

Abstract
Background
µ-Opioid receptor internalization is considered to be critically linked to antinociceptive tolerance. Although µ-opioid receptor agonists have been administered simultaneously with other drugs to control pain, little information is available regarding opioid–opioid interactions. Therefore, the present study was designed to further investigate the utility of a new G protein-biased ligand for µ-opioid receptors, TRV130, which has an antinociceptive effect without β-arrestin-dependent µ-opioid receptor internalization, and its combination with fentanyl using µ-opioid receptor-expressing cells and mice.
Results
In the present study, we confirmed that fentanyl produced a profound increase in β-arrestin-2 recruitment accompanied by µ-opioid receptor internalization, whereas TRV130 did not induce either the recruitment of β-arrestin-2 or µ-opioid receptor internalization in µ-opioid receptor-expressing cells. Under these conditions, β-arrestin-2 recruitment accompanied by µ-opioid receptor internalization induced by fentanyl was abolished by TRV130, whereas TRV130 did not alter the reduction of cyclic adenosine monophosphate formation by fentanyl in µ-opioid receptor-expressing cells. In a behavioral assay, TRV130 exerted an antinociceptive effect in a hot-plate test in mice. In a combination test, the antinociceptive effect of TRV130 was synergistically increased by fentanyl. Fentanyl induced antihyperalgesia and development of its tolerance under a neuropathic pain-like state following sciatic nerve ligation. However, treatment of mice with an antinociceptive dose of TRV130 did not induce the rapid development of tolerance to its antihyperalgesic effect under a neuropathic pain-like state. Furthermore, the rapid development of tolerance to the antihyperalgesic effect induced by fentanyl plus TRV130 in mice with sciatic nerve ligation was not observed, unlike in the case of fentanyl alone.
Conclusions
These findings provide evidence that activation of the G protein-biased pathway through µ-opioid receptors can alter signaling in the β-arrestin-2 pathway linked to the stimulation of µ-opioid receptors. Furthermore, the combination of G protein-biased and β-arrestin-biased ligands of µ-opioid receptors exerts an ideal antinociceptive effect without the rapid development of antinociceptive tolerance.
Keywords: TRV130, fentanyl, G protein-coupled signaling pathway, β-arrestin recruitment pathway
Introduction
The field of pharmacology has advanced due to research on neurotransmitters and their functions along with the discovery and development of G protein-coupled receptor (GPCR) agonists and antagonists. GPCRs have been targeted by pharmacological agents under the assumption of a simple “on/off” system (there is also a concept of partial or inverse agonists). However, recent findings have led to the new perspective that diverse signal transductions serve as internal machinery to transmit signals, even with the stimulation of just one type of GPCR. Such diverse signaling pathways may trigger different pharmacological actions by regulating protein–protein interaction cascades. Based on this viewpoint, novel pharmacological agents, referred to as biased ligands, have been developed.1
Recent studies have shown that µ-opioid receptor agonists regulate at least two pathways after the stimulation of µ-opioid receptors: a G protein-coupled signaling pathway and a β-arrestin recruitment pathway.2,3 Recently, the µ-opioid receptor G protein-biased ligands, [(3-methoxythiophen-2-yl)methyl]({2-[(9R)-9-(pyridin-2-yl)-6-oxaspiro-[4.5]decan-9-yl]ethyl})amine (TRV130) and PZM21, which selectively activate a G protein-coupled signaling pathway after the activation of µ-opioid receptors, have been synthetized.4,5 Interestingly, TRV130 and PZM21 both produce a potent antinociceptive effect in mice without inducing severe constipation or respiratory dysfunction,4,5 indicating that such biased ligands could be safer therapeutic tools for controlling pain.
Clinically, after a patient is given repeated administrations of opioids, the dosage usually needs to be increased.6 Adaptive changes after the chronic application of opioids, so-called antinociceptive tolerance, are related to changes in cellular and molecular pathways. Although several mechanisms for this phenomenon have been proposed, including the downregulation of receptors, desensitization of receptor signaling, and upregulation of drug metabolism, µ-opioid receptor internalization after the recruitment of β-arrestin-2 toward µ-opioid receptors through a β-arrestin signaling pathway is a strong candidate for explaining antinociceptive tolerance in response to opioids.7
Several prescribed opioids are coadministered to titrate and/or control pain. To the best of our knowledge, there is no previous report regarding the regulation of intracellular events as well as pharmacological effects through the simultaneous stimulation of β-arrestin signaling and G protein-dependent signaling pathways by ligands. In this study, we demonstrated that the combination of TRV130 and fentanyl (a preferential β-arrestin-biased ligand7) dramatically influenced intracellular events and prevented the antinociceptive tolerance induced by fentanyl. Our findings indicate that the combination of G protein-biased and β-arrestin-biased ligands for µ-opioid receptors produces an ideal antinociceptive effect without the development of antinociceptive tolerance.
