

Abstract
A new secondary metabolite verbenanone (1) with a unique (4aS,8aS)-octahydro-5H-chromen-5-one moiety has been obtained from the endophytic fungus FT431, which was isolated from the native Hawaiian plant Verbena sp. The structure of compound 1 was characterized based on NMR and MS spectroscopic analysis. The absolute configuration (AC) of compound 1 was determined by Mosher acids. Compound 1 was tested against A2780 and A2780cisR, but it was inactive.
Keywords: Hawaii, Verbena, endophytic fungi
Graphical abstract
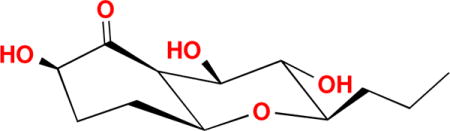
Fungi have been a great source of many biologically active compounds for drug development.1,2 For examples, the famous antibiotic penicillin G was obtained from many Penicillium strains; the immunosuppressant medication cyclosporine was isolated from Tolypocladium inflatum Gams;3 the cholesterol lowering agent mevastatin (ML-236B) is produced by the fungus Penicillium citrinum.4 Endophytic fungi, which live symbiotically within cells of higher plants, produce structurally diverse and biologically active compounds.5–10 During an ongoing search for new and bioactive compounds from Hawaiian endophytic fungi, about 5,000 semi-pure fractions from fungal culture extracts were screened against cisplatin-sensitive A2780 (A2780, human ovarian cancer) and cisplatin-resistant A2780 (A2780cisR, Stat3 abnormally activated) cell lines. The results showed that one semi-pure fraction produced by an endophytic fungus FT431 was active against A2780 and A2780cisR at 20 μg/mL. From one FT431 fraction, compound 1 (Figure 1) was obtained, which was inactive against A2780 and A2780cisR.
Figure 1.
Structure of compound 1
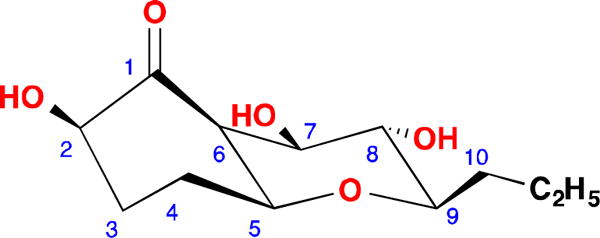
Compound 111 was isolated as a colorless solid. Its molecular formula was determined to be C12H20O5 by HR-ESIMS (m/z 261.1336, calcd for [M + H2O − H]− 2 261.1338), with three degrees of unsaturation. The IR spectrum showed the existence of a ketone carbonyl (1723 cm−1) and hydroxyl (3335 cm−1) groups. A detailed analysis of 1H and 13C NMR spectra (Table 1) demonstrated the presence of one methyl signal, four methylenes, six methines including five oxygenated, and a carbonyl carbon with no hydrogen attached. In the 1H-1H COSY spectrum of 1, only one spin system was readily identified, from H-2 all the way to H3-12, CH-CH2-CH2-CH-CH-CH-CH-CH-CH2-CH2-CH3 (Figure 2). Since there was no double bond in 1, the three degrees of unsaturation must be due to the ketone and two rings in the molecule. In the HMBC spectrum of 1, H-5 showed correlation to C-9 and H-9 correlated to C-5 (Figure 2), indicating that a tetrahydro-2H-pyran ring was formed between C-5 and C-9 since the 13C chemical shifts of C-5 and C-9 were δC 79.4 and 81.9 ppm, respectively (Table 1). The 13C chemical shifts of C-2, C-7 and C-8 were δC 76.5, 75.6 and 72.7 ppm, respectively, so these three positions must be oxygenated. In the HMBC spectrum, H-2, H-3, H-5, H-6 and H-7 correlated to C-1 (212.4), indicating a carbonyl carbon between C-2 and C-6. H-7 showed a weak HMBC correlation to C-2 (δC 76.5), due to a four-bond w-shaped coupling12. Hence, the planar structure of 1 was determined.
