

Summary
Deposition of amyloid is a common aging‐associated phenomenon in several aging‐related diseases. Osteoarthritis (OA) is the most prevalent joint disease, and aging is its major risk factor. Transthyretin (TTR) is an amyloidogenic protein that is deposited in aging and OA‐affected human cartilage and promotes inflammatory and catabolic responses in cultured chondrocytes. Here, we investigated the role of TTR in vivo using transgenic mice overexpressing wild‐type human TTR (hTTR‐TG). Although TTR protein was detected in cartilage in hTTR‐TG mice, the TTR transgene was highly overexpressed in liver, but not in chondrocytes. OA was surgically induced by destabilizing the medial meniscus (DMM) in hTTR‐TG mice, wild‐type mice of the same strain (WT), and mice lacking endogenous Ttr genes. In the DMM model, both cartilage and synovitis histological scores were significantly increased in hTTR‐TG mice. Further, spontaneous degradation and OA‐like changes in cartilage and synovium developed in 18‐month‐old hTTR mice. Expression of cartilage catabolic (Adamts4, Mmp13) and inflammatory genes (Nos2, Il6) was significantly elevated in cartilage from 6‐month‐old hTTR‐TG mice compared with WT mice as was the level of phospho‐NF‐κB p65. Intra‐articular injection of aggregated TTR in WT mice increased synovitis and significantly increased expression of inflammatory genes in synovium. These findings are the first to show that TTR deposition increases disease severity in the murine DMM and aging model of OA.
Keywords: aging, amyloid, cartilage, osteoarthritis, transthyretin
Introduction
Osteoarthritis (OA) is the most prevalent human joint disease with age being the main risk factor (Zhang & Jordan, 2010; Losina et al., 2013). There are currently no established approaches to prevent or slow the progression of OA (Zhang & Jordan, 2010) and over half of adults with knee OA undergo knee replacement (Weinstein et al., 2013). A large number of therapeutic targets have been identified and successfully tested in animal models (Yu & Hunter, 2015). However, thus far clinical trials targeting these pathways have failed (Hunter & Hellio Le Graverand‐Gastineau, 2009). Changes in articular cartilage appear to be the earliest event in disease initiation and are likely to be the main drivers of disease progression, but all the joint tissues are affected by the disease process (Loeser et al., 2012). Age‐related changes in cartilage have been characterized, but the mechanisms that mediate the effect of age on OA are unknown.
In studies of articular cartilage, menisci, and synovium from arthritic joints, the prevalence of amyloid deposits was between thirty and one hundred percent of joints examined (Goffin, 1983; Ladefoged, 1986; Solomon et al., 2006; Gobezie et al., 2007; Niggemeyer et al., 2011). Amyloid deposits in OA synovium were ranged from 8% (Takanashi 2013) to 25% (Gu et al. 2014) of the patients. The most common precursor is the thyroxin (T4) and retinol transporter protein transthyretin (TTR) (Goffin, 1983). TTR is mainly produced by hepatocytes, choroid plexus epithelium, and retinal pigment epithelium. It is found in blood plasma, the cerebrospinal fluid, and the vitreous humor of the eye. The protein is composed of four identical subunits (Blake et al., 1978). TTR amyloid formation requires tetramer dissociation into monomers that misfold and aggregate to initiate the amyloidosis cascade (Hurshman et al., 2004). In contrast with the less common forms of autosomal‐dominant familial amyloidotic polyneuropathy (FAP) and cardiomyopathy (FAC), the more prevalent senile systemic amyloidosis (SSA) is caused by the deposition of amyloid derived from wild‐type TTR (Buxbaum et al., 2008). It occurs mainly in elderly males, with its clinically dominant manifestations related to deposits of wild‐type TTR in the myocardium (Rapezzi et al., 2013). It may be accompanied by carpal tunnel syndrome with TTR deposition in the carpal ligament or gastrointestinal bleeding due to the involvement of blood vessels (Sekijima et al., 2011).
We have previously reported that all human cartilage samples collected at the time of joint replacement surgery were positive for amyloid by Congo red staining and TTR by immunohistochemistry and Western blotting (Akasaki et al., 2015). In addition, we showed in studies of primary cultured chondrocytes that exposure to amyloidogenic TTR affected chondrocyte survival and induced the expression of OA‐related genes (Akasaki et al., 2015). These findings raise the question of whether the deposits contribute to the process of cartilage degradation. It is also possible that the damaged tissues create an environment, which supports TTR tetramer dissociation, misfolding of the released monomers, and subsequent aggregation to oligomers and fibrils, which in turn could amplify the OA process. The objective of this study was to investigate the role of TTR in vivo in mice transgenic for 90‐100 copies of the wild‐type human TTR (hTTR‐TG mice) using an experimental OA and aging model.
Results
Pattern of TTR deposition in mouse knee joints and sources of TTR
As hTTR‐TG mice had not previously been examined for developmental changes in the skeleton, analysis of whole skeleton preparations of newborn (P2) mice was performed. They did not reveal any apparent developmental abnormalities and the long bone lengths were not significantly different from WT mice (Fig. S1).
As previously reported, Western blotting showed strong signals for human TTR in serum and liver from 9‐month‐old hTTR‐TG mice (Teng et al., 2001). In articular cartilage from 9‐month‐old hTTR‐TG mice, TTR was present at a much lower concentration than in serum or liver. However, no TTR protein was detected in the chondrocytes of these mice (Fig. S2A). There was no TTR in the cartilage, chondrocytes, liver, or serum from mTTR‐KO mice. PCR analysis showed a detectable but very low level of hTTR mRNA in cartilage from hTTR‐TG mice compared to liver (Fig. S2B). In 2‐month‐old WT mice, there were similarly low levels of mTTR mRNA in joint tissues compared to liver (Fig. S2C). These results indicate that the TTR protein found in cartilage and synovium comes from systemic rather than local sources.
Immunohistochemical analysis revealed TTR deposits in bone marrow and around blood vessels in the kidney of hTTR‐TG mice (Fig. 1A). TTR deposits were also evident in the synovium of 6‐, 12‐, and 18‐month‐old mice and 6‐month‐old mice with surgically induced OA (Fig. 1B). However, TTR deposition in cartilage was not detected in 6 (n = 5)‐ and 12‐month‐old (n = 14) hTTR‐TG mice, whereas 69% of the 18‐month‐old mice (9/13), and 100% of the mice (5/5) with surgically induced OA had TTR staining in cartilage. The predominant areas of TTR deposition were the regions of fibrillated cartilage in the 18‐month‐old hTTR‐TG mice and in the younger mice subjected to destabilization of the medial meniscus (Fig. 1C). The antibody used in Fig. 1 recognizes human but not mouse TTR. With an antibody that recognizes TTR in human and mouse, TTR was detected in bone marrow and blood vessels in 6‐month‐old hTTR and WT mice and at the articular cartilage surface in 18‐month‐old hTTR‐TG mice but not in WT and mTTR‐KO mice (Fig. S5).
Figure 1.
