

Abstract
Hypertension (HT) is a complex systemic disease involving transcriptional changes in multiple organs. Here we systematically investigate the pan-tissue transcriptional and genetic landscape of HT spanning dozens of tissues in hundreds of individuals. We find that in several tissues, previously identified HT-linked genes are dysregulated and the gene expression profile is predictive of HT. Importantly, many expression quantitative trait loci (eQTL) SNPs associated with the population variance of the dysregulated genes are linked with blood pressure in an independent genome-wide association study, suggesting that the functional effect of HT-associated SNPs may be mediated through tissue-specific transcriptional dysregulation. Analyses of pan-tissue transcriptional dysregulation profile, as well as eQTL SNPs underlying the dysregulated genes, reveals substantial heterogeneity among the HT patients, revealing two broad groupings – a Diffused group where several tissues exhibit HT-associated molecular alterations and a Localized group where such alterations are localized to very few tissues. These two patient subgroups differ in several clinical phenotypes including respiratory, cerebrovascular, diabetes, and heart disease. These findings suggest that the Diffused and Localized subgroups may be driven by different molecular mechanisms and have different genetic underpinning.
Keywords: hypertension, GTEx, differential expression, eQTL, pan-tissue, complex disease, systemic disease
HYPERTENSION (HT) affects about one-third of the adults worldwide (Fields et al. 2004). It is a chronic condition with elevated blood pressure in the arteries, clinically characterized as a persistent systolic blood pressure of at least 140 mm Hg and/or diastolic blood pressure of at least 90 mm Hg (Chobanian et al. 2003). HT increases the risk of cardiovascular disease, renal failure, and stroke (Sowers et al. 2001; Ezzati et al. 2002; Kearney et al. 2005; Statement 2011). Hence, strategies to control blood pressure can lower the burden of such diseases (Veterans Administration Cooperative Study Group on Antihypertensive Agents 1967; SHEP Cooperative Research Group 1991; Lawes et al. 2008).
HT is a complex trait, likely to be caused by the interplay of multiple genetic and environmental factors (Carretero and Oparil 2000). Furthermore, it is a systemic condition, involving multiple tissues and organs, consistent with the fact that HT is a major risk factor for several other diseases: coronary events (Franklin et al. 2001; Psaty et al. 2001; Chobanian et al. 2003; Aronow et al. 2011, 2016a,b), stroke (Aronow et al. 1996, 2011, 2016a; Psaty et al. 2001; Chobanian et al. 2003; Aronow and Frishman 2004), congestive heart failure (Levy et al. 1996; Aronow et al. 1999, 2011, 2016a; Chobanian et al. 2003), peripheral arterial disease (Stokes et al. 1987; Ness et al. 2005; Aronow et al. 2009, 2011, 2016b), renal dysfunction (DiBona 1992, 2002; Hall 2003; Grassi et al. 2012), and diabetes (Barnett 1994; Cheung and Li 2012). Likewise, conditions such as obesity (Garrison et al. 1987; Kaufman et al. 1997; Hall et al. 2001, 2002; Hall 2003; Droyvold et al. 2005; Dua et al. 2014), insulin resistance (Reaven 1990; Soleimani 2015), high alcohol intake (MacMahon 1987), high salt intake (Blaustein 1977), stress (Kulkarni et al. 1998), low potassium intake, low calcium intake (Kromhout et al. 1985) etc. contribute to HT. Understanding the mechanisms underlying HT across the different tissues is thus an important challenge.
Disruption in the normal gene expression profile is a major proximal cause of common diseases (Nadler et al. 2000; Moffatt et al. 2007; Neueder and Bates 2014). Furthermore, complex and systemic diseases, such as HT, are likely to involve dysregulation in multiple tissues and organs affecting multiple genes and pathways (Sacks and McDonald 1996; Dermitzakis 2008; Leonardson et al. 2008). For example, type II diabetes involves transcriptional dysregulation in pancreatic islets, adipose tissue, and the muscle-skeletal (Olokoba et al. 2012). Systemic lupus erythematosus is an autoimmune disease in which the body’s immune system mistakenly attacks healthy tissues of the skin, joints, kidneys, brain, heart, and lung (Mirabelli et al. 2015). However, for most complex diseases it is difficult to ascertain all the tissues involved and necessitates investigation of tissue-specific dysregulation of gene expression.
Until recently it has not been possible to study HT from the perspective of multi-tissue transcriptional dysregulation due to lack of large-scale pan-tissue transcriptional profiles. Thus most previous transcriptomic studies of HT in human have relied on whole blood gene expression data (Bull et al. 2004; Huan et al. 2015a,b). To gain insights into tissue-specific transcriptomic changes in HT, a few studies have utilized rodent models. A meta-analysis of these studies (Marques et al. 2010) identified 143 genes with altered expression in kidney, adrenal gland, heart, and artery of hypertensive rats, and these genes were enriched for gene ontology terms involved in energy production, fatty acid and lipid metabolism, oxidation, and transport. However, a similar study in human has not yet been performed due to lack of appropriate data. A recently published data set, GTEx (The GTEx Consortium 2015), fills this critical gap. GTEx includes transcriptomes in 53 tissues and organs across 572 individuals along with information on 173 phenotypes/diseases (including HT), thus enabling a multi-tissue characterization of a systemic multi-tissue disease such as HT.
Our analyses (Figure 1) are based on the transcriptomic profiles in 29 tissues from 565 individuals from the GTEx data set (details in Supplemental Material, File S2), with known HT status (not every individual has transcriptome data available for every tissue). For each tissue we perform three analyses:
Figure 1.

Overview of the analysis. In each of the tissues for which a sufficient number of samples from individuals with and without HT is available in GTEx, we identify genes that are differentially expressed in hypertensive individuals relative to nonhypertensive individuals and perform a number of downstream analyses to characterize the roles of such genes in HT.
We assess, for each tissue, the extent to which the tissue-specific transcriptome can distinguish HT from non-HT individuals, using LASSO (Friedman et al. 2010). In each tissue, we also identify genes that are differentially expressed (DE) between HT and non-HT individuals; below we use the term “differential” in the context of HT vs. non-HT individuals in a tissue-specific fashion. We then perform a variety of functional enrichment analyses (relative to biological processes, pathways, known HT-related genes) for the identified DE genes.
GTEx also provides the genome-wide genotypes of each individual, which have previously been used to provide an eQTL map of polymorphisms (eSNP) associated with the expression variability for each gene in each tissue. We use the published eQTL map to assess whether the eSNPs associated with DE genes are directly associated with HT in an independent data set; this suggests the mediating role of transcriptome in the genotype–phenotype association.
Finally, we stratify HT individuals based on two independent metrics, the tissue-specific HT model prediction score (HT-score, indicating tissue-specific transcriptional dysregulation) and the eSNPs underlying tissue-specific DE genes in the HT-predictive tissues, to study whether the emerging HT subgroups have distinct clinical manifestations.
Materials and Methods
Classification of HT vs. non-HT samples based on tissue-specific expression profiles
We first assessed our ability to classify HT vs. non-HT using tissue-specific gene expression profile. As a feature selection strategy, for each gene in a tissue, the Wilcoxon rank sum test is performed between its expression in HT and non-HT samples. Separately for each of the 11 tissues (artery-tibial, nerve-tibial, colon-transverse, pancreas, adipose-subcutaneous, stomach, muscle-skeletal, breast-mammary tissue, artery-aorta, colon-sigmoid, and cells-transformed fibroblasts; kidney-cortex not included due to insufficient data), genes are selected based on their differential expression (Wilcoxon test, False Discovery rate ≤ 0.1). Here FDR is computed across all the genes for each tissue separately. The tissue-specific expressions of the top DE genes (FDR ≤ 0.1) were used for classifying HT and non-HT individuals using LASSO (Friedman et al. 2010) from “glmnet” package in R. In the package, the default family “Gaussian” is used, with α being one, i.e., LASSO penalty is used. We performed fivefold cross-validation, and averaged the classification accuracies (area under the receiver operating curve, AUC-ROC) on 20 independent trials in each tissue. As a control we choose the same number of random genes as in the foreground from the non-DE genes (FDR > 0.5) and performed the classification. The prediction accuracies for all the tissues using significant (FDR ≤ 0.1) DE genes and random control genes sets are shown in Figure 2A. For these 11 tissues, we performed a Wilcoxon test using AUC-ROC values obtained using significant DE and random genes. The Wilcoxon P-values for the AUC-ROC corresponding to the DE gene set to be higher than the random set are 1.18E−34, 1.38E−12, 1.26E−02, 7.44E−03, 2.36E−02, 9.99E−02, 8.72E−05, 2.75E−02, 5.95E−02, 4.48E−01, and 1.55E−01, respectively, for the above-mentioned 11 tissues. Among these tissues, expression profiles in artery-tibial, nerve-tibial, colon-transverse, pancreas, adipose-subcutaneous, muscle-skeletal, and breast-mammary tissue have significantly higher predictability than control.
