

Abstract
Here we describe a case of a prenatal diagnosis of a male fetus that inherited the unstable allele from his full mutation mosaic mother (29, 160, >200 CGG repeats) reduced to a normal size range (19 CGG repeats). Haplotype analysis showed that the fetus 19 CGG repeats allele derived from the maternal unstable allele which was inherited from his maternal grandmother. No size mosaicism was detected by testing the DNA from in vitro cultured samples, including seventh passage culture as well as from two amniocentesis samples. Sequence analysis confirmed that the allele was 19 CGG repeats long. Methylation assay showed no methylation. Although none of the techniques used in this study can provide with absolute certainty the diagnosis, the results strongly indicate the presence in the fetus of an allele with a CGG repeat number in the normal range. Because this is a prenatal diagnosis case, the crucial question is whether the 19 CGG allele derived from the maternal unstable expanded allele, which contracted to the normal range, became a normal stable allele or a normal unstable allele which could expand in the next generation. It is also possible that allele size mosaicism of the FMR1 gene that went undetected exists. Because this is a prenatal diagnosis case, we cannot with certainty exclude the presence of an undetected expanded allele of the FMR1 gene, in addition to the 19 CGG allele derived from an unstable expanded allele, which contracted to the normal range.
Keywords: prenatal diagnosis, FMR1 gene, instability, expansion, contraction
Introduction
Fragile X syndrome (FXS) is the most common inherited cause of intellectual disability and the most common monogenic cause of Autism Spectrum Disorders (Harris et al., 2008). The FXS is caused by a dynamic mutation in the FMR1 gene, which is located at Xq27.3. A normal gene carries up to 54 CGG repeats in the 5′ UTR while a premutation allele carries 55–200 CGG repeats and is considered unstable during transmission to offspring. The full mutation allele harbors more than 200 CGG repeats and shows somatic mosaicism in 12–41% cases (Nolin et al., 1994; Rousseau et al., 1994; Merenstein et al., 1996). Full mutation alleles are usually methylated and transcriptionally silent so that no FMR1 protein (FMRP) is produced (Kremer et al., 1991; Oberle et al., 1991; Yu et al., 1991; Verkerk et al., 1993; Allingham-Hawkins et al., 1999). Almost 100% of the males who inherited a full mutation show FXS that usually involves intellectual disability, while only about 60% of the females with a full mutation show mild to severe clinical features. It is generally accepted that the FMR1 instability occurs when the CGG repeats expanded beyond 55 CGG repeats; however, instability in the normal range has also been documented (Nolin et al., 2013). The full mutation occurs only when it is transmitted from the mother; and it is rare for a contraction to occur when the unstable expanded allele is passed on by the mother (Lozano et al., 2016). Here, we described a rare case of prenatal diagnosis of male fetus that inherited an allele that contracted to a normal repeat size from the mother with the full mutation.
Materials and Methods
Subjects: Written informed consent was obtained from the studied subjects for publication of this manuscript.
Allele categories were as described in Maddalena et al. (2001) (normal 5–54 CGG, premutation alleles 55–200 CGG repeats, full mutation alleles >200 CGG repeats).
A 40-year-old full mutation woman known to carry size mosaicism for the presence of a premutation and a full mutation alleles (29, 160, >200 CGG repeats) in the FMR1 gene was referred to our laboratory for prenatal diagnosis of her 24-week fetus. It was a spontaneous pregnancy after a long history of failure in preimplantation genetic diagnosis (PGD) pregnancies. The laboratory received 30 ml of amniotic fluid, which were divided into two different syringes, and 5 ml was used for the direct DNA extraction while the remaining amount was cultured for karyotyping and DNA extraction.
Amniocytes Culture
The amniocytes were centrifuged and half of the pellet suspended in CHANG medium (Irvine Scientific, Santa Ana, CA, United States) while the other half was suspended in BioAMF-2 (Biological Industries, Beit-HaEmek, Israel) and cultured in 25 cm flasks for 10–14 days. When cells reached confluence, they were either harvested for DNA extraction or trypsinized for the next passage. Cells harvested at the fifth and at seventh; by the eighth passage, the culture failed.
