

Abstract
Positron emission tomography (PET) imaging study of fluorine-18 labeled biomolecules is an emerging and rapidly growing area for preclinical and clinical research. The present review focuses on recent advances in radiochemical methods for incorporating fluorine-18 into biomolecules via ‘direct’ or ‘indirect’ bioconjugation. Recently developed prosthetic groups and pre-targeting strategies, as well as representative examples in 18F-labeling of biomolecules in PET imaging research studies are highlighted.
Keywords: positron emission tomography, fluorine 18, biomolecules, bioconjugation, click chemistry
1. Introduction
Positron emission tomography (PET) is a sensitive imaging modality which enables the visualization and quantification of small molecules and biomolecules in a living subject or animal. PET imaging provides valuable in vivo functional information via targeted radiolabeled molecules with high sensitivity complementary to other imaging modalities where anatomic information is the primary focus.[1] Biomolecules such as nucleotides, peptides, aptamers and antibodies can display excellent binding specificities to targets related to normal and diseased states in neurology, oncology and cardiology.[2] The spatial and temporal information of biomolecules[3] in vivo are important for research areas such as clinical diagnostic studies,[4] monitoring biochemical processes,[5] and evaluation of the pharmacological properties of pharmaceutical candidates in support of drug discovery programs.[6] Fluorine-18 (18F, 97% β+, t1/2= 109.7 min) is the most commonly used radionuclide for PET due to its ideal chemical, physical and nuclear properties. Longstanding efforts in the development of fluorine-18 incorporation into biomolecules provide strong impetus to translate these 18F-labeled biomolecules to PET imaging studies in peripheral organs[7] or in the central nervous system in cases where the blood brain barrier is compromised.[8] Based on significant advances in the labeling methodologies for syntheses of 18F-labeled biomolecules, in this article we aim to highlight the recent breakthroughs and provide readers with a general overview of 18F-labeling strategies for biologics, to complement the established literature in this field.[9] Herein we consider fluorine-18 incorporation into biomolecules as broadly falling into two categories: 1) direct methods where fluorine-18 is incorporated directly to biomolecules of interest through C–18F, B–18F and Si–18F bond formation or via chelation with Al–18F complexes; and 2) indirect methods where the synthesis of prosthetic groups labeled with fluorine-18 is carried out before conjugation with biomolecules.
2. Direct Methods for 18F-labeling of Biomolecules
Direct methods for 18F-labeling feature direct and one-step incorporation of fluorine-18 into complex biomolecules in the ultimate or penultimate step of the radiosynthesis. The relatively short half-life of fluorine-18 imposes limitations on the time-frame to achieve labeling reactions as well as the methods that can be employed; therefore, it's highly desirable to minimize synthetic steps following the initial incorporation of fluorine-18 and time for the purification procedures and formulation of the final 18F-labeled biomolecules. The direct methods for fluorine-18 incorporation of biomolecules have advantages in this sense and have found usages in a wide range of applications.
2.1 18F-Radiolabeling Through C–18F Bond Formation
One commonly used method for introducing the fluorine-18 radionuclide into complex biomolecules is through nucleophilic substitution reactions between [18F]fluoride and biomolecule precursors bearing suitable pre-installed leaving groups. Nucleophilic aromatic [18F]fluorination reactions have been extensively studied and aromatic compound bearing a trimethylammonium (TMA) leaving group was first evaluated as a labeling precursor for the synthesis of small molecule radiotracers, including 1-[2-[(4-[18F]fluorophenyl)(phenyl)methoxy)ethyl]-4-(3-phenylpropyl)piperazine in 1989 by Haka et al.[10] Several electron-withdrawing groups such as trifluoromethyl, cyano, and fluorine have been introduced onto aromatic rings to facilitate nucleophilic aromatic substitution. Subsequently Becaud et al. reported a procedure where biomolecule bearing a trimethylammonium (TMA) leaving group is directly replaced by [18F]fluoride (Figure 1A).[11] These reactions were carried out under mild conditions, for example, 50 °C in DMSO, wherein azeotropically dried [18F]fluoride in combination with Kryptofix222 and K2CO3 were reacted with biomolecules bearing TMA group. By employing this method, a series of radiolabeled peptides such as 3-cyano-4-[18F]fluorobenzoyl-Val-Ala-Arg-Gly-NH2 (2), 3-cyano-4-[18F]fluorobenzoyl-Ava-Gln-Trp-Ala-Val-Gly-His-FA-Leu-NH2 (4) were obtained with up to 90% fluorine-18 incorporation from the corresponding TMA precursors 1 and 3, respectively. In addition, the authors achieved a 24% radiochemical conversion (RCC) for peptides bearing a free amino group in lysine which are otherwise difficult to label using conventional labeling conditions. Peptides containing histidine, tryptophan, and arginine amino acids were also labeled with fluorine-18 via this direct approach. As a commonly used leaving group for nucleophilic aromatic substitution, the nitro group is also found to be useful for [18F]fluoride displacement reactions to label biomolecules. Jacobsen et. al. radiolabeled monomeric and dimeric cyclic RGD peptides by employing precursors containing 4-nitro-3-trifluoromethyl substituted aromatic rings via this direct approach (Figure 1B).[12] The [18F]fluoride displacement reaction on nitro precursors 5 and 7 was conducted in the presence of K2CO3 and Kryptofix222 in DMSO. Both monomeric peptide 6 and dimeric peptide 8 were isolated in 9-19% (decay corrected) radiochemical yield (RCY) with high molar activity (79 GBq/μmol; 2.1 Ci/μmol).
Figure 1.

Direct18F-Labeling Through C–18F Bond Formation. Radiochemical conversion, RCC; radiochemical yield, RCY.
In addition to the 18F-aryl bond formation strategy, methods involving aliphatic C–F bond formation have also found their application in incorporating fluorine-18 into biomolecules. Roehn et. al. described a one-step labeling of peptides via ring-opening of activated aziridines by [18F]fluoride (Figure 2).[13] Effects of different activating groups on the aziridine ring were investigated in this study. 2,4,6-Triisopropylphenylsulfone, as shown in the transformation from 9 to 10, was identified as the optimal activating group to facilitate regioselective aziridine ring-opening by [18F]fluoride. A model tetrapeptide 11, octapeptide 12 and a protected thymidine derivative 13 bearing activated aziridine moiety were demonstrated in their work. Tetrapeptide 11 and octapeptide 12 were labeled in 16% and 7% RCCs, respectively, whereas under similar conditions thymidine derivative 13 was prepared in 87% RCC. In general, these approaches for 18F-labeling require the use of polar organic solvents. Thus it is crucial that the biomolecules are stable to survive polar medium under moderate to high temperature (50–135 °C).
Figure 2.
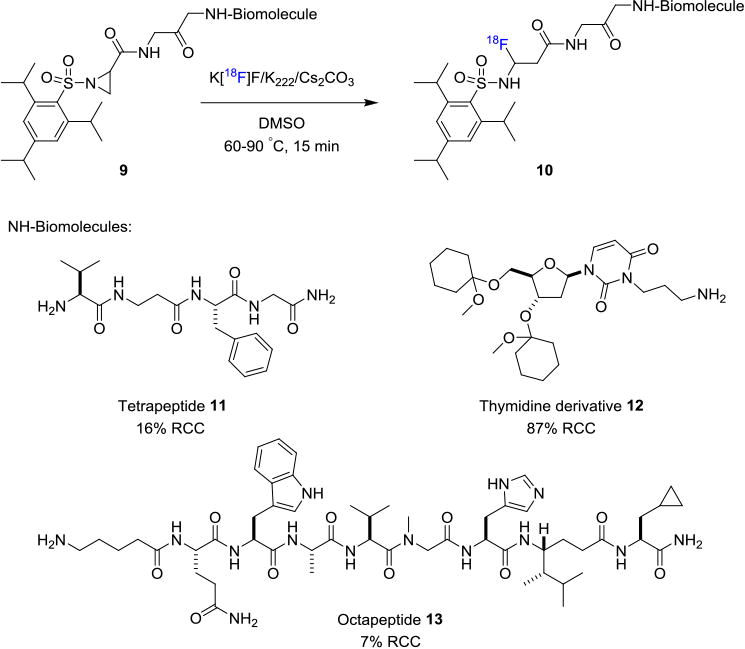
Direct18F-Labeling Through Aziridine Ring-Opening and C–18F Bond Formation.
Furthermore, biocatalytic transformation which takes advantage of the naturally occurring fluorinase enzyme in an SN2 type transhalogenation reaction has also been used for labeling peptides at ambient conditions.[14] A direct evolution of fluorinase for improved fluorination efficiency was also recently reported.[15] For instance, arginylglycylaspartic acid (RDG) peptide 14 bound to 5′-chloro-5′-deoxy-2-ethynyladenosine (ClDEA) through a tetraethylene glycol (TEG) linker could be labeled with fluorine-18 for late-stage modification (Figure 3). Specifically, an aqueous solution of [18F]fluoride, directly from the cyclotron target, was reacted with RDG-TEG-CIDEA in the presence of L-SeMet at room temperature for 30 min to afford the radiolabeled product 15 in 12% RCY (non-decay corrected).
Figure 3.
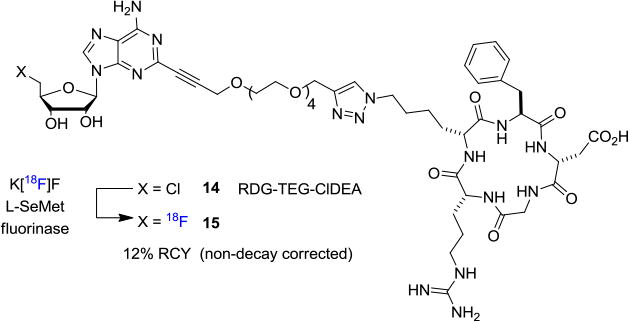
Biocatalytic [18F]Fluorination of Peptide Using Fluorinase.
2.2 18F-labeling Through B–18F Bond Formation
Due to the strong bond dissociation energy of B–F bond, boron based fluorination has been used to enable fluorine-18 incorporation into small molecules since the early 1960s.[16] However, it was not until 2005 that B-18F chemistry of biomolecules started to gain increased attention.[17] In 2005, Ting and Perrin et. al. reported the first example of 18F-labeling of biomolecules in aqueous conditions based on boron-fluorine chemistry.[18] In this method, pinacol phenylboronate diester was conjugated to biotin via an amide bond and the biotinylated arylboronic ester 16 was reacted with aqueous [18F]KHF2 (Figure 4A) and the resulting 18F-labeled aryltrifluorborate 17 precipitated from the reaction medium, thereby greatly facilitating product isolation. The authors also investigated the hydrolytic stability of arytrifluoroborates at pH=7.5 in aqueous conditions mimicking physiological conditions. It was found that there was no significant release of fluorine-18 from the labeled compounds and they showed considerable stability over 60 min. This pioneering work was further extended by the same group whereby the authors demonstrated that biotinylated arylboronic ester 18 can be labeled with moderately high molar activity despite adding 19F–KHF2 as a carrier to facilitate the formation of trifluoroborate 19 (Figure 4B).[19]
Figure 4.
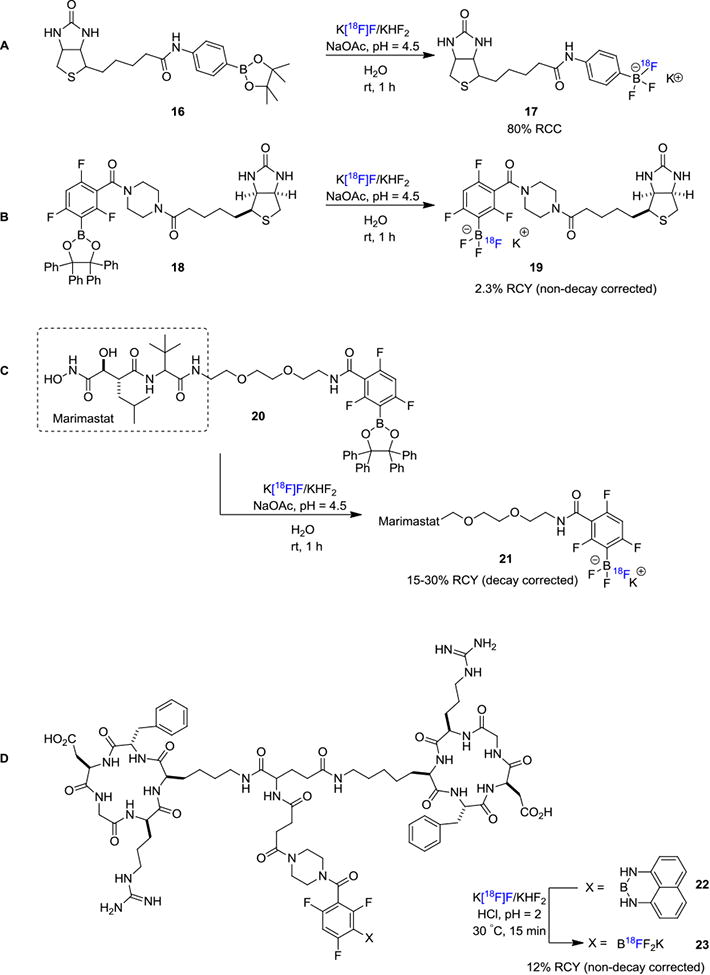
Direct 18F-Labeling Through B–18F Bond Formation.
Keller et. al. reported the 18F-labeling of Marimastat (Figure 4C), a noncovalent MMP inhibitor via the boronic ester approach.[20] They exploited an arylboronic ester conjugate 20 as a captor for aqueous [18F]fluoride for the formation of tracer 21 in a rapid one-step reaction at room temperature. Li and Perrin et. al. reported the usage of arylborimidine 22 as the precursor for 18F-labeling of dimeric-cylcoRGD (23; Figure 4D).[21] From a distinct mechanistic point of view, [18F]trifluorborate based labeling reagents could also be obtained through isotopic exchange with 19F-trifluorborates. These authors also conducted a kit-like 18F-labeling strategy for RGD peptide where RGD–ArBF3 (24) was reacted with fluorine-18 in acidic conditions (Figure 5A).[22] After HPLC purification, the RGD–[18F]ArBF3 (25) was isolated in 65% RCY (non-decay corrected) with high molar activity of 518 GBq/μmol (14 Ci/μmol).
Figure 5. Direct 18F-Labeling Through 19F–18F Isotope Exchange.
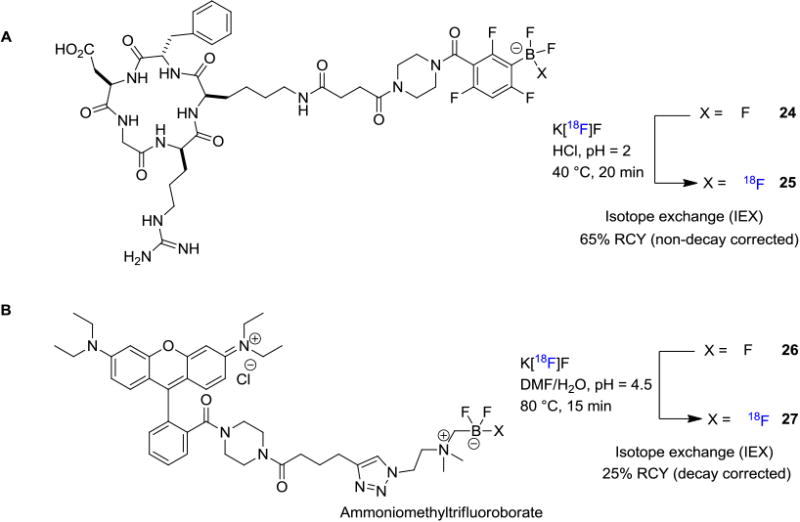
In line with the isotope exchange strategy, the Perrin laboratory further reported an advanced method by developing a novel zwitterionic organotrifluoroborate 26 which could be readily incorporated into clinically relevant peptides through bioconjugation.[23] In this method, [18F]fluoride drying is not required before reacting with the alkylammoniomethyltrifluoroborate (AMBF3) precursor, and an aqueous solution of [18F]fluoride can be used directly (Figure 5B). The radiolabeling of AMBF3-rhodamine afforded a dual-mode fluorescent and PET tracer 27 in 25 min with approximately 150 GBq/μmol (4 Ci/μmol) molar activity. Recently, Liu, Chen and co-workers also utilized the isotope exchange strategy for the synthesis of 18F-labeled boramino acids to serve as surrogates for amino acids to target amino acid transporters.[24] The synthesis involves a simple one-step procedure that does not require azeotropic drying of the [18F]fluoride or HPLC purification. The representative boramino acids were isolated in good radiochemical yields (>60% RCY, non-decay corrected) with high molar activities greater than 37 GBq/μmol (1 Ci/μmol). PET imaging studies with these radiotracers showed high amino acid transporters-mediated tumor uptake and rapid clearance from normal organs and tissues. The B-18F method for labeling biomolecules necessitates relatively acidic conditions (pH 2-4.5); therefore, stability of the biomolecules must be assessed.
