

Abstract
The HIV-1-envelope (Env) spike is a conformational machine that transitions between prefusion (closed, CD4-bound, CCR5-bound) and postfusion states to facilitate HIV-1 entry. Although the prefusion-closed conformation is a potential target for inhibition, development of small molecule leads has been stymied by difficulties in obtaining structural information. Here, we report crystal structures at 3.8-Å resolution of HIV-1-Env trimer with BMS-378806 and its derivative, BMS-626529, for which a prodrug version is currently in Phase III-clinical trials. Both lead candidates recognized an induced-binding pocket, which was mostly excluded from solvent and comprised of Env elements from a conserved helix and the β20-21-hairpin. In both structures, the β20-21-region assumed a conformation distinct from prefusion-closed and CD4-bound states. Together with biophysical and antigenicity characterizations, the structures illuminate the allosteric and competitive mechanisms whereby these small-molecule leads inhibit CD4–induced structural changes in Env.
Introduction
Structure-based drugs have had remarkable impact on the treatment of HIV-1 infection. Since the mid-1990s, when the first structure-based drugs against HIV-1 protease entered clinical use, the prognosis for an HIV-1 infection treated with antiviral therapy has progressed from a less than 50% 10-year survival to an average life-expectancy almost indistinguishable from that of the general population1–3. In 2015, 16 million people were treated with antiviral therapy against HIV-1, for which there are currently over 40 licensed therapeutics. These target HIV-1 enzymes (protease, reverse transcriptase and integrase) and the gp41-envelope glycoprotein (Supplementary Results, Supplementary Figure 1). Currently, however, no FDA-licensed therapeutic directly targets the HIV-1 gp120-envelope glycoprotein.
Three gp120-envelope glycoproteins, along with three gp41-transmembrane subunits, make up the heterodimeric envelope (Env) trimer, a type 1 fusion machine that facilitates HIV-1 entry through a multi-step process involving structural rearrangements of both gp120 and gp41 subunits. First, the prefusion-closed conformation of the assembled Env trimer binds a single CD44, which stabilizes an intermediate state of Env. Binding to additional CD4 molecules induces the formation and exposure of a site on gp120 recognized by co-receptor, either CCR5 or CXCR4. Binding to co-receptor induces further conformational changes, especially in gp41, which result in formation of a 6-helix bundle and the fusion of the virus and host-cell membranes5,6.
HIV-1-entry inhibitors have been developed that include the FDA-approved Enfuvirtide that blocks gp41 conformational changes needed for fusion7,8 and Maraviroc that binds to the CCR5 co-receptor and prevents the formation of the Env-CCR5 complex9. A number of antibodies have also been identified that neutralize over 90% of HIV-110–13; these primarily recognize the prefusion-closed state of Env and block receptor attachment or conformational changes required for entry. CD4-mimetic small molecules and miniproteins have been developed that target an interfacial cavity, known as the ‘Phe43’ cavity14, which forms in the CD4-bound state of gp12015–18.
An especially promising family of low molecular-weight HIV-1 entry inhibitors, identified using a viral infection-based screen19, includes BMS-378806 (Bristol-Myers Squibb) and related compounds19–22. Clinical assessment of BMS-378806 was abandoned for improved versions23,24, and currently, BMS-663068, the prodrug of BMS-626529 (also known as Temsavir (GSK2616713), now being developed by ViiV Healthcare), is the top lead25,26. BMS-663068 has improved in vitro and pharmacokinetic properties compared with other family members, including an improved potency, a higher barrier for resistance, and a good safety profile in humans27–30. BMS-663068 is currently being assessed in a Phase III-therapeutic clinical trial. Here we report the structures of small molecules, BMS-378806 and BMS-626529, in complex with a soluble mimic of HIV-1-Env trimer, BG505 SOSIP, held in a prefusion conformation by antibodies PGT122 and 35O2231. The structures reveal an induced binding pocket under the β20–21 loop, distinct from the Phe43 cavity induced by CD4, suggest an allosteric mechanism of inhibition, and provide atomic-level details for inhibitor optimization.
RESULTS
Neutralization and binding of BMS-378806 and BMS-626529
We performed neutralization assays to assess the potency of BMS-378806 and BMS-626529 against two BG505 pseudoviruses. BMS-378806 and BMS-626529 neutralized BG505 pseudovirus in vitro with IC50s of 1190 and 14 nM, respectively, for BG505, and 790 and 14 nM, respectively, for BG505 T332N. We also assessed the neutralization of BMS-378806 and BMS-626529 against a panel of pseudoviruses and observed IC50 values in the range of <1 to 20,000 nM (0.0001–9.5 μg/ml), indicating highly variable sensitivities of diverse HIV-1 strains to these small molecules (Supplementary Tables 1–2). We note that, in this panel, BMS-378806 and BMS-626529 neutralized BG505 pseudovirus in vitro with IC50s of 170 and 9 nM, respectively (Supplementary Table 1), thus indicating some variation in the assay. The data also confirmed BMS-626529 to be more potent and broad than BMS-378806 with 91% of the viruses neutralized by BMS-626529 compared to 78% by BMS-378806 for an IC50<10 μg/ml and 80% of the viruses neutralized with BMS-626529 compared to 61% with BMS-378806 for an IC50<1 μM (Supplementary Tables 1–2). We note that BMS-626529 neutralized 59% viruses with an IC50<0.01 μg/ml (corresponding to less than 21.1 nM for BMS-626529 and less than 66.7 pmol for the IgG), while coverage by broadly neutralizing antibodies is generally lower at this concentration (Supplementary Table 2).