Materials and Methods
cAMP assay
GloSensor cyclic adenosine monophosphate (cAMP) assay (Promega, Madison, WI, USA) was used to measure G protein activation according to the manufacturer’s protocol. Briefly, HEK293-MOR-FLAG cells that stably expressed μ-opioid receptors were plated at 1.5 × 104 cells/µl in 96-well plates in high-glucose Dulbecco’s modified Eagle medium containing 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 700 µg/ml G418 disulfate aqueous solution (Nacalai Tesque, Kyoto, Japan). Next day, cells were transiently transfected with GloSensor cAMP Plasmid (Promega) using the FuGENE HD Transfection Reagent (Promega). Twenty-four hours after transfection, medium is changed to equilibration medium (88% CO2-independent medium, 10% FBS, 2% GloSensor cAMP Reagent stock solution), and then cells were incubated for 2 h. After 5-min application with forskolin (130 µM), drugs were treated to the well for 30 min. Chemiluminescence in each step was measured by a microplate reader (GLOMAX, Promega).
β-arrestin-2 recruitment assay
The PathHunter enzyme complementation assay (DiscoveRx) was used to assess β-Arrestin-2 recruitment according to the manufacturer’s protocol. Briefly, CHO-K1 Opioid Receptor Mu 1 (OPRM1) express cells that stably overexpressed µ-opioid receptors were seeded to the 96-well plates. Next day, agonists (prepared in HESS, 20 mM 4-(2-hydroxyethyl)-1- piperazineethanesulfonic acid or N-2- hydroxyethylpiperazine-N'-2-ethanesulfonic acid (HEPES), pH 7.4) were added to the cells and incubated for 90 min. The detection reagents were also added to the cells. After 60 min, chemiluminescence per well was measured on the microplate reader (GLOMAX, Promega).
Internalization assay
For the receptor internalization assay, we obtained cDNA for N-terminal HaloTag®-fused human µ-opioid receptors from Kazusa DNA Research Institute (Kisaragi, Chiba, Japan). The clone was stably expressed in human embryonic kidney (HEK) 293 cells, and these cells were used for the receptor internalization assay. Cells were washed with Krebs-Ringer HEPES buffer and then stained with 0.5 µM HaloTag® Alexa Fluor 488, which is a membrane-impermeable dye for HaloTag® that binds irreversibly, for 15 min. Stained cells were washed with Krebs-Ringer HEPES and then stimulated with several µ-opioid receptor ligands for 60 min. The intensities of pixels of green-stained HaloTag®-µ-receptors before and after stimulation with several ligands were captured by confocal microscopy (Carl Zeiss Japan, Tokyo, Japan). The proportion of internalized receptors was analyzed quantitatively.
Quantitative analysis of receptor internalization
HaloTag®-µ-receptor internalization was quantified using a single confocal image that included the nucleus and a large area of the cytoplasm.
A line was drawn around the outside of the cell, and total cell fluorescence (cell membrane + cytoplasm) was measured as the total intensity of pixels with a fixed cell preparation. To measure the cell membrane and cytoplasmic receptors separately, we drew a second line 2 µm from the first line inside the cell membrane and measured the intensity of pixels at the same fluorescence density between the two lines. The percentage of fluorescence in the cytoplasm was calculated as the fluorescence intensity in the cytoplasmic region (pixels measured second) divided by the total fluorescence intensity (pixels measured first) as Δ % internalization.8 Fluorescence intensities obtained from 15 to 20 cells were calculated in independent experiments.
Identification of internalized µ-opioid receptor-induced spots
The HaloTag® pH Sensor Ligand, which is a membrane-impermeable fluorescent probe that is chemically optimized for sensing acidic intravesicular pH values, was also used to investigate receptor internalization. In HEK293 cells expressing N-terminal HaloTag®-fused human µ-opioid receptors, HaloTag® pH Sensor Ligand binds to HaloTag® protein on the cell membrane but does not emit fluorescence under the conditions of Excitation/Emission: 534/562 nm. When HaloTag® pH Sensor Ligand-bound receptors internalize to endosomes by endocytosis, HaloTag® pH Sensor Ligand elicits red fluorescence in acidic vesicles. Therefore, the fluorescent intensity reflects the amount of the internalized-µ opioid receptor.