2.3 18F-labeling Through Si–18F Bond Formation
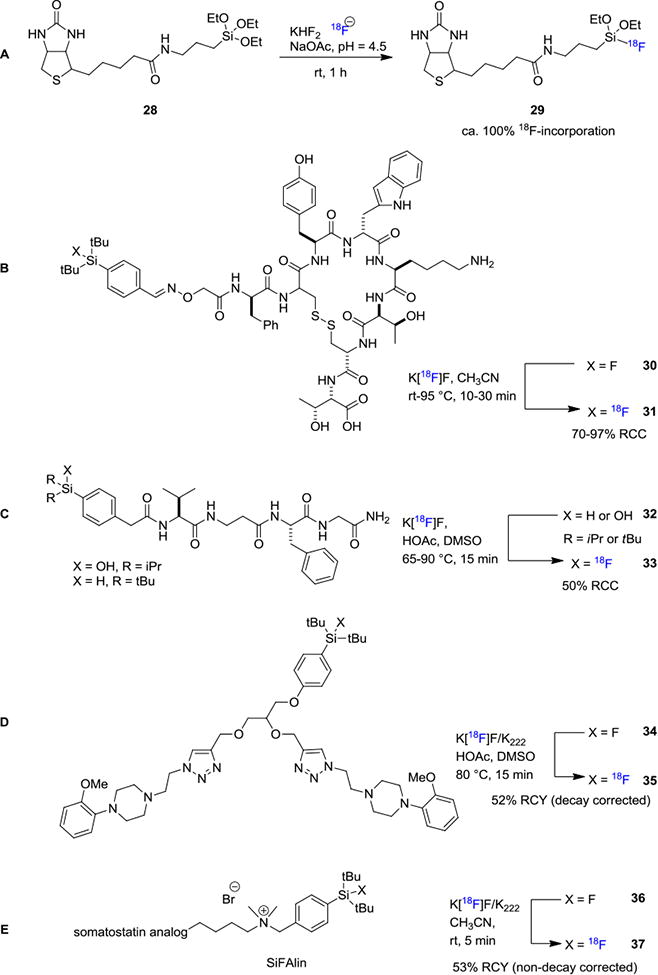
[18F]Fluorosilanes were first reported by Rosenthal et al. in 1985 where chlorotrimethylsilane was reacted with [18F]fluoride to generate corresponding [18F]fluorosilane.[25] However, subsequent in vivo evaluation revealed that [18F]fluorotrimethylsilane was prone to undergo fast hydrolysis accompanied by high bond uptake, making it unsuitable as a labeling synthon. There is an unmet need for the development of a 18F-labeled silicon-based synthon with considerable in vivo stability. Similar to radiolabeling strategies involving boron–fluorine bond formation, the application of silicon–fluorine bond formation in biomolecule radiolabeling was initially reported by Ting et al. where they used an alkyltriethoxysilane conjugated biotin 28 as the precursor for 18F-labeling of 29 (Figure 6A).[18] Fluorination efficiency was reported to approach 100% but the 18F-labeled tetrafluorosilicate was only moderately stable and slowly decomposed in aqueous solution. Concurrently, Schirrmacher et. al. reported the syntheses of substituted [18F]fluorosilanes using organochlorosilanes as labeling precursors and an alternative labeling approach employing di-tert-butylphenyl fluorosilane, an efficient silicon-based fluoride acceptor (SiFA compound) via 18F-[19]F isotope exchange (Figure 6B).[26] The di-tert-butylphenyl based 18F-compounds showed excellent in vitro stability in human serum over 60 min. An application of this method is the 18F-labeling of an aminooxy-derivatized Tyr[3]-octreotate 31 bearing triorganofluorosilane functionality. In this reaction, the authors treated SiFA based precursor 30 with [18F]fluoride in CH3CN and the fluorine-18 incorporation was determined to be 95%. It is interesting to note that this method is also compatible with aqueous conditions, and high 18F-incorporation (70–90%) could be achieved within 30 min at 95°C (Figure 6B). Mu et. al. developed a new type of silicon-based precursors for direct 18F-labeling of biomolecules by nucleophilic displacement of an alkoxy, hydroxy, or hydride leaving group (Figure 6C).[27] Tetrapepeptides 32 with pendant dialkylsilanol or dialkylsilane building blocks were treated with [18F]fluoride under acidic conditions to afford the corresponding tracer 33 in ca. 50% RCCs from either silanol or silane precursors. Radiolabeling of bombesin derivatives could be achieved using di-tert-butyl silyl group as the labeling precursor producing hydrolytically stable 18F-labeled radioligands with 13% RCY (decay corrected) and 62 GBq/μmol (1.6 Ci/μmol) molar activity.[28] Yasui et. al. reported a resin-supported di-tert-butylphenylsilyl chloride system which could enable 18F-labeling of unprotected peptides.[29] This method allows for efficient release of the radiolabeled product with high molar activity 67–92 GBq/μmol (1.8–2.5 Ci/μmol). Mishra et. al. reported the application of silicon based 18F-labeling of dimeric radioligands by utilizing an isotope exchange strategy. For example, [18F]BMPPSiF (35), which is used to image serotonin receptors, was prepared in 52% RCY (decay corrected) with 480 GBq/μmol (12.9 Ci/μmol) molar activity from 34 (Figure 6D).[30] The lipophilic feature of silicon functionalities may increase non-specific binding; therefore, structural modifications including PEGylation and/or zwitterion formation have been developed to reduce the lipophilicity of 18F-Si labeled compounds.[31] Recently, Niedermoser, Schirrmacher and Wangler et al. have developed a new precursor for isotope exchange to achieve 18F-labeling of somatostatin receptor analogs (Figure 6E).[32] In this work, the newly developed SiFAlin-derivatized somatostatin analog 37 was synthesized and radiolabeled with 53% RCY (non-decay corrected) and 44–63 GBq/μmol (1.3–1.7 Ci/μmol) molar activity from SiFAlin 36.
Figure 6.
Direct 18F-Labeling Through Si–18F Bond Formation.
2.4 18F-Radiolabeling Through Al–18F Complex Chelation
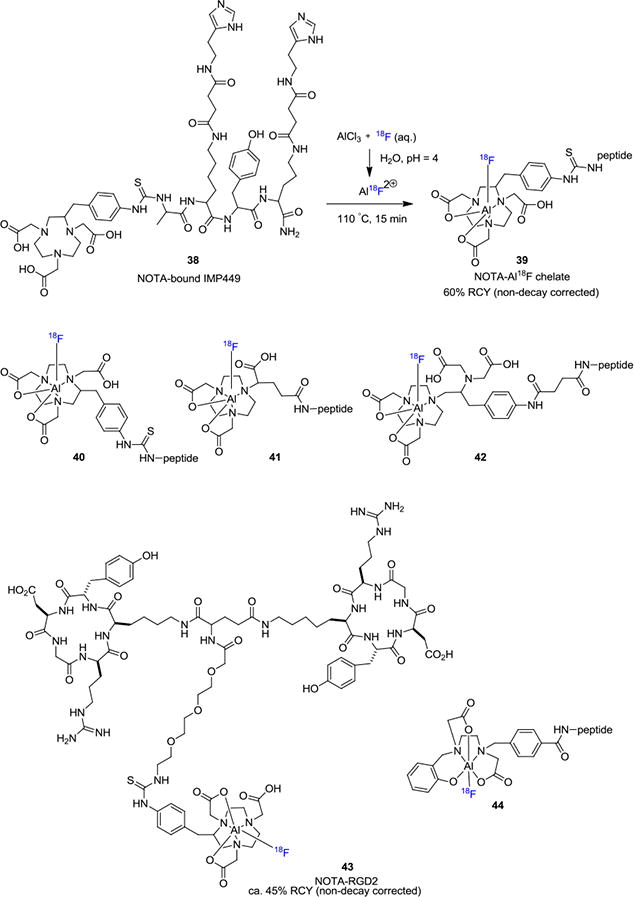
The recent development of 18F-labeling through chelation has greatly enriched the landscape of biomolecule modification for PET imaging. This strategy takes advantage of the fast kinetics of chelation chemistry and can potentially eliminate the need for HPLC purification. McBride et. al. reported a method where fluorine-18 forms an aluminum-18F (Al-18F) complex, which was then bound to a chelating group attached to a peptide, forming a stable Al-18F-chelate-peptide complex in a one-pot process (Figure 7).[33] In their initial report, the authors found that an Al-18F complex could bind tightly to a 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA) chelating ligand. The NOTA ligand contains three nitrogen atoms and three carboxylic acid functional groups which serve as a hexadentate ligand for the Al-18F complex. As a closely relevant labeling strategy, DOTA (1,4,7,10-tetraazacyclododecane-N,N',N'',N'''-tetraacetic acid) has also been widely used for gallium-68 labeling of biomolecules through similar chelation chemistry. However, there are several considerations when choosing Al-[18F]F or gallium-68 based peptide radiolabeling: 1) Fluorine-18 (t1/2= 109.7 min) has a longer half-life than gallium-68 (t1/2= 67.7 min) and can enable longer imaging times and shipment of the tracers to multiple sites; 2) Fluorine-18 has higher positron yield than gallium-68 (96.86% vs. 88.88%), which generally results in better imaging resolution;[34] 3) Gallium-68 is generator-produced while fluorine-18 is generated in a cyclotron, thereby facilitating widespread use of 68Ga. The peptide IMP449 38 bearing the NOTA ligand was treated with a pre-formed Al-18F aqueous solution and approximately 60% of Al-18F was incorporated into the peptide 39 and the molar activity was determined to be 48 GBq/μmol (1.3 Ci/μmol). Following this work, the same group continued to develop several structural variations 40-42 of the NOTA ligand.[35] Peptides were labeled using this method, for example, IMP467 was prepared in 87% RCY (non-decay corrected) with high molar activity up to 115 GBq/μmol (3.1 Ci/μmol) within 30 min without chromatographic purification.
Figure 7.
Direct 18F-Labeling Through Al–18F Chelation.
The Al–18F chelation approach has enriched the landscape for biomolecule radiolabeling and there are numerous applications in literature utilizing this strategy. For example, Lang et. al. reported the 18F-labeling of dimeric cyclic RGDyK peptide in 5–25% RCY (non-decay corrected), which was used for targeting the αβ3 integrin receptor.[36] Laverman et. al. disclosed their findings with the NOTA ligand in the 18F-labeling of NOTA-octreotide (IMP466; 43).[37] These authors investigated the effects of temperature, buffer solutions and solvent on the efficiency of the radiolabeling. The RCYs were ca. 45% (non-decay corrected) when aqueous buffers or CH3CN/buffer was used as the solvent. In sodium acetate buffer, the molar activity was in the range of 23–32 GBq/μmol (0.6–0.7 Ci/μmol). The use of chelation chemistry allows the radiotracer to be prepared without the need of HPLC purification, while maintaining reasonable molar activities. More recently, Chen and co-workers have used the NOTA chelation method to prepare a folate-NOTA-Al18F radiotracer for the diagnosis of folate-receptor (FR) expressing cancers.[38] In this work, the desired 18F-labeled tracer was prepared with a decay corrected RCY of 19% and 69 GBq/μmol (1.9 Ci/μmol) molar activity. Since most Al-18F methods which use the NOTA or NODA chelators require elevated reaction temperatures (100–120 °C), it imposes limitations for 18F-labeling of heat-sensitive biomolecules. In 2016, Cleeren et. al. developed a new polydentate ligand 44 for the complexation of Al-18F at 40 °C.[39] This new Al-18F ligand was used for radiolabeling of peptide Glu-NH-CO-NH-Lys(Ahx)L3 in 25% RCY (non-decay corrected) and 27 GBq/μmol (729 mCi/μmol) molar activity. Similar to the B-18F method, 18F-labeling of biomolecules via Al-18F complex takes place in relatively acidic medium and special care is necessary for acid sensitive molecules.
3. Indirect Methods for 18F-Radiolabeling of Biomolecules
As discussed in the previous section, direct nucleophilic fluorination is usually carried out under relatively harsh conditions for biomolecules, such as low pH conditions (pH ≤ 4-5) and high temperatures (≥100°C). These non-physiological conditions are not well tolerated by many biomolecules and are subject to increased likelihood of hydrolysis and/or degradation during radiolabeling. One attempt to circumvent this problem is to utilize indirect methods, which typically involve the utilization of 18F-labeled prosthetic groups that contain orthogonally reactive functionalities to conjugate with biomolecules. Thus, such radiolabeling takes place over several steps whereby the prosthetic group is attached to a biomolecule via conjugation reactions, including alkylation, amidation, acylation, imidation, oxime or hydrazone bond formation and glycosylation.[40] In addition to traditional prosthetic groups, several prosthetic groups have been developed that allow the employment of ‘click chemistry’ strategy. In the following sections, we aim to highlight recent examples for the 18F-labeling of sensitive biomolecules utilizing traditional or ‘click chemistry’ prosthetic groups. It is worth mentioning that the choice and site of introduction of prosthetic groups are critical factors as these variables can alter the pharmacological and physiological properties of the biomolecule. Therefore, pharmacological testing of non-radioactive analogs must be carried out to ensure that no significant changes to target specificity and selectivity are observed.
3.1 18F-Radiolabeling via Amino-selective Prosthetic Groups
Prosthetic groups are usually bifunctional small molecules that can be modified at one position with fluorine-18 and the second functional group allows for conjugation with the active site in an unlabeled biomolecule, such as an amino or a thiol group, under suitably mild conditions. The most common amino-selective fluorine-18 prosthetic group to date for such bioconjugation is N-succinimidyl-4-[18F]fluorobenzoate ([18F]SFB, 45; Figure 8).[41] [18F]SFB is an acylating agent that conjugates to biomolecules via a reactive amine group, such as the primary amino group in lysine. Its synthesis was first reported by Zalutsky and Vaidyanathan in 1992 via a multi-step process that resulted in 25% decay corrected RCY (synthesis time 100 min).41a Following this synthesis, these authors applied [18F]SFB to the radiolabeling of a monoclonal antibody F(ab')2 fragment and obtained the radiotracer in 40-60% RCY (decay corrected) in 20 min. Other syntheses of [18F]SFB have since been reported that have improved the RCY up to 55% (decay corrected).41a, 42 Carboxylic acids and related derivatives including 4-[18F]fluorobenzoic acid (46),[43] N-succinimidyl o-(di-tert-butyl[18F]fluorosilyl)-benzoate ([18F]SiFB, 47)[44] and 6-[18F]fluoronicotinic acid tetrafluorophenyl ester ([18F]F-Py-TFP, 48)[45] are also used as amine-reactive prosthetic groups. It is noteworthy that the [18F]F-Py-TFP prosthetic group can be prepared in one step from the trimethylammonium precursor[46] and it has been utilized in the fluorine-18 labeling of albumin and c(RGDfK).[47]
Figure 8. Examples of NH2-selective 18F-Prosthetic Groups.
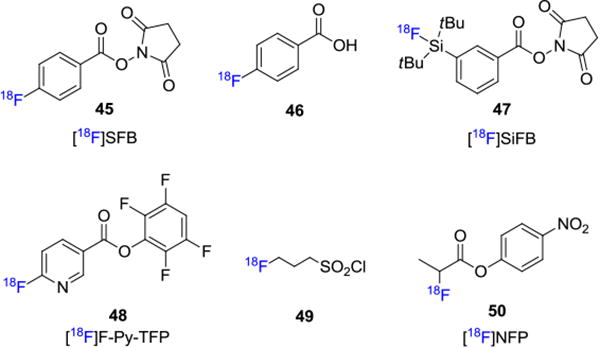
3-[18F]Fluoropropanesulfonyl chloride 49 was reported by Li et. al. for conjugation with amino groups in biomolecules via a sulfonamide bond.[48] An improved synthesis of this prosthetic group was reported and conjugation with model peptides showed that high RCYs could be achieved with this group under neutral conditions.[49] In addition, 4-nitrophenyl-2-[18F]fluoropropionate ([18F]NFP, 50) has also been employed as an efficient agent to acylate amines. This approach has been utilized to label an RGD (Arg-Gly-Asp) sequence to target αvβ3-integrins in cancer patients.[50] Other examples involving photoconjugation of 3-azido-5-nitrobenzyl-[18F]fluoride[51] and 4-([18F]fluoromethyl)phenyl isothiocyanate are also reported for the fluorine-18 labeling of oligonucleotides.[52]
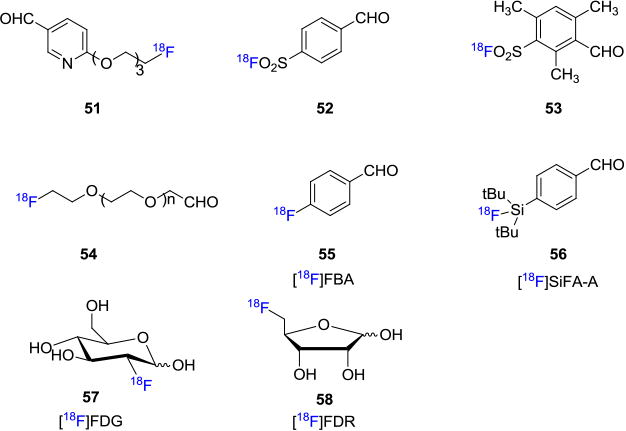
Bioconjugation reactions featuring oximes formation have also drawn extensive attention in the field and are widely used for labeling aminooxy-functionalized peptides with fluorine-18. A variety of 18F-containing aldehydes/hemi-acetal prosthetic groups such as 6-(2-[18F]fluorohexyloxy)nicotinaldehyde (51),[53] 4-formylbenzene-1-sulfonyl [18F]fluoride (52),[54] 3-formyl-2,4,6-trimethylbenzene-1-sulfonyl [18F]fluoride (53),[54] 2-(2-(2-[18F]fluoroethoxy)ethoxy)acetaldehyde (54),[55] 4-[18F]fluorobenzaldehyde ([18F]FBA, 55),55b p-(di-tert-butyl[18F]fluorosilyl)benzaldehyde ([18F]SiFA-A, 56),[56] 2-deoxy-2-[18F]fluoroglucose ([18F]FDG, 57)[57] and 5-deoxy-5-[18F]fluororibose([18F]FDR, 58)[58] have been developed for 18F-labeling of biomolecules (Figure 9).
Figure 9.
Fluorine-18 Containing Prosthetic Groups for Oxime Formation.