Genomic DNA Extraction
Genomic DNA (gDNA) was extracted from peripheral blood samples collected from the mother, the grandmother, and from the father using QIAsymphony DNA Midi Kit (96)-931255 and the QIAsymphony machine according to the manufacturer’s instructions. Isolation of gDNA from cultures cells was carried out using the QIAamp DNA blood Mini Kit-51104 (Qiagen, Hilden, Germany) and the QIAsymphony machine according to the manufacturer’s instructions. DNA was extracted from amniotic fluid using the QIAamp DNA blood Micro Kit-56304 (Qiagen, Hilden, Germany), manually or using the QIAcube machine according to the manufacturer’s instructions.
Triple-Primed FXS PCR
Genomic DNA (gDNA) (40–60 ng) was amplified with the Amplidex FMR1 PCR assay (Asuragen, Austin, TX, United States) as previously described (Nahhas et al., 2012) and accordingly to the manufacturer’s instructions. Samples were analyzed by the 3130xl Genetic Analyzer (Applied Biosystems Inc., ABI, Foster City, CA, United States) and electropherograms were analyzed using GeneMapper 4.0 (4.1 for 3500xL data) (Filipovic-Sadic et al., 2010).
FMR1 5′ UTR Sequencing
Genomic DNA (gDNA) was amplified in 20 μl reaction using forward primer FMR-C: 5′-GCTCAGCTCCGTTTCGGTTTCACTTCCGGT-3′ and reverse primer FMR-F: 5′-AGCCCCGCACTTCCACCACCAGCTCCTCCA-3′ (SIGMA). PCR products were sequenced with BigDye Terminator v1.1 Cycle Sequence Kit – Applied Biosystems – chemistry on a 3130xl Genetic Analyzer (Life Technologies). Electropherograms were analyzed using GeneMapper 4.0 (4.1 for 3500xL data).
Haplotype Analysis
Maternity studies were performed using Elucigene QSTR plus V2 kit. A segregation study was performed on the grandmother, the mother, and the fetus using the following polymorphic makers: DXS106, DXS8028, DXS998, FMR1: 146993469–147032647, DXS106, DXS8069, DXS8061, DXS1073: and florescent primers (Richards et al., 1991; Verkerk et al., 1993). Alleles were resolved on capillary gel electrophoresis using ABI 3130xl apparatus. Electropherograms were analyzed using GeneMapper 4.0 (4.1 for 3500xL data).
Methylation Analysis
Methylation analysis was carried out using the Amplidex FMR1 mPCR FMR1 kit (Asuragen, Austin, TX, United States) as previously described by Chen et al. (2014). Briefly, two aliquots of 40 ng gDNA isolated from the fetus, from a methylated male control and from a normal male were prepared for methylation assessment. One aliquot was mixed with 4 μL Digestion Enzyme Mix (containing HhaI and AvaI), and the other with 4 μL Digestion Control Mix (containing digestion buffer). Aliquoted samples were incubated at 37°C overnight. Following digestion, aliquots from each sample analyzed according to Asuragen’s instruction. The results were analyzed using the 3130xl Genetic Analyzer (Applied Biosystems Inc., ABI, Foster City, CA, United States). Electropherograms were analyzed using GeneMapper 4.0 (4.1 for 3500xL data) as described by Filipovic-Sadic et al. (2010).
Results
Capillary electrophoresis analysis of two different direct amniocentesis samples of DNA revealed the presence of two peaks: one less abundant [3000 relative fluorescent units (RFU)] corresponding to 29 CGG repeat allele and the other much more abundant peak (9000 RFU) corresponding to 19 CGG repeat allele (Figure 1). Karyotype analysis confirmed a male fetus.
FIGURE 1.

FMR1 mutation – length test. Electropherogram of the Asuragen test results showing the fetus (A) and the mother (B) CGG repeats length results. The fetus results show presence of a pronounced peak at 19 CGG repeats (A) and the mother results (B) it shows normal allele with 29 CGG repeats and the unstable allele with mosaicism, a premutation allele of 160 CGG repeats and full mutation greater than 200 CGG repeats.
