

Abstract
Digenic Connexin26 (Cx26, GJB2) and Cx30 (GJB6) heterozygous mutations are the second most frequent cause of recessive deafness in humans. However, the underlying deafness mechanism remains unclear. In this study, we created different double Cx26 and Cx30 heterozygous (Cx26+/−/Cx30+/−) mouse models to investigate the underlying pathological changes and deafness mechanism. We found that double Cx26+/−/Cx30+/− heterozygous mice had hearing loss. Endocochlear potential (EP), which is a driving force for hair cells producing auditory receptor current, was reduced. However, unlike Cx26 homozygous knockout (Cx26−/−) mice, the cochlea in Cx26+/−/Cx30+/− mice displayed normal development and had no apparent hair cell degeneration. Gap junctions (GJs) in the cochlea form two independent networks: the epithelial cell GJ network in the organ of Corti and the connective tissue GJ network in the cochlear lateral wall. We further found that double heterozygous deletion of Cx26 and Cx30 in the epithelial cells did not reduce EP and had normal hearing, suggesting that Cx26+/−/Cx30+/− may mainly impair gap junctional functions in the cochlear lateral wall and lead to EP reduction and hearing loss. Most of Cx26 and Cx30 in the cochlear lateral wall co-expressed in the same gap junctional plaques. Moreover, sole Cx26+/− or Cx30+/− heterozygous mice had no hearing loss. These data further suggest that digenic Cx26 and Cx30 mutations may impair heterozygous coupling of Cx26 and Cx30 in the cochlear lateral wall to reduce EP, thereby leading to hearing loss.
Keywords: gap junction, connexin, Cx26, Cx30, endocochlear potential, deafness, cochlea
Introduction
Gap junction (GJ) connexin gene mutations induce high incidence of nonsyndromic hearing loss. Connexin26 (Cx26, GJB2) and Cx30 (GJB6) are two major deafness genes and predominantly express in the cochlea [1]. Either Cx26 or Cx30 mutations can induce hearing loss [1–3]. However, digenic heterozygous mutations for Cx26 and Cx30 are also often found clinically and are the second most frequent cause of nonsyndromic hearing loss in different human populations [4–16]. The phenotypes in these patients heterozygous for Cx26 and Cx30 mutations are various; hearing loss can be either prelingual or postlingual, ranging from mild or moderate to severe or profound deafness [2,11,14, 17,18].
GJs exist extensively in the inner ear, including the spiral limbus, supporting cells in the organ of Corti, and the cochlear lateral wall, and form two independent networks: epithelial cell network in the cochlear epithelium and connective tissue network in the cochlear lateral wall (Fig. 1f). However, there is no GJ and connexin expression in auditory sensory hair cells [19–22]. Cx26 and Cx30 are predominant isoforms in the cochlea [23]. Cx26 and Cx30 in the cochlea have overlapping distributions [19,21,24–26] and can form functional heterotypic/heteromeric (hybrid) GJ channels [27], which possess a rectified transjunctional gating and can induce a directional passage [27,28].
Fig. 1.

Gap junction network in the cochlea and Cx26 and Cx30 expression in the Cx26+/−/Cx30+/− mouse cochlea. a–d: Immunofluorescent staining for Cx26 (green) and Cx30 (red) in the cochlea in Cx26+/−/Cx30+/− and WT mice. Both Cx26 and Cx30 labeling is visible in the Cx26+/−/Cx30+/− mouse cochlea but their intensities are weaker in comparison with those in WT mice. Scale bar: 50 μm. e: Quantitative measure of Cx26 and Cx30 labeling in the organ of Corti (OC) and the cochlear lateral wall (LW) in WT mice and Cx26+/−/Cx30−/+ mice. The intensity of labeling was normalized to the average value in WT mice. The expression of Cx26 and Cx30 in Cx26+/−/Cx30+/− mice was significantly reduced. **: P > 0.001, t-test. f: Schematic drawing of two gap junctional (GJ) networks in the cochlea: epithelial cell GJ (ECGJ) network in the cochlear sensory epithelium and connective tissue GJ (CTGJ) network in the cochlear lateral wall. Hair cells have neither gap junction nor connexin expression. SV, scala vestibuli; SM, scala media; ST, scala tympani.
Mouse models show that Cx26 deletion can induce deafness [1]. Cx26 deficient mice show cochlear developmental disorders, hair cell degeneration, endocochlear potential (EP) reduction, and active cochlear amplification reduction [1]. In the clinic, Cx26 mutation induced hearing loss can be divided into two groups, congenital deafness and late-onset hearing loss. Congenital deafness induced by Cx26 deficiency mainly results from cochlear developmental disorders, whereas progressive, late-onset hearing loss induced by Cx26 deficiency is associated with the reduction of cochlear active amplification [1, 30–35]. However, the hypothesized K+-recycling impairment is not a primary deafness mechanism for Cx26 deficiency-induced hearing loss [36].
Previous Cx30 knockout (KO) mice also showed hearing loss [29,30]. However, it has been found that Cx26 expression in this Cx30 KO mouse line is also reduced [37]. A recent experiment showed that a Cx30 KO mouse created by Cre-FloxP technique have no hearing loss until 2-month old [38].
It has been reported in brief that double Cx26 and Cx30 heterozygous deficient mice have hearing loss [39]. However, the detailed pathological changes remain unclear; the underlying deafness mechanism also remains undetermined. In this study, the detailed underlying pathological changes in the cochlea were investigated. We also created different mouse models to investigate the mechanisms underlying digenic heterozygous deficiency of Cx26 and Cx30 induced hearing loss. We found that double Cx26 and Cx30 heterozygous (Cx26+/−/Cx30+/−) mice have hearing loss, which mainly results from EP reduction likely caused by impairment of GJ heterotypic coupling function in the cochlear lateral wall.
Materials and Methods
Double Cx26 and Cx30 heterozygous KO mouse generation and genotyping
Two lines of double Cx26 and Cx30 heterozygous (Cx26+/−/Cx30+/−) mice were generated by different Cx26 conditional KO (cKO) mice crossing with Cx30 KO mice. In the first line, Cx26 cKO mice were generated by crossing Cx26loxP/loxP mice (EM00245, European Mouse Mutant Archive) with the Pax2-Cre mouse line (the Mutation Mouse Regional Center, Chapel Hill, NC) [31]. Then, Cx26 cKO mice were further crossed with Cx30 KO mice (EM00323, European Mouse Mutant Archive) [30] to create Cx26 and Cx30 heterozygous (Cx26+/−/Cx30+/−) mice. The Cx26 floxed allele and Pax2-Cre transgene were detected on tail genomic DNA by PCR amplification using the following primers: Cx26F: 5′-CTT TCC AAT GCT GGT GGA GTG-3′ and Cx26R: 5′-ACA GAA ATG TGT TGG TGA TGG-3′ for the Cx26 floxed allele; Pax2-CreF: 5′-GCC TGC ATT ACC GGT CGA TGC AAC GA- 3′ and Pax2-CreR: 5′-GTG GCA GAT GGC GCG GCA ACA CCA TT- 3′ for Pax2-Cre transgene. Cx26loxP/loxP and WT mice generated 400 and 300 bps bands, respectively. The band of Pax2-Cre was 700 bps. Primer pairs for detecting Cx30 KO were Cx30 KO-1 (LACZ e Neo): 5′-GGT ACC TTC TAC TAA TTA GCT TGG -3′; Cx30 KO2 (LACZ e Neo): 5′-AGG TGG TAC CCA TTG TAG AGG AAG -3′; Cx30 KO-3 (LACZ e Neo) 5′-AGC GAG TAA CAA CCC GTC GGA TTC -3′. The bands of Cx30 KO and WT mice are located at 460 and 544 bps, respectively.