Glaser et. al. reported the radiosynthesis of [18F]fluciclatide by reacting [18F]FBA (55) with hydroxyl amine derived-RGD peptide in ammonium acetate buffer at 70°C. The desired [18F]fluciclatide (60) was obtained in 23% RCY (decay corrected, synthesis time 3h) with 76–170 GBq/μmol (2.0–4.6 Ci/μmol) molar activity from precursor 59 (Figure 10).55b Alternatively, Schirrmacher and co-workers disclosed a SiFA-based aldehyde prosthetic group [18F]SiFA-A (56) which takes the advantage of 19F/18F isotope exchange chemistry. In this work, the authors were able to label the N-terminal amino-oxy derived peptide Tyr[3]-octreotate 62 at room temperature with 50% RCY (decay corrected, synthesis time 40 min) from hydroxylamino peptide 61.[56] Furthermore, Inkster et. al. developed several sulfonyl based bifunctional fluorine-18 attached prosthetic groups for fluorine-18 labeling of biomolecules.[54]
Figure 10. Fluorine-18 labeling of Biomolecules involving Oxime Formation.
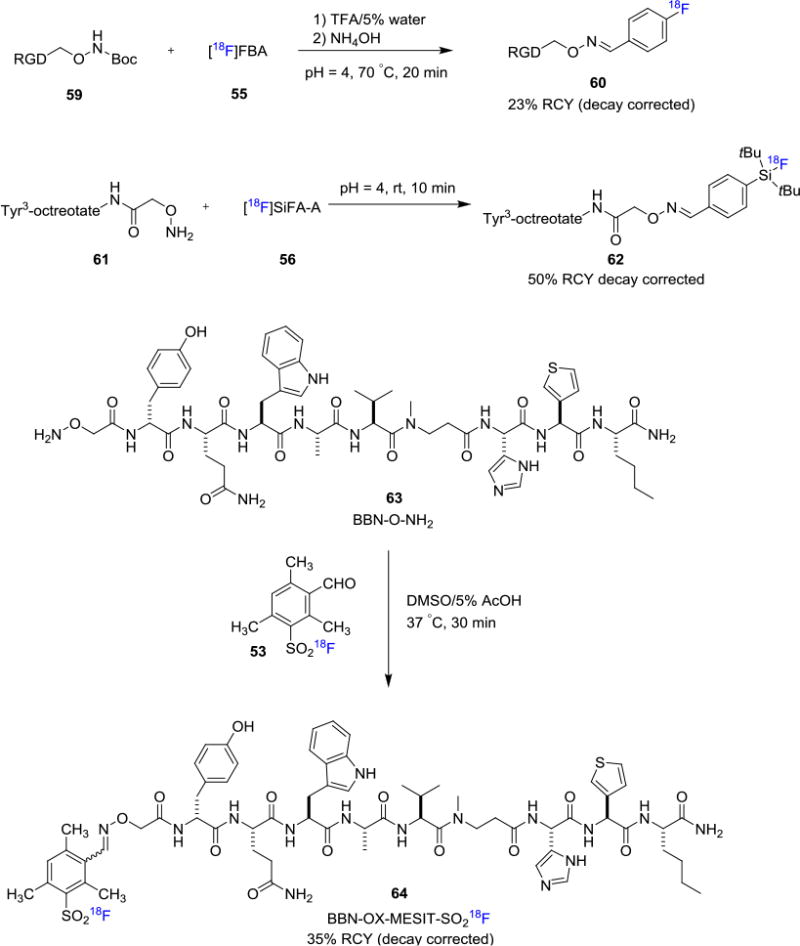
These bifunctional prosthetic groups include 4-formyl-, 3-formyl-, 4-maleimido- and 4-oxylalkkynl-arylsulfonyl [18F]fluorides and they can be conveniently prepared from the corresponding sulfonyl chlorides with Cs[18F]F/Cs2CO3. The authors have demonstrated the utility of sulfonyl [18F]fluoride 53 in the fluorine-18 labeling of a bombesin analogue 64 via oxime linkage with 35% RCY (decay corrected, synthesis time 105 min) from precursor 63. Subsequently, Matesic and co-workers investigated the suitability of various aryl sulfonyl [18F]fluorides under microfuidic fluorine-18 labeling conditions.[59] Moreover, Fiel et. al. reported the use of magnetic droplet microfluidics platform to prepare sulfonyl fluoride prosthetic group for fluorine-18 labeling of biomolecules.[60]
[18F]FDG, the most commonly used fluorine-18 radiotracer, could also be employed as a prosthetic group for biomolecule labeling through oxime formation (Figure 11). Hultsch et. al. reported the fluorine-18 labeling of aminooxyacetyl-conjugated cyclic RGD peptide c(RGDfK)(Aoa-Boc) 65 with [18F]FDG in DMSO/water at 120°C for 20 min to achieve the desired [18F]FDG-RGD (66) in 56-93% RCYs (decay corrected, synthesis time ca. 70 min).[57] As a closely related prosthetic group, [18F]FDR was also found to be efficient in bioconjugation reactions with peptides. For example, Li and co-workers reported the labeling of glutathione attached alkoxyamine 68 with [18F]FDR in more than 95% RCC from precursor 67.[58] In addition, hydrazine derived peptides could be conjugated with [18F]FBA to enable fluorine-18 labeling through hydrazone formation. This strategy has been demonstrated in the fluorine-18 labeling of RGD peptides.[61]
Figure 11.
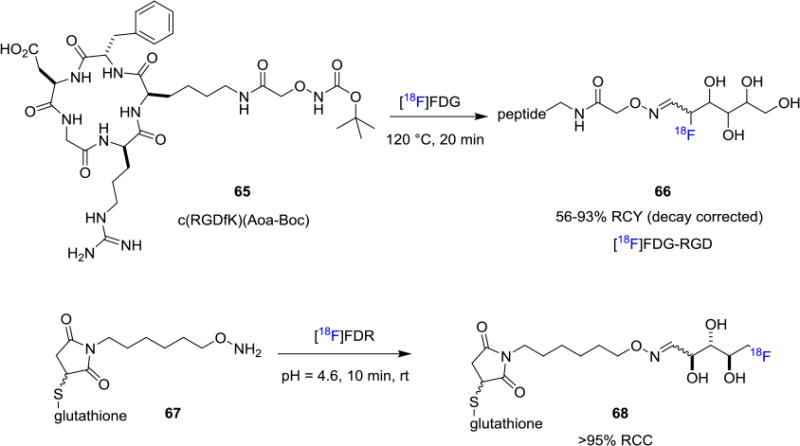
Fluorine-18 labeling of Biomolecules using [18F]Fluorosugars.
3.2 Fluorine-18 Radiolabeling via Thiol-selective Prosthetic Groups
While [18F]SFB could also be employed in a cysteine (cys) thiol acylation to afford fluorine-18 labeled peptides through conjugation with the 4-fluorobenzoyl moiety,[62] alternative strategies amino-targeting prosthetic groups. For example, N-(4-fluorobenzyl)-2-bromoacetamide ([18F]FBBA, 69),[63] and 2-bromo-N-[3-(2-[18F]fluoropyridin-3yloxy)propyl]acetamide ([18F]FPyBrA, 70)[64] were used in thiol-alkylation reactions of α-bromo ketone derivatives (Figure 12A). In addition, fluorine-18 labeled maleimide analogues have been developed for reaction with thiols.[65] Maleimides undergo chemoselective reactions with thiol groups via Michael addition, and are utilized to radiolabel peptides that contain a cysteine or methionine residue. Figure 12B depicts a recent collection of 18F-labeled maleimides from a variety of reliable labeling methodologies that demonstrated the diversity of these thiol-selective prosthetic groups. Maleimide based prosthetic groups such as 4-((4-[18F]fluorobenzylidene) aminooxybutyl) maleimide ([18F]FBAM, 71),[66] 1-[3-(2-[18F]fluoropyridin-3-yloxy)propyl]pyrrole-2,5-dione ([18F]FPyME, 72),[67] N-[2-(4-[18F]fluorobenzamido)ethyl]maleimide ([18F]FBEM, 73),[68] [18F]FDG-maleimidehexyloxime ([18F]FDG-MHO, 74),40a N-5-[18F]fluoropentylmaleimide ([18F]FPenM, 75),[69] N-(2-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)ethyl)-6-[18F]fluoronicotinamide ([18F]FNEM, 76),[70] [18F]SiFApMaleimide (77),[71] 4-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)benzenesulfonyl fluoride (78)[54] and [18F]fluorobutyl ethacrynic amide ([18F]FBuEA, 79)[72] have been used to couple with biomolecules via thiol-selective Michael additions (Figure 12B).
Figure 12.
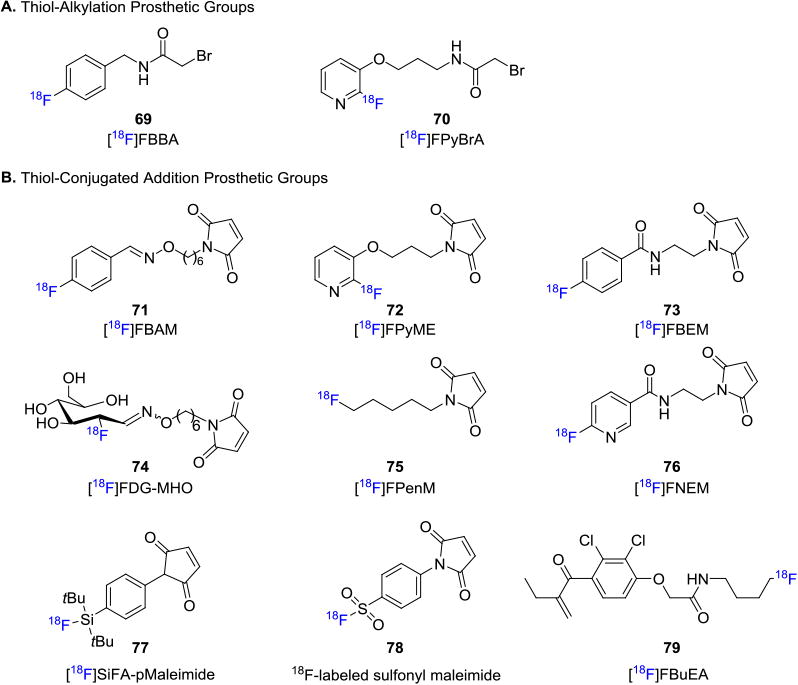
Thiol-selective Prosthetic Groups.
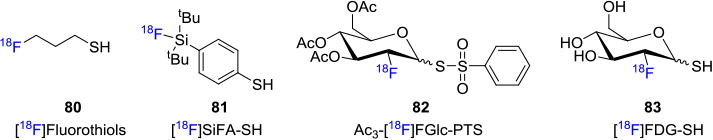
Other fluorine-18 labeling methods which involve sulfur-containing fluorine-18 prosthetic groups have also been developed for thiolation reactions, including disulfide formation. For example, [18F]fluorothiols 80,[73] [18F]SiFA-SH (81)[74] are developed as radiosynthons for peptide labeling. 3,4,5-Tri-O-acetyl-2-[18F]fluoro-2-deoxy-d-glucopyranosyl 1-phenylthiosulfonate (Ac3-[18F]FGlc-PTS, 82)[75] and thiolated-[18F]FDG 83[76] are used for fluorine-18 glycosylation of peptides (Figure 13).
Figure 13. Fluorine-18 Labeling Methods Involving Sulfur-containing Prosthetic Groups.
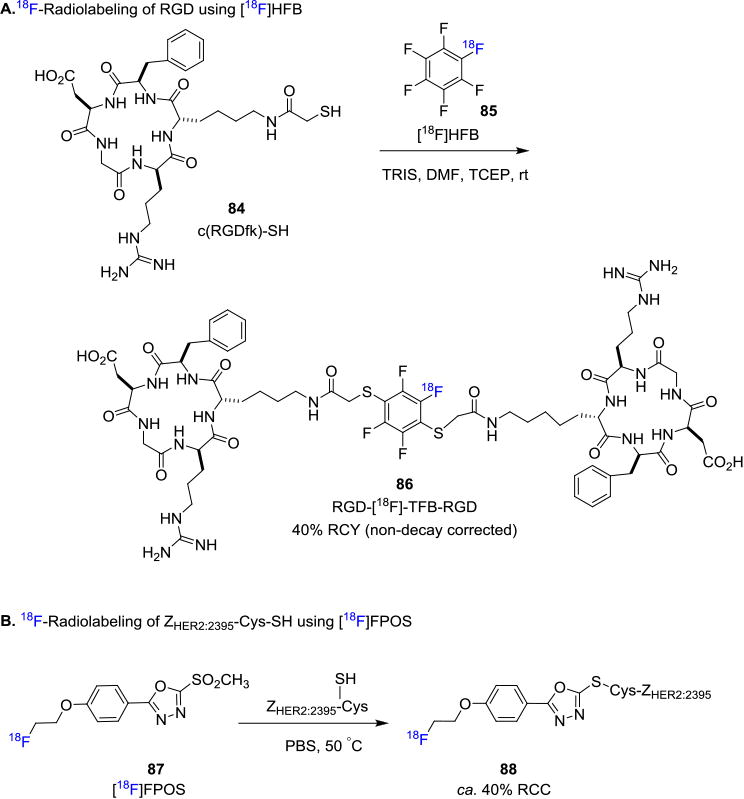
Recently Jacobsen et. al. disclosed a novel approach to produce dimeric [18F]RGD-tetrafluorobenzene peptide by employing hexafluorobenzene (HFB) as the prosthetic group.[77] Specifically [18F]HFB (85) was synthesized by fluorine exchange of hexafluorobenzene with K[18F]F/K222, and the resulting [18F]HFB was engaged in a thiol-substitution reaction in the presence of a thiolated c(RGDfK) peptide 84, TRIS base and tris(2-carboxyethyl)phosphine hydrochloride (TCEP) at room temperature to produce RGD-[18F]-TFB-RGD (86) in 40% RCY (non-decay corrected) with 0.5 GBq/μmol (15 mCi/μmol) molar activity (Figure 14A). There is also a recent progress to address the issue that the reversible nature of maleimide-thiol linkage could sometimes induce the loss of the radiolabeled prosthetic group in vivo. Chiotellis et. al. reported the synthesis of a new prosthetic group based on a fluorine-18 labeled methylsulfone derivative, [18F]FPOS (87).[78] This approach has been exploited to label an affibody, ZHER2:2395-Cys, in PBS buffer at 50°C to produce the fluorine-18 labeled ZHER2:2395-Cys 88 in ca. 40% RCC with 17 GBq/μmol (459 mCi/μmol) molar activity (Figure 14B). One unique feature of this strategy is that the resulting conjugates are more stable under physiological conditions in comparison to the corresponding analogues obtained from maleimide-based reactions. The tumor targeting properties of the resulting fluorine-18 labeled radiotracer was studied in vivo by PET imaging in CD1 nude mice bearing Her2-positive SCOV3 xenografts. The PET scans showed high accumulation of the radiotracer in the tumor with no [18F]fluoride leaching in the bones.
Figure 14. Recent Thiol-Selective Fluorine-18 Radiolabeling.
Fluorine-18 labeled N-terminal cysteine-bearing peptides were synthesized by the coupling of a labeled cyanobenzothiazole 89 with cysteine and cysteine-terminated peptides in good RCY under mild conditions (Figure 15).[79] The fluorine-18 containing cyanobenzothiazole 89 was synthesized from the corresponding tosylate precursor in 20% RCY (decay corrected). A fluorine-18 labeled dimeric cyclic RGD peptide (91, 8% RCY, non-decay corrected) from precursor 90 and fluorine-18 labeled RLuc8 protein (93, 12% RCY, decay corrected) from 92 were successfully synthesized via this methodology in a site-specific manner. Inkster et. al. recently reported the synthesis of a fluorine-18 labeled pyridyl prosthetic group with a cyanobenzothiazole moiety 94 to selectively label cysteine terminated peptides and proteins.79b This reagent was used to label a cyclic RGD peptide with a terminal cysteine group to obtain the purified radiotracer with a decay corrected RCY of 7% (synthesis time ca. 120 min).
Figure 15.
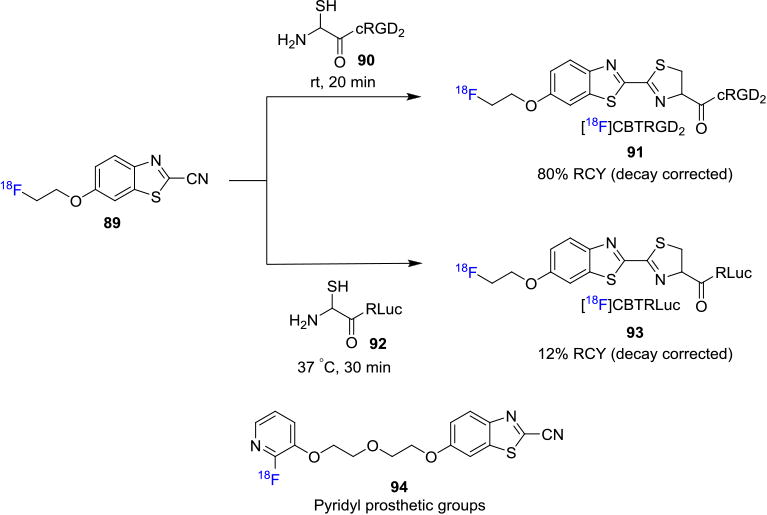
Cyanobenzothiazole Prosthetic Groups for Fluorine-18 labeling of Cysteine-containing Biomolecules.
3.3 Fluorine-18 Radiolabeling via Csp2-X Bond (X = C or N) Formation with Prosthetic Groups
Gao and co-workers recently reported a new prosthetic group, 4-[18F]fluorophenylboronic acid (95, Figure 16A), which was coupled via the Suzuki-Miyaura coupling reaction with peptides and proteins in the presence of a palladium catalyst.[80] It is important to note here that the biomolecule had to be modified to contain an aryl iodide at the desired radiolabeling site in order for the reaction to proceed. The decay corrected RCYs obtained by this procedure are moderate (∼30%) and the prosthetic group is also synthesized, albeit in low RCY over three steps.