We used isothermal titration calorimetry (ITC) to determine the affinities of both compounds to a soluble version of the prefusion Env trimer (BG505 SOSIP)32 as well as to a stabilized version (DS-SOSIP)4, which binds CD4 with an asymmetric single CD4 per Env trimer and is not triggered by sCD4. BMS-378806 bound to the BG505 SOSIP trimer with ~14-fold higher affinity than to monomeric gp120 of the same HIV-1 strain (180 versus 2600 nM, respectively) and bound with slightly weaker affinity to DS-SOSIP (210 nM) (Supplementary Figure 2a). Compared with BMS-378806, BMS-626529 bound with ~2.5-fold higher affinity to BG505 SOSIP and DS-SOSIP with KDs of 73 and 87 nM, respectively (Supplementary Figure 2b). The enthalpy and entropy changes associated with binding were low, similar to what has been described previously33, indicating that, unlike CD4, little conformational change occurs in the Env glycoprotein when these compounds bind (Supplementary Figure 2a, b). Finally, analysis of binding stoichiometry indicated one BMS-378806 molecule to bind to each protomer of the Env trimer (three BMS-small molecules per Env trimer). These experiments showed BMS-378806 and BMS-626529 to bind the HIV-1 Env trimer much tighter than monomeric gp120 and to recognize similarly SOSIP and DS-SOSIP versions of the trimer (Supplementary Fig. 2).
Structures of BMS-378806 and BMS-626529 with HIV-1 Env
We individually soaked small-molecule inhibitors into crystals of the HIV-1 Env trimer. The crystals comprised trimeric BG505 SOSIP bound by antibodies PGT122, a 332-glycan dependent antibody34, and 35O22, a gp120-gp41 interface antibody35, as described previously31. The crystals diffracted to 3.8-Å resolution with both BMS-378806 and BMS-626529. Diffraction was anisotropic, and we used a diffraction anisotropy server (http://services.mbi.ucla.edu/anisoscale/) to determine resolution cut-off in each direction36. Three sigma data extended to 3Å along the c axis and to less than 4Å along a and b axes (see Methods regarding details on resolution). Molecular replacement with the drug-free version of the trimer (trimer-PGT122-35O22 with no drug) (PDB ID 4TVP31) was used to determine phases for the structure, and refinement led to Rwork/Rfree values of 0.2453/0.2888 and 0.2723/0.3254 for crystals of with BMS-378806 and BMS-626529, respectively (Supplementary Table 3).
These crystal structures revealed the BMS molecules to bind a surface-accessible pocket, at the interface between the inner and outer domain of gp120, under the β20-21 loop, and to interact with the C-terminus of the α1-helix (Figure 1a, Supplementary Figure 3a). This pocket is on the other side of β20-21 relative to the Phe43 cavity where CD4 binds, and only minimally surface accessible (Figure 1b, 1c, Supplementary Figure 3b). Because of the moderate resolution, it was difficult to position unambiguously all atoms of the small molecule, particularly those in the oxoacetamide and piperazine moieties. However, the addition of the methyl-triazole ring in BMS-626529 compared to no ring in BMS-378806 allowed for an unequivocal determination of overall drug orientation, with the benzoyl ring inserting deep in the conserved drug-induced pocket and the methoxy-azaindole ring and triazole ring (for BMS-626529) positioned toward the outside of the cavity (Figures 2 and 3).
Figure 1. HIV-1-Env recognition by small molecule entry inhibitor BMS-626529.
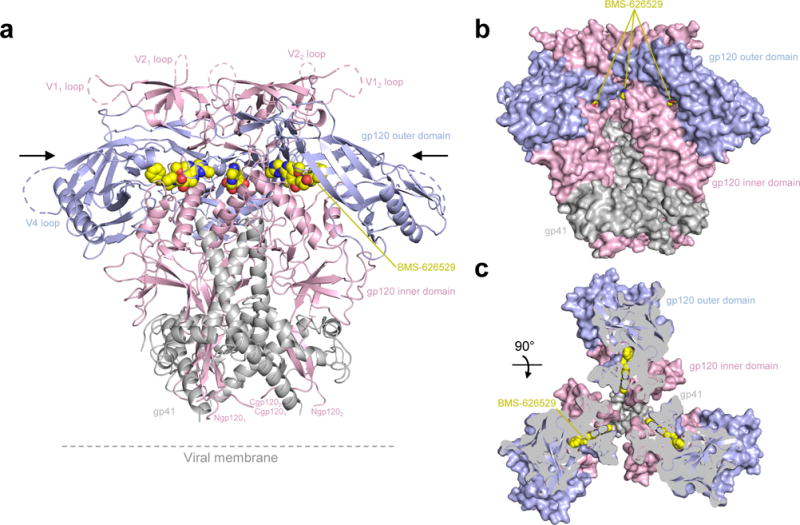
(a) Structure of BG505 SOSIP in complex with BMS-626529. The antibodies PGT122 and 35O22, present in the complex, have been removed for clarity. The HIV-1 Env protomers are shown as ribbon representation. gp120 inner domain is colored pink and gp120 outer domain is in blue, gp41 is shown in grey. BMS-626529 is shown in yellow in sphere representation. (b) Protomers colored as in (a) shown in surface representation with the location of BMS-626529 binding indicated in yellow. (c) 90° rotation from (a) and (b) with protomers shown in surface representation. To visualize the location of BMS-626529 binding, a clip has been done as indicated by the black arrows in (a).
Figure 2. Detailed interactions of BMS-626529 with HIV-1 Env gp120.
