5.Ali M, Neumann R, Ensinger W. ACS Nano. 2010;4:7267–7274. doi: 10.1021/nn102119q. [DOI] [PubMed] [Google Scholar]
- 6.Duan C, Alibakhshi MA, Kim DK, Brown CM, Craik CS, Majumdar A. ACS Nano. 2016;10:7476–7484. doi: 10.1021/acsnano.6b02062. [DOI] [PubMed] [Google Scholar]
- 7.Wang X, Smirnov S. ACS Nano. 2009;3:1004–1010. doi: 10.1021/nn900113x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karnik R, Castelino K, Fan R, Yang P, Majumdar A. Nano Lett. 2005;5:1638–1642. doi: 10.1021/nl050966e. [DOI] [PubMed] [Google Scholar]
- 9.Cui Y, Wei Q, Park H, Lieber CM. Science. 2001;293:1289–1292. doi: 10.1126/science.1062711. [DOI] [PubMed] [Google Scholar]
- 10.Patolsky F, Zheng G, Hayden O, Lakadamyali M, Zhuang X, Lieber CM. Proc Natl Acad Sci U S A. 2004;101:14017–14022. doi: 10.1073/pnas.0406159101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng G, Patolsky F, Cui Y, Wang WU, Lieber CM. Nat Biotechnol. 2005;23:1294–1301. doi: 10.1038/nbt1138. [DOI] [PubMed] [Google Scholar]
- 12.Lin Y, Lu F, Tu Y, Ren Z. Nano Lett. 2004;4:191–195. [Google Scholar]
- 13.Yemini M, Reches M, Rishpon J, Gazit E. Nano Lett. 2005;5:183–186. doi: 10.1021/nl0484189. [DOI] [PubMed] [Google Scholar]
- 14.Liu Y, Yobas L. Nano Lett. 2014;14:6983–6990. doi: 10.1021/nl5032524. [DOI] [PubMed] [Google Scholar]
- 15.Stern E, Wagner R, Sigworth FJ, Breaker R, Fahmy TM, Reed MA. Nano Lett. 2007;7:3405–3409. doi: 10.1021/nl071792z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stern E, Vacic A, Reed MA. IEEE Trans Electron Devices. 2008;55:3119–3130. [Google Scholar]
- 17.Stern E, Vacic A, Rajan NK, Criscione JM, Park J, Ilic BR, Mooney DJ, Reed MA, Fahmy TM. Nat Nanotechnol. 2010;5:138–142. doi: 10.1038/nnano.2009.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elnathan R, Kwiat M, Pevzner A, Engel Y, Burstein L, Khatchtourints A, Lichtenstein A, Kantaev R, Patolsky F. Nano Lett. 2012;12:5245–5254. doi: 10.1021/nl302434w. [DOI] [PubMed] [Google Scholar]
- 19.Gao N, Gao T, Yang X, Dai X, Zhou W, Zhang A, Lieber CM. Proc Natl Acad Sci U S A. 2016;113:14633–14638. doi: 10.1073/pnas.1625010114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao N, Zhou W, Jiang X, Hong G, Fu TM, Lieber CM. Nano Lett. 2015;15:2143–2148. doi: 10.1021/acs.nanolett.5b00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yeh HC, Wang M, Chang CC, Yang RJ. Isr J Chem. 2014;54:1533–1555. [Google Scholar]
- 22.Chen Z, Wang Y, Wang W, Li Z. Appl Phys Lett. 2009;95:102105. [Google Scholar]
- 23.Hakim MMA, Lombardini M, Sun K, Giustiniano F, Roach PL, Davies DE, Howarth PH, de Planque MRR, Morgan H, Ashburn P. Nano Lett. 2012;12:1868–1872. doi: 10.1021/nl2042276. [DOI] [PubMed] [Google Scholar]
- 24.Umehara S, Karhanek M, Davis RW, Pourmand N. Proc Natl Acad Sci U S A. 2009;106:4611–4616. doi: 10.1073/pnas.0900306106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karnik R, Castelino K, Duan C, Majumdar A. Nano Lett. 2006;6:1735–1740. doi: 10.1021/nl061159y. [DOI] [PubMed] [Google Scholar]
- 26.Rubinstein I, Shtilman L. Journal of the Chemical Society, Faraday Transactions 2: Molecular and Chemical Physics. 1979;75:231–246. [Google Scholar]
- 27.Probstein RF. Physicochemical hydrodynamics: an introduction. Wiley-Interscience; New York: 1994. [Google Scholar]
- 28.Pu Q, Yun J, Temkin H, Liu S. Nano Lett. 2004;4:1099–1103. [Google Scholar]
- 29.Kim SJ, Wang YC, Lee JH, Jang H, Han J. Phys Rev Lett. 2007;99:044501. doi: 10.1103/PhysRevLett.99.044501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rubinstein SM, Manukyan G, Staicu A, Rubinstein I, Zaltzman B, Lammertink RGH, Mugele F, Wessling M. Phys Rev Lett. 2008;101:236101. doi: 10.1103/PhysRevLett.101.236101. [DOI] [PubMed] [Google Scholar]
- 31.Kim SJ, Ko SH, Kwak R, Posner JD, Kang KH, Han J. Nanoscale. 2012;4:7406–7410. doi: 10.1039/c2nr32467a. [DOI] [PubMed] [Google Scholar]
- 32.Rubinstein I, Zaltzman B. Physical Review E. 2000;62:2238. doi: 10.1103/physreve.62.2238. [DOI] [PubMed] [Google Scholar]
- 33.Wang YC, Stevens AL, Han J. Anal Chem. 2005;77:4293–4299. doi: 10.1021/ac050321z. [DOI] [PubMed] [Google Scholar]