Figure 16.
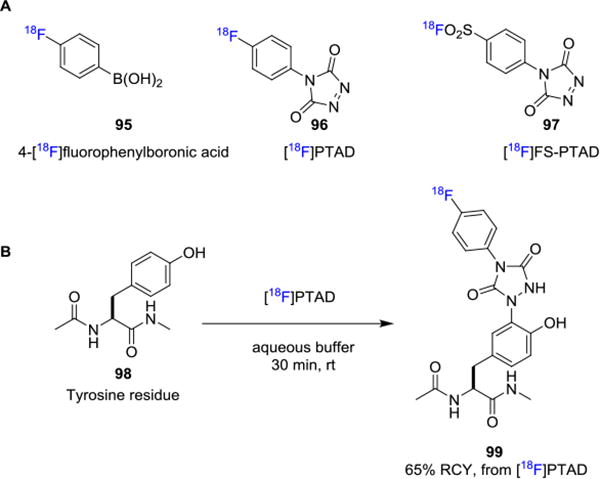
Fluorine-18 Labeling via X-Csp2Bond Formation with Prosthetic Groups.
In 2010, Ban et. al. reported their seminal work on the click-like reaction for tyrosine bioconjugation under aqueous conditions.[81] In this study, a reactive electrophile 4-phenyl-3H-1,2,4-triazole-3,5(4H)-dione (PTAD) was employed to participate in an ene-type reaction with the phenol moiety of tyrosine residues. This site-specific bioconjugation method has been applied to a wide range of biomolecules. For example, Hu and co-workers take advantage of the tyrosine-selective transformation to the synthesis of a well-defined conjugate anti-candidiasis vaccine glycosylated at tyrosines.[82] Ban et. al. developed the PEGlyated PTAD and used this novel agent for the selective PEGylation of chymotrypsinogen and antibody-drug candidate.[83] Subsequently, Nilo et. al. focused on tyrosine-directed conjugation of Group B Streptococcus capsular polysaccharides to streptococcal pilus protein.[84] In this context, Jessica et. al. and Al-Momani et. al. developed 4-(4-[18F]fluorophenyl)-1,2,4-triazole-3,5-dione ([18F]PTAD, 96)[85] and 4-(p-([18F]fluorosulfonyl)phenyl)-1,2,4-triazoline-3,5-dione ([18F]FS-PTAD, 97),[86] respectively, as prosthetic groups, which could be used for selective conjugation with tyrosine residues 98 to afford tyrosine-selective adduct 99 in biomolecules (Figure 16B).
3.4 Cu-Catalyzed Alkyne-Azide Cycloaddition
First described in 1893,[87] the Huisgen 1,3-dipolar cycloaddition reaction between an alkyne and an azide received new attention as the copper-catalyzed variant in 2002.[88] Independently reported by the groups of Sharpless[89] and Meldal,[90] the copper catalyzed alkyne-azide cycloaddition (CuAAC) yields the 1,4-regioisomer exclusively and can be performed at ambient temperatures. As a result of these favorable properties, the CuAAC has been extensively used in diverse fields of chemistry, including biochemistry and radiochemistry. The resulting 1,2,3-triazole linker is stable in vivo and is considered to be a bioisostere to peptide bonds, which has led to the utilization of the CuAAC reaction in the synthesis of several biomolecules.[91] In seeking to develop radiolabeled biomolecules for targeted imaging, several reports have been published detailing the design, synthesis and application of radiolabeled prosthetic groups for CuAAC with the radiolabel both in the alkyne moiety and the azide moiety.
The first application of CuAAC in fluorine-18 labeling of biomolecules was reported in 2006 by Marik and Sutcliffe who synthesized three fluorine-18 containing alkynes 100 of varying chain lengths that were coupled with azide-functionalized peptides (Figure 17).[92] The radiolabeled alkynes were synthesized from the tosylate precursors and purified by co-distillation with CH3CN. The corresponding radiotracers, including a folic acid derivative, were obtained in 10-20 mins after reaction at room temperature in good RCYs (30-97%, non-decay corrected) using solid phase extraction (SPE).[93] The volatility of the fluorine-18 labeled aliphatic alkyne moieties led to the discovery of fluorine-18 labeled PEGylated alkynes 101 and 102 (Figure 17).[94] In contrast with the aliphatic alkynes, these compounds are non-volatile, thermally stable and the corresponding bioconjugates show higher blood clearance. The radiolabeling of various biomolecules, including deoxythymidine (97%, non-decay corrected RCY) and RGD peptide dimer (55%) have been reported utilizing fluorine-18 labeled PEGylated alkynes in two-step, one-pot synthesis.[95]
Figure 17.
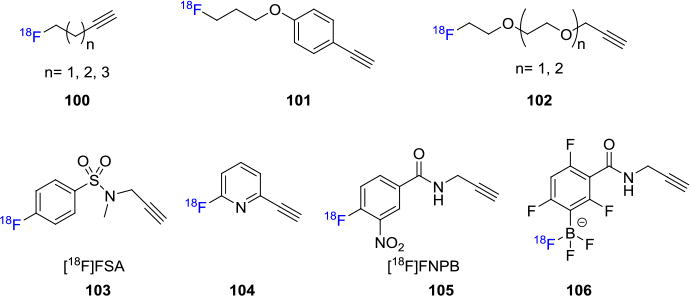
Fluorine-18 Labeled Alkyne Building Blocks.
[18F]Fluoroaryl alkynyl precursors have also been used to radiolabel biomolecules. A wide variety of reactions available to radiolabel aryl compounds in combination with improved metabolic stability of Csp2-F bond make these building blocks favorable for radiolabeling of biomolecules. The first [18F]fluoroaryl alkynyl precursor is a sulfonamide based alkyne 4-[18F]fluoro-N-(prop-2-ynyl)benzamide ([18F]FSA; 103) prepared from trimethylammonium precursor 107.[96] [18F]FSA was then applied in the radiolabeling of an azide functionalized neurotensin, human serum albumin (HSA, 108, Figure 18A),[97] phisphopeptide and an Loligonucleotide.[98] First synthesized in 2008, pyridine-based fluorine-18 alkyne precursors 104 have been utilized to successfully radiolabel a DNA analog (25% decay corrected RCY, synthesis time ca. 270 min) and peptides (5-20%).[99] Li et. al. reported the synthesis of a novel fluorine-18 labeled aromatic alkyne, 4-[18F]fluoro-3-nitro-N-2-prop-1-ynylbenzamide ([18F]FNPB, 105) as a stable prosthetic group for efficient click labeling of peptides.[100] [18F]FNPB was synthesized in 58% overall yield and in vitro studies showed no radiodefluorination in mouse plasma over 2h, suggesting stability in vivo as well. A novel approach was described by using an alkyne functionalized fluorine-18 labeled trifluoroborate building block 106 as the coupling partner to azido containing biomolecules such as bombesin and RGD peptide (Figure 18B).[101] As stated earlier, arylboronate 109 can capture [18F]fluoride easily in aqueous media and subsequent copper-mediated coupling with the azido moieties resulted in the synthesis of the radiotracers 110 in 30-40% non-decay corrected RCY with high molar activities up to 444 GBq/μmol (12 Ci/μmol).
Figure 18.
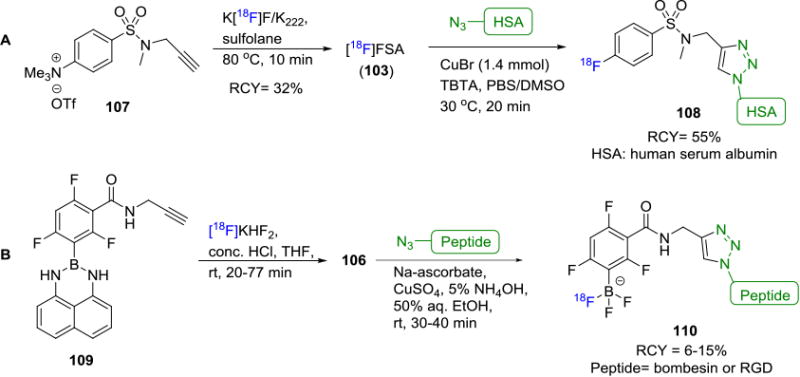
Fluorine-18 Labeling of Biomolecules via CuAAC Between Azide Containing Biomolecules and Labeled Alkynes.
Although a large array of azides have been utilized in the synthesis of non-radiolabeled biomolecules, only a few of them have been applied for PET imaging studies. Fluorine-18 labeled fluoroethylazide ([18F]FEA, 111) is the first aliphatic azide synthesized in good yields and in most cases is obtained by co-distillation with CH3CN (Figure 19A).[102] [18F]FEA has been used widely in the synthesis of fluorine-18 containing radiotracers such as fluorine-18 deoxyuridine (112),103 18F-MitoPhos_01 (113),104 18F-ICMT-11 (114),104-105 18F-RGDfK (115)[106 and 18]F-RGD-PEG analog (116)[107] (Figure 19B). It is also worth noting that 18F-labeling with [18F]FEA has been shown to result in poor yields in some cases. This may be attributed to the reduction of [18F]FEA to 18F-fluoroethylamine in the presence of copper wire in acidic conditions as reported by Glaser et. al.[108] Despite its high volatility, [18F]FEA is one of the most widely used building blocks for fluorine-18 labeling via CuAAC. Other representative aliphatic azides include [18F]fluorobutylazide (117, Figure 19C),[109] a pyrazine derivative [18F]AFP (118),[110] [18F]fluoro-PEG3-azide (119),[111] a glucose derivative [18F]FDG-β-Az (120)[112] and an [18F]SiFA derivative (121).[113]
Figure 19.
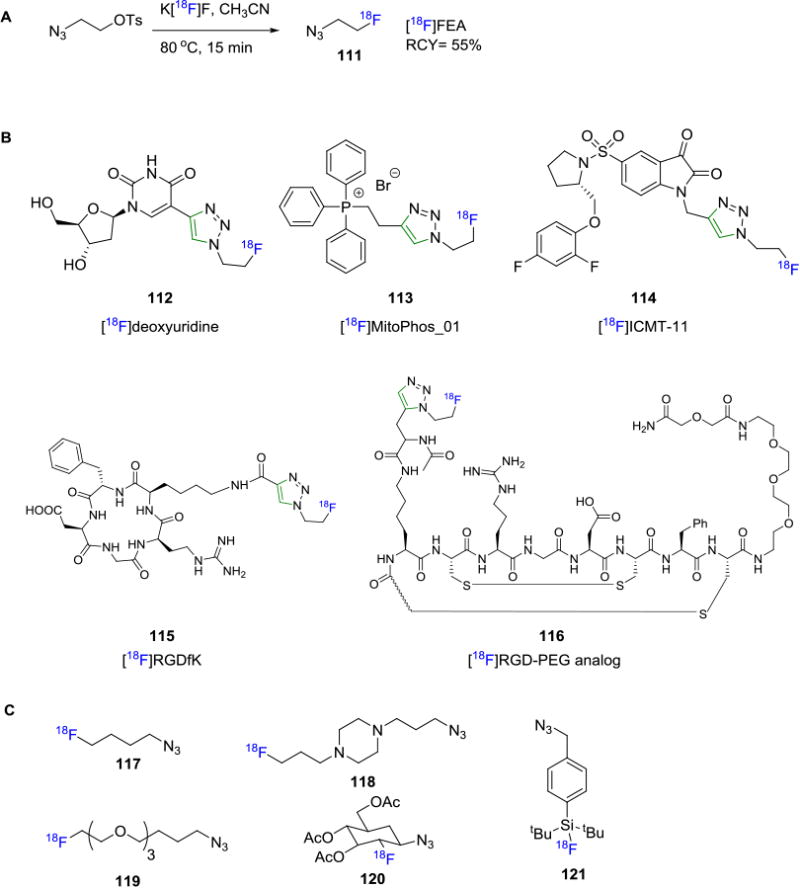
A) Synthesis of [18F]FEA;B) Examples of Radiotracers Synthesized with [18F]FEA Building Block; C) Some Representative Fluorine-18 labeled Azides in Click Chemistry.
Another fluorine-18 azido building block that has been studied is 4-[18F]fluorobenzylazide (123), the first azido aryl-18F click agent used in PET imaging studies. This agent was initially synthesized via a multi-step process in 75 min with 34% RCY (decay corrected),[114] or in a single-step reaction from the respective diaryliodonium salt using a microfluidic device over 94-188s in 35-45% RCCs.[115] 4-[18F]Fluorobenzylazide (123) has since been used for the fluorine-18 labeling of peptides and SiRNA derivatives.[114, 116] Our laboratory recently reported the one-step synthesis of 4-[18F]fluorobenzylazide (123) by reaction of [18F]fluoride with a novel spirocyclic iodonium(III) ylide precursor 122. The 4-[18F]fluorobenzyl azide (123) was isolated with high molar activities of 129.5-148 GBq/μmol (3.5-4 Ci/μmol) in ca. 30% RCY (non-decay corrected, synthesis time 45 min) and applied in the fluorine-18 labeling of single-stranded DNA (ssDNA) aptamers.117 In the same report, a novel ortho-stabilized fluorine-18 azido building block 125 was synthesized from ylide precursor 124 that overcomes the challenges posed by previously used fluorine-18 labeled azido compounds (Figure 20).118 The radiolabeling of ssDNA aptamers 126, including TsC (49% RCY based on 4-[18F]fluorobenzylazide) and sgc8 (48% RCY) were demonstrated, along with the SPAAC reaction of this prosthetic group with a phenylalanine derivative. A similar reaction with [18F]SFB yielded the labeled ssDNA aptamer TsC in only 1.5% RCY (based on [18F]SFB). PET imaging studies of the fluorine-18 containing sgc8-aptamer tracer was performed in mice bearing HCT116 tumors to map expression of the protein PTK-7. The images showed modest tumor uptake of 0.71 ± 0.07% ID/g at various time points, high tumor-to-muscle ratio of 3.54 ± 0.7 (1h) and 4.15 ± 0.26 (2h), and exhibited rapid renal clearance with minimal [18F]fluoride leaching.
Figure 20.
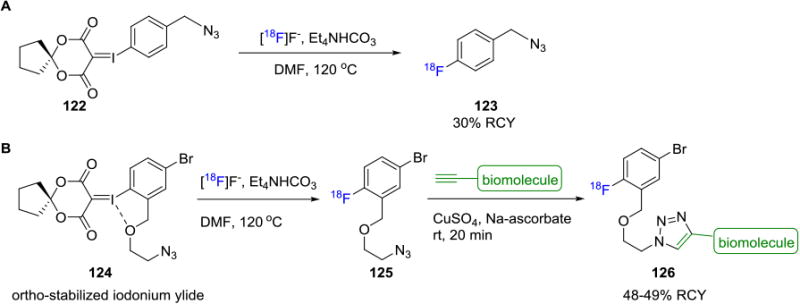
Fluorine-18 Radiolabeling of Biomolecules via Spirocyclic Iodonium Ylides.
Fluorine-18 incorporation to silicon under mild conditions has been discussed earlier, and this method was exploited in the synthesis of a silicon-18F based azide 128 from precursor 127 (Figure 21).[119] Subsequent fluorine-18 labeling of a model peptide was carried out at ambient temperature and the resulting radiotracer 129 was obtained in 75% RCY (decay corrected, based on the fluorine-18 azide). Unfortunately, the radiotracer was shown to undergo hydrolytic cleavage of the Si–18F bond (t1/2 < 10 min) in water (pH=7.5) and acidic buffers (pH <4) due to the lack of stability afforded by the isopropyl groups on the silicon.
Figure 21.
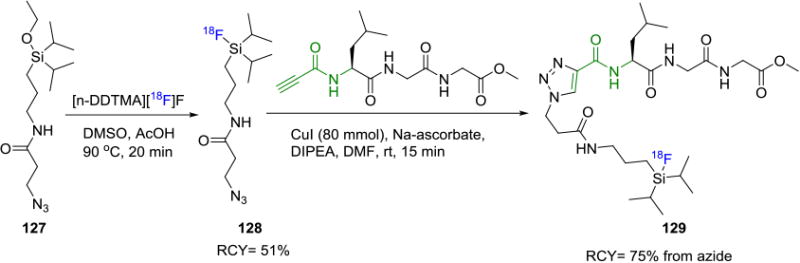
Silicon–18F Based Building Block for Biomolecule Labeling via CuAAC.
3.5 Strain-Promoted Azide Alkyne Click Reaction
First demonstrated by Blomquist and co-workers, the strain-promoted alkyne-azide cycloaddition (SPAAC) reaction is a variant of the traditional Huisgen cycloaddition.[120] It takes advantage of the ring-strain of a cyclooctyne ring, due to its bent triple bond, which then reacts in the absence of any catalyst with an azide to afford the 1,2,3-triazole. Like the copper-mediated Huisgen cycloaddition reaction, the SPAAC is also bioorthogonal and has been employed extensively in the synthesis of biomolecules.[121] It also overcomes one of the major drawbacks of the CuAAC coupling reaction, i.e. the use of copper, which can complicate separation protocols due to its ability to chelate to biomolecules. Two approaches can be considered for its utilization in the radiolabeling of biomolecules – one wherein the radiolabel is on the cyclooctyne moiety and the other where the radiolabel is on the azide. Bouvet et. al. synthesized a fluorine-18 labeled azadibenzocyclooctyne (ADIBO) agent 131 from the commonly used prosthetic group [18F]SFB (85% decay corrected RCY, based on [18F]SFB) and amine 130, which upon reaction with the corresponding azide terminated biomolecules yielded the radiotracers 132-134 in 69-98% RCCs (Figure 22).[122] The in vivo stability of the fluorine-18 labeled alkyne was studied in mice and showed that 60% of the tracer was intact after 1 hour of injection. Carpenter et. al. described the synthesis of another fluorine-18 labeled ADIBO agent with six carbon linker in high efficiency, wherein the two possible regioisomers of the SPAAC were observed.[123]
Figure 22. Fluorine-18 Labeled ADIBO Prosthetic Groups for SPAAC.