Figure 2.

Classifying hypertensive individuals based on tissue-specific gene expression in various tissues. (A) The figure shows the accuracy of predicting hypertensive individuals (in terms of area under ROC i.e., AUC-ROC) using the tissue-specific significant (FDR ≤ 0.1) DE genes using LASSO. The prediction is obtained for fivefold cross-validation in 20 independent trials, where DE genes are identified using only the training samples. For each tissue, a pair of boxplots is shown: the colored boxplot corresponds to the AUC using significant (FDR ≤ 0.1) DE genes and the white boxplot shows the accuracy using random genes (keeping the number of random genes same as the number of DE gene in the corresponding tissue) from FDR > 0.5 DE genes, as a control. Seven tissues – artery-tibial, nerve-tibial, colon-transverse, pancreas, adipose-subcutaneous, muscle-skeletal, and breast-mammary tissue (marked by asterisks) – have prediction accuracy of HT significantly higher than the prediction using random genes. (B) Heatmap of the predicted HT-score for each HE individual (columns) in the model based on each of the six tissues (rows) reveals stratification of HT individuals into two distinct groups, i.e., Localized and Diffused groups.
We have also explored the prediction accuracy using DE genes identified at four FDR thresholds: 0.05, 0.1, 0.2, and 1 (i.e., all genes) (shown in Figure S1 in File S1). For prediction, LASSO (from “glmnet” package in R) is used, and AUC-ROC is computed in a cross-validation manner (fivefold and 20 independent trials). To check the difference in prediction accuracies for DE genes at different FDR thresholds, a paired Wilcoxon test is calculated between the prediction scores for FDR ≤ 0.1 relative to those for FDR ≤ 0.2 and 1 (i.e., all genes) across the 11 tissues. Also, we have estimated the prediction accuracy at different values of glmnet parameter α (0.1–1.0 at 0.1 increments), at FDR < 0.1 DE genes, and found that relative to α = 1, there is no significant difference in prediction accuracy at the other nine α values in all the tissues.
Notably, in each fold of the classification task, the DE genes are identified based only on the training set and they are then used to classify HT vs. non-HT in the left-out test set. However, in the downstream analysis involving DE genes, all individuals’ data were used to identify DE genes.
Classify HT and non-HT after controlling for confounders
We estimated the HT prediction accuracy controlling for age, BMI, race, gender, and other confounders (genetic, transcriptional-gene expression, and splicing) for all 11 tissues. From the training set we selected genes whose expression significantly (FDR ≤ 0.1) contribute to the phenotype in addition to all the confounders using the log likelihood ratio test (in R package), and used those genes in the test set for prediction [using LASSO (Friedman et al. 2010), “glmnet” from R package] of HT vs. non-HT. The prediction scores (in terms of AUC-ROC) are obtained for fivefold CV and 20 independent trials for the 11 tissues (shown in Figure S2 in File S1).
NetWAS analysis
Greene et al. (2015) reported using the NetWAS method to estimate a score for each gene measuring its association with HT based on multiple lines of evidence including genome-wide association studies (GWAS) and tissue-specific inferred regulatory networks (Greene et al. 2015). In this analysis, we assess enrichment of NetWAS-identified HT genes among our identified tissue-specific DE genes, separately for OE and UE genes (Table 2). We performed a Wilcoxon test between the top 1000 and bottom 1000 NetWAS HT genes (obtained from NetWAS ranks) based on their tissue-specific DE effect size (referred to as “DE value”) for OE and UE cases separately (Table 2). The DE value for a gene is the absolute log2 (mean expression value across HT individuals/mean expression value in non-HT people). The effect size is quantified as the median DE value for the top 1000 NetWAS genes divided by the median DE value for the bottom 1000 NetWAS genes. For each tissue we performed two Wilcoxon tests using OE and UE genes, resulting in 22 P-values for 11 tissues, which were then corrected for multiple testing using Benjamini and Hochberg’s method (Benjamini and Hochberg 1995).
Table 2. Differential expression of previously known and predicted HT-associated genes.
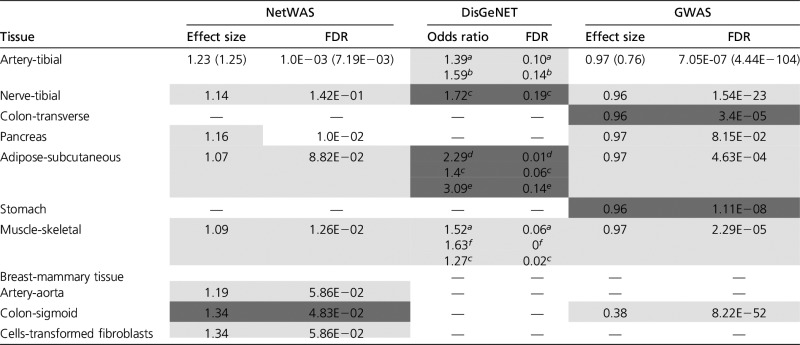 |
NetWAS: Shown are the Wilcoxon test results comparing DE effect size, defined as ratio of median of abs(log2 gene expression of HT) and median of abs(log2 non-HT gene expression) of OE and UE genes for top 1000 and bottom 1000 ranked NetWAS genes. DisGeNET: For DisGeNET, which provides a list of HT genes, the table shows the Fisher test results based on two categorizations – “OE (or UE) vs. non-DE” and “HT vs. non-HT.” Odds ratio >1 indicates differential expression of DisGeNET genes. GWAS: For all eSNPs underlying DE genes the GWAS association score with blood pressure was obtained. For each tissue, a Wilcoxon test was performed between GWAS scores of the eSNPs associated with each of the top OE (or UE) gene (FDR ≤ 0.1) and the eSNPs associated with non-DE genes (FDR > 0.5). The effect size is based on GWAS P-values and hence a smaller effect size indicates a greater association with GWAS. The cases with <0.2 FDR are shaded. For all these analyses, significant tests for OE and UE are shaded with light and dark gray, respectively, and tissues with significant results for both OE and UE are unshaded and are written in the format of OE (UE).
Pulmonary HT.
Idiopathic pulmonary HT.
HT.
Essential HT.
Portal HT.
Blood pressure finding.
DisGeNET analysis
DisGeNET includes 429,111 associations connecting 17,181 genes and 14,586 diseases, disorders, clinical or abnormal human phenotypes, etc., based on both curated experimental data as well as high-confidence computational predictions from multiple resources. It includes 23 different “disease” names that contain the terms “hypertension” or “blood pressure.” We considered the categories with a sufficient number of associated genes for our analyses; the list and the number of genes they are associated with are provided in Table S4a. The genes that do not belong to any of the categories are considered as non-HT genes in DisGeNET and are used as the background for this analysis. For each of the categories separately, using the foreground (HT-associated gene) and the background genes (remaining non-HT-associated genes), we perform a Fisher test. The 2 × 2 contingency table for the Fisher test is as follows:
| Foreground genes | Background genes | |
|---|---|---|
| DE | A | B |
| Non-DE | C | D |
where A, B are the number of DE genes (FDR ≤ 0.1) belonging to the foreground and background, and C, D are the number of non-DE genes (FDR > 0.5) in the foreground and background. Note that the DE genes consist of OE and UE genes, and the test is performed for both these categories separately. Thus 22 P-values are obtained, which are then used to calculate the corresponding FDRs (results shown in Table 2).
Associating eQTL and GWAS of DE genes
Here we assessed whether the HT-linked SNPs from an independent GWAS of blood pressure are enriched among the eSNPs of tissue-specific HT-linked genes, i.e., DE genes. In each tissue, separately for the significant (FDR ≤ 0.1) UE (underexpressed in HT relative to non-HT individuals) and OE (overexpressed in HT relative to non-HT individuals) genes, we obtained all the SNPs that were highly associated with the gene’s expression variability, based on previously reported eQTL (The GTEx Consortium 2015). Using the ranking based on SNP-gene expression association scores in the analysis by The GTEx Consortium (2015), we only considered the SNPs whose association scores with a DE gene were significant (FDR ≤ 0.05). Likewise, as a control, we obtained the SNPs associated with the expression variability of genes that were least differentially expressed (FDR > 0.5) between HT and non-HT individuals in the tissue.