The pH Sensor Ligand was used as follows: HEK293 cells expressing N-terminal HaloTag®-fused human µ-opioid receptors were washed with Krebs-Ringer HEPES buffer and then stained with 0.5 µM HaloTag® pH Sensor Ligand for 15 min. The HaloTag® pH Sensor Ligand-bound cells were washed with Krebs-Ringer HEPES buffer and then stimulated with several agonists for µ-opioid receptor for 60 min. The intensities of pixels of red-stained HaloTag®-µ-receptors in the cells before and after stimulation with several agonists were captured by confocal microscopy (Carl Zeiss Japan, Tokyo, Japan). Quantification of the levels of receptor internalization was achieved by counting red fluorescence spots per cell with a Cellomics ArrayScan VTI (Thermo Fisher Scientific, Waltham, MA, USA).
Animals
Male Institute of Cancer Research (ICR) mice (20–25 g) (Tokyo Laboratory Animals Science Co., Ltd, Tokyo, Japan) were used. Food and water were available ad libitum in their home cages. All animals were housed in a room maintained at 22 ± 1℃ with a 12-h light-dark cycle (light on 8:00 a.m. to 8:00 p.m.). The present study was conducted in accordance with the Guiding Principles for the Care and Use of Laboratory Animals at Hoshi University, as adopted by the Committee on Animal Research of Hoshi University, which is accredited by the Ministry of Education, Culture, Sports, Science, and Technology of Japan. Every effort was made to minimize the numbers and any suffering of animals used in the following experiments.
Hot-plate test
The antinociceptive response was evaluated by recording the latency to paw licking or tapping in the hot-plate test (55 ± 0.5°; Muromachi Kikai Co., Ltd, Tokyo, Japan) as described previously.9 Antinociceptive effects were measured after the administration of TRV130 (0.5 or 1 mg/kg, s.c.) and/or fentanyl (60 µg/kg, s.c.). Each animal served as its own control, and the latency to a response was measured both before and after drug administration. Antinociception was calculated as a percentage of the maximum possible effect (% antinociception) according to the following formula: % antinociception = (test latency − predrug latency)/(cut-off time − predrug latency) × 100. The antinociceptive response represents the mean ± SEM of % antinociception. To prevent tissue damage, we established a 30-s cut-off time.
Neuropathic pain model and measurement of thermal hyperalgesia
The mice were anesthetized with 3% isoflurane. We produced a partial sciatic nerve injury by tying a tight ligature with an 8-0 silk suture around approximately one-third to one-half the diameter of the sciatic nerve on the right side (ipsilateral side) under a light microscope (SD30, Olympus, Tokyo, Japan) as described previously.10,11 To assess the sensitivity to thermal stimulation, each of the hind paws of mice was tested individually using a thermal stimulus apparatus (UGO-BASILE, Biological Research Apparatus, VA, Italy). Briefly, the intensity of the thermal stimulus was adjusted to achieve an average baseline paw-withdrawal latency of approximately 9–12 s in naive mice. Only quick hind paw movements (with or without licking of the hind paws) away from the stimulus were considered to be a withdrawal response. Paw movements associated with locomotion or weight-shifting were not counted as a response. The paws were measured alternating between the left and right with an interval of more than 3 min between measurements. The latency of paw withdrawal after the thermal stimulus was determined as the average of three measurements per paw.
Drugs
The drugs used in the present study were TRV130 (synthesized by us), morphine hydrochloride (Daiichi-Sankyo Co., Ltd, Tokyo, Japan), and fentanyl citrate (Daiichi Sankyo Propharma, Co. Ltd, Tokyo, Japan). All drugs were dissolved in saline and administered in a volume of 10 ml/kg.
Statistical analysis
Data are expressed as the mean ± SEM of 6–10 animals or samples. The statistical significance of differences between groups was assessed by the Mann–Whitney test. A one-way or two-way analysis of variance followed by the Bonferroni multiple comparisons test was used for the statistical analysis. All statistical analyses were performed using Prism software (Version 5.0a; GraphPad Software, Inc., La Jolla, CA, USA). A p value of 0.05 was considered to reflect significance.