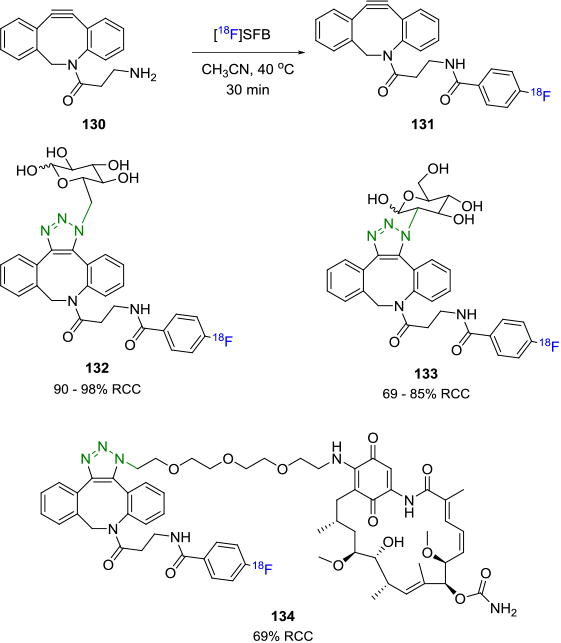
Arumugan et. al. reported the reaction between N-terminal-azido-modified peptides with a novel fluorine-18 ADIBO prosthetic group 135 to produce the tracer 136 via SPAAC in 21% overall yield in a fully automated manner.[122] The reaction was also carried out in ethanol as solvent, thereby allowing for the development of a “kit-like” peptide labeling approach (Figure 23A). Hausner and co-workers reported the synthesis of an fluorine-18 labeled radiotracer that selectively targets the integrin αvβ6-receptor.[123] An azide-terminated peptide containing a PEG7 spacer, A20FMDV2 was coupled with an [18F]fluorobenzyl-ADIBO building block 137 to yield the desired radiotracer 138 which was then subjected to in vivo imaging studies in mice (Figure 23B). Even though the synthesized peptides have been shown to possess good binding to the integrin αvβ6 receptors, in this case, the radiotracer showed no significant binding to the target. This dramatic change in the pharmacokinetics may be attributed to the increased lipophilicity of the radiotracer due to the presence of the dibenzocyclooctyltriazole moiety.[124]
Figure 23.
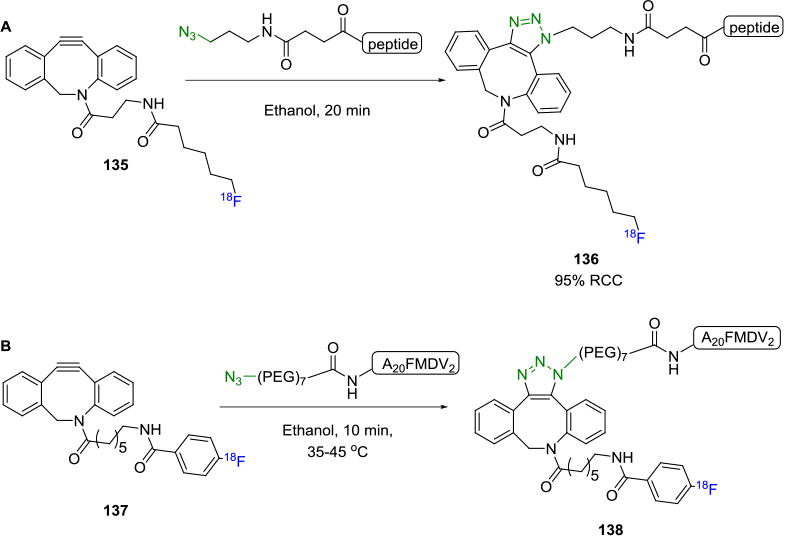
Fluorine-18 Labeling of Azide-Terminated Peptide Containing PEG Linker via SPAAC.
Other groups have also reported the radiolabeling of biomolecules using SPAAC wherein the fluorine-18 label is applied to the azide and the biomolecule has been modified to contain an alkyne. Campbell-Verduyn and co-workers reported the labeling of bombesin derivatives 139 containing an ADIBO group with various azide terminated fluorine-18 containing prosthetic groups, such as [18F]fluorobenzyl azide (123) and [18F]fluorobutylazide (117). The corresponding radiotracers 140 and 141 were obtained in 19-37% RCYs based on the fluorine-18 azide precursors (Figure 24).[109] [18F]Fluoroethylazide and [18F]fluoroPEGx azides have also been utilized as precursors to ADIBO and dibenzocyclooctyne containing biomolecules. Interestingly, Sachin et. al. concurrently devised a method to radiolabel and formulate fluorine-18 labeled peptides as directly injectable solutions at the end of synthesis.[125] This was achieved using a scavenger azide that was attached to a resin that successfully removed all traces of the remaining alkyne containing biomolecule without necessitating any HPLC purification.
Figure 24.

Alkyne-ADIBO Building Block for Fluorine-18 Labeling of Biomolecules.
3.6 Staudinger Ligation
The Staudinger reaction was first reported in 1919 as the reaction between an azide and a phosphine resulting in an iminophosphorane or aza-ylide that rapidly hydrolyzes to yield the primary amine and the corresponding phosphine oxide.[126] The reaction between the phosphine and azide takes place rapidly at room temperature with high yields of the product. Only one application of the non-traceless Staudinger ligation for fluorine-18 labeling of biomolecules has been reported to date. [18F]FDG derivatives for PET imaging of cancers were synthesized by the reaction between a fluorine-18 labeled phosphane and an azide containing sugar or amino acid.[127]In contrast, several fluorine-18 labeled biomolecules have been synthesized utilizing the traceless variant of the Staudinger ligation. The first among these was reported by Mamat et. al. in 2009.[128] In this work, fluorine-18 labeled phosphines were synthesized in three steps starting from para-substituted benzoyl chloride or benzoic acid, via Pd-catalyzed cross-coupling with diphenylphosphine, followed by reaction with 3-[18F]fluoroethyl tosylate. The labeled phosphine was treated with several azide terminated carbohydrates to yield the desired radiotracer in ca. 20% decay corrected RCY. Following this, Pretze et. al.[129] performed a one-step synthesis of the fluorine-18 labeled phosphine prosthetic group 143 based on aliphatic phosphine derivatives containing a 5-carbon long aliphatic chain terminated with a tosyl group as the precursor (142; Figure 25). The ligation between the 6-[18F]fluorohexanoyl phosphane moiety 143 and azides (144-146) provided the products 147-149 with decay corrected RCYs of ca. 12-31% at the end of synthesis.
Figure 25.
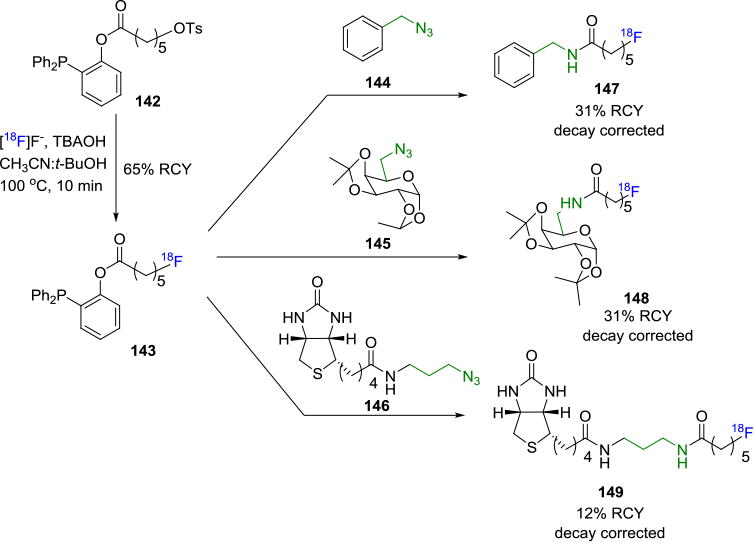
Traceless Staudinger Ligation to Radiolabel Biomolecules.
The common prosthetic group, [18F]FEA (112) was used as the azide component by Carroll et. al.[130] Several amino acid derivatives were functionalized with diarylphosphine based thioesters 150 and the corresponding radiotracers 151 and 152 were obtained in >95% RCC, based on conversion from [18F]FEA (Figure 26). While the Staudinger ligation does not require a metal catalyst and resulting radiotracers can be purified with ease, its major limitation is the relatively slow kinetics (2.0 × 10−3M−1 s−1)[131] of the reaction, which is often accelerated by high reaction temperature.
Figure 26.
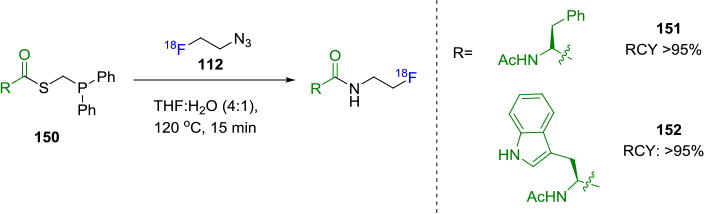
Synthesis of Fluorine-18 Labeled Amino Acid Derivatives from Diarylphosphine Based Thioesters.
3.7 Inverse Electron Demand Diels-Alder Click Reactions
The inverse electron demand Diels-Alder reaction (IEDDA) between 1,2,4,5-tetrazines with strained alkenes, such as trans-cyclooctene (TCO), cyclooctyne and norbornene has found several applications as a bioorthogonal reaction.[132] The extremely rapid reaction kinetics (k2= 2000 M−1 s−1 in 9:1 MeOH/water) for the reaction between tetrazine and TCO has enabled the development of several prosthetic groups with short-lived radioisotopes for radiolabeling. In addition to its fast rate, the IEDDA proceeds in the absence of a metal catalyst, and shows an increase in reaction rate in the presence of water in the reaction medium. This is particularly attractive in the synthesis of biomolecules. Mechanistically, the reaction proceeds via an IEDDA reaction followed by a retro Diels-Alder reaction to eliminate nitrogen gas as the only by-product. While norbornene has also been used as a dienophile in this reaction, higher reaction rates are achieved when TCO is used, and is likely attributed to the high ring-strain of TCO.[132] In 2010, Li, Fox and co-workers demonstrated the utility of this coupling reaction to radiolabel biomolecules.[133] Their initial attempt to radiolabel a tetrazine fragment with fluorine-18 failed due to the instability of the labeled tetrazine under the direct radiofluorination conditions. Thus, a fluorine-18 labeled TCO derivative 154 was synthesized from the nosylate precursor 153 (Figure 27A) and coupled with a sample tetrazine molecule in the initial reaction. The success of the trial reaction led to the radiolabeling of a cyclic RGD peptide 155 with a tetrazine-RGD conjugate and 154 under ambient conditions in less than 5 minutes yielding the radiotracer 156 in >95% RCC (Figure 27B).[134] In comparison, reactions of the RGD peptide in higher concentration with [18F]SFB yielded only 30% RCC by HPLC analysis. Small animal PET imaging studies with this radiotracer in a U87MG xenograft model showed a tumor uptake of 4.4% ID/g after 1h. Blocking experiments showed specific binding to integrin αvβ3. Using a similar strategy, other RGD peptides and the GLP agonist exendin were radiolabeled and used in subsequent PET imaging studies.
Figure 27.
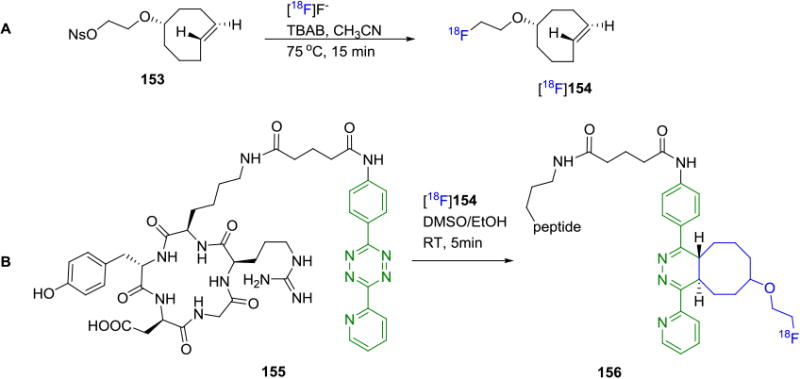
A) Synthesis of Fluorine-18 Labeled TCO, [18F]16. B) Fluorine-18 Labeling of Cyclic-RGD peptide via Tetrazine Ligation.
The efficiency of the tetrazine-click reaction was demonstrated by Weissleder and co-workers in the synthesis of fluorine-18 labeled PARP1 inhibitors by both direct fluorination of a tosyl precursor 159 and indirect labeling by tetrazine ligation of 157 with a suitably substituted tetrazine.[135] By modifying the structure of the PARP1 inhibitor with the addition of a tetrazine moiety in the side chain, they were able to successfully obtain a suitable radiotracer in 60% decay corrected RCY by coupling with the fluorine-18 labeled cyclooctene [18F]154. In contrast, direct 18F-labeling of a derivative of the PARP1 inhibitor 160 resulted in low (1%) radiochemical yield (Figure 28).
Figure 28.
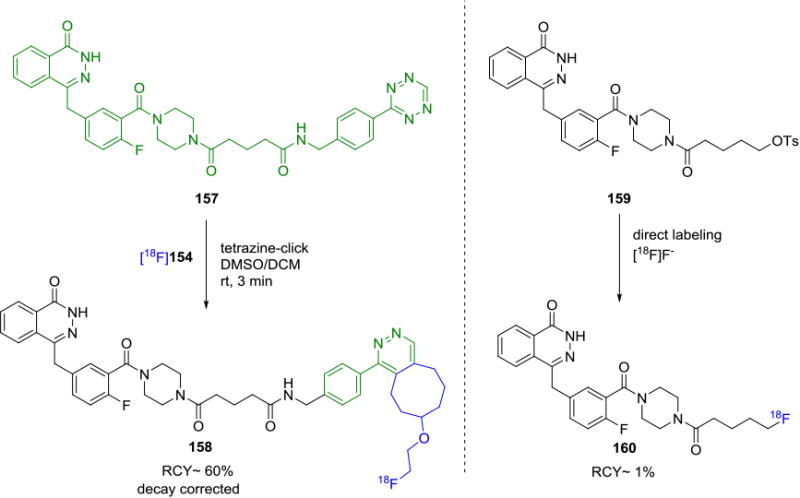
Comparison of Fluorine-18 Labeling by Tetrazine Ligation and Conventional Labeling Strategies.
In 2015, Li and Fox et. al. developed a fluorine-18 labeling system for PET probe construction based on a CF3-substituted diphenyl-s-tetrazine derivative of biomolecules.[136] In this work, c(RGDyK) was tagged by a fluorine-18 labeled TCO via reacting with the tetrazine moiety. The resulting tracer has shown improved metabolic stability. The [18F]TCO derivative was further improved by the same groups to radiolabel a cyclic RGD-conjugate 162 with a TCOPEG analog 161 to improve solubility and in vivo distribution of the resulting probe (Figure 29).[137] The tracer 163 was obtained in 99% RCY relative to the fluorine-18 TCO derivative 161 with high molar activity of 33.7± 7.4 GBq/μmol (0.91 ± 0.20 Ci/μmol). PET imaging of human U87MG tumor bearing mice showed good tumor uptake of 5.3 ± 0.2% ID/g at 1h post injection of the radiotracer and increased to 8.9 ± 0.5% ID/g at 2h.
Figure 29.
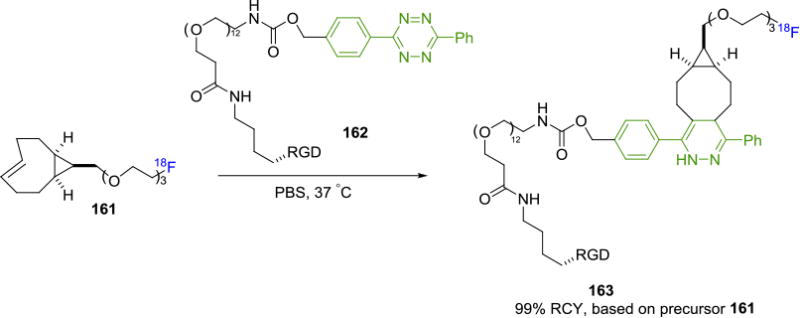
A Fluorine-18 labeled PEG-TCO Prosthetic Group with Improved Pharmacokinetics.
Likewise, Li et. al. developed a bicyclononyne-based prosthetic group for the fluorine-18 labeling of oligonucleotide (Figure 30).[138] In this approach, fluorine-18 bound tobicyclo[6.1.0]nonyne ([18F]BCN, 164) participated in an IEDDA reaction with a tetrazine-bound microRNA 165 to afford 18F-anti-microRAN 166 resulting in nearly quantitative RCC in 3 min and molar activity of 52–61 GBq/μmol (1.4–1.6 mCi/μmol). A tetrazine moiety was also labeled with 18F recently, for example, Pieve and co-workers reported a unique approach where they conjugated the ZHER3:8698 affibody molecule with the NOTA-Al18F ligand bearing a tetrazine functional group.139 The resulting fluorine-18 labeled bioconjugate could be used to target human epidermal growth factor receptor 3 (HER3). It is worth noting that structural rearrangement of adduct dihydropyridazine and aromatization, as identified by chromatography, could occur during IEDDA reactions.[137, 139-140]
Figure 30.
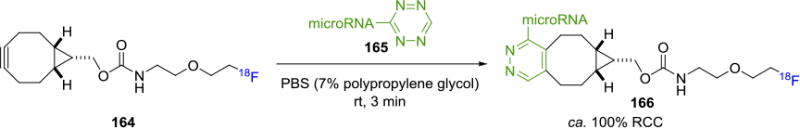
Bioconjugation through Tetrazine-Alkyne IEDDA Reactions.