In parallel, we obtained association signals from a previous study by the International Consortium for Blood Pressure Genome-Wide Association Studies (2011) involving 200,000 individuals and 2,461,325 SNPs. Here we restricted ourselves only to the significant GWAS associations (P-values ≤ 0.01). For each SNP, a chi-square test was performed in the study by the International Consortium for Blood Pressure Genome-wide Association Studies (2011) for major and minor alleles in disease and normal cases. Thus the extent of association of every SNP with HT is measured by a chi-square statistic and the P-value.
Given the top eSNPs for the DE (separately for OE and UE) and control genes, we used the GWAS association (P-values) of those SNPs with blood pressure and tested whether SNPs associated with DE genes are more strongly associated with HT, using a one-sided Wilcoxon test. The significant OE and UE genes are considered separately as the foreground and the least DE genes (FDR > 0.5) as control.
Classification of HT vs. non-HT individuals using SNP information
The HT and non-HT individuals are classified using their eSNPs corresponding to significant (FDR ≤ 0.1) DE genes from seven tissues (artery-tibial, nerve-tibial, colon-transverse, pancreas, adipose-subcutaneous, muscle-skeletal, and breast-mammary tissue). For each of the above tissues we obtain the DE genes (genes that are differentially expressed between HT and non-HT individuals) and their corresponding eSNPs (FDR ≤ 0.05) from the GTEx Consortium (The GTEx Consortium 2015) resulting in 129,442, 45,798, 31,666, 2872, 141,849, 72,612, and 13 SNPs for the artery-tibial, nerve-tibial, colon-transverse, pancreas, adipose-subcutaneous, muscle-skeletal, and breast-mammary tissues, respectively. For each of the seven tissues we used the genotype (ranging from 0 to 2) of eSNPs of the DE genes for 443 individuals and computed the principal components (PCs). To classify HT from non-HT individuals in each of the seven tissues we used the number of PCs (90, 47, 43, 8, 100, 72, and 2, respectively), which explains at least 50% of the variability of the data. The classification is based on LASSO (Friedman et al. 2010) with fivefold cross-validation and 50 independent trials, and the prediction accuracy is assessed in terms of AUC-ROC. The AUC-ROC for the above seven tissues and all seven tissues taken together are 0.55, 0.59, 0.52, 0.53, 0.55, 0.55, 0.54, and 0.56, respectively. The results are shown in Figure S5 in File S1.
Stratification of HT individuals based on tissue-specific HT prediction score and association with various phenotypes
Here we describe the method for stratifying hypertensive individuals using their tissue-specific HT prediction and association of different diseases with the subgroups of HT individuals. For each of the six tissues (namely artery-tibial, nerve-tibial, colon-transverse, pancreas, adipose-subcutaneous, and muscle-skeletal) that are most predictive of HT [based on LASSO (Friedman et al. 2010) using significant FDR ≤ 0.1 genes, Figure 2A], we assign an HT prediction score, henceforth referred as the “HT-score” based on LASSO machine learning method. The score is measured based on fivefold cross-validation and 20 independent trials. Thus, each individual gets an “HT-score” from each of these six tissues, where a score denotes the fraction of trials an individual is classified as HT. We then rank the individuals in each tissue separately based on the HT-score. Based on the HT-score of the HT individuals in these six tissues, we clustered (using hierarchical clustering, with Euclidean distance function) them into two groups, namely G1 (having 60 individuals) and G2 (93 individuals) (see Figure 2B). In general, the individuals in the G1 group have high HT-scores in multiple tissues (we term this the Diffused group), while the G2 group is characterized by smaller HT-scores (we term this the Localized group). Note that in the above HT group finding analysis, we used only the top six out of the seven tissues, which are significant, and did not use breast-mammary tissue. This is because adding this tissue reduces the sample size (HT individuals) to 107, which in turn reduces our power for the downstream analysis.
We then performed a Fisher’s exact test between groups G1 and G2 for presence and absence of various phenotypes. The 2 × 2 contingency table for the Fisher’s exact test is as follows:
| Phenotype present | Phenotype absent | |
|---|---|---|
| Group G1 | A | B |
| Group G2 | C | D |
For each phenotype, A and B are the numbers of individuals in G1 (Diffused group) having and not having the phenotype, and C and D are the numbers of individuals in G2 (Localized group) having and not having the phenotype, respectively.
Additionally, we performed a Fisher’s exact test to assess the association between HT and a phenotype using the following contingency table:
| Phenotype present | Phenotype absent | |
|---|---|---|
| HT | A | B |
| Non-HT | C | D |
Here, A and B are the numbers of HT individuals having and not having a phenotype, and C and D are the number of non-HT individuals with and without the phenotype, respectively.
Genetic underpinning of HT individual stratification and associations with additional phenotypes
Here we describe our approach for investigating underlying genetic heterogeneity among the HT individuals and their link with other clinical phenotypes. For each of the six tissues (artery-tibial, nerve-tibial, colon-transverse, pancreas, adipose-subcutaneous, muscle-skeletal), we identified significant DE genes (all Wilcoxon FDR value ≤ 0.1). For each gene, we obtained their tissue-specific eSNP (FDR ≤ 0.05) as previously identified by the GTEx Consortium (2015), resulting in a total of 424,239 SNPs (129,442, 45,798, 31,666, 2872, 141,849, 72,612 SNPs from the above-mentioned tissues, respectively). Each HT individual is then represented by a vector of length 424,239 [significant DE gene of the above six tissues had significant (FDR ≤ 0.05) eSNPs]. We performed principal component analysis (PCA) (Gower 1966) for the 246 HT individuals, and retained the top nine PCs that together explained ∼15% of the data variability. For each of the top nine PCs, we scored each HT individual based on the individual’s genotype. HT individuals in GTEx have 28 phenotypes with adequate data (at least 12 in each category, for categorical phenotypes): COHORT, GENDER, LBCMVTAB (CMV total antibody), MHABNWBC (abnormal WBC), MHARTHTS (arthritis), MHASTHMA (asthma), MHBLDDND (past blood donation denied), MHCANCERNM (history of nonmetastatic cancer), MHCOCAINE5 (cocaine use within 5 years), MHCOPD (chronic respiratory disease), MHCVD (cerebrovascular disease), MHDLYSIS (dialysis treatment), MHDPRSSN (major depression), MHHRTATT (heart attack, acute myocardial infarction, acute coronary syndrome), MHHRTDIS (ischemic heart disease), MHHRTDISB (heart disease), MHLVRDIS (liver disease), MHOPNWND (open wounds), MHORGNTP (received tissue organ transplant), MHPNMNIA (pneumonia acute respiratory infection affecting the lung), MHPNMIAB (pneumonia), MHRNLFLR (renal failure), MHT2D (diabetes mellitus type 2), MHT1D (diabetes mellitus type 1), age, BMI (body mass index), WEIGHT, and RACE. We excluded LBCMVTAB (since it reflects infection status), MHPNMIAB (as it is largely redundant with MHPNMNIA), MHBLDDND (since its reasons are not clear), MHOPNWND, MHDLYSIS, MHCANCERNM, and MHCOCAINE5 (a behavioral trait confounded by socio-economic factors), yielding 20 phenotypes. In addition to the 20 phenotypes in GTEx, we included the Diffused–Localized grouping of HT individuals revealed by our own analysis. For each of the 21 phenotypes (20 + 1) we estimated the association of the PC scores with the phenotype across individuals. If the phenotype is categorical, we compared the PC scores between the individuals in the two categories using the Wilcoxon test. If the phenotype was noncategorical (BMI, WEIGHT, and age), we estimated the Pearson’s correlation between the PC scores and the phenotype.
Data availability
All gene expression and genotype data used are GTEx version 6 obtained from https://gtexportal.org.
Results
Tissue-specific transcriptomes can differentiate HT and non-HT samples
We first assessed the extent to which gene expression profiles in various tissues can differentiate between samples from HT and non-HT individuals. We used 28 tissues for which at least 50 individuals from each HT and non-HT category are available; the number of samples for each tissue is provided in Table S1. For each tissue, we measured the cross-validation HT classification accuracy based on the expression of significantly (FDR ≤ 0.1) DE genes as features (Materials and Methods). Figure 2A shows the cross-validation accuracies based on LASSO (Friedman et al. 2010) for the 11 tissues that have at least 15 DE genes (Materials and Methods). For each of the 11 tissues, we compared the prediction accuracy based on DE genes with that for the same number of randomly selected genes. Among these 11 tissues, we found that in seven tissues (artery-tibial, nerve-tibial, colon-transverse, pancreas, adipose-subcutaneous, muscle-skeletal, breast-mammary tissue), the gene expression profiles can individually distinguish HT from non-HT samples with accuracy (in terms of AUC-ROC, i.e., area under the receiver operating characteristic curve) significantly higher than random genes (Figure 2A). The prediction accuracy for the top two most predictive tissues is 0.75 and 0.63 (Figure 2A). Comparisons of prediction accuracies using DE genes at other FDR thresholds (0.05, 0.1, 0.2, and 1) do not show a significant difference relative to that at FDR = 0.1 (Figure S1 in File S1; details in Materials and Methods). We find that DE genes are largely tissue-specific; on average only 0.04 fraction of DE genes overlap between any two tissues (Methods in File S2).