Table 1.
NMR Spectroscopic Data for 1 in MeOH-d4
| 1
|
|||
|---|---|---|---|
| no. | δH, J (Hz)a | δCb | ROESY correlations |
| 1 | 212.4 | ||
| 2 | 4.24, dd, 12.0, 6.7 | 76.5 | 6 |
| 3 | 1.90, m 2.14, m |
32.8 | |
| 4 | 2.03, m | 29.2 | 11a, 11b, 12 |
| 5 | 3.97, d, 2.6 m | 79.4 | 4, 6, 7, 9 |
| 6 | 3.15, br s | 55.2 | 2, 4, 5, 7 |
| 7 | 3.49, dd, 9.3, 5.0 | 75.6 | 5, 6, 9 |
| 8 | 3.63, dd, 9.3, 9.3 | 72.7 | |
| 9 | 3.08, ddd, 11.7, 9.3, 2.6 | 81.9 | 5, 7, 10a, 10b |
| 10 | 1.38, m 1.78, m |
35.3 | |
| 11 | 1.35, m 1.49, m |
19.7 | 11a-3 |
| 12 | 0.92, t, 7.2 | 14.6 | 4, 11a, 11b |
Spectra recorded at 400 MHz.
Spectra recorded at 100 MHz. Data based on 1H, 13C, HSQC, and HMBC experiments.
Figure 2.
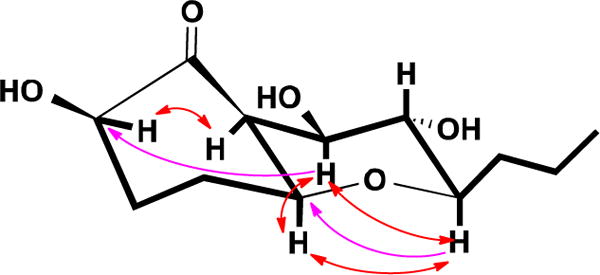
COSY (Bold), key HMBC (Single headed) and ROESY (Double headed) correlations of 1
The large coupling constants (J = 9.3 Hz) of H-8 with H-7 and H-9 meant that H-7, H-8 and H-9 must be in axial positions. The ROESY correlations between H-5 and H-7, H-5 and H-9, H-7 and H-9 indicated that H-5 was at the same side of the ring as H-7 and H-9, and it was also in an axial position. H-6 must be in an equatorial position due to its small coupling constants (br s), and it was also at the same side of ring with H-5 and H-7. The ROESY correlation between H-2 and H-6 suggested a β-. orientation of the hydroxyl group at 1-position. Hence the relative configuration of compound 1 was determined as shown in Figure 1.
To determine the absolute configuration, compound 1 was converted to the two Mosher esters 1a and 1b with (S)- and (R)-MTPA-Cl.11,13 The resulting esters 1a and 1b (Figure 3) were subjected to NMR analysis. The chemical shift differences ΔδSR were significant (Figure 3), which made it possible to conclude that 1 had the R-configuration at C-7. Based on the relative configuration determined above, the configuration at C-2, C-5, C-6, C-7, C-8 and C-9 was determined to be R, S, S, R, S, and R, respectively.
Figure 3.