DNA from eight amniocyte cultures was also tested: two directly from cultured amniocytes, two from the first passage, two from the fifth passage, and two from the seventh passage. All samples showed presence of only the 19 CGG repeat allele as observed in DNA isolated from the amniotic fluid but without the less abundant peak corresponding to the 29 CGG repeat allele. The less abundant peak observed from DNA isolated from amniotic fluid points to low level of mother contamination, which can been detected by the Amplidex PCR (Asuragen) as it has been proven to be much more sensitive than other techniques (Filipovic-Sadic et al., 2010; Tassone, 2015). None of the samples tested showed evidence of an expanded allele excluding the possibility of mosaicism.
Sequence analysis confirmed the presence of a 19 CGG repeat allele in both DNA isolated from amniotic fluid and DNA isolated from cultured amniocytes after seven passages (Figure 1). No AGG interruptions were observed within the allele.
Testing the origin of the FMR1 allele by haplotype analysis using the DNA derived from the mother, the fetus, and from the maternal grandmother showed that the unstable allele was transmitted from the grandmother to her daughter who transmitted it to her male fetus; thus confirmed maternity (Figure 2).
FIGURE 2.
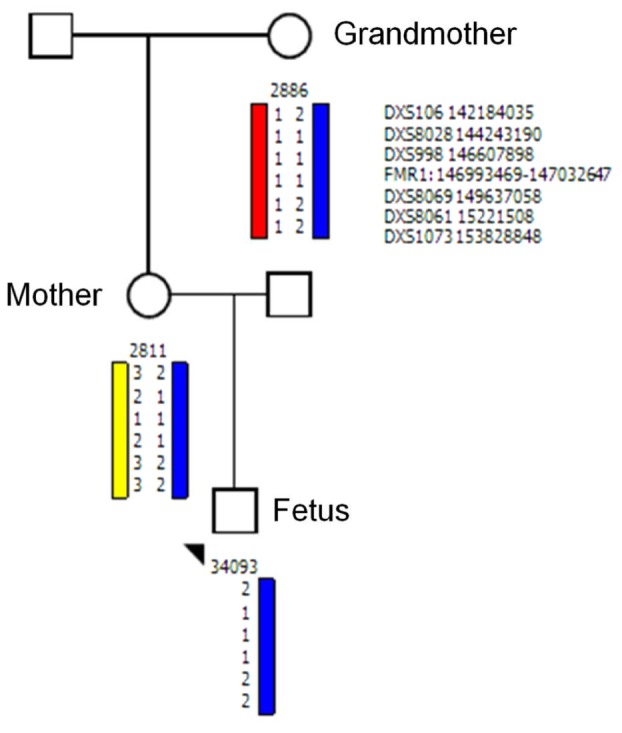
Haplotype analysis for allele discrimination and tracing the origin of FMR1 fetal allele. Haplotype analysis using the polymorphic makers: DXS106, DXS8028, DXS998, FMR1: 146993469–147032647, DXS106, DXS8069, DXS8061, DXS1073 shows that the unstable allele transmitted from the grandmother to the mother and to her fetus.
Methylation assay of the FMR1 gene using AvaI and HhaI showed no methylation in the fetus cells while it showed a normal methylation in both FMR1 alleles of the mother suggesting that methylation assay is not reliable for fetal cells.
Discussion
The case described here represents a rare case of transmission from a full mutation mother to her offspring with an allele pattern different from what we would have expected in such cases. It is a prenatal diagnosis case of a male fetus that inherited an allele with a CGG repeats number in the normal size range (19 CGG repeats) derived from the unstable expanded maternal allele (160, >200 CGG repeats). It is well known that the majority (near to 100%) of the male fetuses that inherited unstable FMR1 allele from mosaic mothers are expected to inherit a full mutation allele. Contraction of a maternal high premutation (>100 CGG repeats)/full mutation FMR1 allele during transmission has been rarely described (Yrigollen et al., 2014; Miranda et al., 2015; Tabolacci et al., 2016; Maia et al., 2017).