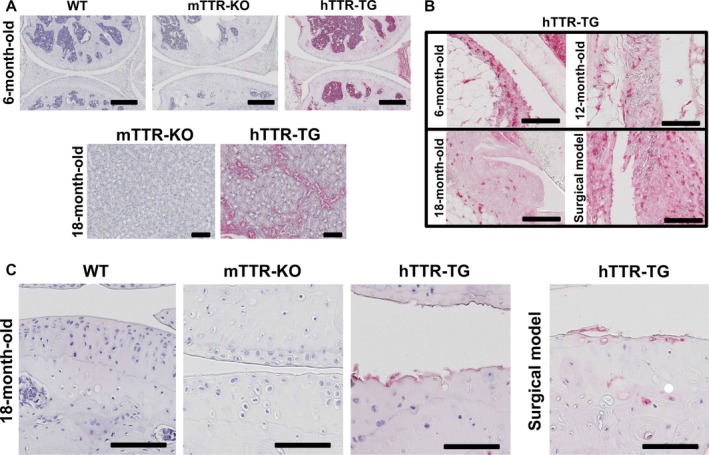
Immunohistochemistry for transthyretin (TTR) in bone marrow, kidney, and synovium. (A) TTR was detected in bone marrow and blood vessels of 6‐month‐old hTTR‐TG but not in wild‐type (WT) and mTTR‐KO mice, and around blood vessels in the kidney of 18‐month‐old hTTR‐TG mice (upper: scale bars = 400 μm, lower: scale bars = 100 μm). (B) TTR was detected in the synovium of 6‐, 12‐, and 18‐month‐old hTTR‐TG mice and hTTR‐TG mice with surgical OA (scale bars = 100 μm). (C) TTR was detected in fibrillated cartilage in 18‐month‐old hTTR‐TG mice and hTTR‐TG mice with surgical OA (scale bars = 100 μm). WT and mTTR‐KO mice showed no TTR staining.
hTTR‐TG mice show severe cartilage damage, synovitis and subchondral bone changes in an experimental OA model and in the aging model
To investigate the effect of hTTR deposition on cartilage, we used the experimental OA model induced by surgical destabilization of the medial meniscus (DMM) in 4‐month‐old male WT, hTTR‐TG, and mTTR‐KO mice. Ten weeks after DMM surgery, cartilage scores were significantly higher in hTTR‐TG mice (n = 26) than in mTTR‐KO mice (n = 22) and WT mice (n = 20) (Fig. 2A,B). Synovial scores were also significantly higher in hTTR‐TG mice than in mTTR‐KO or WT mice (Fig. 2C,D). Surprisingly, scores were also significantly higher in the mTTR‐KO compared to the WT mice but far lower than in the hTTR‐TG animals.
Figure 2.
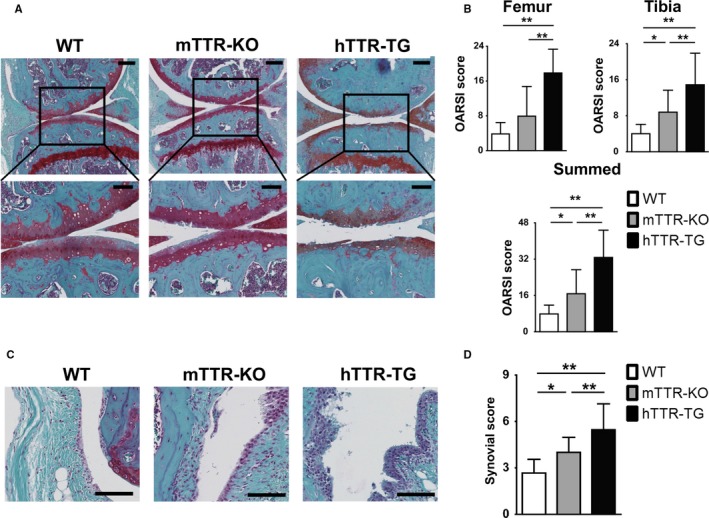
Cartilage and synovial lesions in surgically induced osteoarthritis (OA) in wild‐type (WT), mTTR‐KO, and hTTR‐TG mice. (A) Histological features of knee cartilage 10 weeks after OA surgery (safranin O/fast green; upper: 10×, scale bars = 200 μm, lower: 20×, scale bars = 100 μm). OA was surgically induced by destabilizing the medial meniscus in the right knee joints of 4‐month‐old male mice. (B) Histological OA grade was based on Osteoarthritis Research Society International (OARSI) scoring system. Cartilage scores were significantly higher in hTTR‐TG mice (n = 26) 10 weeks after surgery than in WT (n = 20) and mTTR‐KO mice (n = 22). (C) Histological changes in synovium 10 weeks after OA surgery (scale bars = 100 μm). (D) The synovial score was obtained with Krenn's synovitis scoring system. Synovial scores were significantly higher in hTTR‐TG mice (n = 26) 10 weeks after surgery than in WT (n = 20) and mTTR‐KO mice (n = 22). **P < 0.01 and *P < 0.05.
In the absence of surgical meniscus destabilization, hTTR‐TG mice showed increased severity of age‐related cartilage changes, synovitis, and subchondral bone changes at 18 months, which were not seen at 12 months (Fig. 3).
Figure 3.
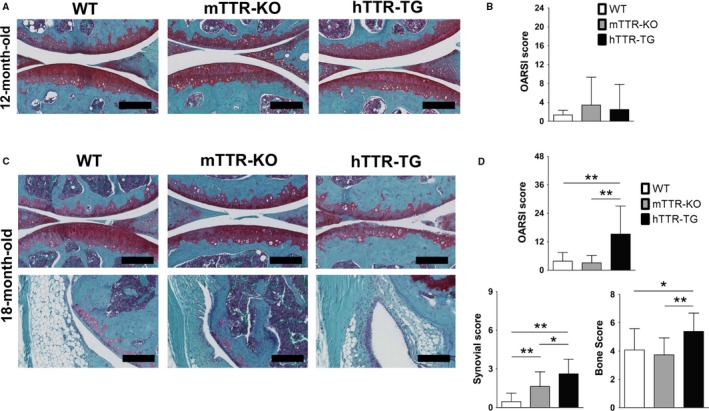
Cartilage, synovial, and subchondral bone histological changes in the aging model of wild‐type (WT), mTTR‐KO, and hTTR‐TG mice. (A) Histological features of cartilage in 12‐month‐old mice (safranin O/fast green; 20×, scale bars = 200 μm). (B) Histological osteoarthritis (OA) grades were based on OARSI scoring system. Cartilage scores were not significantly between WT (n = 12), mTTR‐KO mice (n = 14), and hTTR‐TG mice (n = 14) at 12 months of age. (C) Histological features of cartilage and synovium in 18‐month‐old mice (scale bars = 200 μm). (D) The synovial scores were obtained with Krenn's synovitis scoring system. Cartilage scores were significantly higher in hTTR‐TG mice (n = 18) than in WT (n = 13) and mTTR‐KO mice (n = 22) at 18 months of age. Synovial scores and subchondral bone scores were significantly higher in hTTR‐TG than in WT and mTTR‐KO mice. **P < 0.01 and *P < 0.05.
Markers and mediators of OA pathogenesis
To further examine the mechanism underlying accelerated cartilage damage in the hTTR‐TG mice, markers and mediators of cartilage degradation were examined. Cartilage cell numbers were significantly lower in hTTR‐TG mice at 6 months of age and 10 weeks after DMM surgery as compared to WT and mTTR‐KO mice (Fig. S3A,B).
Real‐time PCR of mRNA from knee cartilage revealed that transcription of the catabolic genes Adamts‐4 and 5, Mmp9, Mmp13, Nos2, and the cytokine Il‐6 was significantly elevated in 6‐month‐old hTTR‐TG mice compared to mTTR‐KO and WT mice (Fig. 4A). Immunohistochemistry showed higher intensity staining for the cartilage degradation marker CTX‐II in 6‐month‐old hTTR‐TG mice, and the number of cells positive for IL‐6, ADAMTS‐4, and MMP13 was significantly increased in 6‐month‐old hTTR‐TG mice (Fig. 4B). In addition, synovium of 6‐month‐old hTTR‐TG mice stained more intensely with antibodies for IL‐6, ADAMTS‐4, and MMP13 (Fig. S4). Furthermore, TUNEL staining showed that apoptotic cells in 12‐month‐old hTTR‐TG and mTTR‐KO mice were increased as compared to WT mice (Fig. 5A). Consistent with immunohistochemistry, in Western blots, the levels of ADAMTS‐4 and MMP‐13 were higher in hTTR‐TG mice (Fig. 5B).