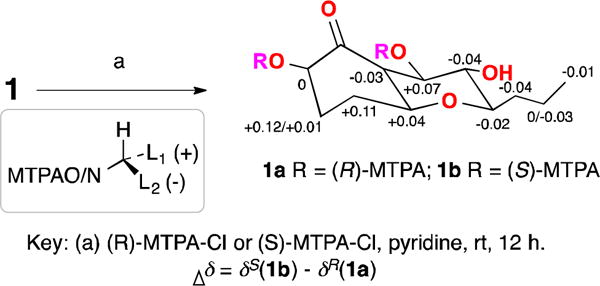
Reactions of compound 1 with Mosher esters
In order to validate the proposed assignment, and intrigued by the chemical shift of C-6 (more deshielded than expected for any regular methine), we undertook quantum chemical calculations of NMR shifts. This approach represents a useful and simple strategy for the elucidation of complex organic molecules,14 and has been extensively employed in the recent past to settle structural issues of a wide variety of natural products.14,15 As shown in Table 2, the chemical shifts of compound 1 computed at the PCM/mPW1PW91/6-31+G**//PCM/B3LYP/6-31G* level of theory (using methanol as solvent) nicely matched our experimental findings. The overall agreement was high, with CMAE (corrected mean average error, defined as Σn|δsc − δexp|/n) values of 1.3 ppm (13C) and 0.09 ppm (1H), and CMaxErr (corrected maximum error, defined as max|δsc − δexp|) of only 2.7 ppm (13C) and 0.19 ppm (1H).
Table 2.
Calculated 1H and 13C NMR shifts of 1
| 1
|
||||
|---|---|---|---|---|
| no. | Exp δH | Calc δH | Exp δC | Calc δC |
| 1 | 212.4 | 212.0 | ||
| 2 | 4.24 | 4.32 | 76.5 | 73.7 |
| 3 | 1.90 | 2.03 | ||
| 2.14 | 2.25 | 32.8 | 32.3 | |
| 4 | 2.03 | 2.02 | 29.2 | 28.6 |
| 5 | 3.97 | 4.02 | 79.4 | 77.2 |
| 6 | 3.15 | 3.00 | 55.2 | 51.7 |
| 7 | 3.49 | 3.38 | 75.6 | 73.8 |
| 8 | 3.63 | 3.43 | 72.7 | 70.4 |
| 9 | 3.08 | 3.21 | 81.9 | 77.6 |
| 10 | 1.38 | 1.32 | ||
| 1.78 | 1.79 | 35.3 | 34.1 | |
| 11 | 1.35 | 1.36 | ||
| 1.49 | 1.58 | 19.7 | 20.3 | |
| 12 | 0.92 | 0.90 | 14.6 | 14.1 |
Despite the experimental NMR observations discussed above provided a strong evidence to support the stereochemistry suggested for 1, we also computed the NMR shifts for all the remaining 31 possible diastereoisomers of 1 to strengthen the confidence in our assignment (Isomers 2–32, see the SI). To our delight, we noticed that in such cases the agreement between experimental and calculated was not as good as in the case of isomer 1 (with all the configurations indicated for 1). For instance, the CMAE values of isomers 2-32 ranged 1.5–3.7 ppm (13C) and 0.09–0.29 ppm (1H), higher than those computed for isomer 1 (1.3 ppm and 0.09 ppm, respectively), showing higher CMaxErr values as well (3.0–12.2 ppm for carbon data, 0.21–0.99 ppm for proton data). With this data in hand, we finally computed the DP4+ probability,16 among the preferred strategies to assess the most likely structure when only one set of experimental data is available.14,16 As expected, the DP4+ values strongly suggested isomer 1 as the correct candidate in high confidence (>99.9%).
During the analysis of the MS (−ve) spectrum of compound 1, we were puzzled by the ion peaks at 245 and 261. We proposed that the ketone at 1-position of 1 was hydrated when the molecule was pushed into the spectrometer. Loss of an OH from the hydrated 1 would generate m/z 245 (Figure 4).
Figure 4.