- 34.Kim SJ, Ko SH, Kang KH, Han J. Nat Nanotechnol. 2010;5:297–301. doi: 10.1038/nnano.2010.34. [DOI] [PubMed] [Google Scholar]
- 35.Hlushkou D, Tallarek U, Crooks R, Knust K, Anand R. Chem Ing Tech. 2014;86:1447–1447. [Google Scholar]
- 36.Kwak R, Kim SJ, Han J. Anal Chem. 2011;83:7348–7355. doi: 10.1021/ac2012619. [DOI] [PubMed] [Google Scholar]
- 37.Cheng LJ, Chang HC. Biomicrofluidics. 2011;5:046502. doi: 10.1063/1.3657928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Senapati S, Slouka Z, Shah SS, Behura SK, Shi Z, Stack MS, Severson DW, Chang HC. Biosens Bioelectron. 2014;60:92–100. doi: 10.1016/j.bios.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Slouka Z, Senapati S, Yan Y, Chang HC. Langmuir. 2013;29:8275–8283. doi: 10.1021/la4007179. [DOI] [PubMed] [Google Scholar]
- 40.Kelly KC, Miller SA, Timperman AT. Anal Chem. 2008;81:732–738. doi: 10.1021/ac802209e. [DOI] [PubMed] [Google Scholar]
- 41.Wang H, Nandigana VVR, Jo KD, Aluru NR, Timperman AT. Anal Chem. 2015;87:3598–3605. doi: 10.1021/ac5019638. [DOI] [PubMed] [Google Scholar]
- 42.Shao L, Zheng M, Wang W. Appl Phys Lett. 2015;106:093105. [Google Scholar]
- 43.Lei Y, Xie F, Wang W, Wu W, Li Z. Lab Chip. 2010;10:2338–2340. doi: 10.1039/c004758a. [DOI] [PubMed] [Google Scholar]
- 44.Lei Y, Wang W, Wu W, Li Z. Appl Phys Lett. 2010;96:263102. [Google Scholar]
- 45.Sang J, Du H, Wang W, Chu M, Wang Y, Li H, Zhang HA, Wu W, Li Z. Biomicrofluidics. 2013;7:024112. doi: 10.1063/1.4802936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ouyang W, Wang W, Zhang H, Wu W, Li Z. Nanotechnology. 2013;24:345401. doi: 10.1088/0957-4484/24/34/345401. [DOI] [PubMed] [Google Scholar]
- 47.Zhao W, Wang B, Wang W. Lab Chip. 2016;16:2050–2058. doi: 10.1039/c6lc00416d. [DOI] [PubMed] [Google Scholar]
- 48.Ko SH, Song YA, Kim SJ, Kim M, Han J, Kang KH. Lab Chip. 2012;12:4472–4482. doi: 10.1039/c2lc21238b. [DOI] [PubMed] [Google Scholar]
- 49.Kim M, Jia M, Kim T. Analyst. 2013;138:1370–1378. doi: 10.1039/c2an36346a. [DOI] [PubMed] [Google Scholar]
- 50.Zhou J, Ren K, Zheng Y, Su J, Zhao Y, Ryan D, Wu H. Electrophoresis. 2010;31:3083–3089. doi: 10.1002/elps.201000113. [DOI] [PubMed] [Google Scholar]
- 51.Choi E, Kwon K, Kim D, Park J. Lab Chip. 2015;15:168–178. doi: 10.1039/c4lc01031k. [DOI] [PubMed] [Google Scholar]
- 52.Ko SH, Kim SJ, Cheow LF, Li LD, Kang KH, Han J. Lab Chip. 2011;11:1351–1358. doi: 10.1039/c0lc00349b. [DOI] [PubMed] [Google Scholar]
- 53.Lee JH, Cosgrove BD, Lauffenburger DA, Han J. J Am Chem Soc. 2009;131:10340–10341. doi: 10.1021/ja902594f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee JH, Song YA, Han J. Lab Chip. 2008;8:596–601. doi: 10.1039/b717900f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee JH, Song YA, Tannenbaum SR, Han J. Anal Chem. 2008;80:3198–3204. doi: 10.1021/ac800362e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schoch RB, Van Lintel H, Renaud P. Physics of Fluids (1994-present) 2005;17:100604. [Google Scholar]
- 57.Schoch RB, Cheow LF, Han J. Nano Lett. 2007;7:3895–3900. doi: 10.1021/nl0724788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martins DC, Chu V, Conde JP. Biomicrofluidics. 2013;7:034111. doi: 10.1063/1.4811277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cannas A, Goletti D, Girardi E, Chiacchio T, Calvo L, Cuzzi G, Piacentini M, Melkonyan H, Umansky SR, Lauria FN. Int J Tuberc Lung Dis. 2008;12:146–151. [PubMed] [Google Scholar]
- 60.Macaya RF, Schultze P, Smith FW, Roe JA, Feigon J. Proc Natl Acad Sci U S A. 1993;90:3745–3749. doi: 10.1073/pnas.90.8.3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oshannessy DJ, Brighamburke M, Soneson KK, Hensley P, Brooks I. Anal Biochem. 1993;212:457–468. doi: 10.1006/abio.1993.1355. [DOI] [PubMed] [Google Scholar]
- 62.de Jager W, Prakken BJ, Bijlsma JWJ, Kuis W, Rijkers GT. J Immunol Methods. 2005;300:124–135. doi: 10.1016/j.jim.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 63.Hennig C, Rink L, Fagin U, Jabs WJ, Kirchner H. J Immunol Methods. 2000;235:71–80. doi: 10.1016/s0022-1759(99)00206-9. [DOI] [PubMed] [Google Scholar]
- 64.Anderson NL, Anderson NG. Mol Cell Proteomics. 2002;1:845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.