There is likely to be complex, nonlinear, and multi-way relationships between gene expression, complex diseases, and various demographic and environmental attributes, such as gender, race, BMI, and age (Carretero and Oparil 2000). Our goal here is to identify genes whose expressions are associated with HT, even if some of those expression variations are contributed by these other factors. Therefore, we did not explicitly control for those attributes when identifying HT-associated genes. However, as shown in Figure S2 in File S1, we found that even if HT-associated genes are selected after controlling for age, BMI, race, gender, and other confounding factors (see Materials and Methods), those genes can still predict HT with a comparable accuracy.
Previous studies have assessed predictability of HT based on genotype (Abraham et al. 2013; Zhou et al. 2013; Wheeler et al. 2014). To assess the extent to which genotype can improve HT prediction accuracy, we built a model using genotype and DE genes’ expression (Methods in File S2). The median prediction accuracy using significant DE gene expression alone, and using gene expression as well as genotype, does not reveal a significant difference (paired Wilcoxon P-value = 0.57), suggesting that the genotype does not substantially contribute to the prediction accuracy in addition to the gene expression (Figure S3 in File S1). To further rule out a potential confounding effect of race, we estimate the prediction accuracy (Methods in File S2) only among Whites (African-Americans do not constitute a sufficiently large sample to perform this analysis). Comparing the accuracy obtained using all races with that when only White individuals were used, we found that the two prediction accuracies were significantly different in none of the tissues (all Wilcoxon P-values > 0.1).
Overall, these results suggest tissue-specific transcriptional disruptions across multiple tissues in HT.
Functional analysis of tissue-specific HT signature genes
Next, we performed a functional enrichment analysis for the DE genes in each tissue. However, to account for different mechanisms underlying the overexpressed and the underexpressed genes, we further distinguish the tissue-specific DE genes into two categories: those that are overexpressed (OE) in HT vs. non-HT individuals and those that are underexpressed (UE). Significant (FDR ≤ 0.1) OE and UE genes for each tissue are provided in Table S2. We performed functional enrichment analysis separately for significant OE and UE genes in each tissue, based on GO biological processes and KEGG pathways from DAVID tool (Huang et al. 2008, 2009), using the top 50% most expressed genes in a tissue as the background. Table 1 and Table S3a present the enriched KEGG pathways (GO term enrichment is provided in Table S3b). Many of the enriched pathways have been implicated in HT etiology, either directly or indirectly through other associated phenotypes. For instance, fatty acid metabolism (Fuh et al. 1987), insulin signaling pathway (Saad et al. 2004), peroxisome proliferator-activated receptor (PPAR) (Chen et al. 2008; Usuda and Kanda 2014), valine, leucine and isoleucine degradation (Chen et al. 2015), and NF-κ B signaling pathway (Henke et al. 2007) are enriched in adipose-subcutaneous tissue; platelet activation (Alexandru et al. 2011), cGMP signaling pathway (Tsai and Kass 2009), and adipocytokine signaling pathway (Cao 2014) are enriched in artery-tibial tissue. A detailed discussion is provided in Text S1 in File S1.
Table 1. Pathway enrichment in tissue-specific DE genes.
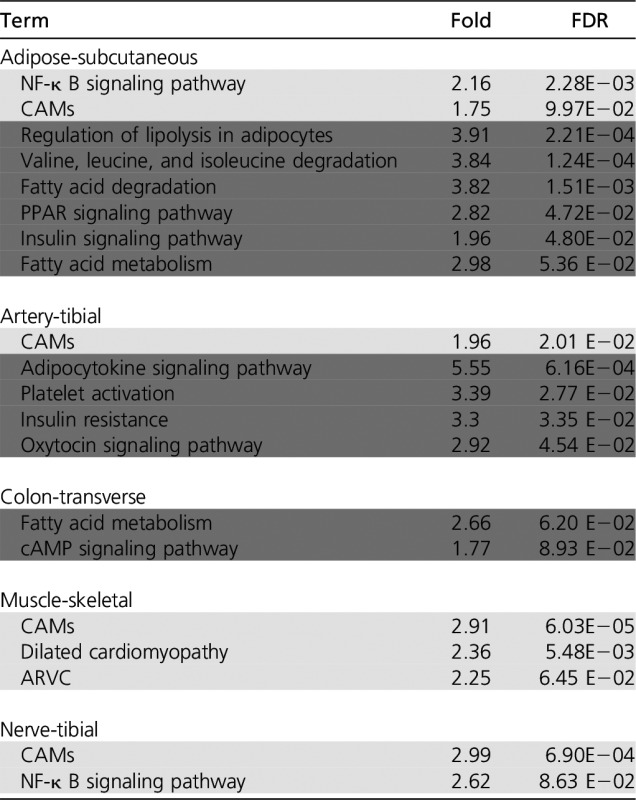 |
Only the relevant enriched KEGG pathway (FDR ≤0.1) among the most significant (FDR ≤ 0.1) overexpressed (light gray) and underexpressed (dark gray) genes in each tissue are listed. All the enriched (FDR ≤ 0.1) KEGG pathways and GO biological processes are shown in Table S3a and b, respectively. PPAR, peroxisome proliferator-activated receptor; CAMs, cell adhesion molecules; ARVC, Arrhythmogenic right ventricular cardiomyopathy.
Known HT-linked genes tend to be DE in specific tissues of HT individuals
We compiled three independent sets of previously identified HT-linked genes. For each set, we provide a tissue map where each gene is most dysregulated in the HT individuals.
First, Greene et al. have reported a NetWAS method that ranks all genes according to their association with HT based on GWAS and tissue-specific inferred regulatory networks (Greene et al. 2015). We compared tissue-specific differential expression effect sizes of the top 1000 and bottom 1000 NetWAS-ranked genes (see Materials and Methods), separately for OE and UE genes. As shown in Table 2, 8 out of 11 tissues show significantly greater differential expression for the top 1000 ranked NetWAS genes relative to the negative control.
Second, we tested whether the DE genes are enriched for 1728 HT-linked genes from the DisGeNET, which is a curated compilation from multiple experimental resources, including orthology-based inference in mouse experiments, as well as computational prediction (Griffith et al. 2013). DisGeNET classifies HT into several categories. We used the Fisher’s exact test (see Materials and Methods) to assess the relative enrichment of DisGeNET HT genes of different HT types among OE and UE genes in each tissue, and the results are presented in Table 2. Overall the known HT genes are enriched among tissue-specific DE genes in the GTEx data set.
Finally, using a rat model, a previous study identified 143 genes with altered expression at the onset of HT (Marques et al. 2010); however, their meta-analyses involved multiple tissues and as such the altered genes were not identified in a tissue-specific fashion. We found that the human orthologs of 82 of the 143 genes were among the significant (FDR ≤ 0.1) DE genes in the seven tissues. Table S5 shows the 82 genes distributed among the seven tissues. For many of the genes we provide literature-based evidence supporting their role in HT (Text S2 in File S1). Figure S4 in File S1 shows a heatmap of genes’ tissue-specific differential expression for all the tissues highlighting the significantly differentially expressed genes.
SNPs associated with the expression of DE genes are linked with blood pressure in an independent cohort
Next, to assess potential causative role of the HT-associated differential expression, we assessed whether the SNPs associated with blood pressure variance in the human populations exert their influence via gene expression changes. In each tissue, for each of the significant OE and UE genes (FDR ≤ 0.1), we obtained all the SNPs that were highly associated with the gene’s expression variability, based on previously reported eQTL (The GTEx Consortium 2015). eSNPs associated with nondifferential (FDR > 0.5) genes were used as control. In parallel, we used association scores (the P-value) for 2,461,325 SNPs with blood pressure from a previous GWAS study (International Consortium for Blood Pressure Genome-Wide Association Studies 2011). We tested whether eSNPs associated with OE and UE genes are more associated with blood pressure, using a one-sided Wilcoxon test (see Materials and Methods). As shown in Table 2, we found this to be broadly true. An alternative Fisher’s exact test (Methods in File S2) showed consistent results (Table S4b). The most significant (P-value < 1E−07) SNPs from the GWAS study that are associated with the significant OE and UE genes in any of the seven tissues are provided in Table S4c. As shown, the DE genes associated with these GWAS SNPs have been previously linked to HT.