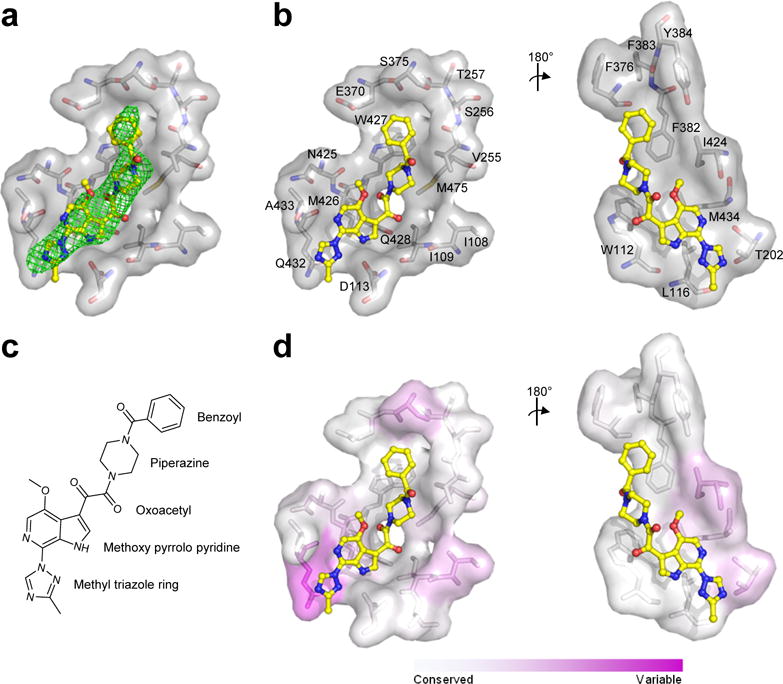
(a) Fo–Fc electron density at 3σ of simulated annealing omit map around BMS-626529 (yellow) shown as green mesh. (b) View from bottom of BMS-626529 and view from the top after 180° rotation. Residues that are within 5 Å of BMS-626529 (yellow) are labeled and shown as sticks and surface representation. (c) Chemical formula for BMS-626529. (d) Sequence variability of the binding pocket is shown in the same two orientations as (b).
Figure 3. Detailed interactions of BMS-378806 with HIV-1 Env gp120.

(a). Fo–Fc electron density at 3σ of simulated annealing omit map around BMS-378806 (orange) shown as green mesh. (b) Chemical formula for BMS-378806. (c) View from bottom of BMS-378806 and view from the top after 180° rotation. Residues that are within 5 Å of BMS-378806 (orange) are labeled and shown as sticks and surface representation.
Details of inhibitor interaction with HIV-1 gp120
Structures of BMS-378806 and BMS-626529 in complex with HIV-1 Env were virtually superimposable, with the methyl-triazole ring of BMS-626529 extending towards the trimer axis. Both compounds bound gp120 in a similar manner (Figures 2b, 2c, 3c), with BMS-626529 having additional interactions through its methyl-triazole ring with Thr202 and Gln432 of gp120 (Figure 2b). The side chain of Gln432 appeared to rearrange compared to the BMS-378806-bound conformation (Figure 3c) to allow for a better fit of the methyl-triazole ring. The buried surface area between BMS-378806 and gp120 totaled 870 Å2 and between BMS-626529 and gp120 totaled 1020 Å2, respectively. BMS-378806 buried a total of 570 Å2 while BMS-626529 buried a total of 630 Å2. Almost all of the accessible surface areas of the compounds were buried; 99 % for BMS-378806 and 94 % for BMS-626529 (Supplementary Tables 4, 5).
The BMS compounds interacted with gp120 mainly through hydrophobic interactions with Trp112, Asp113, Leu116, Val255, Phe382, Ile424, Met426, Gln 432, Trp427, Met434 and Met475, and with Thr202 for BMS-626529 only, each of which contributed more than ~10 Å2 of buried surface area (Figures 2b, 3c, Table 1, Supplementary Table 4). In addition to purely hydrophobic interactions, aromatic-stacking and hydrogen-bonding interactions also contributed. For example, the benzoyl group of the BMS compounds made parallel and offset π-stacking interaction with Phe382 and Trp427of gp120, respectively. Other interactions included a hydrogen bond between the backbone NH of Trp427 and one of the oxoacetamide carbonyls of the compound, and the side chain of Asp113 accepted a hydrogen bond from the azaindole NH group of the compound (Figures 2b, 2c, 3b, 3c, Supplementary Tables 4, 5).
Table 1.
Structural basis for resistance mutations.