Figure 4.
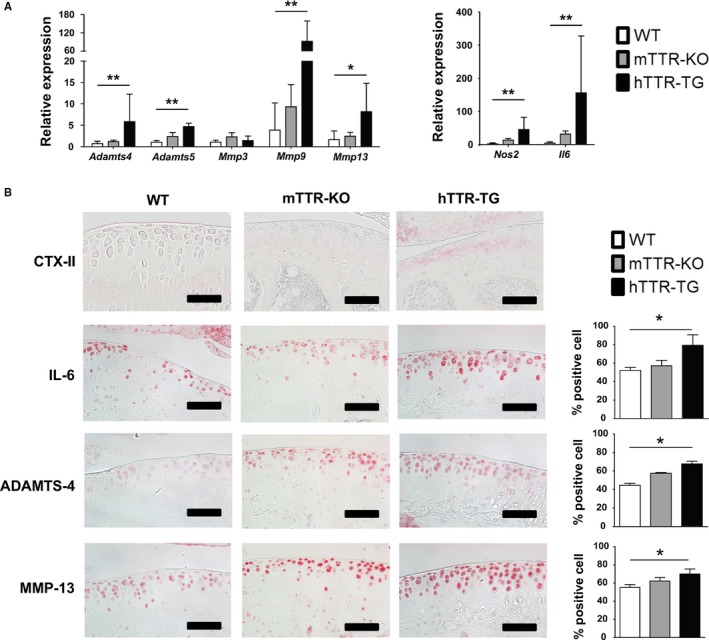
Mediators and markers of osteoarthritis (OA) in wild‐type (WT), mTTR‐KO, and hTTR‐TG mice. (A) RNA was isolated from knee articular cartilage from 6‐month‐old mice for RT‐qPCR. OA‐related genes were significantly elevated in cartilage from TTR‐TG mice (WT n = 6, mTTR‐KO n = 6, hTTR‐TG n = 5). The expression values were relative to Gapdh. **P < 0.01 and *P < 0.05. (B) Immunohistochemistry for CTX‐II, IL‐6, ADAMTS‐4, and MMP13 was performed on joint sections from 6‐month‐old mice (WT n = 4; mTTR‐KO n = 4; hTTR‐TG n = 4). Cells positive for IL‐6, ADAMTS‐4, and MMP13 were significantly elevated in 6‐month‐old hTTR‐TG mice (scale bars = 100 μm). The ratio of positive cells was counted using three sections from four mice for each genotype. *P < 0.05.
Figure 5.
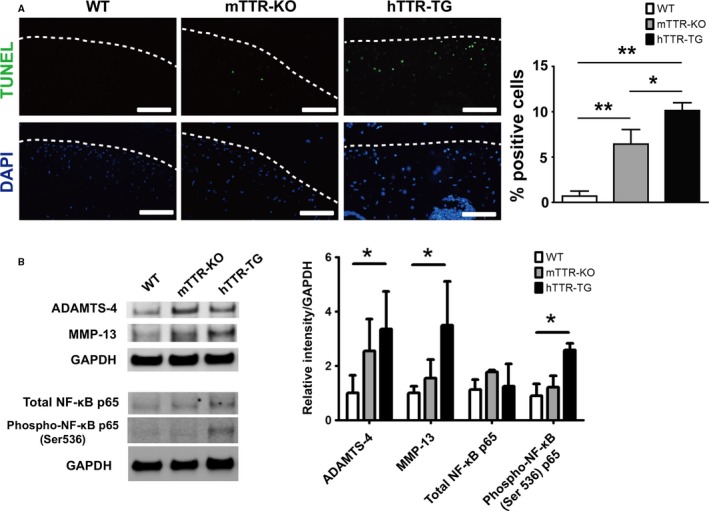
Apoptotic cells, MMP‐13, ADAMTS‐4 protein expression, and NF‐κB activation in cartilage from in WT, mTTR‐KO, and hTTR‐TG mice. (A) TUNEL staining (scale bars = 100 μm) and quantitative analysis of the TUNEL‐positive cell counts per field in the central weight‐bearing region of the medial tibial plateau in 12‐month‐old mice (n = 4, each). White dot shows the surface of cartilage. **P < 0.01. (B) Western blotting was performed on protein extracts from cartilage in 6‐month‐old WT, mTTR‐KO, and hTTR‐TG mice. Quantitative analysis of band intensity was measured using Image Studio (LI‐COR, USA). GAPDH served as a control. ADAMTS‐4, MMP‐13, and NF‐κB p65 were significantly elevated in hTTR‐TG mice (n = 4, each). *P < 0.05.
The NF‐κB pathway regulates the production of inflammatory mediators and cartilage matrix‐degrading enzymes (Lauder et al., 2007). We found that in the 6‐month‐old hTTR‐TG mice, the protein level of NF‐κB p65 phosphorylated at serine 536 was significantly increased compared with control mice (Fig. 5B).
Intra‐articular injection of TTR in WT mice
To examine the effect of aggregated TTR on the knee joint, intra‐articular injections of TTR were performed in WT mice. Injection of aggregated V122I led to increased synovitis (Fig. 6A,B) in a similar magnitude as IL‐1β, which was included as a positive control for a stimulus that is known to induce catabolic and inflammatory mediators in joint tissues (van de Loo et al. 1995). Real‐time PCR of mRNA from synovium also showed that inflammatory genes Nos2 and IL‐6 were significantly elevated in mice injected with aggregated TTR V122I compared to mice without injection or injection of nonaggregated TTR (Fig. 6C).
Figure 6.
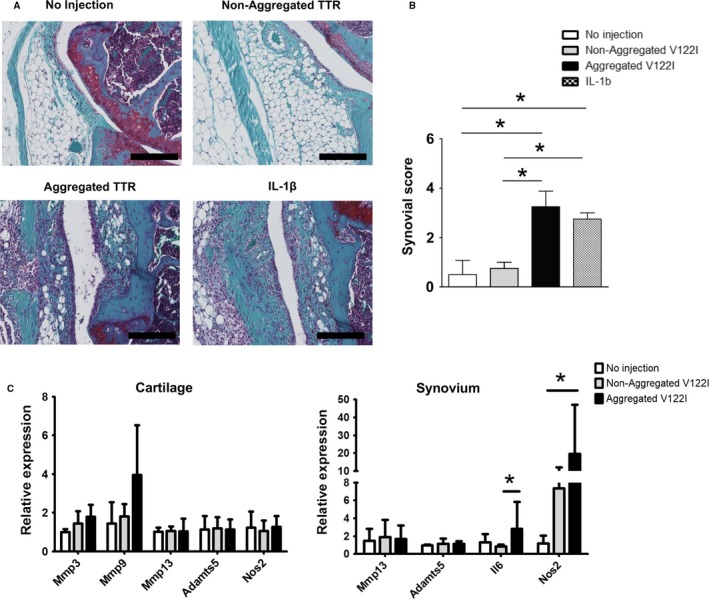
intra articular injection of aggregated transthyretin (TTR) in wild‐type (WT) mice. (A) Histological changes in synovium of 4‐month‐old WT mice 5 days after intra‐articular injections of aggregated TTR, nonaggregated V122 l TTR, or IL‐1β (scale bars = 200 μm). (B) Synovial scores were significantly higher in joints injected with aggregated V122I TTR and IL‐1β than nonaggregated V122I TTR or joints not receiving injections (each n = 4). *P < 0.05. (C) RNA was isolated from knee articular cartilage 1 week after injection. Inflammatory genes were significantly elevated in synovium from joints injected with aggregated V122I TTR (n = 5) than in joints injected with nonaggregated V122I TTR (n = 6) or not injected (n = 4). The values of mRNA expression were relative to Gapdh. *P < 0.05.