Given our results above, we assessed whether the eSNPs associated with the tissue-specific DE genes can jointly distinguish HT from non-HT individuals. We quantified (see Materials and Methods) the cross-validation prediction accuracies based on eSNPs for the significant DE genes (FDR ≤ 0.1) in seven HT-predictive tissues (artery-tibial, nerve-tibial, colon-transverse, pancreas, adipose-subcutaneous, muscle-skeletal, and breast-mammary tissue) and based on union of all those SNPs also. As shown in Figure S5 in File S1, the eSNPs underlying DE genes distinguish HT from non-HT individuals, with a modest (although significant) accuracy (Materials and Methods).
Overall, our results suggest that the mechanisms linking genotype to blood pressure involve, in part, transcriptomic dysregulation in multiple tissues.
Transcriptomic and genetic heterogeneity among HT individuals is associated with distinct clinical phenotypes
Next, we explored the multi-tissue transcriptomic and genetic heterogeneity among HT individuals and whether the heterogeneity is linked with other phenotypes. Our transcriptome-based prediction model provides a tissue-specific “HT-score” for each individual (see Materials and Methods). As shown in Figure 2B, HT individuals exhibit distinct profiles of tissue-specific HT-scores across the six HT-predictive tissues, and reveal two broad patterns: individuals that have substantial transcriptional dysregulation (high HT-scores) in multiple tissues (we call this group the Diffused group) and another subgroup (referred to as Localized) that contains individuals with fewer and more tissue-specific transcriptional alterations. Next, we assessed the associations of the two major subgroups with 17 clinical and demographic phenotypes (Table 3). We found that Diffused and Localized groups are not associated with age or BMI. However, the Diffused group is biased toward males and people with African-American ancestry. Furthermore, the Diffused group has a significant association with other phenotypes, such as abnormal WBCs, chronic respiratory disease, and major depression, while the Localized group is significantly associated with cerebrovascular disease, pneumonia, and diabetes mellitus type II.
Table 3. Comparison of different phenotypes between the diffused subgroup (G1) vs. the rest (G2), based on tissue-specific HT-scores (see text).
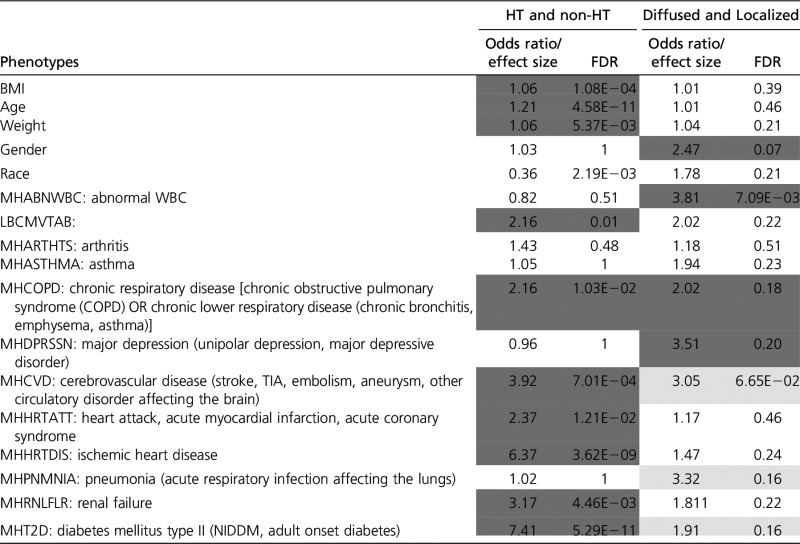 |
HT-scores for only six of the tissues that are most predictive of HT were used for grouping. For BMI, age, and weight, a Wilcoxon test between the groups G1 (Diffused group) and G2 (Localized group) was performed and their corresponding effect sizes defined as the mean (G1)/mean(G2) are provided. For the rest of the phenotypes a Fisher test was performed; significant incidence (FDR ≤ 0.2) of the phenotype in G1 is colored light gray and in G2, dark gray. For the enrichment of the phenotypes among HT and non-HT individuals, the Fisher test is performed, and the significant cases where the HT people are enriched with disease status are shaded. For BMI, AGE, and WEIGHT, a Wilcoxon test is performed, with effect size being mean (HT)/mean (non-HT).
Among several reasons for the high mortality and burdens associated with HT is its comorbidity with various diseases, which plays a crucial role. For instance, diabetes (Long and Dagogo-Jack 2011), chronic kidney disease (Triolo et al. 2004), osteoarthritis, and asthma are found to be prevalent in HT individuals. The nature of comorbidity might plausibly indicate the subcategory of HT. For instance, the cooccurrence of diabetes and HT is linked with several cardiovascular diseases, such as coronary artery disease, myocardial infarction, congestive heart failure, stroke, and peripheral vascular disease (Long and Dagogo-Jack 2011). HT is also seen to be prevalent among COPD patients (Divo et al. 2012), and anxiety and depression are closely linked to the presence of COPD in patients (Maurer et al. 2008; Hillas et al. 2015). The Diffused HT subgroups found in our analysis tend to have comorbidities with macrovascular complications, while the Localized group tends to be comorbid with COPD and associated disease such as depression and asthma.
We further tested whether the two groups are genetically distinct. Based on all the significant eSNPs (The GTEx Consortium 2015) (at FDR ≤ 0.05) corresponding to significant (FDR ≤ 0.1) DE genes in the six HT-predictive tissues (artery-tibial, nerve-tibial, colon-transverse, pancreas, adipose-subcutaneous, muscle-skeletal), we first performed a PCA, pooling all samples from Diffused and Localized groups and using the top 20 PCs, we assessed the classification accuracy between the two subgroups using LASSO (see Methods in File S2). We found the mean cross-validation accuracy (fivefold across 50 randomized iterations) between the two groups to be 0.57, suggesting a somewhat different genetic architecture underlying the two subgroups.
Additionally, independently for each of the top nine PCs identified above, we assessed association of the PC score across HT individuals with each of 21 additional phenotypes provided by GTEx (see Materials and Methods). As shown in Figure 3, we found that the various PCs are significantly correlated with 12 phenotypes (11 phenotypes in GTEx plus the HT canonical groups) (P-value < 0.05) including Asthma (Salako and Ajayi 2000), Chronic respiratory disease (Girgis and Mathai 2016), Major depression (Rubio-Guerra et al. 2013), Ischemic heart disease (Špinar 2012), Diabetes (Epstein and Sowers 1992), CMV total ab, Cerebrovascular disease (Heistad and Baumbach 1992; Johansson 1999), Heart attack (Pedrinelli et al. 2012), Arthritis, and Race. Notably, genotypic heterogeneity is not associated with gender or age. PC1 is associated with race as well as with CMV total ab, chronic respiratory disease, cerebrovascular disease, and heart attack. A direct comparison of race with the above four phenotypes reveals significant association with three of them, suggesting that racial bias may contribute to the observed association of PC1 with these phenotypes. To ensure that the above associations are confounded by race, we repeated the above analysis among only White individuals. In comparison to the mixed race case, all phenotypes (except Chronic respiratory disease) were still significant (results shown in Table S4d). Additionally, with regards to Diffused and Localized subgroups, we also checked whether Whites are disproportionately distributed among the two groups, and did not find this to be the case.
Figure 3.

Associations between heterogeneity among HT individuals and additional phenotypes. Based on the eSNPs associated with the top DE genes in HT-predictive tissues (six), the top nine PC were estimated. The figure shows significant associations between each one of the nine PCs and a phenotype; each panel shows the distributions of the PC scores for the two groups of HT individuals based on the phenotype, termed “cases” (individuals having the phenotype) and “control” (the complement). Wilcoxon test P-value comparing the two sets of PC scores is shown.
Overall, our analysis reveals a grouping of HT individuals based on their pan-tissue transcriptome, and also reveals genetic heterogeneity among HT individuals and importantly, links the genetic heterogeneity with other clinical indicators.
Discussion
We have presented the first pan-tissue analysis of HT-associated transcriptomic variations in human. Our results show distinctive expression differences between the HT and non-HT individuals in several tissues, revealing tissue-specific pathways linked with HT. Previously reported HT-associated genes show tissue-specific differential expression in HT individuals. Beyond associations, our results suggest that HT-associated SNPs are likely to exert their phenotypic influence via transcriptional disruptions in multiple tissues. Together, these results suggest a pan-tissue transcriptional dysregulation, mediated by genotypic variance, as an important basis for HT. Pan-tissue transcriptional dysregulation profiles of HT individuals reveal two groups, Diffuse and Localized, that differ in their clinical attributes. Finally, SNPs underlying the expression variances of DE genes reveal patterns of heterogeneity among HT individuals that cosegregate with several additional physiological phenotypes, suggesting the heterogeneity of the endo-phenotypic profile of high blood pressure.