Most recently, the tetrazine-click reaction has been investigated for in vivo bioorthogonal processes (in vivo pre-targeting), although no clinically-relevant radiotracer has yet been discovered for this application.9h Towards this end, several research groups are working on radiolabeling the tetrazine moiety with fluorine-18 since these molecules, when suitably modified, have been shown to maintain their reactivity in vivo. The first of these was reported by Devaraj et. al. where they showed that in vivo tetrazine/TCO [4+2]cycloaddition was enabled by polymer modified tetrazines (PMTs); fluorine-18 labeled PMT was synthesized and applied in LS174T tumor xenograft model.[141] Denk et. al. reported a novel synthesis of a low-molecular-weight fluorine-18 labeled tetrazine 168 (from tosylate 167) that was successfully coupled with a TCO moiety to yield the targeted radiotracer in decay corrected RCYs up to 18%.[142] The coupling reaction was also carried out in vivo and dynamic PET imaging showed no radiodefluorination. In 2015, a fluorine-18 labeled TCO was successfully coupled with a tetrazine terminated VHH (the variable domain of a camelid heavy-chain only antibody) in a rapid, and site-specific manner and used for the PET imaging of inflammation in xenogenic and syngenic tumor models with high specificity.[143] In another example, our colleagues reported the synthesis of fluorine-18 labeled antibodies by the cycloaddition of a tetrazine-[18F]FDG adduct 170 (from precursor 169) with TCO functionalized proteins in 25% RCY (non-decay corrected).[144] Subsequently, our laboratory reported the synthesis of another fluorine-18 labeled tetrazine moiety that was synthesized by the reaction between [18F]SFB and a suitable tetrazine precursor 171. The fluorine-18 labeled tetrazine moiety 172 was then coupled to a TCO group containing biomolecule to produce fluorine-18 labeled anti-Class-II and anti-CD11b dimers that were applied in preclinical PET imaging studies (Figure 31).[145]
Figure 31.
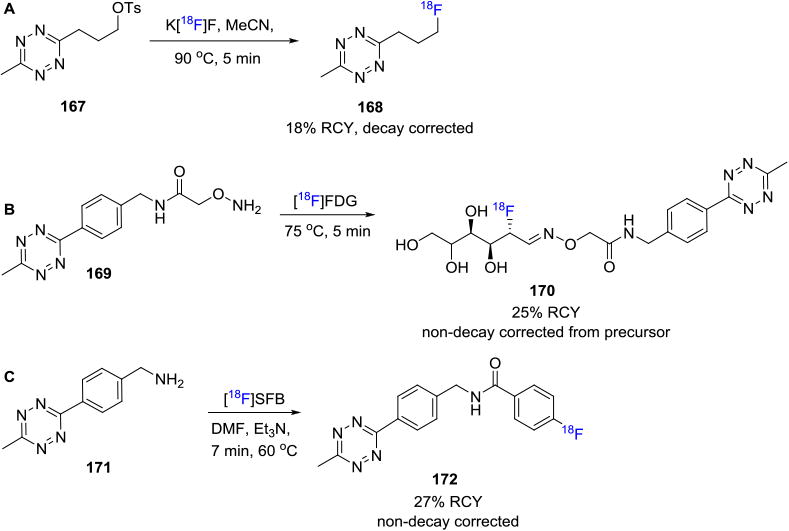
Synthesis of Fluorine-18 Labeled Tetrazine Building Blocks.
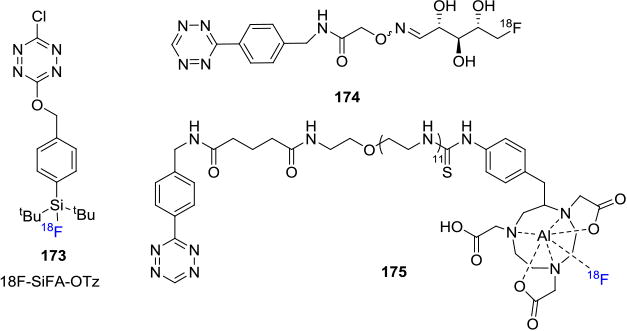
Several groups are also working on development of fluorine-18 labeled tetrazine derivatives for the synthesis of fluorine-18 labeled biomolecules with favorable pharmacokinetics. Schirrmacher and co-workers developed an [18F]SiFA-tetrazine derivative 173 that reacted with a model cyclooctene to yield the fluorine-18 labeled cycloaddition product in 78% isolated RCY.[113] Keinänen et. al. reported the synthesis of a fluorine-18 labeled tetrazine derived from fluorine-18 labeled ribose 174 that was shown to have favorable pharmacokinetics for in vivo biorthogonal IEDDA reactions with suitably substituted cyclooctenes. The labeled tetrazine showed excellent enzymatic stability in mouse serum and minimal radiodefluorination.[146] Another interesting report was summarized by Meyer et. al. wherein they reported the coupling reaction between [18F]NOTA labeled tetrazine 175 and TCO-bearing antibodies to yield the radiotracer in ca. 60% RCY (decay corrected, synthesis time 108 min) for the visualization of CA19.9-expressing BxPC3 pancreatic cancer xenografts.[147]
3.8 Nitrone or Nitrile Oxide-based [3+2] Cycloaddition
In addition to the extensively studied CuAAC and SPAAC, less known bioorthogonal methodologies have also been utilized to label biomolecules with fluorine-18. Zlatopolsky and co-workers in 2012 reported the radiofluorination of maleimides via [3+2] nitrone-alkene cycloaddition. Fluorine-18 labeled C-(4-fluorophenyl)-N-phenyl nitrone reacts with various maleimides to produce corresponding isoxazolidines.[148] Similarly, these authors also investigated reaction of suitable dipolarophiles with 4-[18F]fluorobenzonitrile oxide 177 (prepared from [18F]FBA and oxime 176) and N-hydroxy-4-[18F]fluorobenzimidoyl chloride to yield the corresponding labeled [3+2] cycloaddition product 178 (Figure 33).[149] RCCs >95% were obtained in some cases with sample small molecules. The methodology was extended to synthesize labeled model dipeptides 179-181 in ca.50% RCCs.
Figure 33.
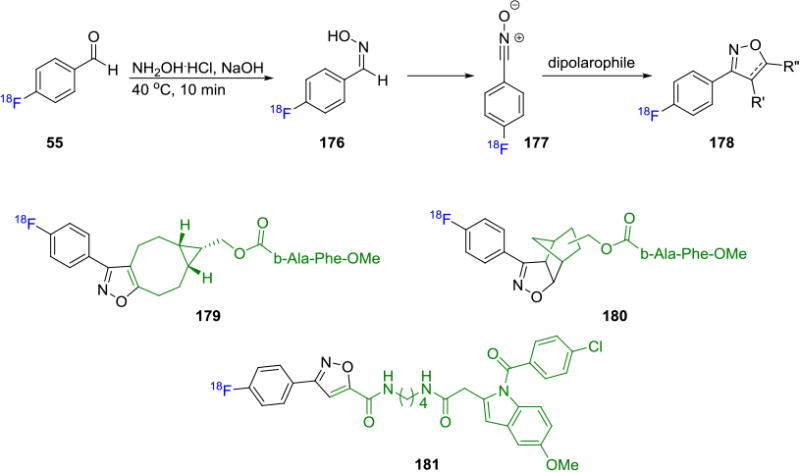
[3+2] Cycloaddition of Dipolarophiles with 4-[18F]Fluorobenzonitrile Oxide.
4. Summary and Outlook
This review provides an overview of the various methodologies developed to radiolabel biomolecules with fluorine-18. With respect to the radiolabeling of biomolecules, however, we still have a way to go as many of the current methods have stalled and very few tracers developed have advanced to clinical trials, in part, due to various limitations such as low molar activities, non-physiological reaction conditions, and large lipophilic residual footprints to name a few. Innovative chemistry such as the use of ‘click chemistry’-type reactions in vitro or in vivo provides an opportunity to solve many of these issues and have streamlined the synthesis of these biomolecules with the recent advances of site-specific chemical modifications of biomolecules.[150] We believe that future work should not only aim to develop new chemistry methodologies that allow for facile and selective radiolabeling of biomolecules by addressing the limitations of current methods, but also to apply existing chemistry and radiolabeled biomolecules to ultimately improve clinical outcomes and to accelerate clinical translation of radiolabeled biomolecules.
Figure 32.
Radiolabeling of TCO-terminated Biomolecules in vivo with Fluorine-18 Labeled Tetrazine Building Blocks.
Acknowledgments
We would like to thank the staff at the Radiochemistry Program, Massachusetts General Hospital and Harvard Medical School for their support. N.V. acknowledges the National Institute on Ageing of the NIH for funding (R01AG054473). S.H.L is a recipient of NIH career development award (DA038000) and Early Career Award in Chemistry of Drug Abuse and Addiction (ECHEM, DA043507) from the National Institute on Drug Abuse.
Biographies
Hema Krishnan studied synthetic chemistry and methodology development for organofluorination, at the University of Southern California (PhD, 2015). Her methodology was applied to synthesize fluorinated analogues of the anti-malarial drug, Artemisinin™. As a Research Fellow in the Radiochemistry Program at Massachusetts General Hospital and at Harvard Medical School, her current research interests include the development of medicinal chemistry libraries, new methodologies for radiolabeling and PET radiotracer development for neurodegenerative diseases.

Longle Ma studied chemistry (B.Sc.2010) at Nankai University, China. He performed his graduate studies in Chemistry at Rutgers University and obtained his Ph.D. in 2015. His doctoral research focused on the development of redox-neutral C–H bond functionalization methodologies for cyclic amines. As a Research Fellow in the Radiochemistry Program at Massachusetts General Hospital and at Harvard Medical School, he has been studying carbon-11 and fluorine-18 radiochemistry and neuroimaging using positron emission tomography since 2015.

Neil Vasdev obtained undergraduate degrees and a PhD in Radiochemistry from McMaster University, followed by a NSERC postdoctoral fellowship at Berkeley National Laboratories. In 2004, he started his independent research career at the University of Toronto and CAMH. In 2011, he was recruited as an Associate Professor at Harvard Medical School and is the Director of Radiochemistry at Massachusetts General Hospital. He has developed a radiopharmaceutical chemistry research program aimed at synthesizing new radiotracers and translating these for human imaging studies.

Steven H. Liang obtained his BSc at Tianjin University in 2003, followed by his PhD in Chemistry with Professor Marco Ciufolini in the University of British Columbia in 2010. Then he started as a NSERC fellow with Professor EJ Corey at Harvard University. In 2012, Dr. Liang accepted an Instructor position in the Department of Radiology, Harvard Medical School and Massachusetts General Hospital. In 2013, he was promoted to Assistant Professor. Steven's research ranges from PET imaging biomarker development to novel radiochemistry methods with 11C and 18F.

References
- 1.Ametamey SM, Honer M, Schubiger PA. Chem Rev. 2008;108:1501–1516. doi: 10.1021/cr0782426. [DOI] [PubMed] [Google Scholar]
- 2.a) Gambhir SS. J Nucl Med. 2006;47:557–558. [PubMed] [Google Scholar]; b) Jacobson O, Chen X. Pharmacol Rev. 2013;65:1214–1256. doi: 10.1124/pr.113.007625. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kuhnast B, de Bruin B, Hinnen F, Tavitian B, Dollé F. Bioconjugate Chem. 2004;15:617–627. doi: 10.1021/bc049979u. [DOI] [PubMed] [Google Scholar]; d) Shi H, He X, Wang K, Wu X, Ye X, Guo Q, Tan W, Qing Z, Yang X, Zhou B. Proc Natl Acad Sci. 2011;108:3900–3905. doi: 10.1073/pnas.1016197108. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Vaidyanathan G, Zalutsky MR. Nucl Med Biol. 1997;24:171–178. doi: 10.1016/s0969-8051(96)00211-9. [DOI] [PubMed] [Google Scholar]; f) Wester HJ, Hamacher K, Stöcklin G. Nucl Med Biol. 1996;23:365–372. doi: 10.1016/0969-8051(96)00017-0. [DOI] [PubMed] [Google Scholar]; g) Kühnast B, Dollé F, Terrazzino S, Rousseau B, Loc'h C, Vaufrey F, Hinnen F, Doignon I, Pillon F, David C. Bioconjugate Chem. 2000;11:627–636. doi: 10.1021/bc990183i. [DOI] [PubMed] [Google Scholar]
- 3.a) Hoffman JM, Gambhir SS. Radiology. 2007;244:39–47. doi: 10.1148/radiol.2441060773. [DOI] [PubMed] [Google Scholar]; b) Adam MJ, Wilbur DS. Chem Soc Rev. 2005;34:153–163. doi: 10.1039/b313872k. [DOI] [PubMed] [Google Scholar]; c) Mankoff DA, Eary JF, Link JM, Muzi M, Rajendran JG, Spence AM, Krohn KA. Clin Cancer Res. 2007;13:3460–3469. doi: 10.1158/1078-0432.CCR-07-0074. [DOI] [PubMed] [Google Scholar]
- 4.Fowler JS, Wolf AP. Acc Chem Res. 1997;30:181–188. [Google Scholar]
- 5.Phelps ME. Proc Natl Acad Sci. 2000;97:9226–9233. doi: 10.1073/pnas.97.16.9226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hargreaves RJ, Rabiner EA. Neurobiol Dis. 2014;61:32–38. doi: 10.1016/j.nbd.2013.08.017. [DOI] [PubMed] [Google Scholar]
- 7.a) McParland BJ, Miller MP, Spinks TJ, Kenny LM, Osman S, Khela MK, Aboagye E, Coombes RC, Hui AM, Cohen PS. J Nucl Med. 2008;49:1664–1667. doi: 10.2967/jnumed.108.052126. [DOI] [PubMed] [Google Scholar]; b) Gaertner F, Kessler H, Wester HJ, Schwaiger M, Beer A. Eur J Nucl Med Mol Imaging. 2012;39:126–138. doi: 10.1007/s00259-011-2028-1. [DOI] [PubMed] [Google Scholar]; c) Cai H, Conti PS. J Labelled Compd Radiopharm. 2013;56:264–279. doi: 10.1002/jlcr.2999. [DOI] [PubMed] [Google Scholar]
- 8.van de Haar HJ, Burgmans S, Hofman PA, Verhey FR, Jansen JF, Backes WH. Neurosci Biobehav Rev. 2015;49:71–81. doi: 10.1016/j.neubiorev.2014.11.022. [DOI] [PubMed] [Google Scholar]
- 9.a) Schirrmacher R, Wangler C, Schirrmacher E. Mini-Rev Org Chem. 2007;4:317–329. [Google Scholar]; b) Glaser M, Robins EG. J Labelled Compd Radiopharm. 2009;52:407–414. [Google Scholar]; c) Pretze M, Pietzsch D, Mamat C. Molecules. 2013;18:8618–8665. doi: 10.3390/molecules18078618. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Jacobson O, Kiesewetter DO, Chen X. Bioconjugate Chem. 2014;26:1–18. doi: 10.1021/bc500475e. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Richter S, Wuest F. Molecules. 2014;19:20536–20556. doi: 10.3390/molecules191220536. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Reiner T, Zeglis BM. J Labelled Comp Radiopharm. 2014;57:285–290. doi: 10.1002/jlcr.3149. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Jacobson O, Kiesewetter DO, Chen X. Bioconjugate Chem. 2015;26:1–18. doi: 10.1021/bc500475e. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Meyer JP, Adumeau P, Lewis JS, Zeglis BM. Bioconjugate Chem. 2016;27:2791–2807. doi: 10.1021/acs.bioconjchem.6b00561. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Chansaenpak K, Vabre B, Gabbai FP. Chem Soc Rev. 2016;45:954–971. doi: 10.1039/c5cs00687b. [DOI] [PubMed] [Google Scholar]
- 10.Haka MS, Kilbourn MR, Leonard Watkins G, Toorongian SA. J Labelled Compd Radiopharm. 1989;27:823–833. [Google Scholar]
- 11.Becaud J, Mu L, Karramkam Mn, Schubiger PA, Ametamey SM, Graham K, Stellfeld T, Lehmann L, Borkowski S, Berndorff D. Bioconjugate Chem. 2009;20:2254–2261. doi: 10.1021/bc900240z. [DOI] [PubMed] [Google Scholar]
- 12.Jacobson O, Zhu L, Ma Y, Weiss ID, Sun X, Niu G, Kiesewetter DO, Chen X. Bioconjugate Chem. 2011;22:422–428. doi: 10.1021/bc100437q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roehn U, Becaud J, Mu L, Srinivasan A, Stellfeld T, Fitzner A, Graham K, Dinkelborg L, Schubiger AP, Ametamey SM. J Fluorine Chem. 2009;130:902–912. [Google Scholar]
- 14.a) Winkler M, Domarkas J, Schweiger LF, O'Hagan D. Angew Chem Int Ed. 2008;47:10141–10143. doi: 10.1002/anie.200804040. [DOI] [PubMed] [Google Scholar]; b) Li XG, Domarkas J, O'Hagan D. Chem Commun. 2010;46:7819–7821. doi: 10.1039/c0cc02264k. [DOI] [PubMed] [Google Scholar]; c) Thompson S, Zhang Q, Onega M, McMahon S, Fleming I, Ashworth S, Naismith JH, Passchier J, O'Hagan D. Angewandte Chemie International Edition. 2014;53:8913–8918. doi: 10.1002/anie.201403345. [DOI] [PubMed] [Google Scholar]