Discussion
The deposition of amyloid in aged and OA‐affected cartilage has been documented in several publications (Goffin, 1983; Ladefoged, 1986; Solomon et al., 2006; Gobezie et al., 2007; Niggemeyer et al., 2011) with TTR being the predominantly identified amyloid precursor (Goffin, 1983). The consequences of the presence of amyloid deposits for tissue integrity and OA pathogenesis have not been explored. In this study, we used transgenic mice overexpressing wild‐type human TTR and mTTR‐KO mice to study the role of TTR deposition in joint tissues. We used mice with transgenic overexpression of human rather than mouse TTR as the mouse protein is kinetically several orders of magnitude more stable than the human protein and hence is not subject to amyloid formation, which depends on tetramer dissociation (Reixach et al., 2008).
TTR deposition was not detected in the cartilage of 6‐ and 12‐month‐old hTTR‐TG mice, but 69% of 18‐month‐old hTTR‐TG mice were positive for TTR deposits in cartilage. All hTTR‐TG mice were positive for TTR immunostaining in cartilage in the surgically induced OA model. However, Congo red staining for amyloid fibrils was negative in all mice tested. Hence, the TTR deposits were nonfibrillar.
The hTTR‐TG mouse strain that we have studied showed nonfibrillar, noncongophilic anti‐human TTR‐positive deposits between 12 and 17 months of age in the kidneys and heart (Teng et al., 2001). In mice over 18 months of age, TTR‐related deposits were found in 84% in the kidneys and 39% in the heart, while Congo red staining was found in 17% of the kidneys and in 16% of the hearts (Teng et al., 2001). Congophilic deposition was rarely seen before 18 months of age (Teng et al., 2001). The difference between the mice showing cardiac deposition and those that do not is whether their livers mount an unfolded protein response (UPR) (Buxbaum et al., 2008) and the nature of the inflammatory response in the heart. As those responses consist of endogenous mouse chaperones and inflammatory molecules, they do not appear to be related to the species specificity of the UPR and other chaperones involved in the organismal response to overexpression of human TTR in mice.
Our previous study on human cartilage revealed Congo red‐positive amyloid deposits in 58% of aged normal cartilage and all OA cartilage samples (Akasaki et al., 2015). TTR was detected in all OA cartilage samples and 83% of aged normal cartilage samples by immunohistochemistry (Akasaki et al., 2015). Collectively studies on wild‐type and mutant TTR indicate that age‐related changes in the tissue environment and/or the precursor protein are likely to be required for the deposition of TTR aggregates. The finding that TTR deposits formed in cartilage from young hTTR mice with surgically induced OA suggests that OA‐related cartilage changes promote TTR deposition, which, in turn, seems to amplify the OA damage.
We examined potential sources of TTR. In WT mice, the levels of TTR mRNA are very low in cartilage as compared to liver, the main source of TTR production (Holmgren et al., 1991, 1993). The level of hTTR mRNA in cartilage was also very low in the hTTR‐TG mice. The hTTR transgene in these mice contains all known regulatory regions responsible for tissue‐specific TTR expression (Teng et al., 2001) and thus recapitulates the physiological tissue distribution of TTR. We reported previously that TTR levels in human synovial fluid were similar to plasma, and there were no differences between OA and normal (Akasaki et al., 2015). Collectively, the prior and current data from the mouse models suggest that it is not a change in TTR production, but changes in the tissue environment and possible posttranslational changes in TTR that are the likely drivers of TTR deposition in arthritic joints.
The main observation from the present study is that hTTR‐TG mice develop more severe cartilage damage and synovitis than WT and mTTR‐KO mice in the surgically induced OA model and aging model. Although these mice were negative for amyloid fibrils as detected by Congo red staining, the observation is consistent with recent evidence from studies on tissues with TTR deposits indicating that inflammation, apoptosis, and reactive oxygen species are present in human carriers of mutant TTR variants and transgenic mice, well before amyloid deposits can be detected (Sousa et al., 2002; Starck & Sutherland‐Smith, 2010). In addition, that we were using, the hearts of 3‐month‐old transgenic mice overexpressing human WT TTR had a significant increase in the transcription levels of genes related to inflammation and immune response, compared to nontransgenic littermates (Buxbaum et al., 2012) even though these transgenic animals do not display cardiac TTR deposition until 18 months of age (Buxbaum et al., 2012). The hTTR‐TG mice did not present abnormalities in skeletal development and there were no differences in joint pathology compared to WT and mTTR‐KO mice by 12 months. However, at 18 months, hTTR‐TG mice developed significantly increased OA degeneration and synovial changes compared to WT and mTTR‐KO mice.
One of the main mechanisms of TTR amyloid pathogenesis is cytotoxicity (Reixach et al., 2004; Akasaki et al., 2015). We showed that amyloidogenic TTR‐induced cell death in cultured chondrocytes (Akasaki et al., 2015) and one of the histological features of the hTTR mice was reduced cartilage cellularity in the DMM model. Thus, it appears that this is also one factor contributing to the increased OA severity in these mice.
It has been demonstrated that TTR activates the receptor for glycation end products (RAGE) and stimulates nuclear translocation of NF‐κB (Sousa et al., 2000). Consistent with those observations, we have previously found that an anti‐RAGE antibody inhibited V122I TTR variant‐induced chondrocyte iNOS gene expression (Akasaki et al., 2015). It is known that RAGE is increased in OA cartilage, apparently mediating the induction of metalloproteinases and inflammatory mediators through the NF‐κB pathway (Loeser et al., 2005; Steenvoorden et al., 2006). The consequences of that activation are increased cartilage degradation, synovitis, and chondrocyte hypertrophy (Cecil et al., 2005; Steenvoorden et al., 2006). Further, our data indicate the cartilage‐degrading enzymes ADAMTS‐4 and MMP‐13, and the cytokine IL‐6 were all increased in the 6‐month‐old hTTR‐TG mice. In addition, NF‐κB p65 phosphorylation was increased in hTTR‐TG mice compared with control mice, suggesting that the increase in inflammatory mediators in hTTR‐TG mice might be related to activation of the NF‐κB pathway. To directly test effects of TTR on joint tissues, we performed intra‐articular injections in WT mice and found that aggregated but not nonaggregated TTR led to synovitis and increased expression of inflammatory genes in synovium. These mice were only observed for 5 days after the TTR injections to establish proof‐of‐concept that TTR aggregates are biologically active in vivo. Longer term injections would be necessary to observe changes in articular cartilage.
An additional mechanism of TTR and inflammation relates to the formation of autoantibodies against TTR, which have been reported in rheumatoid arthritis (Sharma et al. 2014) and juvenile idiopathic arthritis (Clement et al. 2016). Misfolding, aggregation, and oxidation of TTR enhanced its immunogenicity in a transgenic mouse model (Clement et al. 2016) and other posttranslational modifications of TTR were observed in human RA sera (Ni et al. 2013). However, there was no difference in autoantibodies against TTR between OA patients and healthy controls (Sharma et al. 2014).