Results
Effects of opioids on G protein-biased and β-arrestin-biased signal transduction in cells that stably expressed µ-opioid receptor
It is well known that stimulation of µ-opioid receptors reduces cAMP formation. To confirm whether TRV130, like prescribed opioids, could produce signal transduction through the G protein-biased pathway, we examined the effects of µ-opioid receptor agonists on the forskolin-induced accumulation of cAMP in Chinese hamster ovary (CHO) cells that stably expressed µ-opioid receptors. The accumulated cAMP induced by forskolin was dose-dependently suppressed by all of the µ-opioid receptor agonists tested (Figure 1(a)).
Figure 1.

Evaluation of µ-opioid receptor agonist-induced intracellular events in cells that stably overexpressed µ-opioid receptor. Effects of morphine, fentanyl, and TRV130 on the forskolin-induced increase in intracellular cAMP levels in CHO cells that overexpressed human µ-opioid receptors (a). Measurement of β-arrestin-2 recruitment by the PathHunter enzyme complementation assay (b). β-arrestin-2 recruitment was measured in terms of an increase in luminescence after the administration of morphine, fentanyl, or TRV130. Localization of μ-opioid receptors after activation of μ-opioid receptors by μ-opioid receptor agonists in HEK-293 cells that stably overexpressed Halo-µ-opioid receptors (c). Measurement of Δ% of internalization of μ-opioid receptors (d). Analysis of internalized μ-opioid receptors following treatment with μ-opioid receptor agonists in HEK-293 cells that stably overexpressed Halo-µ-opioid receptors by using pH Sensor Ligand (e). Δ of spotted μ-opioid receptors (f) after the administration of opioids. Blue: Hoechst nuclear staining, Green: membrane-localized Halo-µ-opioid receptors, Red: internalized Halo-µ-opioid receptors. Each point and column represents the mean with SEM of three independent experiments.
cAMP: cyclic adenosine monophosphate; HEK: human embryonic kidney.
Next, we determined whether TRV130 and other opioids could induce a β-arrestin-biased pathway by measuring the recruitment of β-arrestin-2. Treatment with fentanyl (10−9–10−6 M) produced a dose-dependent increase in luminescence that reflects the recruitment of β-arrestin-2 in CHO cells that stably expressed µ-opioid receptors, whereas morphine (10−8–10−5 M) produced an intermediate level of β-arrestin-2 recruitment compared to that of fentanyl (Figure 1(b)). In contrast, TRV130 (10−8–10−5 M) scarcely affected the recruitment of β-arrestin-2 (Figure 1(b)). To confirm whether such recruitment of β-arrestin-2 could correspond to the internalization of µ-opioid receptors, we analyzed Halo fluorescence in CHO cells that stably expressed N-terminal-Halo-tagged µ-opioid receptors. In this study, µ-opioid receptors were mainly observed in the cellular membrane and were scarcely observed in the cytoplasm (Figure 1(c) and (e), left panels). The apparent internalization of µ-opioid receptors was observed after treatment with fentanyl (10−6 M), and these effects were significant compared to the findings in nontreated cells (Figure 1(d)). On the other hand, neither TRV130 (10−5 M) nor morphine increased the fluorescence signals in the cytoplasm (Figure 1(c) to (f)). Consistent with these results, fentanyl, but not morphine or TRV130, promoted the significant internalization of µ-opioid receptors as reflected by spot counts inside cells that stably expressed µ-opioid receptors (Figure 1(d)), indicating that signal transductions after the stimulation of µ-opioid receptors by µ-opioid receptor ligands can differ. In particular, TRV130 has a profile that is similar to that of morphine and selectively stimulates the G protein pathway without transmitting a signal through the β-arrestin recruitment pathway. In contrast, fentanyl potently stimulates the β-arrestin recruitment pathway.