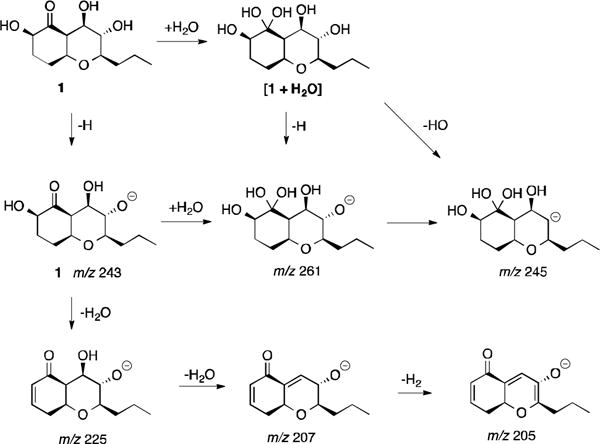
Proposed fragmentation mechanism of compound 1
Biogenetically, 1 could be derived from an unsaturated long chain molecule (i). A 6π electrocyclization event would furnish ii, that after the etherification process indicated in Figure 5 would entail the core chromene-like structure present in iii. Further hydrogenation, hydroxylation, and oxidation of iii could generate compound 1 (Figure 5).
Figure 5.
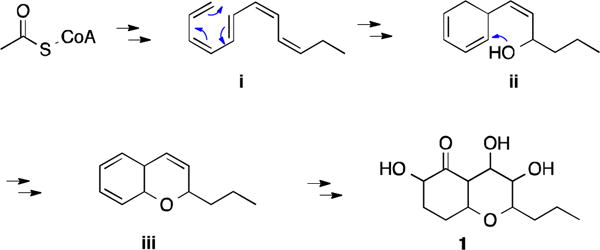
Proposed biosynthetic pathway for compound 1
While octahydro-2H-chromene derivatives are very common, small molecule octahydro-2H-chromenes with hydroxyl groups at 1- and 2-positions are unusual. For examples, compounds 2–417,18 (Figure 6) are hydroxylated octahydro-2H-chromenes, but they are synthesized compounds. Hydroxylated (4aS,8aS)-octahydro-5H-chromen-5-one19 analogs are rarer, even for organically synthetic compounds.
Figure 6.
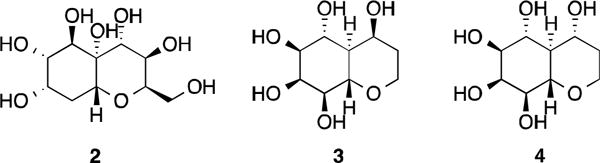
Some multiple hydroxylated octahydro-2H-chromenes
Compound 1 was tested against A2780 and A2780cisR, but it was inactive.
Supplementary Material
Verbenanone with an uncommon structure has not been reported before.
Absolute configuration of verbenanone was determined.
NMR calculation was utilized to determine the stereochemistry of verbenanone.
The biosynthesis of verbenanone has been proposed.
Acknowledgments
This work was financially supported mainly by a start-up funding from University of Hawaii Cancer Center, University of Hawaii at Hilo, and the Victoria S. and Bradley L. Geist Foundation (15ADVC-74420) (to SC) and the National Institutes of Health (NIH)/National Cancer Institute (NCI) Grant CA128865 (JT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Notes: The authors declare no competing financial interest.
Supplementary Material
Supplementary data (NMR spectra of the new compound) associated with this article can be found, in the online version, at www.sciencedirect.com.