In addition to TTR deposition in cartilage and synovium, it is also possible that TTR aggregates may form in subchondral bone marrow in OA‐affected joints and contribute to changes in subchondral bone and cartilage. The close association of changes in subchondral bone and articular cartilage has been recognized in human OA and in animal models (Weinans et al. 2012). Furthermore, systemic increases in inflammatory mediators due to TTR deposition in extraarticular tissues such as liver and serum may contribute to increased joint inflammation and OA severity.
The present study also revealed, somewhat paradoxically, that mice with deletion of the TTR gene had significantly increased OA severity as compared to wild‐type mice in OA model. This implies a previously unknown physiological function of TTR that appears to be required for joint homeostasis independent of the amyloidogenicity of human TTR. Murine TTR is 80% homologous to human TTR. Nonetheless, it is not amyloidogenic because of its greater kinetic stability (Reixach et al., 2008). Our study shows that skeletal development and growth are not significantly different between WT mice and mTTR‐KO mice, even though there are reduced levels of serum retinol, retinol‐binding protein, and thyroid hormone (Palha et al., 1994). However, our results suggest that there may be effects of TTR deficiency on articular cartilage that may result in increased susceptibility to OA in the setting of induced joint damage. The role of thyroid hormones in regulating hypertrophic chondrocyte differentiation is well known (Williams, 2013), and recent studies indicate that thyroxin for which TTR functions as a carrier protein stimulated cartilage formation in mesenchymal stem cells (Lee et al., 2015). However, only limited information is available on the role of thyroxine in articular chondrocytes (Rosenthal et al., 2003). Further analyses are needed to elucidate mechanisms of TTR and thyroxin in articular cartilage homeostasis.
Our findings that TTR amyloid deposition is highly prevalent in aging and OA‐affected joints (Akasaki et al., 2015) and that TTR promotes inflammation and cartilage in mouse models suggest that TTR deposition contributes to the progression of OA. Recent advances in the understanding of mechanisms of TTR aggregation (Eisele et al., 2015) led to the discovery of TTR stabilizers such as tafamidis or diflunisal (Adamski‐Werner et al., 2004; Obici & Merlini, 2014) and tafamidis have been approved for the treatment of familial amyloidotic polyneuropathy caused by amyloid formed by mutant TTR (Coelho et al., 2013; Waddington Cruz & Benson, 2015). Evaluation of these drugs in preclinical models of OA will be the next step in establishing proof‐of‐concept that prevention of TTR aggregation is a potential therapeutic approach for OA.
This study is the first to show that transgenic overexpression of TTR led to deposition on cartilage and increased the severity of cartilage damage and synovitis in the surgically induced murine OA model. Together with prior observations that aging and OA in humans are associated with TTR and amyloid deposition in cartilage, the present findings suggest that reducing TTR amyloid formation can be a new therapeutic approach for OA.
Experimental procedures
Mice
hTTR‐transgenic (hTTR‐TG) and mice lacking endogenous Ttr genes (mTTR‐KO) mice were generated as described previously (Teng et al., 2001), and wild‐type mice of the same Sv129 strain (WT) were used. All procedures were performed according to protocols approved by the Institutional Animal Care and Use Committee (IACUC) at The Scripps Research Institute. All mice were freely allowed to access to food, water, and activity.
Cell culture and Western blotting analysis
Chondrocytes were isolated from the femoral condyle and tibial plateau of postnatal day 1 control and hTTR‐TG mice (n = 5, pooled), as described previously (Gosset et al., 2008). Chondrocytes were seeded on 6‐well plates (2 × 105/well) and incubated for 24 h, and protein was extracted as described (Akasaki et al., 2015). Proteins were separated by NuPAGE (Thermo Fisher Scientific, Waltham, MA, USA) and transferred to nitrocellulose. The following antibodies were used: human TTR (Dako, Santa Clara, CA, USA), ADAMTS‐4 (Abcam, Cambridge, MA, USA), MMP‐13 (Abcam), nuclear factor‐κB (NF‐κB) p65 (Cell Signaling, Danvers, MA, USA), and phospho‐NF‐κB p65 (Ser536) (Cell Signaling). The primary antibodies were added and incubated at 4°C overnight. After secondary antibody application, signal detection was performed using an LI‐COR immunofluorescence detection system as described (Akasaki et al., 2014).
Surgical OA model and aging model in mice
To examine the effect of TTR overexpression on induced cartilage damage, we analyzed hTTR‐TG mice and compared them with WT and mTTR‐KO mice using an experimental OA model (Glasson et al., 2007). OA was surgically induced by destabilizing the medial meniscus (DMM) in the right knee joints of 4‐month‐old male WT, mTTR‐KO, and hTTR‐TG mice. The left knees were subjected to sham surgery. The mice were killed at 10 weeks after surgery, and the entire knee joints were fixed in 10% zinc‐buffered formalin for 2 days and decalcified in TBD‐2 for 24 h. Sections of the mouse knee joints were used for further analysis. The destabilized and sham‐operated joints were processed and analyzed in parallel. To examine the role of hTTR during aging, we compared WT, mTTR‐KO, and hTTR‐TG mice at 12 and 18 months.
Intra‐articular injection of TTR
To examine the direct effect of aggregated TTR on knee joint tissue, intra‐articular injections of aggregated (25 μM), nonaggregated V122 l (25 μM), and IL‐1β (100 ng) were performed in 4‐month‐old WT mice three times on days 0, 2, and 4. Aggregated V122I solution was created using acetate buffer (200 mM sodium acetate/100 mM KCl/1 mM EDTA, pH 4.2) as described previously (Reixach et al., 2008) and incubated at 37°C for 3 days. The precipitate was diluted in Hank's balanced salt solution. One day after final intra‐articular injection, knee joints were collected for histological analysis and RNA isolation.
Immunohistochemistry
Joint sections were deparaffinized, and the slides were washed and blocked with 10% goat serum for 1 h at room temperature. Antigen retrieval was used as necessary. Anti‐human TTR (Dako A0002; 1:400), anti‐mouse and human TTR (Abcam ab9015; 1:100), anti‐CTX‐II (Cloud‐Clone Corp. PAA686Hu01; 1:2000 dilution), anti‐ADAMTS‐4 (Abcam ab28285; 1:100 dilution), anti‐MMP‐13 (Acam ab39012; 1:100 dilution), anti‐IL‐6 (Abcam ab6672; 1:600 dilution), and negative control rabbit IgG (Vector I‐1000; 1 μg/mL) were applied with 0.1% Tween 20 and incubated overnight at 4°C, and followed by secondary antibody using Vectastain ABC‐AP alkaline phosphatase (Vector Laboratories, Burlingame, CA, USA). Slides were washed, and sections were incubated with alkaline phosphatase substrate for 5–10 min. Specificity controls were obtained by replacing the primary antibody with nonimmune rabbit IgG (1 μg/mL), and no staining was observed under these conditions. Hematoxylin was used for counter staining. The percent positive cells was determined as the ratio of the total number of positive cells to the total number of chondrocytes in the section. TUNEL staining was performed using In Situ Cell Death Detection Kit (Roche Applied Science, Indianapolis, IN, USA). The number of positive cells was quantified in tibial cartilage and the percent positive cells was determined as the ratio of the total number of TUNEL‐positive cells to the total number of DAPI‐positive cells in the section.