Investigation of the combined effects of TRV130 and fentanyl on cellular events in cells that overexpressed µ-opioid receptor
It is well known that opioid rotation as well as titration with the simultaneous use of several opioids can be effective for combating pain in clinical situations. Therefore, we hypothesized that the simultaneous stimulation of both the G protein-coupled signaling pathway and the β-arrestin recruitment pathway by each ligand could alter the balance of intracellular signal transduction. To better understand the intracellular consequences after simultaneous stimulation of the G protein-coupled signaling pathway and β-arrestin recruitment pathway, we decided to analyze intracellular events after the stimulation of µ-opioid receptors by a combination of TRV130 and fentanyl. The reduction of the forskolin-induced accumulation of cAMP by fentanyl (100 pM–1 µM) in CHO cells that stably expressed µ-opioid receptor was not changed by the simultaneous administration of TRV130 (1 nM–10 µM) (Figure 2(a)). On the other hand, both the induction of β-arrestin-2 recruitment (Figure 2(b)) as well as µ-opioid receptor internalization (Figure 2(c)) induced by fentanyl (1 nM–1 µM) were completely diminished when fentanyl was combined with TRV130 (10 µM).
Figure 2.

Evaluation of TRV130 plus fentanyl-induced intracellular events in cells that stably overexpressed µ-opioid receptor. Effects of the combination of TRV130 (1 nM–10 µM) and fentanyl (100 pM–1 µM) on the forskolin-induced increase in intracellular cAMP levels in cells (a). Changes in β-arrestin recruitment to μ opioid receptor following treatment with fentanyl (1 nM–1 μM) and/or TRV130 (10 nM–10 μM) using a β-arrestin recruitment assay (b). Localization of μ-opioid receptors after activation by fentanyl (100 nM) or fentanyl (100 nM) plus TRV130 (10 µM) in HEK-293 cells that stably overexpressed Halo-µ-opioid receptors (c). Evaluation of Δ spot counts/cell induced by μ-opioid receptor agonists. Each column or point represents the mean with SEM of three independent experiments.
cAMP: cyclic adenosine monophosphate; HEK: human embryonic kidney.
Effects of the combination of TRV130 and fentanyl in mice
Previous studies have demonstrated that in vivo treatment with TRV130 had several pharmacological effects, especially an antinociceptive effect.4,5 TRV130 promptly produced an antinociceptive effect over a limited time course (data not shown). The combination of TRV130 (0.5 mg/kg) and fentanyl (60 µg/kg) produced a moderate antinociceptive effect, whereas a much greater antinociceptive effect was observed with the combination of a high dose of TRV130 (1.0 mg/kg) and fentanyl (60 µg/kg) (Figure 3(a)). We next examined the ability of TRV130 to induce antinociceptive tolerance. In the present study, partial sciatic nerve-ligated mice exhibited neuropathic pain-like hypersensitivity only on the ipsilateral side at seven days after nerve ligation, and the persistent painful state caused by sciatic nerve ligation lasted for at least 11 days (data not shown). Under a state of neuropathic pain, fentanyl (30 µg/kg) produced an antinociceptive effect against hyperalgesia. The development of tolerance to the antinociceptive effect of fentanyl (30 µg/kg) under neuropathic pain was observed after daily treatment for seven days (Figure 3(b)). In contrast, TRV130 (0.3 mg/kg) plus fentanyl (30 µg/kg) produced an antihyperalgesic effect under sciatic nerve ligation. However, the development of tolerance was not observed with this combination after treatment for seven days (Figure 3(b)).
Figure 3.
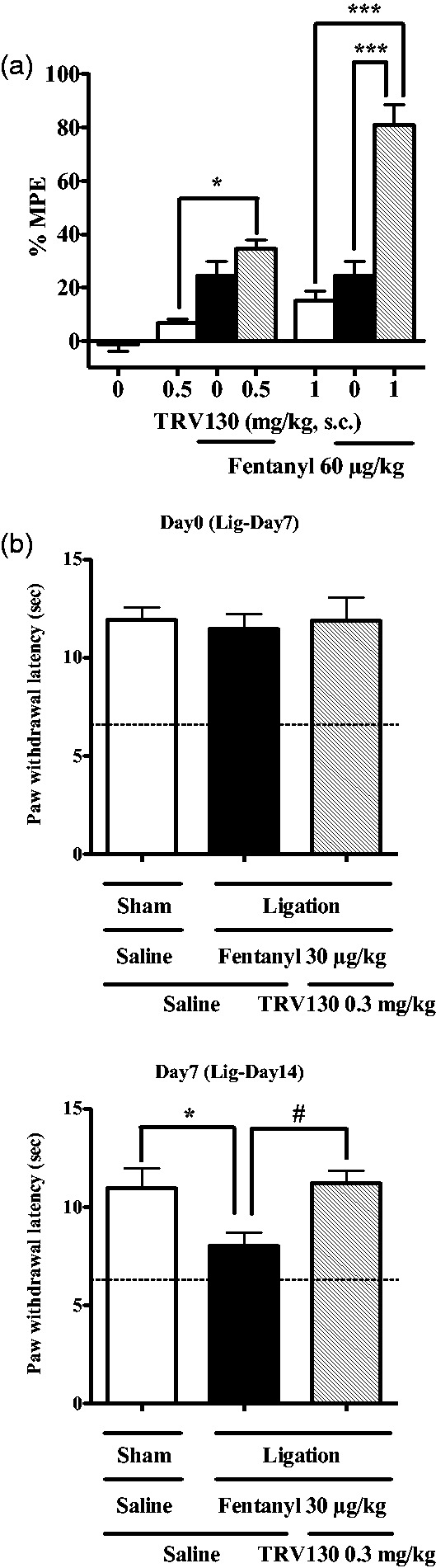