- 15.Sun H, Yeo WL, Lim YH, Chew X, Smith DJ, Xue B, Chan KP, Robinson RC, Robins EG, Zhao H, Ang EL. Angew Chem Int Ed. 2016;55:14277–14280. doi: 10.1002/anie.201606722. [DOI] [PubMed] [Google Scholar]
- 16.a) Askenasy H, Anbar M, Laor Y, Lewitus Z, Kosary I, Guttmann S. Am J Roentgenol Radium Ther Nucl Med. 1962;88:350–354. [PubMed] [Google Scholar]; b) Entzian W, Aronow S, Soloway A, Sweet W. J Nucl Med. 1964;5:542–550. [PubMed] [Google Scholar]
- 17.Perrin DM. Acc Chem Res. 2016;49:1333–1343. doi: 10.1021/acs.accounts.5b00398. [DOI] [PubMed] [Google Scholar]
- 18.Ting R, Adam MJ, Ruth TJ, Perrin DM. J Am Chem Soc. 2005;127:13094–13095. doi: 10.1021/ja053293a. [DOI] [PubMed] [Google Scholar]
- 19.Ting R, Harwig C, auf dem Keller U, McCormick S, Austin P, Overall CM, Adam MJ, Ruth TJ, Perrin DM. J Am Chem Soc. 2008;130:12045–12055. doi: 10.1021/ja802734t. [DOI] [PubMed] [Google Scholar]
- 20.auf dem Keller U, Bellac CL, Li Y, Lou Y, Lange PF, Ting R, Harwig C, Kappelhoff R, Dedhar S, Adam MJ. Cancer Res. 2010;70:7562–7569. doi: 10.1158/0008-5472.CAN-10-1584. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Liu Z, Lozada J, Wong MQ, Lin KS, Yapp D, Perrin DM. Nucl Med Biol. 2013;40:959–966. doi: 10.1016/j.nucmedbio.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 22.Liu Z, Li Y, Lozada J, Wong MQ, Greene J, Lin KS, Yapp D, Perrin DM. Nucl Med Biol. 2013;40:841–849. doi: 10.1016/j.nucmedbio.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 23.a) Liu Z, Pourghiasian M, Radtke MA, Lau J, Pan J, Dias GM, Yapp D, Lin KS, Bénard F, Perrin DM. Angew Chem Int Ed. 2014;53:11876–11880. doi: 10.1002/anie.201406258. [DOI] [PubMed] [Google Scholar]; b) Liu Z, Lin KS, Benard F, Pourghiasian M, Kiesewetter DO, Perrin DM, Chen X. Nat Protoc. 2015;10:1423–1432. doi: 10.1038/nprot.2015.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Z, Chen H, Chen K, Shao Y, Kiesewetter DO, Niu G, Chen X. Sci Adv. 2015;1:e1500694. doi: 10.1126/sciadv.1500694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosenthal M, Bosch A, Nickles R, Gatley S. Appl Radiat Isot. 1985;36:318–319. doi: 10.1016/0020-708x(85)90094-8. [DOI] [PubMed] [Google Scholar]
- 26.Schirrmacher R, Bradtmöller G, Schirrmacher E, Thews O, Tillmanns J, Siessmeier T, Buchholz HG, Bartenstein P, Wängler B, Niemeyer CM. Angew Chem Int Ed. 2006;45:6047–6050. doi: 10.1002/anie.200600795. [DOI] [PubMed] [Google Scholar]
- 27.Mu L, Höhne A, Schubiger P, Ametamey SM, Graham K, Cyr JE, Dinkelborg L, Stellfeld T, Srinivasan A, Voigtmann U. Angew Chem Int Ed. 2008;47:4922–4925. doi: 10.1002/anie.200705854. [DOI] [PubMed] [Google Scholar]
- 28.Höhne A, Mu L, Honer M, Schudiger PA, Ametamey SM, Graham K, Stellfeld T, Borkowski S, Berndorff D, Klar U. Bioconjugate Chem. 2008;19:1871–1879. doi: 10.1021/bc800157h. [DOI] [PubMed] [Google Scholar]
- 29.Yasui N, Kim SH, Carroll V, Zhou D, Dence C, Katzenellenbogen J. J Nucl Med. 2014;55:1167–1167. [Google Scholar]
- 30.Hazari PP, Schulz J, Vimont D, Chadha N, Allard M, Szlosek‐Pinaud M, Fouquet E, Mishra AK. ChemMedChem. 2014;9:337–349. doi: 10.1002/cmdc.201300458. [DOI] [PubMed] [Google Scholar]
- 31.Zhu J, Chin J, Wängler C, Wängler B, Lennox RB, Schirrmacher R. Bioconjugate Chem. 2014;25:1143–1150. doi: 10.1021/bc5001593. [DOI] [PubMed] [Google Scholar]
- 32.Niedermoser S, Chin J, Wängler C, Kostikov A, Bernard-Gauthier V, Vogler N, Soucy JP, McEwan AJ, Schirrmacher R, Wängler B. J Nucl Med. 2015;56:1100–1105. doi: 10.2967/jnumed.114.149583. [DOI] [PubMed] [Google Scholar]
- 33.McBride WJ, Sharkey RM, Karacay H, D'Souza CA, Rossi EA, Laverman P, Chang CH, Boerman OC, Goldenberg DM. J Nucl Med. 2009;50:991–998. doi: 10.2967/jnumed.108.060418. [DOI] [PubMed] [Google Scholar]
- 34.Sanchez-Crespo A. Appl Radiat Isot. 2013;76:55–62. doi: 10.1016/j.apradiso.2012.06.034. [DOI] [PubMed] [Google Scholar]
- 35.McBride WJ, D'Souza CA, Sharkey RM, Karacay H, Rossi EA, Chang CH, Goldenberg DM. Bioconjugate Chem. 2010;21:1331–1340. doi: 10.1021/bc100137x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lang L, Li W, Guo N, Ma Y, Zhu L, Kiesewetter DO, Shen B, Niu G, Chen X. Bioconjugate Chem. 2011;22:2415–2422. doi: 10.1021/bc200197h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laverman P, D'Souza CA, Eek A, McBride WJ, Sharkey RM, Oyen WJ, Goldenberg DM, Boerman OC. Tumor Biol. 2012;33:427–434. doi: 10.1007/s13277-011-0250-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Q, Meng X, McQuade P, Rubins D, Lin SA, Zeng Z, Haley H, Miller P, González Trotter D, Low PS. Mol Pharm. 2016;13:1520–1527. doi: 10.1021/acs.molpharmaceut.5b00989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cleeren F, Lecina J, Billaud EM, Ahamed M, Verbruggen A, Bormans GM. Bioconjugate Chem. 2016;27:790–798. doi: 10.1021/acs.bioconjchem.6b00012. [DOI] [PubMed] [Google Scholar]
- 40.a) Wuest F, Berndt M, Bergmann R, van den Hoff J, Pietzsch J. Bioconjugate Chem. 2008;19:1202–1210. doi: 10.1021/bc8000112. [DOI] [PubMed] [Google Scholar]; b) Maschauer S, Prante O. BioMed Res Int. 2014;2014:16. doi: 10.1155/2014/214748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.a) Vaidyanathan G, Zalutsky MR. Int J Rad Appl Instrum B. 1992;19:275–281. doi: 10.1016/0883-2897(92)90111-b. [DOI] [PubMed] [Google Scholar]; b) Vaidyanathan G, Zalutsky MR. Nat Protoc. 2006;1:1655–1661. doi: 10.1038/nprot.2006.264. [DOI] [PubMed] [Google Scholar]
- 42.a) Tang G, Zeng W, Yu M, Kabalka G. J Labelled Compd Radiopharm. 2008;51:68–71. [Google Scholar]; b) Maeding P, Fuechtner F, Wuest F. Appl Radiat Isot. 2005;63:329–332. doi: 10.1016/j.apradiso.2005.03.005. [DOI] [PubMed] [Google Scholar]; c) Bejot R, Elizarov AM, Ball E, Zhang J, Miraghaie R, Kolb HC, Gouverneur V. J Labelled Compd Radiopharm. 2011;54:117–122. [Google Scholar]
- 43.Sutcliffe-Goulden JL, O'Doherty MJ, Marsden PK, Hart IR, Marshall JF, Bansal SS. Eur J Nucl Med Mol Imaging. 2002;29:754–759. doi: 10.1007/s00259-001-0756-3. [DOI] [PubMed] [Google Scholar]
- 44.a) Kostikov AP, Chin J, Orchowski K, Schirrmacher E, Niedermoser S, Jurkschat K, Iovkova-Berends L, Wängler C, Wängler B, Schirrmacher R. Nat Protoc. 2012;7:1956–1963. doi: 10.1038/nprot.2012.110. [DOI] [PubMed] [Google Scholar]; b) Zeng JL, Wang J, Ma JA. Bioconjugate Chem. 2015;26:1000–1003. doi: 10.1021/acs.bioconjchem.5b00180. [DOI] [PubMed] [Google Scholar]
- 45.Olberg DE, Arukwe JM, Grace D, Hjelstuen OK, Solbakken M, Kindberg GM, Cuthbertson A. J Med Chem. 2010;53:1732–1740. doi: 10.1021/jm9015813. [DOI] [PubMed] [Google Scholar]
- 46.Basuli F, Zhang X, Jagoda EM, Choyke PL, Swenson RE. Nucl Med Biol. 2016;43:770–772. doi: 10.1016/j.nucmedbio.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Basuli F, Zhang X, Woodroofe C, Jagoda EM, Choyke PL, Swenson RE. J Labelled Compd Radiopharm. 2017;60:168–175. doi: 10.1002/jlcr.3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Z, Lang L, Ma Y, Kiesewetter DO. J Labelled Compd Radiopharm. 2008;51:23–27. [Google Scholar]
- 49.Loeser R, Fischer S, Hiller A, Koeckerling M, Funke U, Maisonial A, Brust P, Steinbach J. Beilstein J Org Chem. 2013;9:1002–1011. doi: 10.3762/bjoc.9.115. No. 1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.a) Haubner R, Wester HJ, Burkhart F, Senekowitsch-Schmidtke R, Weber W, Goodman SL, Kessler H, Schwaiger M. J Nucl Med. 2001;42:326–336. [PubMed] [Google Scholar]; b) Haubner R, Wester HJ, Weber WA, Mang C, Ziegler SI, Goodman SL, Senekowitsch-Schmidtke R, Kessler H, Schwaiger M. Cancer Res. 2001;61:1781–1785. [PubMed] [Google Scholar]
- 51.Lange CW, VanBrocklin HF, Taylor SE. J Labelled Compd Radiopharm. 2002;45:257–268. [Google Scholar]
- 52.Hedberg E, Langstrom B. Acta Chem Scand. 1997;51:1236–1240. [Google Scholar]
- 53.Carberry P, Carpenter AP, Kung HF. Bioorg Med Chem Lett. 2011;21:6992–6995. doi: 10.1016/j.bmcl.2011.09.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Inkster JA, Liu K, Ait‐Mohand S, Schaffer P, Guérin B, Ruth TJ, Storr T. Chem Eur J. 2012;18:11079–11087. doi: 10.1002/chem.201103450. [DOI] [PubMed] [Google Scholar]
- 55.a) Olberg DE, Hjelstuen OK. Curr Top Med Chem. 2010;10:1669–1679. doi: 10.2174/156802610793176747. [DOI] [PubMed] [Google Scholar]; b) Glaser M, Morrison M, Solbakken M, Arukwe J, Karlsen H, Wiggen U, Champion S, Kindberg GM, Cuthbertson A. Bioconjugate Chem. 2008;19:951–957. doi: 10.1021/bc700472w. [DOI] [PubMed] [Google Scholar]
- 56.Schirrmacher E, Wängler B, Cypryk M, Bradtmöller G, Schäfer M, Eisenhut M, Jurkschat K, Schirrmacher R. Bioconjugate Chem. 2007;18:2085–2089. doi: 10.1021/bc700195y. [DOI] [PubMed] [Google Scholar]
- 57.Hultsch C, Schottelius M, Auernheimer J, Alke A, Wester HJ. Eur J Nucl Med Mol Imaging. 2009;36:1469–1474. doi: 10.1007/s00259-009-1122-0. [DOI] [PubMed] [Google Scholar]
- 58.Li XG, Dall'Angelo S, Schweiger LF, Zanda M, O'Hagan D. Chem Commun. 2012;48:5247–5249. doi: 10.1039/c2cc31262j. [DOI] [PubMed] [Google Scholar]
- 59.Matesic L, Wyatt NA, Fraser BH, Roberts MP, Pham TQ, Greguric I. J Org Chem. 2013;78:11262–11270. doi: 10.1021/jo401759z. [DOI] [PubMed] [Google Scholar]
- 60.Fiel SA, Yang H, Schaffer P, Weng S, Inkster JA, Wong MC, Li PC. ACS Appl Mater Interfaces. 2015;7:12923–12929. doi: 10.1021/acsami.5b02631. [DOI] [PubMed] [Google Scholar]
- 61.a) Chang YS, Jeong JM, Lee YS, Kim HW, Rai GB, Lee SJ, Lee DS, Chung JK, Lee MC. Bioconjugate Chem. 2005;16:1329–1333. doi: 10.1021/bc050086r. [DOI] [PubMed] [Google Scholar]; b) Lee YS, Jeong JM, Kim HW, Chang YS, Kim YJ, Hong MK, Rai GB, Chi DY, Kang WJ, Kang JH. Nucl Med Biol. 2006;33:677–683. doi: 10.1016/j.nucmedbio.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 62.Rojas S, Nolis P, Gispert JD, Spengler J, Albericio F, Herance JR, Abad S. RSC Adv. 2013;3:8028–8036. [Google Scholar]
- 63.Dolle F, Hinnen F, Vaufrey F, Tavitian B, Crouzel C. J Labelled Compd Radiopharm. 1997;39:319–330. [Google Scholar]
- 64.von Guggenberg E, Sader JA, Wilson JS, Shahhosseini S, Koslowsky I, Wuest F, Mercer JR. Appl Radiat Isot. 2009;67:1670–1675. doi: 10.1016/j.apradiso.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.a) Kiesewetter DO, Jacobson O, Lang L, Chen X. Appl Radiat Isot. 2011;69:410–414. doi: 10.1016/j.apradiso.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kniess T, Kuchar M, Pietzsch J. Appl Radiat Isot. 2011;69:1226–1230. doi: 10.1016/j.apradiso.2011.03.043. [DOI] [PubMed] [Google Scholar]
- 66.a) Toyokuni T, Walsh JC, Dominguez A, Phelps ME, Barrio JR, Gambhir SS, Satyamurthy N. Bioconjugate Chem. 2003;14:1253–1259. doi: 10.1021/bc034107y. [DOI] [PubMed] [Google Scholar]; b) Berndt M, Pietzsch J, Wuest F. Nuclear medicine and biology. 2007;34:5–15. doi: 10.1016/j.nucmedbio.2006.09.009. [DOI] [PubMed] [Google Scholar]; c) Li X, Link JM, Stekhova S, Yagle KJ, Smith C, Krohn KA, Tait JF. Bioconjugate Chem. 2008;19:1684–1688. doi: 10.1021/bc800164d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.a) De Bruin B, Kuhnast B, Hinnen F, Yaouancq L, Amessou M, Johannes L, Samson A, Boisgard R, Tavitian B, Dollé F. Bioconjugate Chem. 2005;16:406–420. doi: 10.1021/bc0497463. [DOI] [PubMed] [Google Scholar]; b) Denholt CL, Kuhnast B, Dollé F, Hinnen F, Hansen PR, Gillings N, Kjær A. J Labelled Compd Radiopharm. 2010;53:774–778. [Google Scholar]
- 68.Cai W, Zhang X, Wu Y, Chen X. J Nucl Med. 2006;47:1172–1180. [PMC free article] [PubMed] [Google Scholar]
- 69.Yue X, Kiesewetter DO, Guo J, Sun Z, Zhang X, Zhu L, Niu G, Ma Y, Lang L, Chen X. Bioconjugate Chem. 2013;24:1191–1200. doi: 10.1021/bc400084u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yue X, Yan X, Wu C, Niu G, Ma Y, Jacobson O, Shen B, Kiesewetter DO, Chen X. Mol Pharm. 2014;11:3875–3884. doi: 10.1021/mp5001857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wängler B, Quandt G, Iovkova L, Jurkschat K, Bartenstein P, Slim M, Schirrmacher E. J Nucl Med. 2008;49:13P–13P. [Google Scholar]
- 72.Huang H, Huang Y, Lee W, Yeh C, Lin K. US Patent Application. 2014
- 73.Glaser M, Karlsen H, Solbakken M, Arukwe J, Brady F, Luthra SK, Cuthbertson A. Bioconjugate Chem. 2004;15:1447–1453. doi: 10.1021/bc0498774. [DOI] [PubMed] [Google Scholar]
- 74.Wängler C, Niedermoser S, Chin J, Orchowski K, Schirrmacher E, Jurkschat K, IovkovaBerends L, Kostikov AP, Schirrmacher R, Wängler B. Nat Protoc. 2012;7:1946–1955. doi: 10.1038/nprot.2012.109. [DOI] [PubMed] [Google Scholar]
- 75.Prante O, Einsiedel J, Haubner R, Gmeiner P, Wester HJ, Kuwert T, Maschauer S. Bioconjugate Chem. 2007;18:254–262. doi: 10.1021/bc060340v. [DOI] [PubMed] [Google Scholar]
- 76.Boutureira O, Bernardes GJ, D'Hooge F, Davis BG. Chem Commun. 2011;47:10010–10012. doi: 10.1039/c1cc13524d. [DOI] [PubMed] [Google Scholar]
- 77.Jacobson O, Yan X, Ma Y, Niu G, Kiesewetter DO, Chen X. Bioconjugate Chem. 2015;26:2016–2020. doi: 10.1021/acs.bioconjchem.5b00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chiotellis A, Sladojevich F, Mu L, Herde AM, Valverde IE, Tolmachev V, Schibli R, Ametamey SM, Mindt TL. Chem Commun. 2016;52:6083–6086. doi: 10.1039/c6cc01982j. [DOI] [PubMed] [Google Scholar]
- 79.a) Jeon J, Shen B, Xiong L, Miao Z, Lee KH, Rao J, Chin FT. Bioconjugate Chem. 2012;23:1902–1908. doi: 10.1021/bc300273m. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Inkster JAH, Colin DJ, Seimbille Y. Org Biomol Chem. 2015;13:3667–3676. doi: 10.1039/c4ob02637c. [DOI] [PubMed] [Google Scholar]