RNA and protein isolation, and real‐time PCR
Articular cartilage was collected from both sides of the femoral head, femoral condyle, and tibial plateau of each mouse at the ages indicated as described previously (Gepstein et al., 2002; Xu et al., 2005). Care was taken to avoid any other tissue such as synovium in the cartilage collection and to resect full thickness articular cartilage without subchondral bone. In intra‐articular injection study, only knee joints were collected for RNA. Synovium was carefully resected and total RNA was extracted using TRIzol (Invitrogen, Burlington, ON), followed by Zymo Direct‐zol RNA MiniPrep kits (Zymo Research, Irvine, CA, USA), according to the manufacturer's instructions. Real‐time PCR was performed on a Light Cycler 480 instrument (Roche Diagnostics, Indianapolis, IN, USA) using the following TaqMan probes: MMP‐3 (Mm00440295_m1), MMP‐9 (Mm00442991_m1), MMP‐13 (Mm01168713_m1), ADAMTS‐4 (Mm00556068_m1), ADAMTS‐5 (Mm01344182_m1), NOS2 (Mm01309897_m1), IL‐6 (Mm00446190_m1). Protein isolation was performed on the residue from the RNA extraction (Akasaki et al., 2014).
Histological analysis
The histological OA scores for medial femoral condyle, the medial tibial plateau, and summed scores of femur and tibia were evaluated using the Osteoarthritis Research Society International (OARSI) cartilage OA histopathology semiquantitative scoring system (score 0–24) (Pritzker et al., 2006). The histological synovial score was evaluated using Krenn's synovitis scoring system (score 0–9) (Krenn et al., 2002). To evaluate the subchondral bone, we used a scoring system (score 0–8) based on previous publications (Botter et al., 2011; Ko et al., 2013) that evaluates trabecular bone structure (mostly cystic: 2, partially cystic: 1), lamellar structure (destroyed: 2, partially destroyed: 1), angiogenesis (number of vessels ≥3: 2, 1–2: 1), and ectopic ossification/articular cartilage absorption in the tibia. (present: 2). Histological grading was performed by two blinded observers.
Skeletal preparation
Whole skeletons of WT mTTR‐KO and hTTR‐TG mice on postnatal day 2 were fixed in 95% ethanol. Alcian blue staining was performed, followed by placement into potassium hydroxide, and alizarin red staining.
Statistical methods
Descriptive statistics are summarized as mean ± standard deviation (SD). An unpaired two‐tailed Student's t‐test or Mann–Whitney U‐test was used to compare differences between two groups. One‐way analysis of variance was used to compare multiple groups, with subsequent pairwise (group) comparisons assessed via Bonferroni's procedure, at an experiment‐wise error level of 0.05. P values less than 0.05 were considered statistically significant.
Funding
This study was supported by National Institutes of Health grant AG007996.
Conflict of interest
The authors declare that they have no competing for financial interests.
Author contributions
MKL had full access to all the data in the study and took responsibility for the integrity of the data and the accuracy of the data analysis. TM, JB, NR, and MKL designed research. TM, MO, YA, OA performed research. TM and MKL analyzed data. All authors involved in writing the paper.
Supporting information
Fig. S1 Whole mouse skeletal preparations. Double‐staining with alizarin red and alcian blue of whole skeletal preparations was performed on P2 WT, mTTR‐KO, and hTTR‐TG mice (scale bars = 500 μm). (B) Length of femur and tibia (scale bars = 2 mm) in P2 control (n = 7), mTTR‐KO (n = 7), and hTTR‐TG (n = 6) mice. N.S. = Non‐Significant.
Fig. S2 TTR mRNA and protein expression in chondrocytes, cartilage, synovium, serum, and liver. (A) Chondrocytes were isolated from the femoral condyle and tibial plateau of postnatal day 1 (P1). Cartilage, liver and serum protein were extracted from 9‐month‐old mice. Human recombinant TTR was used as a positive control. Western blotting showed that TTR was detected in cartilage in 9‐month‐old hTTR‐TG mice, but expressed at low levels in chondrocytes. High TTR expression was observed in liver and serum in hTTR‐TG but not in mTTR‐KO. (B) Real‐time PCR for human Ttr in 2‐month‐old hTTR‐TG mice showed lower expression in cartilage than liver (n = 3, each). (C) Real‐time PCR for mouse Ttr in 2‐month‐old wild‐type C57BL/6 mice showed that cartilage and synovium were lower in primary chondrocytes (n = 6).
Fig. S3 Cartilage cellularity. (A) Representative images medial tibia cartilage of Safranin O stained joint sections. B) Three micrographs of the medial tibial plateau were taken under 40× magnification. Cell numbers per each area were counted in 6‐month‐old mice and mice with surgical OA (scale bars = 400 μm, 6 months: WT n = 20, hTTR‐TG n = 28, mTTR‐KO n = 28, Surgical model: WT n = 20, hTTR‐TG n = 26, mTTR‐KO n = 22). **P < 0.01.
Fig. S4 Expression of OA‐related markers in synovium. Immunohistochemistry for IL‐6, ADAMTS‐4, and MMP13 was performed on joint sections from 6‐month‐old mice (WT n = 4; mTTR‐KO n = 4; hTTR‐TG n = 4, scale bars = 100 μm).
Fig. S5 Immunohistochemistry with antibody that recognizes human and mouse TTR. (A) TTR was detected in bone marrow, blood vessels and at the articular cartilage surface of 6‐month‐old hTTR‐TG and WT mice but not in mTTR‐KO mice (scale bars = 400 μm). (B) TTR was detected in fibrillated cartilage in 18‐month‐old hTTR‐TG mice and in chondrocyte in hTTR‐TG and WT mice. There was no TTR staining in the cartilage in 18‐month‐old mTTR‐KO mice (scale bars = 100 μm).
Acknowledgments
None.