References and notes
- 1.Schueffler A, Anke T. Nat Prod Rep. 2014;31:1425–1448. doi: 10.1039/c4np00060a. [DOI] [PubMed] [Google Scholar]
- 2.Evidente A, Kornienko A, Cimmino A, Andolfi A, Lefranc F, Mathieu V, Kiss R. Nat Prod Rep. 2014;31:617–627. doi: 10.1039/c3np70078j. [DOI] [PubMed] [Google Scholar]
- 3.Cyclosporin:; Petcher TJ, Weber HP, Rüegger A. Helv Chim Acta. 1976;59:1480–1489. doi: 10.1002/hlca.19760590509. [DOI] [PubMed] [Google Scholar]
- 4.Mevastatin:; Endo A, Kuroda M, Tanzawa K. FEBS Lett. 1976;72:323–326. doi: 10.1016/0014-5793(76)80996-9. [DOI] [PubMed] [Google Scholar]
- 5.a) Khaarwar RN, Mishra A, Gond SK, Stierle A, Stiele D. Nat Prod Rep. 2011;28:1208–1228. doi: 10.1039/c1np00008j. [DOI] [PubMed] [Google Scholar]; b) Zhang HW, Song YC, Tan RX. Nat Prod Rep. 2006;23:753–771. doi: 10.1039/b609472b. [DOI] [PubMed] [Google Scholar]
- 6.Huang P, Li CS, Sarotti AM, Turkson J, Cao S. Tetrahedron Lett. 2017;58:1330–1333. doi: 10.1016/j.tetlet.2017.04.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li CS, Ding Y, Yang BJ, Gabriella M, Yin HQ, Walker L, Fenstemacher R, Turkson J, Cao S. Org Lett. 2015;17:3556–3559. doi: 10.1021/acs.orglett.5b01650. [DOI] [PubMed] [Google Scholar]
- 8.Li CS, Yang BJ, Fenstemacher R, Turkson J, Cao S. Tetrahedron Lett. 2015;56:1724–1727. [Google Scholar]
- 9.Li CS, Ding Y, Yang BJ, Hoffman N, Yin HQ, Mahmud T, Turkson J, Cao S. Phytochemistry. 2016;126:41–46. doi: 10.1016/j.phytochem.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 10.Li CS, Ren G, Yang BJ, Miklossy G, Turkson J, Fei P, Ding Y, Walker LA, Cao S. Org Lett. 2016;18:2335–2338. doi: 10.1021/acs.orglett.6b00685. [DOI] [PubMed] [Google Scholar]
- 11.a) General experimental procedures: Optical rotation was measured with a Rudolph Research Analytical AutoPol IV Automatic Polarimeter. UV and IR spectra were obtained with Shimadzu UV-1800 sepctrophotometer and Thermo scientific Nicolet iS50FT-IR spectrometer, respectively. NMR spectra including 1D and 2D experiments were recorded in methanol-d4 on a Bruker 400 MHz NMR; CD spectrum was recorded on Jasco J-815 circular dichroism spectrophotometer in methanol. HPLC was carried out on Thermo scientific Ultimate 3000 LC system, and all solvents were HPLC grade. Column chromatography used Diaion HP-20 (Sigma).b) Isolation and identification of fungal strain: The fungal strain was isolated on PDA medium from a healthy leaf of Hawaiian indigenous plant, Verbena sp., which was collected in Lyon Botanic Gardon in 2014. The strain FT431 was identified as Peyronellaea sp. based on the analysis of the DNA sequence of the nuclear ribosomal internal transcribed spacer, which has been deposited in GenBank with the accession no. KY971272. A voucher specimen was deposited at Daniel K. Inouye College of Pharmacy, University of Hawaii at Hilo, USA (accession no. FT431).c) Cultivation: The fungus was grown under static condition at room temperature for 30 days in 1 L conical flask containing the liquid medium (300 mL/flask) composed of mannitol (20 g/L), sucrose (20 g/L), monosodium glutamate (5 g/L), KH2PO4 (0.5 g/L), MgSO4·7H2O (0.3 g/L), yeast extract (3 g/L), corn steep liquor (2 g/L) at