The second Cx26+/−/Cx30+/− mouse line was created by crossing Prox1-CreERT2 mouse line (Stack No. 022075, The Jackson Lab) with Cx26loxP/loxP transgenic mice to create Tamoxifen-inducible Prox1-Cx26 cKO mice [32]. After intraperitoneal injection with Tamoxifen (T5648, Sigma-Aldrich, St. Louis, MO) with 0.5 mg/10g per day for three days after birth, Cx26 expression in Deiters cells (DCs) and outer pillar cells (OPCs) in the cochlear epithelial cell GJ network is selectively deleted [32]. Then, Prox1-Cx26 cKO mice were further crossed with Cx30 KO mice to establish double Prox1-Cx26+/−/Cx30+/− heterozygous mice. WT littermates were used as controls. All experimental procedures were conducted in accordance with the policies of the University of Kentucky Animal Care & Use Committee.
Auditory brainstem response and distortion product otoacoustic emission measurements
As we previously reported [30–33], mice were anesthetized by intraperitoneal injection with a mixture of Ketamine and Xylazine (a stock solution: 8.5 ml saline+1 ml Ketamine+0.55 ml Xylazine) given at a dose of 0.1 ml/10 g body weight. Body temperature was maintained at 37–38°C by placing anesthetized mice on an isothermal pad (Deltaphase, model 39dp, Braintree Scientific Inc., Massachusetts). Auditory brainstem response (ABR) and distortion product otoacoustic emission (DPOAE) were recorded in a double-wall sound isolated chamber by use of a Tucker-Davis ABR workstation (Tucker-Davis Tech. Alachua, FL). ABR was evoked by both clicks and series of tone bursts (4 – 40 kHz, 10–80 dB SPL, a 5dB-step) with an ES-1 high-frequency speaker (Tucker-Davis Tech. Alachua, FL). The ABR threshold was determined by the lowest level at which an ABR can be recognized. If mice had severe hearing loss, the ABR test at the intensity range of 70 – 110 dB SPL was used.
For DPOAE recording, two plastic tubes were inserted into the external ear canal and sealed with an earplug. Two pure tones (f1 and f2, f2/f1 =1.22) were simultaneously delivered into the ear. The test frequencies were presented by a geometric mean of f1 and f2 [f0 = (f1 × f2)1/2]. The intensity of f1 (I1) was set at 5 dB SPL higher than that of f2 (I2). The responses were averaged by 150 times [32,33].
Endocochlear potential recording
As we previously reported [30,32,40], mice were anaesthetized as described above and the body temperature was maintained at 37–38 °C. The trachea was exposed and cut along the middle line. The tracheal tube was put into the trachea. Then, the cochlea was exposed by a ventral approach and the bone over the spiral ligament was gently picked to form a small hole. A glass pipette filled with a K+-based intracellular solution [41] was inserted into the hole. The DC potential was continually recorded as the electrode pipette penetrated through the lateral wall by use of MultiClamp 700A amplifier (Molecular Devices, CA) and digitized utilizing a Digidata 1322A (Molecular Devices, CA).
Immunofluorescent staining
The cochlear tissue preparation and immunofluorescent staining were performed as we previously described [21,26]. The cochlear cross-sections were fixed with 4% paraformaldehyde in 0.1 M PBS (pH 7.4) for 30 min. After being washed with PBS (0.1 M) 3 times, the tissue was incubated in a blocking solution (10% goat serum and 1% BSA in the PBS) with 0.1% Triton X-100 for 30 min at room temperature. The tissue then was incubated with primary antibody in the blocking solution at 4°C overnight. Monoclonal mouse anti-Cx26 (Cat# 33-5800) and polyclonal rabbit anti-Cx30 (Cat#71-2200, Invitrogen Corp, Carlsbad, CA) were used. After completely washing out the primary antibodies with PBS, the reaction to a 1:600 dilution of secondary Alexa Fluor® 488 or 568 conjugated antibodies (Molecular Probes) in the blocking solution followed at room temperature for 1 hr. After completely washing out, the section was mounted with a fluorescence mounting medium (H-1000, Vector Lab, CA) and observed under a Leica confocal microscope (Leica TCS SP2) or a fluorescence microscope (Nickon T2000).
For quantitative measure of Cx26 and Cx30 expression, the labeled pixels and intensity in the organ of Corti and the cochlear lateral wall were accounted and measured by use of ImageJ software (NIH, Bethesda, USA) [21,33]. The average of labeling intensity was calculated after subtraction of background intensity. Then, the averaged labeling intensities were normalized to those in WT mice.
Data processing and statistical analysis
Data were plotted by SigmaPlot and statistically analyzed by SPSS v18.0 (SPSS Inc. Chicago, IL). Error bars represent s.e.m. and data were expressed as mean ± s.e.m. other than indicated in text.
Results
Hearing loss in double Cx26+/−/Cx30+/− heterozygous mice
Fig. 1 shows Cx26 and Cx30 expression in the cochlea in Cx26+/−/Cx30+/− heterozygous mice. Fluorescent staining shows that expression of Cx26 and Cx30 in the cochlea is visible in Cx26+/−/Cx30+/− heterozygous mice (Fig. 1c&d). However, in comparison with that in WT mice (Fig. 1a&b), the intensity of staining appears weaker. Quantitative measure shows that intensities of Cx26 and Cx30 labeling in the organ of Corti and cochlear lateral wall in Cx26+/−/Cx30+/− heterozygous mice were reduced by ~ 50% (Fig. 1e). The normalized intensities of Cx26 and Cx30 labeling in Cx26+/−/Cx30+/− mice were 50.9±3.8% and 53.7±3.2%, respectively, in the organ of Corti and 53.4±6.4% and 52.7±5.4%, respectively, in the cochlear lateral wall.
ABR recording shows that double Cx26+/−/Cx30+/− heterozygous mice had hearing loss (Fig. 2). The ABR threshold in double Cx26+/−/Cx30+/− heterozygous mice was 46.4±1.26 dB SPL for click stimulations and was significantly increased in comparison with the ABR threshold (29.5±0.56 dB SPL) in littermate WT controls (P<0.001, one-way ANOVA with a Bonferroni correction). The hearing loss occurred in whole-frequency range (Fig. 2c). The ABR thresholds in 8–40 kHz range were increased to 45–70 dB SPL. In comparison with Cx26−/− mice and Cx30−/− mice, which demonstrated complete deafness (ABR thresholds >100 dB SPL, Fig. 2a&b), hearing loss in double Cx26+/−/Cx30+/− heterozygous mice was mild to moderate. However, single Cx26+/− or Cx30+/− heterozygous mice had no hearing loss. The ABR thresholds for click stimulation in sole Cx26+/− mice or Cx30+/− mice were 30.2±0.61 and 30.1±0.75 dB SPL, respectively, similar to that (29.5±0.56 dB SPL) in WT mice (Fig. 2b).