References
- Adamski‐Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW (2004) Diflunisal analogues stabilize the native state of transthyretin. Potent inhibition of amyloidogenesis. J. Med. Chem. 47, 355–374. [DOI] [PubMed] [Google Scholar]
- Akasaki Y, Hasegawa A, Saito M, Asahara H, Iwamoto Y, Lotz MK (2014) Dysregulated FOXO transcription factors in articular cartilage in aging and osteoarthritis. Osteoarthritis Cartilage 22, 162–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akasaki Y, Reixach N, Matsuzaki T, Alvarez‐Garcia O, Olmer M, Iwamoto Y, Buxbaum JN, Lotz MK (2015) Transthyretin deposition in articular cartilage: a novel mechanism in the pathogenesis of osteoarthritis. Arthritis Rheumatol. 67, 2097–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake CC, Geisow MJ, Oatley SJ, Rerat B, Rerat C (1978) Structure of prealbumin: secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 A. J. Mol. Biol. 121, 339–356. [DOI] [PubMed] [Google Scholar]
- Botter SM, van Osch GJ, Clockaerts S, Waarsing JH, Weinans H, van Leeuwen JP (2011) Osteoarthritis induction leads to early and temporal subchondral plate porosity in the tibial plateau of mice: an in vivo microfocal computed tomography study. Arthritis Rheum. 63, 2690–2699. [DOI] [PubMed] [Google Scholar]
- Buxbaum J, Koziol J, Connors LH (2008) Serum transthyretin levels in senile systemic amyloidosis: effects of age, gender and ethnicity. Amyloid 15, 255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JN, Tagoe C, Gallo G, Walker JR, Kurian S, Salomon DR (2012) Why are some amyloidoses systemic? Does hepatic “chaperoning at a distance” prevent cardiac deposition in a transgenic model of human senile systemic (transthyretin) amyloidosis? FASEB J. 26, 2283–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecil DL, Johnson K, Rediske J, Lotz M, Schmidt AM, Terkeltaub R (2005) Inflammation‐induced chondrocyte hypertrophy is driven by receptor for advanced glycation end products. J. Immunol. 175, 8296–8302. [DOI] [PubMed] [Google Scholar]
- Clement CC, Moncrieffe H, Lele A, Janow G, Becerra A, Bauli F, Saad FA, Perino G, Montagna C, Cobelli N, Hardin J, Stern LJ, Ilowite N, Porcelli SA, Santambrogio L (2016). Autoimmune response to transthyretin in juvenile idiopathic arthritis. JCI Insight. 1. [DOI] [PMC free article] [PubMed]
- Coelho T, Maia LF, da Silva AM, Cruz MW, Plante‐Bordeneuve V, Suhr OB, Conceicao I, Schmidt HH, Trigo P, Kelly JW, Labaudiniere R, Chan J, Packman J, Grogan DR (2013) Long‐term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J. Neurol. 260, 2802–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele YS, Monteiro C, Fearns C, Encalada SE, Wiseman RL, Powers ET, Kelly JW (2015) Targeting protein aggregation for the treatment of degenerative diseases. Nat. Rev. Drug Discov. 14, 759–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gepstein A, Shapiro S, Arbel G, Lahat N, Livne E (2002) Expression of matrix metalloproteinases in articular cartilage of temporomandibular and knee joints of mice during growth, maturation, and aging. Arthritis Rheum. 46, 3240–3250. [DOI] [PubMed] [Google Scholar]
- Glasson SS, Blanchet TJ, Morris EA (2007) The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage 15, 1061–1069. [DOI] [PubMed] [Google Scholar]
- Gobezie R, Kho A, Krastins B, Sarracino DA, Thornhill TS, Chase M, Millett PJ, Lee DM (2007) High abundance synovial fluid proteome: distinct profiles in health and osteoarthritis. Arthritis Res. Ther. 9, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin YA (1983) The association of amyloid deposits and osteoarthritis. Arthritis Rheum. 26, 120. [DOI] [PubMed] [Google Scholar]
- Gosset M, Berenbaum F, Thirion S, Jacques C (2008) Primary culture and phenotyping of murine chondrocytes. Nat. Protoc. 3, 1253–1260. [DOI] [PubMed] [Google Scholar]
- Gu YJ, Ge P, Mu Y, Lu JH, Zheng F, Sun XG (2014) Clinical and laboratory characteristics of patients having amyloidogenic transthyretin deposition in osteoarthritic knee joints. J Zhejiang Univ Sci B 15, 92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren G, Steen L, Ekstedt J, Groth CG, Ericzon BG, Eriksson S, Andersen O, Karlberg I, Norden G, Nakazato M, Hawkins PN, Richardson S, Pepys MB. (1991) Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP‐met30). Clin. Genet. 40, 242–246. [DOI] [PubMed] [Google Scholar]
- Holmgren G, Ericzon BG, Groth CG, Steen L, Suhr O, Andersen O, Wallin BG, Seymour A, Richardson S, Hawkins PN, Pepys MB. (1993) Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet 341, 1113–1116. [DOI] [PubMed] [Google Scholar]
- Hunter DJ, Hellio Le Graverand‐Gastineau MP (2009) How close are we to having structure‐modifying drugs available? Med. Clin. North Am. 93, 223–234. xiii. [DOI] [PubMed] [Google Scholar]
- Hurshman AR, White JT, Powers ET, Kelly JW (2004) Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry 43, 7365–7381. [DOI] [PubMed] [Google Scholar]
- Ko FC, Dragomir C, Plumb DA, Goldring SR, Wright TM, Goldring MB, van der Meulen MC (2013) In vivo cyclic compression causes cartilage degeneration and subchondral bone changes in mouse tibiae. Arthritis Rheum. 65, 1569–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenn V, Morawietz L, Haupl T, Neidel J, Petersen I, Konig A (2002) Grading of chronic synovitis – a histopathological grading system for molecular and diagnostic pathology. Pathol. Res. Pract. 198, 317–325. [DOI] [PubMed] [Google Scholar]
- Ladefoged C (1986) Amyloid deposits in the knee joint at autopsy. Ann. Rheum. Dis. 45, 668–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauder SN, Carty SM, Carpenter CE, Hill RJ, Talamas F, Bondeson J, Brennan P, Williams AS (2007) Interleukin‐1β induced activation of nuclear factor‐kappab can be inhibited by novel pharmacological agents in osteoarthritis. Rheumatology (Oxford) 46, 752–758. [DOI] [PubMed] [Google Scholar]
- Lee JK, Gegg CA, Hu JC, Reddi AH, Athanasiou KA (2015) Thyroid hormones enhance the biomechanical functionality of scaffold‐free neocartilage. Arthritis Res. Ther. 17, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeser RF, Yammani RR, Carlson CS, Chen H, Cole A, Im HJ, Bursch LS, Yan SD (2005) Articular chondrocytes express the receptor for advanced glycation end products: potential role in osteoarthritis. Arthritis Rheum. 52, 2376–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeser RF, Goldring SR, Scanzello CR, Goldring MB (2012) Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 64, 1697–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losina E, Weinstein AM, Reichmann WM, Burbine SA, Solomon DH, Daigle ME, Rome BN, Chen SP, Hunter DJ, Suter LG, Jordan JM, Katz JN (2013) Lifetime risk and age at diagnosis of symptomatic knee osteoarthritis in the US. Arthritis Care Res. (Hoboken) 65, 703–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni M, Wei W, Feng Q, Sun XG, Wang YC, Gu YJ, Zheng F (2013) Transthyretin as a potential serological marker for the diagnosis of patients with early rheumatoid arthritis. Clin Exp Rheumatol 31, 394–399. [PubMed] [Google Scholar]
- Niggemeyer O, Steinhagen J, Deuretzbacher G, Zustin J, Ruther W (2011) Amyloid deposition in osteoarthritis of the hip. Arch. Orthop. Trauma Surg. 131, 637–643. [DOI] [PubMed] [Google Scholar]
- Obici L, Merlini G (2014) An overview of drugs currently under investigation for the treatment of transthyretin‐related hereditary amyloidosis. Expert Opin. Investig. Drugs 23, 1239–1251. [DOI] [PubMed] [Google Scholar]
- Palha JA, Episkopou V, Maeda S, Shimada K, Gottesman ME, Saraiva MJ (1994) Thyroid hormone metabolism in a transthyretin‐null mouse strain. J. Biol. Chem. 269, 33135–33139. [PubMed] [Google Scholar]