| BG505 Env Sequence (HXBc2 numbering) | Mutation | Fold inhibition of BMS-378806 (Madani.37 or from this study) (Supplementary Table 6, 7) | Clinically-derived resistance substitution that reduces susceptibility of BMS-62652930 | Buried surface area (rounded) (Å2) with BMS-378806 and BMS-626529 (Supplementary Table 4) | Hydrogen bonds distance (Å) with BMS-378806/BMS-626529 (Supplementary Table 5) | Structural basis explanation |
|---|---|---|---|---|---|---|
| I109 | I109A | This study, >20 | 6/10 | No major effect | ||
| W112 | W112A | >100 | 50 /57 | Loss of hydrophobic interactions | ||
| D113 | D113A | 85 | 9 /27 | 2.6/2.4 | Loss of hydrogen bonds | |
| L116 | L116A | This study, ~20 | 19/21 | No major effect | ||
| T202 | T202K | This study, ~8 | 17 (BMS-626529) | No major effect | ||
| V255 | V255A | This study, ~13 (YU2) | 25/32 | No major effect | ||
| S375 | S375W | >100 | 7/10 | Fill pocket/prevent compound entry | ||
| S375M | Yes | Fill pocket/prevent compound entry | ||||
| S375H | Fill pocket/prevent compound entry | |||||
| S375A | 1.2 | No major effect | ||||
| F376 | 7/10 | No mutational data | ||||
| N377 | N377L | 7.3 | <5 for both | No major effect | ||
| F382 | F382L | >100 | 20/22 | Loss of hydrophobic interactions | ||
| Y384 | Y384E | This study, >100 | <5/6 | Loss of hydrophobic interactions | ||
| I424 | I424A | This study, 78 | 27/21 | Loss of hydrophobic interactions | ||
| N425 | N425A | This study, ~2 | <5/5 | No major effect | ||
| M426 | M426L | >100 | Yes | 32/29 | Loss of hydrophobic interactions | |
| W427 | W427F | 0.5 | 26/42 | 3/3 (main chain) | No major effect | |
| W427V | Loss of binding20 | Loss of hydrophobic interactions | ||||
| W427A | This study, >100 | Loss of hydrophobic interactions | ||||
| W427R | This study, >100 | Loss of hydrophobic interactions | ||||
| Q432 | K432A | 2.3 | 13/36 | No major effect | ||
| M434 | M434I | Yes | 14/15 | <4 (BMS-626529) (main chain) | Loss of hydrophobic interactions | |
| M475 | M475S | This study, 90 | 23/18 | Loss of hydrophobic interactions | ||
| M475I | Yes | Loss of hydrophobic interactions |
Mapping of resistance mutations to BMS-378806, BMS-626529 and other related inhibitors. Listed residues are within 5 Å of the inhibitor and have more than 5 Å2 buried surface area.
Inhibitor-induced conformational changes in Env
The overall conformations of the HIV-1 Env trimers with bound BMS compounds were virtually identical and highly similar to prefusion-closed drug-free or apo conformation with no drug present, but bound to antibodies PGT122 and 35O22 (PDB ID 4TVP31) (Figure 4a). The Cα-root mean square deviation (rmsd) between BMS-378806-bound and BMS-626529-bound structures was 0.4 Å over all gp120 residues. Cα-rmsd between the drug-free 4TVP and BMS-378806-bound was 0.6 Å and between the drug-free 4TVP and BMS-626529-bound structure was 0.7 Å over all gp120 residues. One region involving the β20-21 loop, however, differed more substantially between the drug-free 4TVP and the compound-bound structures, with a Cα-rmsd of 1.5-1.8 Å over 13 residues that comprise the loop (residues 423-435) and a Cα-rmsd of 1.6–2.1 Å over the 8 residues that comprise the majority of the interactions (425–432). Met26 in this loop showed the largest Cα-distance difference (2.9–3.0 Å) between compound-bound and ligand-free structures.
Figure 4. Induced-fit conformational changes.

Comparison of HIV-1 gp120 structures with and without BMS-626529: conformational rearrangement of the loop between β20 and β21. Superposition of gp120 structures in the presence (grey) and the absence (wheat) of BMS-626529 (yellow). (a) Cartoon representation showing the movement in β20–β21 loop. (b) Same orientation as (a) with residues that change conformations shown as sticks on the apo form. Intra-molecule hydrogen bonds are shown in black line. (c) Same orientation as (a) with residues that change conformations shown as sticks in the compound-bound conformation with BMS-626529 present (yellow). Residues that change conformations are labeled. (d) Superposition of (b) and (c). (e) Sequences alignment of BG505 and HXBc2 gp120. Residues that contact BMS-626529 are indicated with filled circles (for backbone interactions only), sunray (for sidechain contacts only) and filled circle with sunray (for backbone and side chain contacts). RMSDs between the BMS-626529 structure and the apo prefusion-closed conformation and between the BMS-626529 structure and the CD4-bound conformation are shown as follows: “1–8” for 1–8 Å difference, “9” for 9 Å or larger change and “X” if residue is not defined. Secondary structure is also shown. Sites of N-linked glycosylation highlighted in green. Residues in the V2 loop of strain BG505 are labeled 185a–h as previously31.
The side chains of residues Trp112 (in the α1 helix), Asn425, Met426, Trp427, Gln428, Arg429 (all in β20-21 loop) and Met 475 rearranged to provide a binding pocket and to interact with the BMS compounds (Figure 4b, c, d, e, Supplementary Figure 4). Thus, a number of local changes in the binding pocket contributed to an induced fit of gp120 for the BMS compounds.
Structural basis for inhibitor resistance
We analyzed the BMS compound-bound and drug-free structures to provide atomic-level explanations for phenotypes observed with resistance mutations. When altered, most of the gp120 residues that interact with the BMS molecules affect drug sensitivity30,37 (Table 1, Supplementary Tables 6, 7) or confer drug resistance38 (Table 1). Clinically-derived resistance substitutions known to reduce BMS-626529 susceptibility include Met426Leu or Ser375Met, with Met434Ile and Met475Ile contributing to a lesser extent30. Met426Leu was also shown to affect BMS-378806 sensitivity, while Met475Ser had minor effects37. BMS-378806 and BMS-626529 buried ~ 30 Å2, with Met426, which is part of the β20-21 loop and packs directly against the face of the compound azaindole ring. The branched side chain of the Met426Leu substitution is predicted to reduce sterically the size of the compound binding site and to affect the dynamic range of the β20-21 loop. Interactions between compound and Met475 buried ~20 Å2 of surface area and the branched side chain of Met475Ile would be expected to reduce the size of the compound-binding site.