Fig. 2.

Hearing loss in double Cx26 and Cx30 heterozygous (Cx26+/−/Cx30+/−) mice. a: ABRs were evoked by click stimulations in WT, Cx26+/−/Cx30+/−, Cx26+/−, Cx30+/−, Cx26−/−, and Cx30−/− mice. b: Hearing loss in Cx26+/−/Cx30+/−, Cx26−/−, and Cx30−/− mice. ABR thresholds evoked by click stimulations were increased in Cx26+/−/Cx30+/−, Cx26−/−, and Cx30−/− mice. c: ABR thresholds in Cx26+/−/Cx30+/− and WT mice evoked by tone bursts. Hearing loss was observed in the whole-tested frequency range in Cx26+/−/Cx30+/− mice. WT littermates served as control. Mice were 6–8 weeks old. **: P<0.001 as determined by one-way ANOVA with a Bonferroni correction.
Reduction of DPOAE in Cx26+/−/Cx30+/− mice
DPOAE in Cx26+/−/Cx30+/− mice was also reduced (Fig. 3). The DPOAE thresholds in Cx26+/−/Cx30+/− mice at 8, 16, and 20 kHz were 38.8±1.30, 30.6±0.95, and 43.4±1.13 dB SPL, respectively (Fig. 3b). In comparison with WT mice, the threshold of DPOAE in Cx26+/−/Cx30+/− mice were significantly increased by 5- 10 dB SPL (P>0.001, one-way ANOVA with a Bonferroni correction).
Fig. 3.
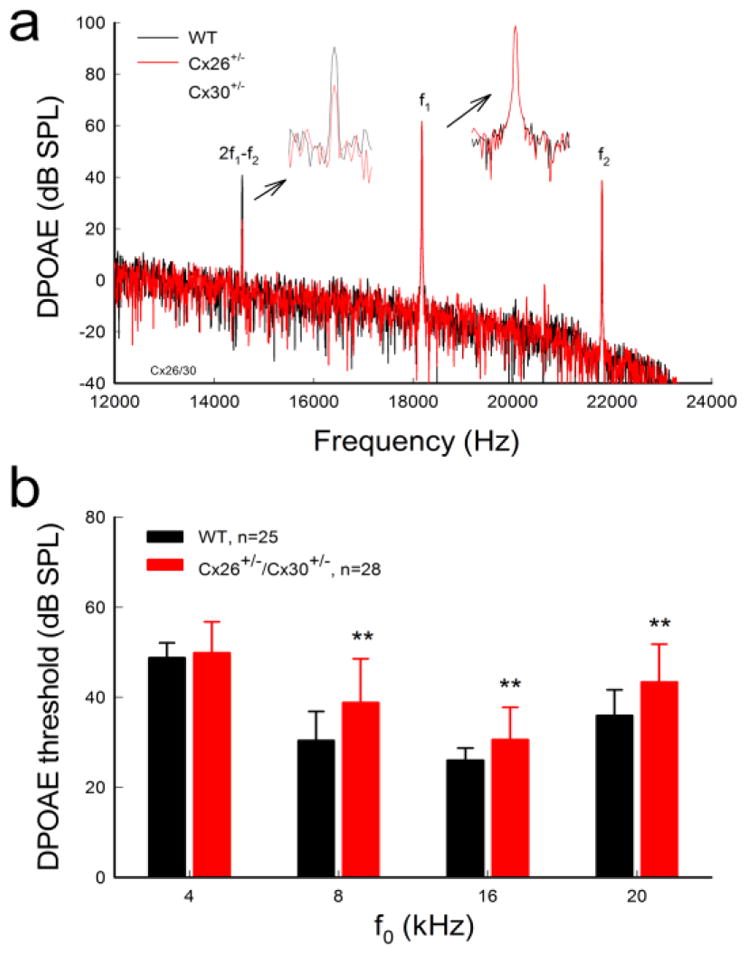
Reduction of DPOAE in Cx26+/−/Cx30+/− mice. a: Spectrum of acoustic emission recorded from Cx26+/−/Cx30+/− and WT mice. Insets: Large scale plotting of 2f1−f2 and f1 peaks. The peak of DPOAE (2f1−f2) in Cx26+/−/Cx30+/− mice was reduced but f1 and f2 peaks remained the same as those in WT mice. f0=20 kHz, I1/I2=60/55 dB SPL. b: DPOAE thresholds in Cx26+/−/Cx30+/− mice were increased. WT littermates served as control. Data are expressed as mean ± S.D. **: P < 0.001 as determined by one-way ANOVA with a Bonferroni correction.
Normal cochlear development and no cell degeneration in Cx26+/−/Cx30+/− mice
Cx26 cKO mice with deletion of Cx26 at birth have cochlear developmental disorders and severe hair cell degeneration [31,35,42]. However, double Cx26+/−/Cx30+/− heterozygous mice had no cochlear developmental disorders (Fig. 4a&b). The cochlea showed normal shape and the cochlear tunnel is open. Also, there was no apparent cell degeneration in Cx26+/−/Cx30+/− mice (Fig. 4). Hair cells also had no apparent loss (Fig. 4c&d).
Fig. 4.

Normal development of the cochlea and no hair cell loss in Cx26+/−/Cx30+/− mice. a–b: Cross-sections of the Cx26+/−/Cx30+/− mouse cochlea. The cochlea demonstrates normal shape and has no apparent cell degeneration. SV, scala vestibuli; SM, scala media; ST, scala tympani. c–d: whole-mounting of the cochlear sensory epithelium in Cx26+/−/Cx30+/− mice. Inner hair cells (IHCs) and outer hair cells (OHCs) were stained with phalloidin-Alexa 488 (green) and the cell nuclei were labeled with propidium iodide (PI, red). Mice were 2–3 months old. Scale bar: 100 μm in a & c, 40 μm in b, and 20 μm in d.
No hearing loss in double Prox1-Cx26+/−/Cx30+/− heterozygous mice
As shown in Fig. 1f, gap junctions in the cochlea form two GJ networks. We previously reported that Prox1-Cx26 cKO mice [32,33] have selective deletion of Cx26 expression in the Deiters cells (DCs) and outer pillar cells (OPCs) in the epithelial cell GJ network (Fig. 5a&b). In this experiment, we further crossed Prox1-Cx26−/− mice with Cx30−/− mice to establish double Prox1-Cx26+/−/Cx30+/− heterozygous mice (Fig. 5). In this Prox1-Cx26+/−/Cx30+/− mouse, only DCs and OPCs in the epithelial cell GJ network were double heterozygous for Cx26 and Cx30. Intensity of labeling for Cx26 and Cx30 in the organ of Corti was reduced almost by half (0.555±0.063 and 0.549±0.053, respectively) in comparison with those in WT littermate mice. However, in the lateral wall, only Cx30 labeling was reduced by half (0.517±0.079), whereas the expression of Cx26 appeared as the same (0.939±0.054) as that in WT mice (Fig. 5c&d). There is no significant difference between them (P=0.289, one-way ANOVA).