- 80.Gao Z, Gouverneur V, Davis BG. J Am Chem Soc. 2013;135:13612–13615. doi: 10.1021/ja4049114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ban H, Gavrilyuk J, Barbas CF., III J Am Chem Soc. 2010;132:1523–1525. doi: 10.1021/ja909062q. [DOI] [PubMed] [Google Scholar]
- 82.Hu QY, Allan M, Adamo R, Quinn D, Zhai H, Wu G, Clark K, Zhou J, Ortiz S, Wang B. Chem Sci. 2013;4:3827–3832. [Google Scholar]
- 83.Ban H, Nagano M, Gavrilyuk J, Hakamata W, Inokuma T, Barbas CF., III Bioconjugate Chem. 2013;24:520–532. doi: 10.1021/bc300665t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nilo A, Allan M, Brogioni B, Proietti D, Cattaneo V, Crotti S, Sokup S, Zhai H, Margarit I, Berti F. Bioconjugate Chem. 2014;25:2105–2111. doi: 10.1021/bc500438h. [DOI] [PubMed] [Google Scholar]
- 85.Jessica F, Corentin W, Sylvestre D, Christian L, André L. RSC Adv. 2013;3:24936–24940. [Google Scholar]
- 86.Al-Momani E, Israel I, Buck AK, Samnick S. Appl Radiat Isot. 2015;104:136–142. doi: 10.1016/j.apradiso.2015.06.021. [DOI] [PubMed] [Google Scholar]
- 87.Dimroth O, Fester G. Ber Dtsch Chem Ges. 1910;43:2219–2223. [Google Scholar]
- 88.Huisgen R. Angew Chem. 1963;75:604–637. [Google Scholar]
- 89.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 90.Tornoe CW, Christensen C, Meldal M. J Org Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 91.Nwe K, Brechbiel MW. Cancer Biother Radiopharm. 2009;24:289–302. doi: 10.1089/cbr.2008.0626. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wangler C, Schirrmacher R, Bartenstein P, Wangler B. Curr Med Chem. 2010;17:1092–1116. doi: 10.2174/092986710790820615. [DOI] [PubMed] [Google Scholar]
- 92.Marik J, Sutcliffe JL. Tetrahedron Lett. 2006;47:6681–6684. [Google Scholar]
- 93.Ross TL, Honer M, Lam PYH, Mindt TL, Groehn V, Schibli R, Schubiger PA, Ametamey SM. Bioconjugate Chem. 2008;19:2462–2470. doi: 10.1021/bc800356r. [DOI] [PubMed] [Google Scholar]
- 94.Sirion U, Kim HJ, Lee JH, Seo JW, Lee BS, Lee SJ, Oh SJ, Chi DY. Tetrahedron Lett. 2007;48:3953–3957. [Google Scholar]
- 95.Li ZB, Wu Z, Chen K, Chin FT, Chen X. Bioconjugate Chem. 2007;18:1987–1994. doi: 10.1021/bc700226v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ramenda T, Bergmann R, Wuest F. Lett Drug Des Discovery. 2007;4:279–285. [Google Scholar]
- 97.Ramenda T, Kniess T, Bergmann R, Steinbach J, Wuest F. Chem Commun. 2009:7521–7523. doi: 10.1039/b916075b. [DOI] [PubMed] [Google Scholar]
- 98.Ramenda T, Steinbach J, Wuest F. Amino Acids. 2013;44:1167–1180. doi: 10.1007/s00726-012-1450-4. [DOI] [PubMed] [Google Scholar]
- 99.a) Inkster JAH, Guerin B, Ruth TJ, Adam MJ. J Labelled Compd Radiopharm. 2008;51:444–452. [Google Scholar]; b) Inkster JAH, Adam MJ, Storr T, Ruth TJ. Nucleosides, Nucleotides Nucleic Acids. 2009;28:1131–1143. doi: 10.1080/15257770903400691. [DOI] [PubMed] [Google Scholar]
- 100.Li Y, Liu Y, Zhang L, Xu Y. J Labelled Compd Radiopharm. 2012;55:229–234. [Google Scholar]
- 101.a) Li Y, Guo J, Tang S, Lang L, Chen X, Perrin DM. Am J Nucl Med Mol Imaging. 2013;3:44–56. [PMC free article] [PubMed] [Google Scholar]; b) Li Y, Liu Z, Harwig CW, Pourghiasian M, Lau J, Lin KS, Schaffer P, Benard F, Perrin DM. Am J Nucl Med Mol Imaging. 2013;3:57–70. [PMC free article] [PubMed] [Google Scholar]
- 102.a) Glaser M, Aarstad E. Bioconjugate Chem. 2007;18:989–993. doi: 10.1021/bc060301j. [DOI] [PubMed] [Google Scholar]; b) Kobus D, Giesen Y, Ullrich R, Backes H, Neumaier B. Appl Radiat Isot. 2009;67:1977–1984. doi: 10.1016/j.apradiso.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 103.Ackermann U, O'Keefe G, Lee ST, Rigopoulos A, Cartwright G, Sachinidis JI, Scott AM, Tochon-Danguy HJ. J Labelled Compd Radiopharm. 2011;54:260–266. [Google Scholar]
- 104.Smith G, Glaser M, Perumal M, Nguyen QD, Shan B, Aarstad E, Aboagye EO. J Med Chem. 2008;51:8057–8067. doi: 10.1021/jm801107u. [DOI] [PubMed] [Google Scholar]
- 105.Glaser M, Goggi J, Smith G, Morrison M, Luthra SK, Robins E, Aboagye EO. Bioorg Med Chem Lett. 2011;21:6945–6949. doi: 10.1016/j.bmcl.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 106.Li J, Shi L, Jia L, Jiang D, Zhou W, Hu W, Qi Y, Zhang L. Bioorg Med Chem. 2012;20:3850–3855. doi: 10.1016/j.bmc.2012.04.037. [DOI] [PubMed] [Google Scholar]
- 107.Glaser M, Solbakken M, Turton DR, Pettitt R, Barnett J, Arukwe J, Karlsen H, Cuthbertson A, Luthra SK, Årstad E. Amino Acids. 2009;37:717–724. doi: 10.1007/s00726-008-0200-0. [DOI] [PubMed] [Google Scholar]
- 108.Glaser M, Arstad E, Gaeta A, Nairne J, Trigg W, Robins EG. J Labelled Compd Radiopharm. 2012;55:326–331. [Google Scholar]
- 109.Campbell-Verduyn LS, Mirfeizi L, Schoonen AK, Dierckx RA, Elsinga PH, Feringa BL. Angew Chem Int Ed. 2011;50:11117–11120. doi: 10.1002/anie.201105547. [DOI] [PubMed] [Google Scholar]
- 110.Pretze M, Kuchar M, Bergmann R, Steinbach J, Pietzsch J, Mamat C. ChemMedChem. 2013;8:935–945. doi: 10.1002/cmdc.201300053. [DOI] [PubMed] [Google Scholar]
- 111.Gill HS, Marik J. Nat Protocols. 2011;6:1718–1725. doi: 10.1038/nprot.2011.390. [DOI] [PubMed] [Google Scholar]
- 112.Maschauer S, Prante O. Carbohydr Res. 2009;344:753–761. doi: 10.1016/j.carres.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 113.Zhu J, Li S, Wangler C, Wangler B, Bruce Lennox R, Schirrmacher R. Chem Commun. 2015;51:12415–12418. doi: 10.1039/c5cc03623b. [DOI] [PubMed] [Google Scholar]
- 114.Thonon D, Kech C, Paris J, Lemaire C, Luxen A. Bioconjugate Chem. 2009;20:817–823. doi: 10.1021/bc800544p. [DOI] [PubMed] [Google Scholar]
- 115.Chun JH, Pike VW. Eur J Org Chem. 2012;2012:4541–4547. doi: 10.1002/ejoc.201200695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mercier F, Paris J, Kaisin G, Thonon D, Flagothier J, Teller N, Lemaire C, Luxen A. Bioconjugate Chem. 2011;22:108–114. doi: 10.1021/bc100263y. [DOI] [PubMed] [Google Scholar]
- 117.Jacobson O, Weiss ID, Wang L, Wang Z, Yang X, Dewhurst A, Ma Y, Zhu G, Niu G, Kiesewetter DO, Vasdev N, Liang SH, Chen X. J Nucl Med. 2015;56:1780–1785. doi: 10.2967/jnumed.115.160960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang L, Jacobson O, Avdic D, Rotstein BH, Weiss ID, Collier L, Chen X, Vasdev N, Liang SH. Angew Chem Int Ed. 2015;54:12777–12781. doi: 10.1002/anie.201505927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Balentova E, Collet C, Lamande-Langle S, Chretien F, Thonon D, Aerts J, Lemaire C, Luxen A, Chapleur Y. J Fluorine Chem. 2011;132:250–257. [Google Scholar]
- 120.Blomquist AT, Liu LH. J Am Chem Soc. 1953;75:2153–2154. [Google Scholar]
- 121.Agard NJ, Prescher JA, Bertozzi CR. J Am Chem Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- 122.Arumugam S, Chin J, Schirrmacher R, Popik VV, Kostikov AP. Bioorg Med Chem Lett. 2011;21:6987–6991. doi: 10.1016/j.bmcl.2011.09.126. [DOI] [PubMed] [Google Scholar]
- 123.Hausner SH, Abbey CK, Bold RJ, Gagnon MK, Marik J, Marshall JF, Stanecki CE, Sutcliffe JL. Cancer Res. 2009;69:5843–5850. doi: 10.1158/0008-5472.CAN-08-4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hausner SH, Carpenter RD, Bauer N, Sutcliffe JL. Nucl Med Biol. 2013;40:233–239. doi: 10.1016/j.nucmedbio.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 125.Sachin K, Jadhav VH, Kim EM, Kim HL, Lee SB, Jeong HJ, Lim ST, Sohn MH, Kim DW. Bioconjugate Chem. 2012;23:1680–1686. doi: 10.1021/bc3002425. [DOI] [PubMed] [Google Scholar]
- 126.a) Nilsson BL, Kiessling LL, Raines RT. Org Lett. 2001;3:9–12. doi: 10.1021/ol006739v. [DOI] [PubMed] [Google Scholar]; b) Staudinger H, Meyer J. Helv Chim Acta. 1919;2:635–646. [Google Scholar]; c) Saxon E, Armstrong JI, Bertozzi CR. Org Lett. 2000;2:2141–2143. doi: 10.1021/ol006054v. [DOI] [PubMed] [Google Scholar]; d) Nilsson BL, Kiessling LL, Raines RT. Org Lett. 2000;2:1939–1941. doi: 10.1021/ol0060174. [DOI] [PubMed] [Google Scholar]; e) Saxon E, Bertozzi CR. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 127.Robillard MS, Gruell H. Compounds, kits and methods for use in medical imaging. Koninklijke Philips Electronics N.V., Neth; 2006. p. 19. Vol. (Ed. U. S. P. A. No) [Google Scholar]
- 128.a) Mamat C, Flemming A, Koeckerling M, Steinbach J, Wuest FR. Synthesis. 2009:3311–3321. [Google Scholar]; b) Mamat C, Franke M, Peppel T, Koeckerling M, Steinbach J. Tetrahedron. 2011;67:4521–4529. [Google Scholar]
- 129.a) Pretze M, Flemming A, Koeckerling M, Mamat C. Z Naturforsch B: J Chem Sci. 2010;65:1128–1136. [Google Scholar]; b) Pretze M, Wuest F, Peppel T, Koeckerling M, Mamat C. Tetrahedron Lett. 2010;51:6410–6414. [Google Scholar]
- 130.Carroll L, Boldon S, Bejot R, Moore JE, Declerck J, Gouverneur V. Org Biomol Chem. 2011;9:136–140. doi: 10.1039/c0ob00564a. [DOI] [PubMed] [Google Scholar]
- 131.Agard NJ, Baskin JM, Prescher JA, Lo A, Bertozzi CR. ACS Chem Biol. 2006;1:644–648. doi: 10.1021/cb6003228. [DOI] [PubMed] [Google Scholar]
- 132.a) Blackman ML, Royzen M, Fox JM. J Am Chem Soc. 2008;130:13518–13519. doi: 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bach RD. J Am Chem Soc. 2009;131:5233–5243. doi: 10.1021/ja8094137. [DOI] [PubMed] [Google Scholar]
- 133.Li Z, Cai H, Hassink M, Blackman ML, Brown RCD, Conti PS, Fox JM. Chem Commun. 2010;46:8043–8045. doi: 10.1039/c0cc03078c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.a) Selvaraj R, Liu S, Hassink M, Huang Cw, Yap Lp, Park R, Fox JM, Li Z, Conti PS. Bioorg Med Chem Lett. 2011;21:5011–5014. doi: 10.1016/j.bmcl.2011.04.116. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu S, Hassink M, Selvaraj R, Yap LP, Park R, Wang H, Chen X, Fox JM, Li Z, Conti PS. Mol Imaging. 2013;12:121–128. [PMC free article] [PubMed] [Google Scholar]
- 135.a) Keliher EJ, Reiner T, Turetsky A, Hilderbrand SA, Weissleder R. ChemMedChem. 2011;6:424–427. doi: 10.1002/cmdc.201000426. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Reiner T, Keliher EJ, Earley S, Marinelli B, Weissleder R. Angew Chem Int Ed. 2011;50:1922–1925. doi: 10.1002/anie.201006579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Selvaraj R, Giglio B, Liu S, Wang H, Wang M, Yuan H, Chintala SR, Yap LP, Conti PS, Fox JM. Bioconjugate Chem. 2015;26:435–442. doi: 10.1021/acs.bioconjchem.5b00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang M, Svatunek D, Rohlfing K, Liu Y, Wang H, Giglio B, Yuan H, Wu Z, Li Z, Fox J. Theranostics. 2016;6:887–895. doi: 10.7150/thno.14742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Li XG, Roivainen A, Bergman J, Heinonen A, Bengel F, Thum T, Knuuti J. Chem Commun. 2015;51:9821–9824. doi: 10.1039/c5cc02618k. [DOI] [PubMed] [Google Scholar]
- 139.Da Pieve C, Allott L, Martins CD, Vardon A, Ciobota DM, Kramer-Marek G, Smith G. Bioconjugate Chem. 2016;27:1839–1849. doi: 10.1021/acs.bioconjchem.6b00259. [DOI] [PubMed] [Google Scholar]
- 140.Selvaraj R, Fox JM. Curr Opin Chem Biol. 2013;17:753–760. doi: 10.1016/j.cbpa.2013.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Devaraj NK, Thurber GM, Keliher EJ, Marinelli B, Weissleder R. Proc Natl Acad Sci. 2012;109:4762–4767. doi: 10.1073/pnas.1113466109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Denk C, Svatunek D, Filip T, Wanek T, Lumpi D, Froehlich J, Kuntner C, Mikula H. Angew Chem Int Ed. 2014;53:9655–9659. doi: 10.1002/anie.201404277. [DOI] [PubMed] [Google Scholar]
- 143.Rashidian M, Keliher EJ, Bilate AM, Duarte JN, Wojtkiewicz GR, Jacobsen JT, Cragnolini J, Swee LK, Victora GD, Weissleder R, Ploegh HL. Proc Natl Acad Sci. 2015;112:6146–6151. doi: 10.1073/pnas.1502609112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rashidian M, Keliher EJ, Dougan M, Juras PK, Cavallari M, Wojtkiewicz GR, Jacobsen JT, Edens JG, Tas JMJ, Victora G, Weissleder R, Ploegh H. ACS Cent Sci. 2015;1:142–147. doi: 10.1021/acscentsci.5b00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rashidian M, Wang L, Eden JG, Jacobsen JT, Hossain I, Wang Q, Victora GD, Vasdev N, Ploegh H, Liang SH. Angew Chem Int Ed. 2016;55:528–533. doi: 10.1002/anie.201507596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Keinänen O, Li XG, Chenna NK, Lumen D, Ott J, Molthoff CFM, Sarparanta M, Helariutta K, Vuorinen T, Windhorst AD, Airaksinen AJ. ACS Med Chem Lett. 2016;7:62–66. doi: 10.1021/acsmedchemlett.5b00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Meyer JP, Houghton JL, Kozlowski P, Abdel-Atti D, Reiner T, Pillarsetty NVK, Scholz WW, Zeglis BM, Lewis JS. Bioconjugate Chem. 2016;27:298–301. doi: 10.1021/acs.bioconjchem.5b00504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zlatopolskiy BD, Kandler R, Mottaghy FM, Neumaier B. Appl Radiat Isot. 2012;70:184–192. doi: 10.1016/j.apradiso.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 149.Zlatopolskiy BD, Kandler R, Kobus D, Mottaghy FM, Neumaier B. Chem Commun. 2012;48:7134–7136. doi: 10.1039/c2cc31335a. [DOI] [PubMed] [Google Scholar]
- 150.a) Chalker JM, Bernardes GJL, Davis BG. Acc Chem Res. 2011;44:730–741. doi: 10.1021/ar200056q. [DOI] [PubMed] [Google Scholar]; b) Spicer CD, Davis BG. Nat Commun. 2014;5 doi: 10.1038/ncomms5740. [DOI] [PubMed] [Google Scholar]; c) Drake CR, Sevillano N, Truillet C, Craik CS, VanBrocklin HF, Evans MJ. ACS Chem Biol. 201611:1587–1594. doi: 10.1021/acschembio.6b00172. [DOI] [PMC free article] [PubMed] [Google Scholar]