Mutagenesis studies show Ser375Trp to affect substantially BMS-378806 sensitivity, while a Ser375Ala substitution results in more muted impact37. Although direct interactions between the compounds and Ser375 were minimal, in the crystal structure, the β-carbon of Ser375 was positioned near the 4-position of the compound’s benzoyl ring (BSA of ~7–10 Å2). While a Ser375Ala mutation would not substantially affect this interaction, larger side-chain substitutions, such as a Trp indole or a His imidazole, would likely prevent the entry or the binding of the BMS compounds. Thus, the structures explained why the drugs do not inhibit entry of HIV-2 or SIV, both of which have a Trp at position 375, as well as subtype CRF01_AE, which has a His at that position39.
In the BMS compound-bound structures, Met434 packed against Trp112, Leu116, Ala204, Phe210, Phe382, Ile424, and the compound azaindole ring. The branched side chain associated with the Met434Ile change would require either rearrangement of the gp120 inner and outer domain interface or alteration in compound orientation for binding. It should be noted that in vitro-selected changes Ala204Asp and Leu116Gln or Pro also impact susceptibility30. Other substantial interactions between the BMS compounds and gp120 occur through large hydrophobic residues, such as with Trp112 and Trp427 (with compound-buried surface area of 25-50 Å2). Decreasing the size of the side chains of these residues would likely result in a loss of hydrophobic interactions with the BMS compounds, diminishing inhibitor potency.
It has been shown that changing Thr257of gp120 to Arg decreases BMS-378806 sensitivity dramatically while Thr257Ala has little effect37. While we did not observe direct contact between Thr257 and the BMS compounds, Thr257 packs against Trp427, which is part of the β20–21 loop and interacts directly with the compounds. It is thus possible that introducing a large hydrophilic side chain would prevent the compound from entering or binding properly, similarly to the Ser375Trp change discussed above.
Additionally, we analyzed neutralization data with BMS-378806 and BMS-626529 from a panel of 208 isolates from diverse clades, respectively, evaluating all amino acids within 10 Å of the small molecule for their contribution to neutralization resistance (Supplementary Figure 5). Three variant sequences, at positions 375, 432 and 475, occurred at least three times in the isolate panel and scored above 0.8 with BMS-378806, indicating the presence of these residues in resistant strains to be 4-times as frequent as the presence of the residues in sensitive strains. Only position 475 scored above 0.8 for BMS-626529. The changes at position Ser375His, Ser375Met and Met475Ile had previously been characterized30, with a fourth change Arg432Leu newly identified by this analysis. Met375 and His375 (for BMS-378806 only) were located in the compound-binding pocket and Ile475 would reduce hydrophobic interactions. While Leu432 would seem to be compatible with BMS compound binding, one possible explanation for its contribution to resistance would be for the Leu side chain to interact with other hydrophobic residues in the compound-binding pocket to block entry. Overall, the resistance analysis described here identified commonly circulating resistance mutations.
Mechanism of inhibition
To understand the mechanism of BMS-378806 and BMS-626529 inhibition, we assessed first their binding to soluble Env trimers by MSD-ECLIA in presence of antibodies and of CD4 (Figure 5a). Both drugs were pre-incubated with Env trimer at a 10-fold excess molar ratio. Interestingly, BMS-626529, which is more potent than BMS-378806, inhibited CD4-induced conformational changes (required to bind CD4i antibodies 17b and 48d as well as V3-directed antibodies 447-52D, 3074 and 2557), while binding to other antibodies was unchanged. The inhibition of CD4-induced conformational changes could also be observed with BMS-378806, but was less pronounced than with BMS-626529. As anticipated, the drugs had no effect on DS-SOSIP antigenicity, as this stabilized trimer is already fixed in a conformation that cannot be triggered by sCD4.
Figure 5. Entry inhibition mechanism for BMS-378806 and BMS-626529 small molecules.