To our knowledge this is the first report of a prenatal diagnosis case showing a pattern of contraction of an expanded FMR1 unstable allele to a normal size allele. Tabolacci et al. (2016) previously described a case of a healthy 10-year-old boy who inherited a contracted unstable FMR1 allele from his mother. The authors described a family with a carrier mother (190 CGG repeats) and her three children. Two of them presented with FXS and had expanded alleles in the full mutation range (265–830 and 430–790 CGG repeats, respectively). In contrast, the healthy boy inherited a contracted allele of 43 CGG repeats. Another study (Maia et al., 2017) described four FXS boys carrying mosaic alleles ranging from the normal (35, 26, 39, and 18 CGG repeats) to the full mutation range (>200 CGG repeats). These studies sharpen the obscure mechanism of expansion of the trinucleotide repeat in the FMR1 gene, indicating that the same allele during the generation might expand or contract and possibly undergo methylation.
Since this is a prenatal diagnosis case, the questions are as follows: Is the unstable allele contracted to the normal range able to become a stable normal allele with no phenotypic consequences? Is it a normal unstable allele that may lead to some clinical involvement? Will it expand during transmission in the following generations? Is it a case of mosaicism through an undetected expanded allele of the FMR1 gene, which may cause features of FXS?
It is of importance to understand the consequence of contraction of high premutation/full mutation FMR1 allele as understanding the mechanism of FMR1 instability can improve genetic counseling of the family.
Although CGG instability within the FMR1 gene occurs mainly from the premutation to the full mutation range, instability on transmission with CGG repeat length, defined as any measurable repeat change from the parent to the offspring, has also been described in allele within the normal range (Nolin et al., 2013). The magnitude of instability correlates to the presence of AGG interruptions (Nolin et al., 2013; Yrigollen et al., 2014). Normal alleles have an AGG interruption that usually occurs after 9 or 10 CGG triplets (Yrigollen et al., 2014). However, it has been found that increased instability seems to be correlated with the presence of the first interruption at the 10th triplet (Gunter et al., 1998) and to the total number of AGGs. The loss of AGGs occurs in a polarized way at the 3′-end, creating a long pure (CGG)n with higher mutability (Kunst and Warren, 1994; Limprasert et al., 2016). In the presented case, the allele was a pure stretch of CGG without any AGG interruption. The AGG interspersions in the CGG repeat region are well known as an important element for stability of the repeats (Ennis et al., 2007; Fernandez-Carvajal et al., 2009; Carolyn et al., 2012; Nolin et al., 2013, 2015; Yrigollen et al., 2014).
Sharony et al. (2012), Wakeling et al. (2014), and also our laboratory (data not shown) experienced instability/mosaicism in the normal–intermediate range (less than 55 CGG repeats). Wakeling et al. (2014) reported that about 0.4% of the normal population tested showed an extra allele. Our experience from the routine FMR1 screening test of normal population as well as the Sharony et al. (2012) study showed a lower frequency of cases with an extra allele (12 out of 13,000 cases; data not shown) in the normal range size. The presence of an extra allele could be attributed to the presence of a cryptic duplication event of the FMR1 locus (Mononen et al., 2007; Vengoechea et al., 2012) of an early postzygotic expansion or contraction of the FMR1 repeat tract or to a somatic instability of the CGG tract. While somatic instability of premutation and full mutation alleles is well documented (Cambouris et al., 1996; Dobkin et al., 1996; Mingroni-Netto et al., 1996), information regarding unstable normal alleles (Sullivan et al., 2005; Nolin et al., 2011) is limited. In contrast to the report of Wakeling et al. (2014), Maia et al. (2017) reported on four FXS mosaic males carrying normal, premutated and full mutation FMR1 alleles. The authors suggested that the normal allele of these patients resulted from postzygotic contraction of the full expansion. The authors study suggested the existence of a predisposing haplotype in the population, which they named “risk lineage-specific haplotype,” more prone to large contraction.
Although the mechanism(s) of the FMR1 instability are still unknown, a mice model including the interplay among components of the base excision repair, mismatch repair, and transcription coupled repair pathways has been proposed (Zhao et al., 2016). The authors indicated that expanded alleles are unstable and can undergo to an expansion or contraction event through different mechanisms. Contraction events have been observed on both paternal and maternal transmission although more frequently on paternal transmission (Nolin et al., 2015). Although FMR1 expansion in human occurred almost exclusively through maternal inheritance, there is no reference to any sex factor. Contractions may derive from simple strand slippage during replication or repair; however, to date, which factors are involved in the processes of repeat instability at the FMR1 locus remain an unanswered question.