Fig. 5.

Connexin expression in Prox1-Cx26+/−/Cx30+/− mice. a: Double immunofluorescent staining for Cx26 and Cx30 in Prox1-Cx26+/−/Cx30+/− mice. b: Schematic drawing shows target-deletion of Cx26 in Deiters cells (DCs) and outer pillar cells (OPCs) in the cochlear sensory epithelium in Prox1-Cx26 KO mice. c–d: Quantitative measure of Cx26 and Cx30 labeling in the organ of Corti (OC) and the cochlear lateral wall (LW) in WT mice and Prox1-Cx26+/−/Cx30−/+ mice. Littermates were used as controls. The intensity of labeling was normalized to the average value in WT mice. The expression of Cx26 and Cx30 in the OC in Prox1-Cx26+/−/Cx30+/− mice was significantly reduced. However, the expression of Cx26 in the lateral wall was not reduced. **: P > 0.001, t-test.
Unlike Cx26+/−/Cx30+/− mice that have hearing loss (Fig. 2), Prox1-Cx26+/−/Cx30+/− mice had no hearing loss as measured by ABR threshold (Fig. 6). In comparison with WT littermate mice, there were no significant difference in ABR thresholds between two groups (P=0.53, one-way ANOVA).
Fig. 6.
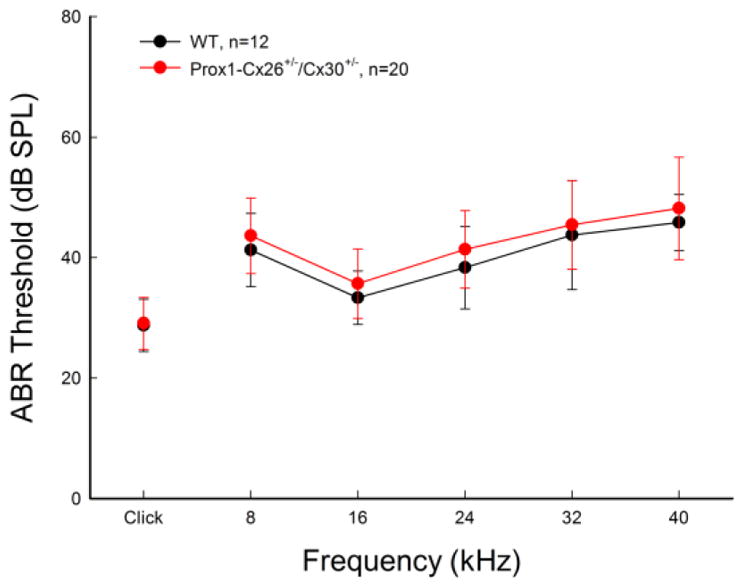
No hearing loss in Prox1-Cx26+/−/Cx30+/− mice. WT littermates served as control. Mice were 4–8 weeks old. Data are expressed as mean ± S.D.
EP reduction in Cx26+/−/Cx30+/− mice but not in Prox1-Cx26+/−/Cx30+/− mice
EP is a driving force required for hair cells generating auditory receptor current and potential and is required for hearing. We found that EP in WT and Cx26+/−/Cx30+/− mice was 98.2±1.41 and 55.7±5.14 mV, respectively (Fig. 7a); EP in Cx26+/−/Cx30+/− mice was significantly reduced (P<0.001, one-way ANOVA with a Bonferroni correction). However, EP in Prox1-Cx26+/−/Cx30+/− mice was 92.6±6.59 mV and was not significantly changed in comparison with EP in WT littermate mice (91.1±4.51 mV, P=0.42, one-way ANOVA). On the other hand, EP in Cx26 homozygous KO mice and Cx30 homozygous KO mice were dramatically reduced to 62.8±6.25 and 2.98±1.62 mV, respectively (Fig. 7b). Positive EP in Cx30 KO mice was almost abolished.
Fig. 7.

EP reduction in Cx26+/−/Cx30+/− mice but not in Prox1- Cx26+/−/Cx30+/− mice. a. The EP was significantly reduced in Cx26+/−/Cx30+/− mice but not in Prox1- Cx26+/−/Cx30+/− mice. WT littermates served as control. **: P < 0.001 as determined by one-way ANOVA with a Bonferroni correction. b. EP reduction in homozygous Cx26−/− KO mice and homozygous Cx30−/− KO mice. Mice were 4–8 weeks old.
EP generation and heterozygous gap junctional coupling in the cochlear lateral wall
Positive EP is generated in the cochlear lateral wall. Based on a widely-accepted “two-cell model” [43–45], EP is generated by marginal cells (cell 1) and the intermediate cells coupled with the basal cells in the stria vascularis and neighboring fibrocytes in the spiral ligament by GJs (cell 2) (Fig. 9). Cx26 and Cx30 are predominant connexin isoforms in the cochlear lateral wall [24–26]. Fig. 8 shows that expression of Cx26 and Cx30 overlapped in most cells in the cochlear lateral wall. In particular, most of Cx26 and Cx30 co-located at the same punctate but at different sides (indicated by empty triangles in Fig. 8b and white error heads in Fig. 8c), indicating that Cx26 and Cx30 may form heterotypic gap junctional channels as previously reported in the cochlear supporting cells in the cochlear sensory epithelium [21,27].
Fig. 9.