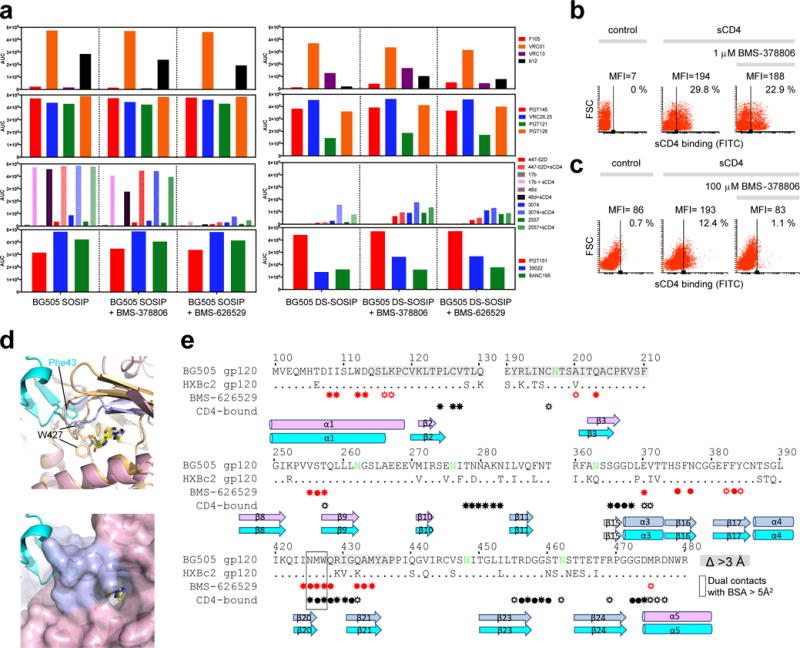
(a) MSD-ECLIA of a panel of antibodies binding to BG505 SOSIP and BG505 DS-SOSIP with and without BMS-378806 and BMS-626529. (b) Flow cytometric analysis of the effect of BMS-378806 on sCD4 binding of the HIV-1 JR-FLΔCT Env expressed on the cell surface. Analysis of sCD4 binding in the presence and absence of BMS-378806. (c). Same as (b) but at high BMS-378806 concentration. The control shows binding of the FITC-conjugated anti-CD4 antibody to the cells in the absence of sCD4. (d) (Top) Structures superposition of BMS-626529 (yellow) bound to gp120 (light pink (inner domain) and light blue (outer domain) in a HIV-1 Env trimeric prefusion-closed conformation and CD4-bound conformation (gp120 in light orange and CD4 in cyan). W427 is shown as sticks in both conformations. (Bottom) gp120 in a prefusion-closed conformation is shown in surface representation. (e) Sequences alignment of BG505 and HXBc2 gp120 regions. Residues in gp120 that make contact with BMS-626529 and CD4 (using the CD4-bound conformation) are indicated with filled circles (for backbone interactions only), sunray (for sidechain contacts only) and filled circle with sunray (for backbone and side chain contacts) in red (for BMS-626529 contacts) and black (for CD4 contacts), under the sequences. Secondary structure is shown. gp120 residues that contact both BMS-626529 and CD4, when gp120 is in the postfusion conformation, are boxed in black. Residues that move more than 3Å between the prefusion and the CD4-bound gp120 conformation are shaded in grey.
Testing the effects of BMS-378806 on the native HIV-1 Env trimer expressed on the cell surface showed that, as previously described33,40,41, at a concentration (1 μM) in the virus-inhibitory range, the compound minimally affected CD4 binding (Figure 5b) while completely blocking the CD4-induced formation/exposure of the gp41 HR1 coiled coil (Supplementary Figure 6). Notably, this analysis specifically identified cells that bound sCD4 in the presence of BMS-378806 but did not bind C34-Ig, a gp41 HR2 peptide construct that detects the HR1 coiled coil (Figure 5b, Supplementary Figure 6). At high concentrations (100 μM), BMS-378806 inhibited the binding of sCD4 (Figure 5c). These results suggest that the mechanism of HIV-1 inhibition by the BMS compounds may be concentration dependent.
To clarify further the mechanism of inhibition, we compared the structure of the drugs bound to soluble trimer to the CD4-bound structure of gp120 in the monomeric shed or ‘postfusion’ conformation that is compatible with co-receptor recognition (PDB ID 3JWD42). While the drugs bound to an Env site distinct from the CD4-binding site, there was overlap in interacting residues: indeed, three residues, Asn425, Met426 and Trp427 contact both BMS compounds and CD4 (burying more than 5Å2 surface area). Thus, the structural data indicate formal competition between binding BMS compounds and CD4 by these residues, which are in the β20-21 loop that changes the most in conformation between the drug-free and the CD4-bound structures. Structurally, the critical Trp427 exhibits different conformations in the drug-bound trimer and the CD4-bound monomer and thus cannot accommodate both ligands simultaneously (Figure 5d, e).
Discussion
The development of a soluble mimic of the viral spike, BG505 SOSIP32, which is amenable to crystallization and atomic-level structural analysis, is allowing for a structural understanding of the mechanisms of action for ligands that inhibit HIV-1 entry. In this study, we sought both a mechanistic understanding of entry inhibition by the BMS-378806 family of small-molecule inhibitors and a crystallization template for structure-based design. With Env locked into a prefusion conformation by antibodies PGT122 and 35O226,31,34,35, we observed that both BMS-378806 and BMS-626529 bound to a conserved pocket under the β20–21 loop, a loop which CD4 also recognizes14. This pocket was previously proposed as a possible binding site for the BMS drugs using mutagenesis data37 and in a recent homology modeling study43, unlike docking studies that place these compounds in the Phe43 cavity44–46.
There is general agreement that BMS-378806 and related compounds work only prior to CD4 binding21,33; the nuance here being that CD4 binding can occur with a number of Env conformations, including binding by a single CD4 to a prefusion-closed trimer4,47, binding within the context of an obligatory “high-FRET” intermediate6, and binding in the monomeric gp120 context14. Indeed, controversy has existed regarding the mechanism of action of the BMS-378806 family of small molecule inhibitors20–22,33,48. Some studies have shown the BMS compounds to bind gp120 and to block its attachment with CD420,22, while other studies have shown simultaneous binding of the BMS compounds and CD4 to HIV-1 membrane-localized Env trimers and have suggested that inhibition occurs at a later step of the entry process21,33. Recently, it has been shown that the BMS compounds stabilize a prefusion-closed conformation of Env6,48 and block Env glycoprotein transitions critical for entry40. Site-directed, in vitro-selected, and clinical-resistant mutations have helped in defining the binding region within gp120 for the BMS compounds, and multiple computer modeling studies have been performed to explore possible binding modes and further elucidate the mechanism of action of these small molecules43,44,49. However, until now, the experimental structure of a BMS compound bound within the Env trimer has remained elusive.