Blood pressure is known to increase with age; the median ages for HT and non-HT individuals in GTEx are, respectively, 59 and 50. However, several lines of evidence suggest that our results are not biased due to age differences between HT and non-HT individuals. First, the significant (FDR ≤ 0.1) tissue-specific OE and UE genes have little overlap with previously detected age-related genes whose tissue-specific expression strongly correlated with age (Yang et al. 2015) with a mean of 0.12 for OE and 0.05 for UE HT genes across tissues. Consistently, gene ranking based on differential expression is poorly correlated with the ranking based on age dependency with mean R2 of 0.02 for both OE and UE genes across 11 tissues (Figure S6 in File S1). Second, as mentioned in our stratification analysis above, age is not associated with individual PCs representing genetic heterogeneity among HT individuals. Finally, when we identify HT-associated genes while controlling for age (as well as many other confounders), those genes can still predict HT individuals with a comparable accuracy (Figure S2 in File S1). Collectively, these results suggest that age alone does not explain our results, and genetic and expression variance among individuals (some of which can also be age-associated) play a significant role in HT.
Our functional enrichment analysis (Table 1 and Table S3, a and b) broadly revealed genes involved in several processes from individual tissues. Imperfections of the functional annotations (incompleteness, inaccuracy, and lack of resolution), complicates the interpretation of the revealed functional associations of the DE genes. We nevertheless found several compelling cases, including fatty acid metabolism (Fuh et al. 1987) in adipose, insulin response (Saad et al. 2004) in multiple tissues including adipose-subcutaneous and artery-tibial, PPAR signaling pathway (Chen et al. 2008; Usuda and Kanda 2014), and regulation of lipolysis in adipocytes (Duncan et al. 2007) in adipose-subcutaneous, NF-κ B signaling pathway (Henke et al. 2007) in adipose-subcutaneous, valine, leucine and isoleucine degradation (Chen et al. 2015) in adipose-subcutaneous, and dilated cardiomyopathy (Takarada et al. 1985) in muscle-skeletal. In artery-tibial tissue, adipocytokine signaling (Cao 2014) genes are DE, which along with insulin signaling regulates vascular endothelial function (Ritchie et al. 2004), while the oxytocin signaling pathway is found to have several roles in cardiovascular systems (Gutkowska and Jankowski 2012), and platelet activation is associated with hypercholesterolemia, a very high level of cholesterol in the blood (Alexandru et al. 2011).
Genetic variants associated with complex diseases largely reside in the noncoding, presumably regulatory, regions of the genome (Zhang and Lupski 2015). Moreover, the causality from genotype to phenotype is largely mediated via gene expression, i.e., genetic variance affects gene expression variance, which in turn affects the phenotypic variance (Kraft and Hunter 2009). We have shown that many of the genes that are DE in HT individuals in specific tissues, may be causal, as their interindividual variance are associated with the same SNPs that are also associated with HT in independent cohorts. Although environment, including age, is likely to play an important role in HT, our result suggests that genetics plays a substantial role, specifically the regulatory variants underlying tissue-specific expression variance.
Given the potential involvement of multiple pathways across multiple tissues in the development of HT, it is likely that different pathways and systems may be affected in different HT individuals, which may in turn reflect different genetic or environmental contexts underlying HT individuals. Our investigation of pan-tissue transcriptomic dysregulation revealed a distinct “Diffused” subgroup of hypertensive individuals characterized by transcriptional dysregulation in multiple tissues, which also show a slight (but significantl) association with gender and differential incidence of several cardiovascular-related diseases that are commonly associated with HT but not with race and age. Although environmental contexts are difficult to ascertain in GTEx, we exploited the SNPs underlying the DE genes’ variance to explore potential genetic heterogeneity among HT individuals. For several clinical phenotypes, as well as race, we found significant associations between the phenotypes and various dimensions of genetic heterogeneity (PCs) among HT individuals. Our analysis may suggest a distinct genotypic profile underlying different HT-centered comorbidity patterns.
While reporting the first pan-tissue transcriptomic landscape of HT and its potential genetic functional underpinnings, our work also represents a novel methodological pipeline applicable to other complex systemic disease.
Supplementary Material
Supplemental material is available online at www.genetics.org/lookup/suppl/doi:10.1534/genetics.117.300280/-/DC1.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grant R01GM100335 and National Science Foundation (NSF) 1564785 (S.H.), and Maryland “MPowering the state” funding (M.B.).
Author contributions: The project was conceived and designed by SH and ER. Data acquisition and analysis was done by MB. Data interpretation was done by SH with help from ER and MB. The manuscript was written by SH and MB with help from ER. MS, AD, NUN, KW, JSL and Y-PCC helped with various aspects of design, data acquisition, and analyses. All authors read and approved the final manuscript. The authors declare that they have no competing interests.
Footnotes
Communicating editor: E. Hauser
Literature Cited
- Abraham G., Kowalczyk A., Zobel J., Inouye M., 2013. Performance and robustness of penalized and unpenalized methods for genetic prediction of complex human disease. Genet. Epidemiol. 37: 184–195. [DOI] [PubMed] [Google Scholar]
- Alexandru N., Popov D., Dragan E., Andrei E., Georgescu A., 2011. Platelet activation in hypertension associated with hypercholesterolemia: effects of irbesartan. J. Thromb. Haemost. 9: 173–184. [DOI] [PubMed] [Google Scholar]
- Aronow W. S., Ahn C., 2016. Risk factors for new coronary events in a large cohort of very elderly patients with and without coronary artery disease. Am. J. Cardiol. 77: 864–866. [DOI] [PubMed] [Google Scholar]
- Aronow W. S., Frishman W. H., 2004. Treatment of hypertension and prevention of ischemic stroke. Curr. Cardiol. Rep. 6: 124–129. [DOI] [PubMed] [Google Scholar]
- Aronow W. S., Ahn C., Gutstein H., 1996. Risk factors for new atherothrombotic brain infarction in 664 older men and 1,488 older women. Am. J. Cardiol. 77: 1381–1383. [DOI] [PubMed] [Google Scholar]
- Aronow W. S., Ahn C., Kronzon I., 1999. Comparison of incidences of congestive heart failure in older African-Americans, Hispanics, and Whites. Am. J. Cardiol. 84: 611–612. [DOI] [PubMed] [Google Scholar]
- Aronow W. S., Ahmed M. I., Ekundayo O. J., Allman R. M., Ahmed A., 2009. A propensity-matched study of the association of peripheral arterial disease with cardiovascular outcomes in community-dwelling older adults. Am. J. Cardiol. 103: 130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronow W. S., Fleg J. L., Pepine C. J., Artinian N. T., Bakris G., et al. , 2011. ACCF/AHA 2011 expert consensus document on hypertension in the Elderly: A report of the American College of Cardiology Foundation Task Force on clinical expert consensus documents developed in collaboration with the American Academy of Neurology, American Geriatrics Society, American Society for Preventive Cardiology, American Society of Hypertension, American Society of Nephrology, Association of Black Cardiologists, and European Society of Hypertension. J. Am. Coll. Cardiol. 57: 2037–2114. [DOI] [PubMed] [Google Scholar]
- Aronow W. S., Ahn C., Kronzon I., Koenigsberg M., 2016a Congestive heart failure, coronary events and atherothrombotic brain infarction in elderly blacks and whites with systemic hypertension and with and without echocardiographic and electrocardiographic evidence of left ventricular hypertrophy. Am. J. Cardiol. 67: 295–299. [DOI] [PubMed] [Google Scholar]