- Pritzker KP, Gay S, Jimenez SA, Ostergaard K, Pelletier JP, Revell PA, Salter D, van den Berg WB (2006) Osteoarthritis cartilage histopathology: grading and staging. Osteoarthritis Cartilage 14, 13–29. [DOI] [PubMed] [Google Scholar]
- Rapezzi C, Quarta CC, Obici L, Perfetto F, Longhi S, Salvi F, Biagini E, Lorenzini M, Grigioni F, Leone O, Cappelli F, Palladini G, Rimessi P, Ferlini A, Arpesella G, Pinna AD, Merlini G, Perlini S (2013) Disease profile and differential diagnosis of hereditary transthyretin‐related amyloidosis with exclusively cardiac phenotype: an Italian perspective. Eur. Heart J. 34, 520–528. [DOI] [PubMed] [Google Scholar]
- Reixach N, Deechongkit S, Jiang X, Kelly JW, Buxbaum JN (2004) Tissue damage in the amyloidoses: transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc. Natl. Acad. Sci. U. S. A. 101, 2817–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reixach N, Foss TR, Santelli E, Pascual J, Kelly JW, Buxbaum JN (2008) Human‐murine transthyretin heterotetramers are kinetically stable and non‐amyloidogenic. A lesson in the generation of transgenic models of diseases involving oligomeric proteins. J. Biol. Chem. 283, 2098–2107. [DOI] [PubMed] [Google Scholar]
- Rosenthal AK, Heinkel D, Gohr CM (2003) Thyroxine stimulates transglutaminase activity in articular chondrocytes. Osteoarthritis Cartilage 11, 463–470. [DOI] [PubMed] [Google Scholar]
- Sekijima Y, Uchiyama S, Tojo K, Sano K, Shimizu Y, Imaeda T, Hoshii Y, Kato H, Ikeda S (2011) High prevalence of wild‐type transthyretin deposition in patients with idiopathic carpal tunnel syndrome: a common cause of carpal tunnel syndrome in the elderly. Hum. Pathol. 42, 1785–1791. [DOI] [PubMed] [Google Scholar]
- Sharma S, Ghosh S, Singh LK, Sarkar A, Malhotra R, Garg OP, Singh Y, Sharma RS, Bhakuni DS, Das TK, Biswas S (2014) Identification of autoantibodies against transthyretin for the screening and diagnosis of rheumatoid arthritis. PLoS One 9, e93905. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Solomon A, Murphy CL, Kestler D, Coriu D, Weiss DT, Makovitzky J, Westermark P (2006) Amyloid contained in the knee joint meniscus is formed from apolipoprotein A‐I. Arthritis Rheum. 54, 3545–3550. [DOI] [PubMed] [Google Scholar]
- Sousa MM, Yan SD, Stern D, Saraiva MJ (2000) Interaction of the receptor for advanced glycation end products (RAGE) with transthyretin triggers nuclear transcription factor kB (NF‐kB) activation. Lab. Invest. 80, 1101–1110. [DOI] [PubMed] [Google Scholar]
- Sousa MM, Fernandes R, Palha JA, Taboada A, Vieira P, Saraiva MJ (2002) Evidence for early cytotoxic aggregates in transgenic mice for human transthyretin Leu55Pro. Am. J. Pathol. 161, 1935–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starck CS, Sutherland‐Smith AJ (2010) Cytotoxic aggregation and amyloid formation by the myostatin precursor protein. PLoS ONE 5, e9170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenvoorden MM, Huizinga TW, Verzijl N, Bank RA, Ronday HK, Luning HA, Lafeber FP, Toes RE, DeGroot J (2006) Activation of receptor for advanced glycation end products in osteoarthritis leads to increased stimulation of chondrocytes and synoviocytes. Arthritis Rheum. 54, 253–263. [DOI] [PubMed] [Google Scholar]
- Takanashi T, Matsuda M, Yazaki M, Yamazaki H, Nawata M, Katagiri Y, Ikeda S (2013) Synovial deposition of wild‐type transthyretin‐derived amyloid in knee joint osteoarthritis patients. Amyloid 20, 151–155. [DOI] [PubMed] [Google Scholar]
- Teng MH, Yin JY, Vidal R, Ghiso J, Kumar A, Rabenou R, Shah A, Jacobson DR, Tagoe C, Gallo G, Buxbaum J (2001) Amyloid and nonfibrillar deposits in mice transgenic for wild‐type human transthyretin: a possible model for senile systemic amyloidosis. Lab. Invest. 81, 385–396. [DOI] [PubMed] [Google Scholar]
- van de Loo AA, Arntz OJ, Bakker AC, van Lent PL, Jacobs MJ, van den Berg WB (1995) Role of interleukin 1 in antigen‐induced exacerbations of murine arthritis. Am J Pathol 146, 239–249. [PMC free article] [PubMed] [Google Scholar]
- Waddington Cruz M, Benson MD (2015) A review of tafamidis for the treatment of transthyretin‐related amyloidosis. Neurol. Ther. 4, 61–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinans H, Siebelt M, Agricola R, Botter SM, Piscaer TM, Waarsing JH (2012) Pathophysiology of peri‐articular bone changes in osteoarthritis. Bone 51, 190–196. [DOI] [PubMed] [Google Scholar]
- Weinstein AM, Rome BN, Reichmann WM, Collins JE, Burbine SA, Thornhill TS, Wright J, Katz JN, Losina E (2013) Estimating the burden of total knee replacement in the United States. J. Bone Joint Surg. Am. 95, 385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GR (2013) Thyroid hormone actions in cartilage and bone. Eur. Thyroid J. 2, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Peng H, Wu D, Hu K, Goldring MB, Olsen BR, Li Y (2005) Activation of the discoidin domain receptor 2 induces expression of matrix metalloproteinase 13 associated with osteoarthritis in mice. J. Biol. Chem. 280, 548–555. [DOI] [PubMed] [Google Scholar]
- Yu SP, Hunter DJ (2015) Emerging drugs for the treatment of knee osteoarthritis. Expert Opin. Emerg. Drugs 20, 361–378. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Jordan JM (2010) Epidemiology of osteoarthritis. Clin. Geriatr. Med. 26, 355–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Whole mouse skeletal preparations. Double‐staining with alizarin red and alcian blue of whole skeletal preparations was performed on P2 WT, mTTR‐KO, and hTTR‐TG mice (scale bars = 500 μm). (B) Length of femur and tibia (scale bars = 2 mm) in P2 control (n = 7), mTTR‐KO (n = 7), and hTTR‐TG (n = 6) mice. N.S. = Non‐Significant.
Fig. S2 TTR mRNA and protein expression in chondrocytes, cartilage, synovium, serum, and liver. (A) Chondrocytes were isolated from the femoral condyle and tibial plateau of postnatal day 1 (P1). Cartilage, liver and serum protein were extracted from 9‐month‐old mice. Human recombinant TTR was used as a positive control. Western blotting showed that TTR was detected in cartilage in 9‐month‐old hTTR‐TG mice, but expressed at low levels in chondrocytes. High TTR expression was observed in liver and serum in hTTR‐TG but not in mTTR‐KO. (B) Real‐time PCR for human Ttr in 2‐month‐old hTTR‐TG mice showed lower expression in cartilage than liver (n = 3, each). (C) Real‐time PCR for mouse Ttr in 2‐month‐old wild‐type C57BL/6 mice showed that cartilage and synovium were lower in primary chondrocytes (n = 6).
Fig. S3 Cartilage cellularity. (A) Representative images medial tibia cartilage of Safranin O stained joint sections. B) Three micrographs of the medial tibial plateau were taken under 40× magnification. Cell numbers per each area were counted in 6‐month‐old mice and mice with surgical OA (scale bars = 400 μm, 6 months: WT n = 20, hTTR‐TG n = 28, mTTR‐KO n = 28, Surgical model: WT n = 20, hTTR‐TG n = 26, mTTR‐KO n = 22). **P < 0.01.
Fig. S4 Expression of OA‐related markers in synovium. Immunohistochemistry for IL‐6, ADAMTS‐4, and MMP13 was performed on joint sections from 6‐month‐old mice (WT n = 4; mTTR‐KO n = 4; hTTR‐TG n = 4, scale bars = 100 μm).
Fig. S5 Immunohistochemistry with antibody that recognizes human and mouse TTR. (A) TTR was detected in bone marrow, blood vessels and at the articular cartilage surface of 6‐month‐old hTTR‐TG and WT mice but not in mTTR‐KO mice (scale bars = 400 μm). (B) TTR was detected in fibrillated cartilage in 18‐month‐old hTTR‐TG mice and in chondrocyte in hTTR‐TG and WT mice. There was no TTR staining in the cartilage in 18‐month‐old mTTR‐KO mice (scale bars = 100 μm).