The BMS compound-Env trimer structures reported here are in agreement with the location of resistance-associated Env changes and explain how the drugs inhibit HIV-1 entry. In concordance with prior published studies21,22,33, our data indicate BMS-378806 and related compounds to use multiple mechanisms to inhibit HIV-1 infection. At lower concentration where the BMS compounds are first observed to inhibit HIV-1 entry, they stabilize a prefusion conformation of the Env trimer and interfere with Env-conformational changes induced by CD4, such as formation and exposure of the gp41 HR1-coiled coil. We note that inhibition by conformational stabilization likely involves alteration of the kinetic transitions the prefusion spike spontaneously undergoes6, and such inhibition can have an out-sized effect: with BMS-378806 and BMS-626529, we observed a 2.5-fold difference in binding affinity to the BG505 SOSIP trimer and a 19–85-fold difference in neutralization of BG505 virus. Consistent with this, BMS-626529 binding to the BG505 SOSIP.664 trimer is accompanied by an unfavorable entropy change, indicating that BMS-626529 binding introduces more order into the Env trimer than BMS-378806 binding. At higher concentrations where the BMS compounds are observed to inhibit CD4 binding, a mechanism related to that proposed by Langley et al.43 may apply. We observed the BMS compounds to bind under and on the opposite side of the β20-21 loop from that recognized by CD4, thereby allosterically blocking CD4 attachment. In contrast, the β20-21 loop in the postfusion conformation (PDB ID 3JWD42) is organized and stabilized by CD4 in a way that places Trp427 in the Phe43 binding pocket50; this eliminates the BMS compound-binding site and rearranges the parallel β2-3-21-20 bridging sheet of the prefusion conformation into the antiparallel β3-2-21-20 conformation observed in the CD4-bound postfusion state. Overall, our findings clarify both binding mode and mechanism of action. Thus BMS-626529 and related compounds preferentially recognize a prefusion-closed trimer over monomeric gp120, can only interact with gp120 prior to CD4 binding, and, at low concentrations, stabilize a prefusion conformation of the Env trimer, or, at high concentrations, allosterically interfere with CD4 binding, with three gp120 residues having dual contact with drug and CD4 in their respective high affinity Env-bound structures.
Online Methods
Proteins expression and purification
Proteins were purified as described previously31. Briefly, BG505 SOSIP was cotransfected with furin plasmid into Gnti−/− cells (ATCC (Cat No. CRL-3022)). Supernatant was purified over 2G12 affinity column followed by size exclusion chromatography in 5mM Hepes, 150 mM NaCl, 0.02 % azide. BG505 was also expressed in Gnti- cells, purified over 2G12 affinity column and S200 in the same buffer. PGT122 and 35O22 Fabs were made as described31. Heavy and light chain of each IgG was co-transfected in Gnti- cells. Supernatants were purified over protein A, antigen binding fragments (Fab) were obtained by digesting IgG with HRV3C enzyme and the Fabs were purified through S200.
Small molecule inhibitors
BMS-378806 and BMS-626529 were purchased from APEXxBIO, catalog # B1533 and # A3253, respectively, with a purity of 98.00 % and 98.53 %, respectively.
Isothermal titration calorimetry
The binding of BMS-378806 and BMS-626529 to HIV-1 Env trimer, BG505 SOSIP and DS-SOSIP, and BG505 gp120 monomer was studied by isothermal titration calorimetry (ITC) using a VP-ITC from MicroCal/Malvern Instruments Ltd. (Northampton, MA). The proteins were dialyzed against PBS, pH 7.5, prior to the experiments. The titrations were performed at 25°C by injecting aliquots of the inhibitor solution into the calorimetric cell (volume ~ 1.4 mL) containing either one of the two proteins prepared at ~ 3 μM in buffer with 2 % DMSO. In the case of the Env trimer, the protein concentration corresponds to that of the protomer. BMS-378806 and BMS-626529 were first dissolved in 100 % DMSO at concentrations of 10 or 20 mM which were then diluted into PBS with additional DMSO to their desired concentrations in the presence of 2 % DMSO. The concentration of inhibitor in the syringe was 60 and 120 μM for the titrations of the Env trimer and the gp120 monomer, respectively, and the solution was injected in aliquots of 10 μL. Results shown are representative of those obtained in three experiments.
HIV-1 neutralization assays
Neutralization was measured using single-round-of-infection HIV-1 Env-pseudoviruses and TZM-bl target cells, as described previously51,52. Briefly, pseudoviruses were incubated with serial dilutions of antibodies or drug compounds, then added to TZM-bl target cells which have a luciferase reporter gene. After 2 days, infection of target cells was measured by luciferase activity. Assays with drug compounds were performed with 1% DMSO in all wells. Neutralization curves were fit by nonlinear regression using a 5-parameter hill slope equation as previously described52. The sensitivity of a panel of HIV-1HXBc2 and HIV-1YU2 mutants to inhibition by BMS-378806 was determined as described in the legend to Supplementary Table 6.