- Aronow W. S., Sales F. F., Etienne F., Lee N. H., 2016b Prevalence of peripheral arterial disease and its correlation with risk factors for peripheral arterial disease in elderly patients in a long-term health care facility. Am. J. Cardiol. 62: 644–646. [DOI] [PubMed] [Google Scholar]
- Barnett A. H., 1994. Diabetes and hypertension. Br. Med. Bull. 50: 397–407. [DOI] [PubMed] [Google Scholar]
- Benjamini Y., Hochberg Y., 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57: 289–300. [Google Scholar]
- Blaustein M. P., 1977. Sodium ions, calcium ions, blood pressure regulation, and hypertension: a reassessment and a hypothesis. Am. J. Physiol. 323: C165–C173. [DOI] [PubMed] [Google Scholar]
- Bull T. M., Coldren C. D., Moore M., Sotto-Santiago S. M., Pham D. V., et al. , 2004. Gene microarray analysis of peripheral blood cells in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 170: 911–919. [DOI] [PubMed] [Google Scholar]
- Cao H., 2014. Adipocytokines in obesity and metabolic disease. J. Endocrinol. 220: T47–T59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carretero O. A., Oparil S., 2000. Essential hypertension: part I: definition and etiology. Circulation 101: 329–335. [DOI] [PubMed] [Google Scholar]
- Chen H.-H., Tseng Y. J., Wang S.-Y., Tsai Y.-S., Chang C.-S., et al. , 2015. The metabolome profiling and pathway analysis in metabolic healthy and abnormal obesity. Int. J. Obes. 39: 1241–1248. [DOI] [PubMed] [Google Scholar]
- Chen R., Liang F., Moriya J., Yamakawa J., Takahashi T., et al. , 2008. Peroxisome proliferator-activated receptors (PPARs) and their agonists for hypertension and heart failure: are the reagents beneficial or harmful? Int. J. Cardiol. 130: 131–139. [DOI] [PubMed] [Google Scholar]
- Cheung B. M. Y., Li C., 2012. Diabetes and hypertension: is there a common metabolic pathway? Curr. Atheroscler. Rep. 14: 160–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chobanian A. V., Bakris G. L., Black H. R., Cushman W. C., Green L. A., et al. , 2003. Seventh report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Hypertension 42: 1206–1252. [DOI] [PubMed] [Google Scholar]
- Dermitzakis E. T., 2008. From gene expression to disease risk. Nat. Genet. 40: 492–493. [DOI] [PubMed] [Google Scholar]
- DiBona G. F., 1992. Sympathetic neural control of the kidney in hypertension. Hypertension 19: I28–I35. [DOI] [PubMed] [Google Scholar]
- DiBona G. F., 2002. Sympathetic nervous system and the kidney in hypertension. Curr. Opin. Nephrol. Hypertens. 11: 197–200. [DOI] [PubMed] [Google Scholar]
- Divo M., Cote C., de Torres J. P., Casanova C., Marin J. M., et al. , 2012. Comorbidities and risk of mortality in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 186: 155–161. [DOI] [PubMed] [Google Scholar]
- Droyvold W. B., Midthjell K., Nilsen T. I. L., Holmen J., 2005. Change in body mass index and its impact on blood pressure: a prospective population study. Int. J. Obes. Relat. Metab. Disord. 29: 650–655. [DOI] [PubMed] [Google Scholar]
- Dua S., Bhuker M., Sharma P., Dhall M., Kapoor S., 2014. Body mass index relates to blood pressure among adults. N. Am. J. Med. Sci. 6: 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan R. E., Ahmadian M., Jaworski K., Sarkadi-Nagy E., Sul H. S., 2007. Regulation of lipolysis in adipocytes. Annu. Rev. Nutr. 27: 79–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein M., Sowers J. R., 1992. Diabetes mellitus and hypertension. Hypertension 19: 403–418. [DOI] [PubMed] [Google Scholar]
- Ezzati M., Lopez A. D., Rodgers A., Vander Hoorn S., Murray C. J. L., 2002. Selected major risk factors and global and regional burden of disease. Lancet 360: 1347–1360. [DOI] [PubMed] [Google Scholar]
- Fields L. E., Burt V. L., Cutler J. A., Hughes J., Roccella E. J., et al. , 2004. The burden of adult hypertension in the United States 1999 to 2000: a rising tide. Hypertension 44: 398–404. [DOI] [PubMed] [Google Scholar]
- Franklin S. S., Larson M. G., Khan S. A., Wong N. D., Leip E. P., et al. , 2001. Does the relation of blood pressure to coronary heart disease risk change with aging?: the Framingham heart study. Circulation 103: 1245–1249. [DOI] [PubMed] [Google Scholar]
- Friedman J., Hastie T., Tibshirani R., 2010. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33: 1–22. [PMC free article] [PubMed] [Google Scholar]
- Fuh M. M.-T., Shieh S.-M., Wu D.-A., Chen Y.-D. I., Reaven G. M., et al. , 1987. Abnormalities of carbohydrate and lipid metabolism in patients with hypertension. Arch. Intern. Med. 147: 1035–1038. [PubMed] [Google Scholar]
- Garrison R. J., Kannel W. B., Stokes J., III, Castelli W. P., 1987. Incidence and precursors of hypertension in young adults: the Framingham offspring study. Prev. Med. (Baltim) 16: 235–251. [DOI] [PubMed] [Google Scholar]
- Girgis R. E., Mathai S. C., 2016. Pulmonary hypertension associated with chronic respiratory disease. Clin. Chest Med. 28: 219–232. [DOI] [PubMed] [Google Scholar]
- Gower J. C., 1966. Some distance properties of latent root and vector methods used in multivariate analysis. Biometrika 53: 325–338. [Google Scholar]
- Grassi G., Bertoli S., Seravalle G., 2012. Sympathetic nervous system: role in hypertension and in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 21: 46–51. [DOI] [PubMed] [Google Scholar]
- Greene C. S., Krishnan A., Wong A. K., Ricciotti E., Zelaya R. A., et al. , 2015. Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 32: 453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith M., Griffith O. L., Coffman A. C., Weible J. V., McMichael J. F., et al. , 2013. DGIdb: mining the druggable genome. Nat. Methods 10: 1209–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutkowska J., Jankowski M., 2012. Oxytocin revisited: its role in cardiovascular regulation. J. Neuroendocrinol. 24: 599–608. [DOI] [PubMed] [Google Scholar]
- Hall J. E., 2000. Pathophysiology of obesity hypertension. Curr. Hypertens. Rep. 2: 139–147. [DOI] [PubMed] [Google Scholar]
- Hall J. E., 2003. The kidney, hypertension, and obesity. Hypertension 41: 625–633. [DOI] [PubMed] [Google Scholar]
- Hall J. E., Hildebrandt D. A., Kuo J., 2001. Obesity hypertension: role of leptin and sympathetic nervous system. Am. J. Hypertens. 14: S103–S115. [DOI] [PubMed] [Google Scholar]
- Hall J. E., Crook E. D., Jones D. W., Wofford M. R., Dubbert P. M., 2002. Mechanisms of obesity-associated cardiovascular and renal disease. Am. J. Med. Sci. 324: 127–137. [DOI] [PubMed] [Google Scholar]
- Heistad D. D., Baumbach G. L., 1992. Cerebral vascular changes during chronic hypertension: good guys and bad guys. J. Hypertens. Suppl. 10: S71–S75. [PubMed] [Google Scholar]
- Henke N., Schmidt-Ullrich R., Dechend R., Park J.-K., Qadri F., et al. , 2007. Vascular endothelial cell-specific NF-κB suppression attenuates hypertension-induced renal damage. Circ. Res. 101: 268–276. [DOI] [PubMed] [Google Scholar]
- Hillas G., Perlikos F., Tsiligianni I., Tzanakis N., 2015. Managing comorbidities in COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 10: 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huan T., Esko T., Peters M. J., Pilling L. C., Schramm K., et al. , 2015a A meta-analysis of gene expression signatures of blood pressure and hypertension. PLoS Genet. 11: e1005035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huan T., Meng Q., Saleh M. A., Norlander A. E., Joehanes R., et al. , 2015b Integrative network analysis reveals molecular mechanisms of blood pressure regulation. Mol. Syst. Biol. 11: 799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D. W., Sherman B. T., Lempicki R. A., 2008. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4: 44–57. [DOI] [PubMed] [Google Scholar]
- Huang D. W., Sherman B. T., Lempicki R. A., 2009. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Consortium for Blood Pressure Genome-Wide Association Studies, . Ehret G. B., Munroe P. B., Rice K. M., Bochud M., et al. , 2011. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 478: 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson B. B., 1999. Hypertension mechanisms causing stroke. Clin. Exp. Pharmacol. Physiol. 26: 563–565. [DOI] [PubMed] [Google Scholar]
- Kaufman J. S., Asuzu M. C., Mufunda J., Forrester T., Wilks R., et al. , 1997. Relationship between blood pressure and body mass index in lean populations. Hypertension 30: 1511–1516. [DOI] [PubMed] [Google Scholar]