pH 6.5.d) Isolation of compound 1: The fermented whole broth (6 L) was filtered through filter paper to separate the supernatant from the mycelia. The supernatant solution was passed through Diaion HP-20 eluted with MeOH–H2O (10%, 30%, 50%, 70% and 90%) to afford five fractions (Fr. A–E). Fr. B (258.60 mg) was further fractionated by preparative HPLC (Phenyl Hexyl column, 100.0 × 21.2 mm; 10 mL/min; with 0.1% formic acid in mobile phases) eluted with 20–50% MeOH–H2O in 30 min to get 30 sub-fractions (B1–B30). B18 (7.20 mg) was purified by semi-preparative HPLC (C18 column, 250.0 × 10.0 mm; 3 mL/min; with 0.1% formic acid in 13% CH3CN/H2O) to obtain compound 1 (2.75 mg, tR 33.4 min). (1) white powder; [α] +9.4 (c 0.32, MeOH); UV (MeOH) λmax 203, 268 nm; IR (film) νmax 3335, 2955, 2870, 2360, 2342, 1723, 1653, 1605, 1559, 1457, 1437, 1397, 1375, 1358, 1253 cm−1; 1H NMR (400 MHz, CD3OD) and 13C NMR (100 MHz, CD3OD): see Table 1; HRESIMS m/z 261.1336 [M+H2O−H]− (calcd for C12H21O6, 261.1338).e) Reaction with Mosher reagents: Acylation of compound 1 (0.7 mg, 2.87 μM each) with S-(+) and R-(−)-α-methoxy-α-(trifluoromethyl) phenyl acetyl chloride (MTPA-Cl)13 (21.43 μM each) yielded 2′-MTPA esters 1a (1.26 mg, 1.87 μM) and 1b (1.32 mg, 1.95 μM), respectively (Fig. 3). The 1H NMR signals of the MTPA esters were assigned on the basis of their COSY spectra, and the δH(S–R) values were then calculated (Fig. 3).13
- 12.Bister B, Bischoff D, Nicholson GJ, Valdebenito M, Schneider K, Winkelmann G, Hantke K, Süssmuth RD. Biometals. 2004;17:471–481. doi: 10.1023/b:biom.0000029432.69418.6a. [DOI] [PubMed] [Google Scholar]
- 13.a) Cao S, Guza RC, Wisse JH, Miller JS, Evans R, Kingston DGI. J Nat Prod. 2005;68:487–492. doi: 10.1021/np049629w. [DOI] [PubMed] [Google Scholar]; b) Hoye TR, Jeffrey CS, Shao F. Nat Proctoc. 2007;2:2451–2458. doi: 10.1038/nprot.2007.354. [DOI] [PubMed] [Google Scholar]
- 14.a) Grimblat N, Sarotti AM. Chem Eur J. 2016;22:12246. doi: 10.1002/chem.201601150. [DOI] [PubMed] [Google Scholar]; b) Lodewyk MW, Siebert MR, Tantillo DJ. Chem Rev. 2012;112:1839. doi: 10.1021/cr200106v. [DOI] [PubMed] [Google Scholar]; c) Zanardi MM, Suárez AG, Sarotti AM. J Org Chem. 2017;82:1873. doi: 10.1021/acs.joc.6b02129. [DOI] [PubMed] [Google Scholar]; d) Zanardi MM, Sarotti AM. J Org Chem. 2015;80:9371. doi: 10.1021/acs.joc.5b01663. [DOI] [PubMed] [Google Scholar]
- 15.a) Grimblat N, Kaufman TS, Sarotti AM. Org Lett. 2016;18:6420. doi: 10.1021/acs.orglett.6b03318. [DOI] [PubMed] [Google Scholar]; b) Novaes LFT, Sarotti AM, Pilli RA. J Org Chem. 2015;80:12027. doi: 10.1021/acs.joc.5b01956. [DOI] [PubMed] [Google Scholar]; c) Sarotti AM, Suárez AG, Spanevello RA. Tetrahedron Lett. 2011;52:3116. [Google Scholar]
- 16.Grimblat N, Zanardi MM, Sarotti AM. J Org Chem. 2015;80:12526. doi: 10.1021/acs.joc.5b02396. [DOI] [PubMed] [Google Scholar]
- 17.Ansari AA, Rajasekaran P, Khan MM, Vankar YD. J Org Chem. 2014;79:1690–1699. doi: 10.1021/jo402574h. [DOI] [PubMed] [Google Scholar]
- 18.Ogawa S, Ohno M, Ohhira T. Heterocycles. 1999;50:57–62. [Google Scholar]
- 19.Georgian V. 2858314 19581028 US. 1958
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.