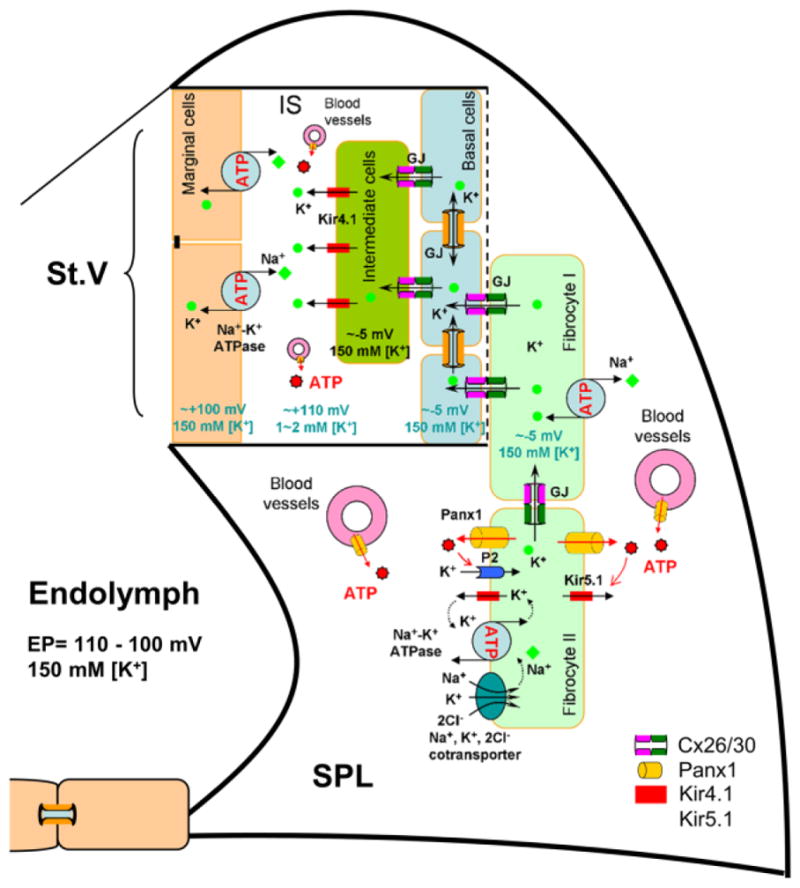
Schematic drawing of EP generation in the cochlear lateral wall. Based on the “two-cell” model, EP is generated by Kir4.1 in the apical membrane of intermediate cells in conjunction with Kir5.1 channels, Na+/K+ ATPases, and Na+, K+, 2Cl−-cotransporters in the fibrocytes through GJ coupling [40,45]. Heterozygous couplings of gap junctions between fibrocute I and II cells in the spiral ligament (SPL), between fibrocyte I cells and base cells in the stria vascularis (St. V), and between base cells and intermediate cells are important for eventually positive EP generation. MC: marginal cell; BC: basal cell; FC: fibrocyte; IC: intermediate cell; IS: intrastrial space; GJ: gap junction.
Fig. 8.

Co-expression of Cx26 and Cx30 and EP generation in the cochlear lateral wall. The cross-section of the mouse cochlear lateral wall was stained for Cx26 (green) and Cx30 (red) in immunofluorescent staining. Empty triangles in panel b indicate heterozygous coupling between stria vascularis (basal cell layer) and spiral ligament (fibrocyte I cells). White errors in panel c indicate co-labeling of Cx26 and Cx30 at the same GJ plaques between fibrocyte cells in the spiral ligament. Most of GJ plaque-punctates have Cx26 and Cx30 co-labeling and show yellow color in the over-lap image. St. V, stria vascularis; SPL, spiral ligament. Scale bar: 20 μm in a & b and 10 μm in c.
Discussion
In this study, we investigated the deafness mechanism underlying digenic Cx26 and Cx30 heterozygous mutations by creating double Cx26+/−/Cx30+/− heterozygous deficient mice. We found that double Cx26+/−/Cx30+/− heterozygous mice had hearing loss (Fig. 1). The ABR and DPOAE thresholds were increased (Figs. 2&3). The cochlea displayed normal development and had no apparent hair cell loss (Fig. 4). EP in Cx26+/−/Cx30+/− mice was reduced (Fig. 7). These data are consistent with a previous report that double Cx26 and Cx30 heterozygous mice had EP reduction and hearing loss [39].
The positive EP is generated in the cochlear lateral wall. As mentioned above, the cochlear gap junctions form two independent networks (Fig. 1f): epithelium GJ network in the cochlear epithelium and connective tissue GJ network in the cochlear lateral wall. In this study, we further created Prox1-Cx26+/−/Cx30+/− mice, in which Cx26 expression only in DCs and OPCs in the epithelial cell GJ network was selectively reduced [32,33]. Prox1-Cx26+/−/Cx30+/− mice displayed normal hearing (Fig. 6) and normal EP (Fig. 7a). These data further suggest that Cx26+/−/Cx30+/− may mainly impair connective tissue GJ network function in the cochlear lateral wall leading to EP reduction and hearing loss, similar to our previous finding that the reduction of EP by deletion of Panx1 reducing ATP release induces hearing loss [40].
Positive EP is a driving force for hair cells producing auditory receptor current and is required for hearing. As described by two-cell model for EP generation [40,45], fibrocyte cells in the spiral ligament are depolarized to approximately -5 mV by coordination of ATP-dependent Kir5.1 K+ channels, Na+, K+-ATPases, and Na+, K+, 2Cl− cotransporters, which consequently leads to depolarization of the intermediate cells in the stria vascularis to ~ −5 mV through gap junctional coupling (Fig. 9). Due to low K+ concentration (1–2 mM) in the intrastrial space (IS), ATP-dependent Kir4.1 K+ channels located at the apical membrane of the intermediate cells further generate 105–110 mV transmembrane potential between the intracellular space and the intrastrial space. Thus, with respect to normal extracellular space, the intrastrial space becomes positive 110–115 mV. This positive potential eventually leads to positive EP (+100–110 mV) in the endolymph in the scala media [40,45]. Apparently, gap junctional coupling plays a critical role in EP generation (Fig. 9). Also, K+-flowing from the fibrocytes to the intermediate cells to the intrastrial space is following a K+-ion concentration gradient but against the voltage gradient (moving cationic ions from negative voltage to positive voltage). Thus, the rectified gating between basal cells and intermediate cells or between the fibrocytes and the basal cells may be required for the directional passage of K+-ions and the efflux of K+-ions to the intrastrial space (Fig. 9), just like rectified-outward K+-channels acting in the efflux of K+-ions after cell depolarization. Moreover, both Cx26 and Cx30 deletion can reduce EP (Fig. 7b). This indicates that either Cx26 or Cx30 deletion cannot be compensated by each other for EP generation, i.e., Cx26/Cx30 heterotypic channels are required for the EP generation.
Indeed, Cx26 and Cx30 can form heterotypic gap junctional channels both in vitro [46] and in vivo, which possess a rectified gating property [27] and can induce a directional voltage gradient and passage [28]. In this experiment (Fig. 8a–c) and also in our previous study [26], we found that most of Cx26 and Cx30 in the cochlear lateral wall co-localize at the same GJ plaque, which may be able to assemble to functional heterotypic gap junctional channels [25,27]. Digenic Cx26 and Cx30 mutations may compromise heterotypic gap junctional function in the cochlear lateral wall, thereby leading to EP reduction and hearing loss. Indeed, it has been found that some Cx26 deafness-associated mutants, such as p.V84L and p.V95M, can form functional homotypic gap junctional channels but cannot form functional heterotypic channels [47–50], indicating that these Cx26 mutants may specifically impair heterotypic channel function in vivo. The current study may also provide a deafness mechanism underlying these Cx26 mutants. In this experiment, we found that Cx26 and Cx30 expression in Cx26+/−/Cx30+/− heterozygous mice reduced but not completely abolished (Fig. 1). Moreover, sole Cx26+/− heterozygous or Cx30+/− heterozygous mice have no hearing loss (Fig. 2b). These data further suggest that digenic Cx26 and Cx30 mutations may mainly impair heterotypic GJ channel function leading to hearing loss.