Although PCR-based approach and the sequencing tests used in the present study have shown to be very reliable, we cannot categorically exclude the presence of an expanded allele. However, the results demonstrate the presence of a normal size allele with no mosaicism, most likely leading to a normal phenotype as the case described by Tabolacci et al. (2016).
Our hypothesis is that the maternal premutation/full mutation allele contracted pre-zygotically either in the mitotic or in the meiotic stage resulting in a normal size allele in the fertilized egg with no syndromic consequence.
Ethics Statement
All subjects gave written informed consent in accordance with the Declaration of Helsinki.
Author Contributions
EM: Performed design for this study, management, and wrote the manuscript. AJ: Performed Fragile tests and Fragile methylation. NM: Performed Haplotype testing and confirmed the results. AK: Performed medical management and clinical evaluation of the case and the family. RH: Worked as a part of Genetic advisory and edited the article. FT: Performed the evaluation of the results and edited the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Allingham-Hawkins D. J., Babul-Hirji R., Chitayat D., Holden J. J., Yang K. T., Lee C., et al. (1999). Fragile X premutation is a significant risk factor for premature ovarian failure: the International Collaborative POF in fragile X study. Am. J. Med. Genet. 83 322–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambouris M., Snow K., Thibodeau S., Bluhm D., Green M., Feldman G. L. (1996). Segregation of the fragile X mutation from a male with a full mutation: unusual somatic instability in the FMR-1 locus. Am. J. Med. Genet. 64 404–407. [DOI] [PubMed] [Google Scholar]
- Carolyn Y., Blythe D.-J., Louise G., David N., Randi H., Paul H., et al. (2012). AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with fragile X syndrome. Genet. Med. 14 729–736. 10.1038/gim.2012.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Hadd A., Sah S., Houghton J. F., Filipovic-sadic S., Zhang W., et al. (2014). High-resolution methylation polymerase chain reaction for fragile X analysis: evidence for novel FMR1 methylation patterns undetected in Southern blot analyses. Genet. Med. 13 528–538. 10.1097/GIM.0b013e31820a780f [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobkin C. S., Nolin S. L., Cohen I., Sudhalter V., Bialer M. G., Ding X. H., et al. (1996). Tissue differences in fragile X mosaics: mosaicism in blood cells may differ greatly from skin. Am. J. Med. Genet. 64 296–301. [DOI] [PubMed] [Google Scholar]
- Ennis S., Murray A., Brightwell G., Morton N. E., Jacobs P. A. (2007). Closely linked cis-acting modifier of expansion of the CGG repeat in high risk FMR1 haplotypes. Hum. Mutat. 28 1216–1224. 10.1002/humu.20600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Carvajal I., Lopez Posadas B., Pan R., Raske C., Hagerman P. J., Tassone F. (2009). Expansion of an FMR1 grey-zone allele to a full mutation in two generations. J. Mol. Diagn. 11 306–310. 10.2353/jmoldx.2009.080174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipovic-Sadic S., Sah S., Chen L., Krosting J., Sekinger E., Zhang W., et al. (2010). A novel FMR1 PCR method for the routine detection of low abundance expanded alleles and full mutations in fragile X syndrome. Clin. Chem. 56 399–408. 10.1373/clinchem.2009.136101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter C., Paradee W., Crawford D. C., Meadows K. A., Newman J., Kunst C. B., et al. (1998). Re-examination of factors associated with expansion of CGG repeats using a single nucleotide polymorphism in FMR1. Hum. Mol. Genet. 7 1935–1946. 10.1093/hmg/7.12.1935 [DOI] [PubMed] [Google Scholar]