Combined effects of fentanyl and TRV130 on antinociception in mice. Enhancement of the antinociceptive effect produced by fentanyl (60 µg/kg, s.c.) and/or TRV130 (0.5 and 1.0 mg/kg, s.c.) using a hot-plate test. Antinociception is expressed as the % maximum possible effect (MPE). Each column represents the mean ± SEM of six to nine mice. *p < 0.05 and ***p < 0.001 versus saline control (a). Changes in the latency of paw withdrawal in response to a thermal stimulus in mice after sciatic nerve ligation after repeated treatments with fentanyl (30 µg/kg, s.c.) or fentanyl (30 µg/kg, s.c.) plus TRV130 (0.3 mg/kg, s.c.) for seven days. Each column represents the mean ± SEM of 6–10 mice. The dotted line indicates the mean control latency of paw withdrawal in sciatic nerve-ligated mice. *p < 0.05 versus sham-saline-saline group. #p < 0.05 versus ligation-fentanyl-TRV130 group.
Discussion
Stimulation of µ-opioid receptors by ligands regulates multiple downstream pathways, and at least two biased pathways have been proposed to date (i.e., a β-arrestin signaling pathway and a G protein-dependent signaling pathway).3 The antinociceptive effect induced by morphine is enhanced in β-arrestin-2 knockout mice,12 whereas constipation and respiratory depression induced by morphine are suppressed in these mice.13 These findings raise the possibility that a G protein-biased ligand that shows poor β-arrestin-2 recruitment may offer prominent analgesia without inducing severe constipation or respiratory depression.4,5 Our present study also confirmed that the selective G protein-biased µ-opioid receptor agonist TRV130 induced nearly undetectable levels of β-arrestin-2 recruitment and internalization of µ-opioid receptors in CHO cells that stably expressed µ-opioid receptors, whereas in vivo treatment with TRV130 produced a robust antinociceptive effect in mice.
As mentioned above, the morphine-induced antinociceptive effect is enhanced in β-arrestin-2 knockout mice.12 The GTPγS binding induced by the µ-opioid receptor agonist DAMGO in the periaqueductal gray membrane was potently increased in β-arrestin-2 knockout mice,12 indicating that the G protein pathway could be dominantly activated by µ-opioid receptor ligands when the β-arrestin pathway is inactivated. In the present study, we found that the profound induction of β-arrestin-2 recruitment and the µ-opioid receptor internalization induced by fentanyl were abolished by the simultaneous administration of TRV130. In contrast, the reduction of the forskolin-induced accumulation of cAMP by fentanyl was not suppressed by the simultaneous administration of TRV130. These findings imply that combined treatment with TRV130 may abolish the effect of fentanyl on intracellular signaling from the β-arrestin-dependent pathway without changing the G protein-biased pathway.
The key in vivo finding of the present study was that TRV130 enhanced the antinociceptive effect of fentanyl without leading to the development of antinociceptive tolerance in mice, suggesting that TRK130 could be a new tool for use in combination therapy to combat pain. We have previously shown that repeated treatment with fentanyl, but not morphine or oxycodone, produced a rapid development of tolerance to its antinociceptive effect in mice with sciatic nerve ligation.14 Opioid tolerance has been considered to result from an imbalance between the desensitization, internalization, and resensitization of µ-opioid receptors,15,16 and it is widely believed that β-arrestin-biased signaling followed by the internalization of µ-opioid receptors plays a critical role in the development of tolerance to opioids. Taken together, these results suggest that in vivo treatment with TRV130 may inhibit the recruitment of β-arrestin-2 by fentanyl as well as µ-opioid receptor internalization, leading to suppression of the development of antinociceptive tolerance to fentanyl under a neuropathic pain-like state.