In the clinic, the most common genotype for digenic Cx26 and Cx30 deafness mutations is Cx30 truncate deafness mutations (GJB6-D13S1830 and GJB6-D13S1854) with either homozygous, or heterozygous in trans with recessive Cx26 (GJB2) mutations [2,3]. It has been reported the existence of a distance cis-regulatory region that controls both Cx26 and Cx30 expression [18,51–53]. The truncate deletion may involve this co-regulatory element (region) and prevents the expression in cis of the normal GJB2 gene. If co-existence of GJB2 (Cx26) mutations in trans, this can lead to the complete absence of Cx26 expression and function. Indeed, it has been reported that Cx26 and Cx30 expressions in mutated GJB2 allele in trans with large deletion in Cx30 (GJB6-D13S1830 and GJB6-D13S1854) dramatically reduced or abolished Cx26 expression at the transcription level [18,52,53]. However, the further pathological changes and the underlying deafness mechanism still remain unclear. Recently, we found that Cx26 deficiency can disrupt miRNA intercellular transfer in the cochlea and cause cochlear developmental disorders, thereby leading to deafness [35,54]. It has been reported that expression of Cx26 in the Cx30 KO mice and Cx30 T5M deafness mutant knockin mice is also reduced, which may lead to hearing loss [37,55]. As mentioned above, Cx26 deficiency can induce cochlear developmental disorders and hair cell degeneration [31,35]. However, Cx26+/−/Cx30+/− heterozygous mice have neither cochlear developmental disorders nor severe hair cell degeneration (Fig. 4). Thus, hearing loss in Cx26+/−/Cx30+/− heterozygous mice does not solely result from the reduction of Cx26 expression.
In this study, we found that EP reduction is a determining factor for hearing loss in double Cx26+/−/Cx30+/− heterozygous mice (Fig. 7). EP was also reduced or abolished in Cx26 KO or Cx30 KO mice (Fig. 7b, also see [29,30,56]). However, we previously found that EP reduction and hair cell degeneration are not primary causes for Cx26 deletion induced congenital deafness [30,31]; EP reduction is also not associated with Cx26 deficiency induced progressive, late-onset hearing loss [34]. These data further indicate that unlike monogenic Cx26 or Cx30 mutations, heterozygous Cx26/Cx30 mutations have different underlying mechanisms for deafness and may be associated with impairment in Cx26/30 heterotypic channel function, which need to be further studied in future.
Our recent experiments also demonstrated that the hypothesized impairment of K+-recycling in the cochlea is not a mechanism for Cx26 deficiency induced hearing loss [1,34,36]. The hypothesis of K+-recycling impairment proposes that cochlear GJs will be impaired due to Cx26 or Cx30 deficiency and causes K+-recycling in the cochlea impaired, which consequently causes K+-ions accumulated in the extracellular space around hair cells and eventually leads to hair cell degeneration and hearing loss. In this experiment, double Cx26+/−/Cx30+/− heterozygous mice displayed normal cochlear development and had no hair cell loss (Fig. 4). These data provide further evidence that the K+-recycling hypothesis is not a mechanism for Cx26 and Cx30 deficiency induced hearing loss.
Highlights.
Digenic Cx26 and Cx30 mutation is the 2nd cause for recessive deafness in humans
Double Cx26+/−/Cx30+/− heterozygous mice have hearing loss
There is EP reduction but no hair cell loss in Cx26+/−/Cx30+/− mice
Sole Cx26+/− or Cx30+/− heterozygous mice have no hearing loss
Hearing loss may result from heterotypic-GJ impairment in the cochlear lateral wall
Acknowledgments
This work was supported by NIH (R01) DC 05989 and R56 DC 015019 to HBZ, the National Natural Science Foundation of China (No. 81600795) to LZ and (No. 81500791) to JC.
Footnotes
Conflict interest: The authors declare no competing financial interests.
Author contributions: HBZ designed experiments. LM, LZ, JC, YZ, CL, and HBZ performed experiments. LM. LZ, JC, and HBZ analyzed data. RJ wrote and revised paper. HBZ wrote paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wingard JC, Zhao HB. Cellular and Deafness Mechanisms Underlying Connexin Mutation-Induced Hearing Loss - A Common Hereditary Deafness. Front Cell Neurosci. 2015;9:202. doi: 10.3389/fncel.2015.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Castillo FJ, Castillo I. The DFNB1 subtype of autosomal recessive non- syndromic hearing impairment. Front Biosci. 2011;17:3252–3274. doi: 10.2741/3910. [DOI] [PubMed] [Google Scholar]
- 3.Chan DK, Chang KW. GJB2-associated hearing loss: systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope. 2014;124:E34–53. doi: 10.1002/lary.24332. [DOI] [PubMed] [Google Scholar]
- 4.Lerer I, Sagi M, Ben-Neriah Z, Wang T, Levi H, Abeliovich D. A deletion mutation in GJB6 cooperating with a GJB2 mutation in trans in non-syndromic deafness: A novel founder mutation in Ashkenazi Jews. Hum Mutat. 2001;18:460. doi: 10.1002/humu.1222. [DOI] [PubMed] [Google Scholar]
- 5.Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Tellería D, Menéndez I, Moreno F. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med. 2002;346:243–249. doi: 10.1056/NEJMoa012052. [DOI] [PubMed] [Google Scholar]
- 6.Castillo FJ, Rodríguez-Ballesteros M, Alvarez A, Hutchin T, Leonardi E, de Oliveira CA, Azaiez H, Brownstein Z, Avenarius MR, Marlin S, Pandya A, Shahin H, Siemering KR, Weil D, Wuyts W, Aguirre LA, Martín Y, Moreno-Pelayo MA, Villamar M, Avraham KB, Dahl HH, Kanaan M, Nance WE, Petit C, Smith RJ, Van Camp G, Sartorato EL, Murgia A, Moreno F, del Castillo I. A novel deletion involving the connexin-30 gene, del(GJB6-d13s1854), found in trans with mutations in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. J Med Genet. 2005;42:588–594. doi: 10.1136/jmg.2004.028324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pallares-Ruiz N, Blanchet P, Mondain M, Claustres M, Roux AF. A large deletion including most of GJB6 in recessive non syndromic deafness: a digenic effect? Eur J Hum Genet. 2002;10:72–76. doi: 10.1038/sj.ejhg.5200762. [DOI] [PubMed] [Google Scholar]
- 8.Stevenson VA, Ito M, Milunsky JM. Connexin-30 deletion analysis in connexin-26 heterozygotes. Genet Test. 2003;7:151–154. doi: 10.1089/109065703322146867. [DOI] [PubMed] [Google Scholar]
- 9.Wu BL, Kenna M, Lip V, Irons M, Platt O. Use of a multiplex PCR/sequencing strategy to detect both connexin 30 (GJB6) 342 kb deletion and connexin 26 (GJB2) mutations in cases of childhood deafness. Am J Med Genet A. 2003;121A:102–108. doi: 10.1002/ajmg.a.20210. [DOI] [PubMed] [Google Scholar]