- Kearney P. M., Whelton M., Reynolds K., Muntner P., Whelton P. K., et al. , 2005. Global burden of hypertension: analysis of worldwide data. Lancet 365: 217–223. [DOI] [PubMed] [Google Scholar]
- Kraft P., Hunter D. J., 2009. Genetic risk prediction – are we there yet? N. Engl. J. Med. 360: 1701–1703. [DOI] [PubMed] [Google Scholar]
- Kromhout D., Bosschieter E. B., Coulander C. D., 1985. Potassium, calcium, alcohol intake and blood pressure: the Zutphen Study. Am. J. Clin. Nutr. 41: 1299–1304. [DOI] [PubMed] [Google Scholar]
- Kulkarni S., O’Farrell I., Erasi M. K. M., 1998. Stress and hypertension. WMJ 97: 34–38. [PubMed] [Google Scholar]
- Lawes C. M., Vander Hoorn S., Rodgers A., International Society of Hypertension , 2008. Global burden of blood-pressure-related disease, 2001. Lancet 371: 1513–1518. [DOI] [PubMed] [Google Scholar]
- Leonardson A., Reitman M., Emilsson V., Thorleifsson G., Zhang B., et al. , 2008. Genetics of gene expression and its effect on disease. Nature 452: 423–428. [DOI] [PubMed] [Google Scholar]
- Levy D., Larson M. G., Vasan R. S., Kannel W. B., Ho K. K., 1996. The progression from hypertension to congestive heart failure. JAMA 275: 1557–1562. [PubMed] [Google Scholar]
- Long A. N., Dagogo-Jack S., 2011. The comorbidities of diabetes and hypertension: mechanisms and approach to target organ protection. J. Clin. Hypertens. (Greenwich) 13: 244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMahon S., 1987. Alcohol consumption and hypertension. Hypertension 9: 111–121. [DOI] [PubMed] [Google Scholar]
- Marques F. Z., Campain A. E., Yang Y. H. J., Morris B. J., 2010. Meta-analysis of genome-wide gene expression differences in onset and maintenance phases of genetic hypertension. Hypertension 56: 319–324. [DOI] [PubMed] [Google Scholar]
- Maurer J., Rebbapragada V., Borson S., Goldstein R., Kunik M. E., et al. , 2008. Anxiety and depression in COPD: current understanding, unanswered questions, and research needs. Chest 134: 43S–56S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirabelli G., Cannarile F., Bruni C., Vagelli R., De Luca R., et al. , 2015. One year in review 2015: systemic lupus erythematosus. Clin. Exp. Rheumatol. 33: 414–425. [PubMed] [Google Scholar]
- Moffatt M. F., Kabesch M., Liang L., Dixon A. L., Strachan D., et al. , 2007. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 448: 470–473. [DOI] [PubMed] [Google Scholar]
- Nadler S. T., Stoehr J. P., Schueler K. L., Tanimoto G., Yandell B. S., et al. , 2000. The expression of adipogenic genes is decreased in obesity and diabetes mellitus. Proc. Natl. Acad. Sci. USA 97: 11371–11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness J., Aronow W. S., Newkirk E., McDanel D., 2005. Prevalence of symptomatic peripheral arterial disease, modifiable risk factors, and appropriate use of drugs in the treatment of peripheral arterial disease in older persons seen in a university general medicine clinic. J. Gerontol. A Biol. Sci. Med. Sci. 60: 255–257. [DOI] [PubMed] [Google Scholar]
- Neueder A., Bates G. P., 2014. A common gene expression signature in Huntington’s disease patient brain regions. BMC Med. Genomics 7: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olokoba A. B., Obateru O. A., Olokoba L. B., 2012. Type 2 diabetes mellitus: a review of current trends. Oman Med. J. 27: 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrinelli R., Ballo P., Fiorentini C., Denti S., Galderisi M., et al. , 2012. Hypertension and acute myocardial infarction: an overview. J. Cardiovasc. Med. (Hagerstown) 13: 194–202. [DOI] [PubMed] [Google Scholar]
- Psaty B. M., Furberg C. D., Kuller L. H., Cushman M., Savage P. J., et al. , 2001. Association between blood pressure level and the risk of myocardial infarction, stroke, and total mortality: The cardiovascular health study. Arch. Intern. Med. 161: 1183–1192. [DOI] [PubMed] [Google Scholar]
- Reaven G. M., 1990. Insulin and hypertension. Clin. Exp. Hypertens. A 12: 803–816. [DOI] [PubMed] [Google Scholar]
- Ritchie S. A., Ewart M.-A., Perry C. G., Connell J. M. C., Salt I. P., 2004. The role of insulin and the adipocytokines in regulation of vascular endothelial function. Clin. Sci. 107: 519–532. [DOI] [PubMed] [Google Scholar]
- Rubio-Guerra A. F., Rodriguez-Lopez L., Vargas-Ayala G., Huerta-Ramirez S., Serna D. C., et al. , 2013. Depression increases the risk for uncontrolled hypertension. Exp. Clin. Cardiol. 18: 10–12. [PMC free article] [PubMed] [Google Scholar]
- Saad M. F., Rewers M., Selby J., Howard G., Jinagouda S., et al. , 2004. Insulin resistance and hypertension: the insulin resistance atherosclerosis study. Hypertension 43: 1324–1331. [DOI] [PubMed] [Google Scholar]
- Sacks D. B., McDonald J. M., 1996. The pathogenesis of type II diabetes mellitus. A polygenic disease. Am. J. Clin. Pathol. 105: 149–156. [DOI] [PubMed] [Google Scholar]
- Salako B. L., Ajayi S. O., 2000. Bronchial asthma: a risk factor for hypertension? Afr. J. Med. Med. Sci. 29: 47–50. [PubMed] [Google Scholar]
- Sawicka, K., M. Szczyrek, I. Jastrzębska, M. Prasał, A. Zwolak et al., 2011 Hypertension – the silent killer. J. Pre-Clin. and Clin. Res. 5: 43–46. [Google Scholar]
- SHEP Cooperative Research Group 1991. Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension: final results of the systolic hypertension in the elderly program (shep). JAMA 265: 3255–3264. [PubMed] [Google Scholar]
- Soleimani M., 2015. Insulin resistance and hypertension: new insights. Kidney Int. 87: 497–499. [DOI] [PubMed] [Google Scholar]
- Sowers J. R., Epstein M., Frohlich E. D., 2001. Diabetes, hypertension, and cardiovascular disease: an update. Hypertension 37: 1053–1059. [DOI] [PubMed] [Google Scholar]
- Špinar J., 2012. Hypertension and ischemic heart disease. Cor Vasa 54: 433–438. [Google Scholar]
- Stokes J., III, Kannel W. B., Wolf P. A., Cupples L. A. D. R., 1987. The relative importance of selected risk factors for various manifestations of cardiovascular disease among men and women from 35 to 64 years old: 30 years of follow-up in the Framingham Study. Circulation 75: V65–V73. [PubMed] [Google Scholar]
- Takarada A., Yokota Y., Kumaki T., Toh S., Seo T., et al. , 1985. Hypertensive heart disease simulating dilated cardiomyopathy. J. Cardiogr. 15: 1015–1026. [PubMed] [Google Scholar]
- The GTEx Consortium , 2015. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348: 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triolo L., Cattaruzza M. S., Sicoli R., Ansali F., Malaguti M., et al. , 2004. Blood pressure control and comorbidity in a nephrology clinic. J. Nephrol. 17: 808–812. [PubMed] [Google Scholar]
- Tsai E. J., Kass D. A., 2009. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol. Ther. 122: 216–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veterans Administration Cooperative Study Group on Antihypertensive Agents 1967 Effects of treatment on morbidity in hypertension: results in patients with diastolic blood pressures averaging 115 through 129 mm Hg. JAMA 202: 1028–34. [PubMed] [Google Scholar]
- Usuda D., Kanda T., 2014. Peroxisome proliferator-activated receptors for hypertension. World J. Cardiol. 6: 744–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler H. E., Aquino-Michaels K., Gamazon E. R., Trubetskoy V. V., Dolan M. E., et al. , 2014. Poly-omic prediction of complex traits: OmicKriging. Genet. Epidemiol. 38: 402–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Huang T., Petralia F., Long Q., Zhang B., et al. , 2015. Synchronized age-related gene expression changes across multiple tissues in human and the link to complex diseases. Sci. Rep. 5: 15145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., Lupski J. R., 2015. Noncoding genetic variants in human disease. Hum. Mol. Genet. 24: R102–R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Carbonetto P., Stephens M., 2013. Polygenic modeling with Bayesian sparse linear mixed models. PLoS Genet. 9: e1003264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1967. Effects of treatment on morbidity in hypertension: results in patients with diastolic blood pressures averaging 115 through 129 mm hg. JAMA 202: 1028–1034. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All gene expression and genotype data used are GTEx version 6 obtained from https://gtexportal.org.