- Harris S. W., Hessl D., Goodlin-Jones B., Ferranti J., Bacalman S., Barbato I., et al. (2008). Autism profiles of males with fragile X syndrome. Am. J. Ment. Retard. 113 427–438. 10.1352/2008.113:427-438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer E., Pritchard M., Lynch M., Yu S., Holman K., Baker E., et al. (1991). Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science 252 1711–1714. 10.1126/science.1675488 [DOI] [PubMed] [Google Scholar]
- Kunst C. B., Warren S. T. (1994). Cryptic and polar variation of the fragile X repeat could result in predisposing normal alleles. Cell 77 853–861. 10.1016/0092-8674(94)90134-1 [DOI] [PubMed] [Google Scholar]
- Limprasert P., Thanakitgosate J., Jaruthamsophon K., Sripo T. (2016). Unique AGG interruption in the CGG repeats of the FMR1 gene exclusively found in Asians linked to a specific SNP haplotype. Genet. Res. Int. 2016:8319287. 10.1155/2016/8319287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano R., Azarang A., Wilaisakditipakorn T., Hagerman R. J. (2016). Fragile X syndrome: a review of clinical management. Intractable Rare Dis. Res. 5 145–157. 10.5582/irdr.2016.01048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddalena A., Richards C. S., Mcginniss M. J., Brothman A., Desnick R. J., Grier R. E., et al. (2001). Technical standards and guidelines for fragile X: the first of a series of disease-specific supplements to the standards and guidelines for clinical genetics laboratories of the American college of medical genetics. Genet. Med. 3 200–205. 10.1097/00125817-200105000-00010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia N., Loureiro J. R., Oliveria B., Marques I., Santos R., Jorge P. (2017). Contraction of fully expanded FMR1 alleles to normal range: predisposing or a rare event? J. Hum. Genet. 62 269–275. 10.1038/jhg.2016.122 [DOI] [PubMed] [Google Scholar]
- Merenstein S. A., Sobesky W. E., Taylor A. K., Riddle J. E., Tran H. X., Hagerman R. J. (1996). Molecular-clinical correlations in males with an expanded FMR1 mutation. Am. J. Med. Genet. 64 388–394. [DOI] [PubMed] [Google Scholar]
- Mingroni-Netto R. B., Haddad L. A., Vianna-Morgante A. M. (1996). The number of CGG repeats of the FMR1 locus in premutated and fully mutated heterozygotes and their offspring: implications for the origin of mosaicism. Am. J. Med. Genet. 64 270–273. [DOI] [PubMed] [Google Scholar]
- Miranda P., Miranda P., Jiraanont P., Abrams L. J., Basuta K. K., Youngblom J., et al. (2015). Contraction of a maternal fragile X mental retardation 1 premutation allele. J. Med. Cases 6 547–553. 10.14740/jmc.v6i12.2301 [DOI] [Google Scholar]
- Mononen T., von Koskull H., Airaksinen R.-L., Juvonen V. (2007). A novel duplication in the FMR1 gene: implications for molecular analysis in fragile X syndrome and repeat instability. Clin. Genet. 72 528–531. 10.1111/j.1399-0004.2007.00903.x [DOI] [PubMed] [Google Scholar]
- Nahhas F. A., Monroe T. J., Prior T. W., Botma P. I., Fang J., Snyder P. J., et al. (2012). Evaluation of the human fragile X mental retardation 1 polymerase chain reaction reagents to amplify the FMR1 gene: testing in a clinical diagnostic laboratory. Genet. Test. Mol. Biomark. 16 187–192. 10.1089/gtmb.2011.0128 [DOI] [PubMed] [Google Scholar]
- Nolin S. L., Glicksman A., Ding X., Ersalesi N., Brown W. T., Sherman S. L., et al. (2011). Fragile X analysis of 1112 prenatal samples from 1991 to 2010. Prenat. Diagn. 31 925–931. 10.1002/pd [DOI] [PubMed] [Google Scholar]
- Nolin S. L., Glicksman A., Ersalesi N., Dobkin C., Brown W. T., Cao R., et al. (2015). Fragile X full mutation expansions are inhibited by one or more AGG interruptions in premutation carriers. Genet. Med. 17 358–364. 10.1038/gim.2014.106 [DOI] [PubMed] [Google Scholar]