Conclusion
In conclusion, TRV130 enhanced the antinociceptive effect of fentanyl without inducing the development of antinociceptive tolerance, possibly by abolishing its effect on intracellular signaling from the β-arrestin-dependent pathway without affecting the G protein-biased pathway. Therefore, our findings suggest that TRV130 may be ideal for treating pain when used in combination with other opioids. The present results may contribute to a better understanding of the intracellular mechanisms of µ-opioid receptor-biased ligands and how to control pain.
Author Contributions
NK, MY, and YU designed the study; TA, MN, TK, YM, HK, and KM performed the experiments; MK and JM collected the data; RT and MK analyzed the data; KS, AM, KB, TY, KH and MN synthetized TRV130; TM and MN wrote the manuscript. TM and NK contributed equally to the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by grants from NEXT-Supported Program for the Strategic Research Foundation at Private Universities, 2014–2018, S1411019.
References
- 1.Violin JD, Crombie AL, Soergel DG, et al. Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol Sci 2014; 35: 308–316. [DOI] [PubMed] [Google Scholar]
- 2.Borgland SL, Connor M, Osborne PB, et al. Opioid agonists have different efficacy profiles for G protein activation, rapid desensitization, and endocytosis of mu-opioid receptors. J Biol Chem 2003; 278: 18776–18784. [DOI] [PubMed] [Google Scholar]
- 3.Williams JT, Ingram SL, Henderson G, et al. Regulation of μ-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev 2013; 65: 223–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeWire SM, Yamashita DS, Rominger DH, et al. A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther 2013; 344: 708–717. [DOI] [PubMed] [Google Scholar]
- 5.Manglik A, Lin H, Aryal DK, et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016; 17: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hayhurst CJ, Durieux ME. Differential opioid tolerance and opioid-induced hyperalgesia: a clinical reality. Anesthesiology 2016; 124: 483–488. [DOI] [PubMed] [Google Scholar]
- 7.Cahill CM, Walwyn W, Taylor AM, et al. Allostatic mechanisms of opioid tolerance beyond desensitization and downregulation. Trends Pharmacol Sci 2016; 37: 963–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Minnis JG, Patierno S, Kohlmeier SE, et al. Ligand-induced mu opioid receptor endocytosis and recycling in enteric neurons. Neuroscience 2003; 119: 33–42. [DOI] [PubMed] [Google Scholar]
- 9.Narita M, Takahashi Y, Takamori K, et al. Effects of kappa-agonist on the antinociception and locomotor enhancing action induced by morphine in mice. Jpn J Pharmacol 1993; 62: 15–24. [DOI] [PubMed] [Google Scholar]
- 10.Malmberg AB, Basbaum AI. Partial sciatic nerve injury in the mouse as a model of neuropathic pain: behavioral and neuroanatomical correlates. Pain 1998; 76: 215–222. [DOI] [PubMed] [Google Scholar]
- 11.Seltzer Z, Dubner R, Shir Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain 1990; 43: 205–218. [DOI] [PubMed] [Google Scholar]
- 12.Bohn LM, Lefkowitz RJ, Gainetdinov RR, et al. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 1999; 286: 2495–2498. [DOI] [PubMed] [Google Scholar]
- 13.Raehal KM, Walker JK, Bohn LM. Morphine side effects in beta-arrestin 2 knockout mice. J Pharmacol Exp Ther 2005; 314: 1195–1201. [DOI] [PubMed] [Google Scholar]
- 14.Narita M, Imai S, Nakamura A, et al. Possible involvement of prolonging spinal μ-opioid receptor desensitization in the development of antihyperalgesic tolerance to μ-opioids under a neuropathic pain-like state. Addict Biol 2013; 18: 614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Claing A, Laporte SA, Caron MG, et al. Endocytosis of G protein-coupled receptors: roles of G protein-coupled receptor kinases and beta-arrestin proteins. Prog Neurobiol 2002; 66: 61–79. [DOI] [PubMed] [Google Scholar]
- 16.Gainetdinov RR, Premont RT, Bohn LM, et al. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci 2004; 27: 107–144. [DOI] [PubMed] [Google Scholar]