- 10.Gualandi E, Ravani A, Berto A, Burdo S, Trevisi P, Ferlini A, Martini A, Calzolari E. Occurrence of del(GIB6-D13S1830) mutation in Italian non-syndromic hearing loss patients carrying a single GJB2 mutated allele. Acta Otolaryngol Suppl. 2004;552:29–34. doi: 10.1080/03655230410017166. [DOI] [PubMed] [Google Scholar]
- 11.Bolz H, Schade G, Ehmer S, Kothe C, Hess M, Gal A. Phenotypic variability of non-syndromic hearing loss in patients heterozygous for both c.35delG of GJB2 and the 342-kb deletion involving GJB6. Hear Res. 2004;188:42–46. doi: 10.1016/S0378-5955(03)00346-0. [DOI] [PubMed] [Google Scholar]
- 12.Frei K, Ramsebner R, Lucas T, Baumgartner WD, Schoefer C, Wachtler FJ, Kirschhofer K. Screening for monogenetic del(GJB6-D13S1830) and digenic del(GJB6-D13S1830)/GJB2 patterns of inheritance in deaf individuals from Eastern Austria. Hear Res. 2004;196:115–118. doi: 10.1016/j.heares.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 13.Batissoco AC, Abreu-Silva RS, Braga MC, Lezirovitz K, Della-Rosa V, Alfredo T, Jr, Otto PA, Mingroni-Netto RC. Prevalence of GJB2 (connexin-26) and GJB6 (connexin-30) mutations in a cohort of 300 Brazilian hearing-impaired individuals: implications for diagnosis and genetic counseling. Ear Hear. 2009;30:1–7. doi: 10.1097/AUD.0b013e31819144ad. [DOI] [PubMed] [Google Scholar]
- 14.Chan DK, Schrijver I, Chang KW. Connexin-26-associated deafness: Phenotypic variability and progression of hearing loss. Genet Med. 2010;12:174–181. doi: 10.1097/GIM.0b013e3181d0d42b. [DOI] [PubMed] [Google Scholar]
- 15.Silva-Costa SM, Martins FT, Pereira T, Pomilio MC, Marques-de-Faria AP, Sartorato EL. Searching for digenic inheritance in deaf Brazilian individuals using the multiplex ligation-dependent probe amplification technique. Genet Test Mol Biomarkers. 2011;15:849–853. doi: 10.1089/gtmb.2011.0034. [DOI] [PubMed] [Google Scholar]
- 16.Asma A, Ashwaq A, Norzana AG, Atmadini AM, Ruszymah BH, Saim L, Wahida IF. The association between GJB2 mutation and GJB6 gene in non syndromic hearing loss school children. Med J Malaysia. 2011;66:124–128. [PubMed] [Google Scholar]
- 17.Cama E, Melchionda S, Palladino T, Carella M, Santarelli R, Genovese E, Benettazzo F, Zelante L, Arslan E. Hearing loss features in GJB2 biallelic mutations and GJB2/GJB6 digenic inheritance in a large Italian cohort. Int J Audiol. 2009;48:12–17. doi: 10.1080/14992020802400654. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez-Paris J, Tamayo ML, Gelvez N, Schrijver I. Allele-specific impairment of GJB2 expression by GJB6 deletion del(GJB6-D13S1854) PLoS One. 2011;6:e21665. doi: 10.1371/journal.pone.0021665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kikuchi T, Kimura RS, Paul DL, et al. Gap junctions in the rat cochlea: immunohistochemical and ultrastructural analysis. Anat Embryol. 1995;191:101–118. doi: 10.1007/BF00186783. [DOI] [PubMed] [Google Scholar]
- 20.Zhao HB, Santos-Sacchi J. Auditory collusion and a coupled couple of outer hair cells. Nature. 1999;399:359–362. doi: 10.1038/20686. [DOI] [PubMed] [Google Scholar]
- 21.Zhao HB, Yu N. Distinct and gradient distributions of connexin26 and connexin30 in the cochlear sensory epithelium of guinea pigs. J Comp Neurol. 2006;499:506–518. doi: 10.1002/cne.21113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu N, Zhao HB. Modulation of outer hair cell electromotility by cochlear supporting cells and gap junctions. PLoS One. 2009;4:e7923. doi: 10.1371/journal.pone.0007923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao HB, Kikuchi T, Ngezahayo A, White TW. Gap junctions and cochlear homeostasis. J Memb Biol. 2006;209:177–186. doi: 10.1007/s00232-005-0832-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lautermann J, ten Cate WJF, Altenhoff P, Grümmer R, Traub O, Frank HG, Jahnke K, Winterhager E. Expression of the gap-junction connexins 26 and 30 in the rat cochlea. Cell Tissue Res. 1998;294:415–420. doi: 10.1007/s004410051192. [DOI] [PubMed] [Google Scholar]
- 25.Forge A, Becker D, Casalotti S, Edwards J, Marziano N, Nevill G. Gap junctions in the inner ear: comparison of distribution patterns in different vertebrates and assessement of connexin composition in mammals. J Comp Neurol. 2003;467:207–231. doi: 10.1002/cne.10916. [DOI] [PubMed] [Google Scholar]
- 26.Liu YP, Zhao HB. Cellular characterization of Connexin26 and Connnexin30 expression in the cochlear lateral wall. Cell Tissue Res. 2008;333:395–403. doi: 10.1007/s00441-008-0641-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao HB, Santos-Sacchi J. Voltage gating of gap junctions in cochlear supporting cells: evidence for nonhomotypic channels. J Membr Biol. 2000;175:17–24. doi: 10.1007/s002320001051. [DOI] [PubMed] [Google Scholar]
- 28.Zhao HB. Directional rectification of gap junctional voltage gating between Deiters cells in the inner ear of guinea pig. Neurosci Lett. 2000;296:105–108. doi: 10.1016/s0304-3940(00)01626-8. [DOI] [PubMed] [Google Scholar]
- 29.Teubner B, Michel V, Pesch J, Lautermann J, Cohen-Salmon M, Sohl G, Jahnke K, Winterhager E, Herberhold C, Hardelin JP, Petit C, Willecke K. Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum Mol Genet. 2003;12:13–21. doi: 10.1093/hmg/ddg001. [DOI] [PubMed] [Google Scholar]
- 30.Chen J, Chen J, Zhu Y, Liang C, Zhao HB. Deafness induced by Connexin26 (GJB2) deficiency is not determined by endocochlear potential (EP) reduction but is associated with cochlear developmental disorders. Biochem Biophys Res Commun. 2014;448:28–32. doi: 10.1016/j.bbrc.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang C, Zhu Y, Zong L, Lu GJ, Zhao HB. Cell degeneration is not a primary causer for Connexin26 (GJB2) deficiency associated hearing loss. Neurosci Lett. 2012;528:36–41. doi: 10.1016/j.neulet.2012.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu Y, Liang C, Chen J, Zong L, Chen GD, Zhao HB. Active cochlear amplification is dependent on supporting cell gap junctions. Nat Commun. 2013;4:1786. doi: 10.1038/ncomms2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zong L, Chen J, Zhu Y, Zhao HB. Progressive age-dependence and frequency difference in the effect of gap junctions on active cochlear amplification and hearing. Biochem Biophys Res Commun. 2017;489:223–227. doi: 10.1016/j.bbrc.2017.05.