- Nolin S. L., Glicksman A., Houck G. E., Jr., Brown W. T., Dobkin C. S. (1994). Mosaicism in fragile X affected males. Am. J. Med. Genet. 51 509–512. 10.1002/ajmg.1320510444 [DOI] [PubMed] [Google Scholar]
- Nolin S. L., Sah S., Glicksman A., Sherman S. L., Allen E., Berry-Kravis E., et al. (2013). Fragile X AGG analysis provides new risk predictions for 45-69 repeat alleles. Am. J. Med. Genet. A 161 771–778. 10.1002/ajmg.a.35833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberle I., Rousseau F., Heits D., Kretz C., Devys D., Hanauer A. (1991). Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 252 1097–1102. 10.1126/science.252.5009.1097 [DOI] [PubMed] [Google Scholar]
- Richards R. I., Holman K., Kozman H., Kremer E., Lynch M., Pritchard M. (1991). Fragile X syndrome: genetic localization bi linkage mapping of two microsatellite repeats FRAXAC1 and FRAXC2 which immediately flank the fragile site. J. Med. Genet. 28 818–823. 10.1136/jmg.28.12.818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau F., Heitz D., Tarleton J., Macpherson J., Malmgren H., Dahl N., et al. (1994). A multicenter study on genotype-phenotype correlations in the fragile X syndrome, using direct diagnosis with probe StB12.3: the first 2,253 cases. Am. J. Hum. Genet. 55 225–237. [PMC free article] [PubMed] [Google Scholar]
- Sharony R., Shtorch A., Amiel A., Guetta E., Peleg L., Pras E., et al. (2012). Three peaks in the polymerase chain reaction fragile X analysis. J. Med. Screen. 19 112–115. 10.1258/jms.2012.012029 [DOI] [PubMed] [Google Scholar]
- Sullivan A. K., Marcus M., Epstein M. P., Allen E. G., Anido A. E., Paquin J. J., et al. (2005). Association of FMR1 repeat size with ovarian dysfunction. Hum. Reprod. 20 402–412. 10.1093/humrep/deh635 [DOI] [PubMed] [Google Scholar]
- Tabolacci E., Palumbo F., Nobile V., Neri G. (2016). Transcriptional reactivation of the FMR1 gene. a possible approach to the treatment of the fragile X syndrome. Genes 7:E49. 10.3390/genes7080049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F. (2015). Advanced technologies for the molecular diagnosis of fragile X syndrome. Expert Rev. Mol. Diagn. 15 1465–1473. 10.1586/14737159.2015.1101348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vengoechea J., Parikh A. S., Zhang S., Tassone F. (2012). De novo microduplication of the FMR1 gene in a patient with developmental delay, epilepsy and hyperactivity. Eur. J. Hum. Genet. 20 1197–1200. 10.1038/ejhg.2012.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkerk A. J., de Graaff E., De Boulle K., Eichler E. E., Konecki D. S., Reyniers E., et al. (1993). Alternative splicing in the fragile X gene FMR1. Hum. Mol. Genet. 2 399–404. 10.1093/hmg/2.4.399 [DOI] [PubMed] [Google Scholar]
- Wakeling E. N., Nahhas F. A., Feldman G. L. (2014). Extra alleles in FMR1 triple-primed PCR: artifact, aneuploidy, or somatic mosaicism. J. Mol. Diagn. 16 689–696. 10.1016/j.jmoldx.2014.06.006 [DOI] [PubMed] [Google Scholar]
- Yrigollen C. M., Sweha S., Durbin-Johnson B., Zhou L., Berry-Kravis E., Fernandez-Carvajal I., et al. (2014). Distribution of AGG interruption patterns within nine world populations. Intractable Rare Dis. Res. 3 153–161. 10.5582/irdr.2014.01028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S., Pritchard M., Kremer E., Lynch M., Nancarrow J., Baker E., et al. (1991). Fragile X genotype characterized by an unstable region of DNA. Science 252 1179–1181. 10.1126/science.252.5009.1179 [DOI] [PubMed] [Google Scholar]
- Zhao X. N., Lokanga R., Allette K., Gazy I., Wu D., Usdin K. (2016). A MutSβ-dependent contribution of MutSα to repeat Expansions in fragile X premutation mice? PLOS Genet. 12:e1006190. 10.1371/journal.pgen.1006190 [DOI] [PMC free article] [PubMed] [Google Scholar]