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu Y, Chen J, Liang C, Zong L, Chen J, Jones RO, Zhao HB. Connexin26 (GJB2) deficiency reduces active cochlear amplification leading to late-onset hearing loss. Neuroscience. 2015;284:719–729. doi: 10.1016/j.neuroscience.2014.10.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu Y, Zong L, Mei L, Zhao HB. Connexin26 gap junction mediates miRNA intercellular genetic communication in the cochlea and is required for inner ear development. Sci Rep. 2015;5:15647. doi: 10.1038/srep15647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao HB. Hypothesis of K+-recycling defect is not a primary deafness mechanism for Cx26 (GJB2) deficiency. Front Mol Neurosci. 2017;10:162. doi: 10.3389/fnmol.2017.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahmad S, Tang W, Chang Q, Qu Y, Hibshman J, Li Y, Söhl G, Willecke K, Chen P, Lin X. Restoration of connexin26 protein level in the cochlea completely rescues hearing in a mouse model of human connexin30-linked deafness. Proc Natl Acad Sci U S A. 2007;104:1337–1341. doi: 10.1073/pnas.0606855104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boulay AC, del Castillo FJ, Giraudet F, Hamard G, Giaume C, Petit C, Avan P, Cohen-Salmon M. Hearing is normal without connexin30. J Neurosci. 2013;33:430–434. doi: 10.1523/JNEUROSCI.4240-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Michel V, Hardelin JP, Petit C. Molecular mechanism of a frequent genetic form of deafness. N Engl J Med. 2003;349:716–717. doi: 10.1056/NEJMc030327. [DOI] [PubMed] [Google Scholar]
- 40.Chen J, Zhu Y, Liang C, Chen J, Zhao HB. Pannexin1 channels dominate ATP release in the cochlea ensuring endocochlear potential and auditory receptor potential generation and hearing. Sci Rep. 2015;5:10762. doi: 10.1038/srep10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu Y, Zhao HB. ATP-mediated potassium recycling in the cochlear supporting cells. Purinergic Signal. 2010;6:221–229. doi: 10.1007/s11302-010-9184-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y, Chang Q, Tang W, Sun Y, Zhou B, Li H, Lin X. Targeted connexin26 ablation arrests postnatal development of the organ of Corti. Biochem Biophys Res Commun. 2009;385:33–37. doi: 10.1016/j.bbrc.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wangemann P, Schacht J. Homeostatic mechanisms in the cochlea. In: Dallos P, Popper AN, Fay RR, editors. The Cochlea. Springer-Verlag; New York: 1996. pp. 130–185. [Google Scholar]
- 44.Nin F, Hibino H, Doi K, Suzuki T, Hisa Y, Kurachi Y. The endocochlear potential depends on two K+ diffusion potentials and an electrical barrier in the stria vascularis of the inner ear. Proc Natl Acad Sci USA. 2008;105:1751–1756. doi: 10.1073/pnas.0711463105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen J, Zhao HB. The role of an inwardly rectifying K+ channel (Kir4.1) in the inner ear and hearing loss. Neuroscience. 2014;265:137–146. doi: 10.1016/j.neuroscience.2014.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dahl E, Manthey D, Chen Y, Schwarz HJ, Chang YS, Lalley PA, Nicholson BJ, Willecke K. Molecular cloning and functional expression of mouse connexin-30, a gap junction gene highly expressed in adult brain and skin. J Biol Chem. 1996;271:17903–17910. doi: 10.1074/jbc.271.30.17903. [DOI] [PubMed] [Google Scholar]
- 47.Choung YH, Moon SK, Park HJ. Functional study of GJB2 in hereditary hearing loss. Laryngoscope. 2002;112:1667–1671. doi: 10.1097/00005537-200209000-00026. [DOI] [PubMed] [Google Scholar]
- 48.Thonnissen E, Rabionet R, Arbones ML, Estivill X, Willecke K, Ott T. Human connexin26 (GJB2) deafness mutations affect the function of gap junction channels at different levels of protein expression. Hum Genet. 2002;111:190–197. doi: 10.1007/s00439-002-0750-2. [DOI] [PubMed] [Google Scholar]
- 49.Bruzzone R, Veronesi V, Gomes D, Bicego M, Duval N, Marlin S, Petit C, D’Andrea P, White TW. Loss-of-function and residual channel activity of connexin26 mutations associated with non-syndromic deafness. FEBS Lett. 2003;533:79–88. doi: 10.1016/s0014-5793(02)03755-9. [DOI] [PubMed] [Google Scholar]
- 50.Wang HL, Chang WT, Li AH, Yeh TH, Wu CY, Chen MS, Huang PC. Functional analysis of connexin-26 mutants associated with hereditary recessive deafness. J Neurochem. 2003;84:735–742. doi: 10.1046/j.1471-4159.2003.01555.x. [DOI] [PubMed] [Google Scholar]
- 51.Wilch E, Zhu M, Burkhart KB, Regier M, Elfenbein JL, Fisher RA, Friderici KH. Expression of GJB2 and GJB6 is reduced in a novel DFNB1 allele. Am J Hum Genet. 2006;79:174–179. doi: 10.1086/505333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilch E, Azaiez H, Fisher RA, Elfenbein J, Murgia A, Birkenhäger R, Bolz H, Da Silva-Costa SM, Del Castillo I, Haaf T, Hoefsloot L, Kremer H, Kubisch C, Le Marechal C, Pandya A, Sartorato EL, Schneider E, Van Camp G, Wuyts W, Smith RJ, Friderici KH. A novel DFNB1 deletion allele supports the existence of a distant cis-regulatory region that controls GJB2 and GJB6 expression. Clin Genet. 2010;78:267–274. doi: 10.1111/j.1399-0004.2010.01387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rodriguez-Paris J, Schrijver I. The digenic hypothesis unraveled: the GJB6 del(GJB6-D13S1830) mutation causes allele-specific loss of GJB2 expression in cis. Biochem Biophys Res Commun. 2009;389:354–359. doi: 10.1016/j.bbrc.2009.08.152. [DOI] [PubMed] [Google Scholar]
- 54.Zong L, Zhu Y, Liang R, Zhao HB. Gap junction mediated miRNA intercellular transfer and gene regulation: A novel mechanism for intercellular genetic communication. Sci Rep. 2016;6:19884. doi: 10.1038/srep19884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schütz M, Scimemi P, Majumder P, De Siati RD, Crispino G, Rodriguez L, Bortolozzi M, Santarelli R, Seydel A, Sonntag S, Ingham N, Steel KP, Willecke K, Mammano F. The human deafness-associated connexin 30 T5M mutation causes mild hearing loss and reduces biochemical coupling among cochlear non-sensory cells in knock-in mice. Hum Mol Genet. 2010;19:4759–4773. doi: 10.1093/hmg/ddq402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cohen-Salmon M, Ott T, Michel V, Hardelin JP, Perfettini I, Eybalin M, Wu T, Marcus DC, Wangemann P, Willecke K, Petit C. Targeted ablation of connexin26 in the inner ear epithelial gap junction network causes hearing impairment and cell death. Curr Biol. 2002;12:1106–1111. doi: 10.1016/s0960-9822(02)00904-1. [DOI] [PMC free article] [PubMed] [Google Scholar]