Crystallization and data collection
Crystals of BG505 SOSIP, PGT122 and 35O22 were obtained as described previously31 in 0.2 M LiSO4, 16% isopropanol, 5.32% PEG 1500 and 0.1 M sodium acetate pH 5.5 by the vapor diffusion method. The drugs – BMS-378806 and BMS-626529 - were dissolved in mother liquor solution containing 0.2 M LiSO4, 20% isopropanol, 6.65% PEG1500, 0.1M sodium acetate pH 5.5 at 1 μM concentration. The stabilizing solution containing the drug was then added to the drops containing the crystals for a period of 5 minutes to 1 hour. Oil was then added to the drop and crystals suitable for diffraction were analyzed. Diffraction data was collected at APS ID22 and processed using HKL200053. The overall resolution was determined as the highest resolution for which the completeness was greater than 50% and the I/σI was greater than 2.0. Thus, for both BMS-378806 and BMS-626529 with BG505 SOSIP complexes, the overall resolution was 3.8 Å. The BMS-378806 and BMS-626529 with BG505 SOSIP complexes crystal diffraction data were then assessed for anisotropy through use of the Diffraction Anisotropy Server (http://services.mbi.ucla.edu/anisoscale/)36, which indicates the resolution at which F/sigma drops below 3.0 along a, b, and c axes; for the two lattices, these were 3.8 Å, 3.8 Å, and 3.0 Å. Because we sought to use as much of the data as possible for refinement, we used the overall resolution described above to define the resolution of the a and b axes, with the resolution limit for the c axis defined by the Diffraction Anisotropy Server. (We note that other investigators have determined overall resolutions using the following equation: Res(eff)=(high resolution)(completeness)(−1/3)54, where Res(eff) is effective resolution. For BMS-378806/BG505 SOSIP complex, the untruncated data had a completeness of 81.5%, and the effective resolution is calculated as Res(eff)=(3.0)(0.815)(−1/3) = 3.2 Å. For BMS-626529/BG505SOSIP complex, the untruncated data had a completeness of 85.6%, and the effective resolution is calculated as Res(eff)=(3.0)(0.856)(−1/3) = 3.2 Å. These alternatively defined resolutions are 0.6 Å, respectively, higher than our 50% completeness, 2 I/sigma-defined overall resolutions. Additionally, the effective resolution using the truncated data for the BMS-378806 complex and the BMS-626529 complex, which had a completeness of 51.7% and 54.06, respectively, are Res(eff)=(3.0)(0.517)(−1/3) = 3.7 Å and Res(eff)=(3.0)(0.546)(−1/3) = 3.7 Å, respectively. We also report the CC½ value of the highest resolution shell (0.450 at 3.0 Å for BMS-378806/BG505 SOSIP complex; 0.141 at 3.0 Å for BMS-626529/BG505 SOSIP complex); CC½ values of >0.15 are reported to be significant55.)
Structure solution and refinement
The structure of BMS-378806 and BMS-626529 in complex with BG505 SOSIP/PGT122/35O22 Fab was solved by molecular replacement using Phaser in CCP456 using the non-drug-complex structure as search model, PDB ID 4TVP31. Small molecules were manually fitted using COOT57 and refinement of the structures was done in Phenix58 using optimization of X-ray/stereochemistry and X-ray/ADP weight. CIF files for the small molecules were obtained using ELBOW in Phenix. The refinement statistics are summarized in Supplementary Table 3. For the BMS-378806 structure in complex with BG505 SOSIP/PGT122/35O22 Fabs, the overall score from Molprobity59 was 1.85, with a clash score of 5.59, Ramachadran favored of 92.5% and Ramachadran allowed of 99.3%. For the BMS-626529 structure in complex with BG505 SOSIP/PGT122/35O22 Fabs, the overall score from Molprobity was 1.97, with a clash score of 5.54, Ramachadran favored of 92.1% and Ramachadran allowed of 99.4%. Structural figures were made with Pymol60.
Sequence conservation calculation
Sequence conservation was calculated based on the HIV-1 filtered web alignment of the year 2015, downloaded from the Los Alamos HIV sequence database (https://www.hiv.lanl.gov/). The conservation values for each HXB2 position were obtained applying R, in particular the bio3d package function conserv with the input argument “method” set to “identity”.
Antigenic Analysis of BG505 SOSIP and DS-SOSIP in the presence of the drugs BMS-378806 and BMS-626529 by MSD-ECLIA
Standard 96-well bare MULTI-ARRAY Meso Scale Discovery (MSD) Plates (MSD, Cat# L15XA-3) were coated with a panel of HIV neutralizing (VRC01, b12, VRC13, PGT121, PGT128, 2G12, PGT145, CAP256-VRC26.25, 35O22, 8ANC195, PGT151), non-neutralizing monoclonal antibodies (F105, 17b (+sCD4), 48D (+sCD4) and 447-52D (+sCD4), 3074 (+sCD4), 2557 (+sCD4)) and non-cognate antibodies (anti-influenza antibody CR9114, and anti-RSV antibody, D25) at a concentration of 4 μg/mL in duplicates (30 μL/well), after dilution in 1X PBS. and kept overnight at 4°C. The next day, the plates were washed with 0.05% Tween-20 in 1X PBS and blocked with 150 μL of 5% [W/V] MSD Blocker A (MSD, Cat# R93BA-4)) for 1 hr at room temperature on a vibrational shaker (Heidolph TITRAMAX 100; Cat# P/N: 544-11200-00) at 650 rpm. BG505 SOSIP and DS-SOSIP were pre-incubated for 1 hr with BMS-378806 or BMS-626529 at 1:10 molar ratio of trimer:drug. The trimer or trimer-drug complex were titrated in two-fold serial dilutions starting at 5 μg/mL concentration in assay diluent (1% [W/V] MSD blocker A + 0.05 % Tween-20). For soluble CD4 (sCD4) induction, trimer/trimer-drug complex were combined with sCD4 at a constant molar concentration of 1 μM before being added to the MSD plate. After the incubation with blocking buffer was complete, the plates were washed and the diluted trimer was transferred (25 μl/well) to the MSD plates and incubated for 2 hrs on the vibrational shaker at 650 rpm. After the 2 hr incubation with trimer, plates were washed again and 2G12 antibody labeled with MSD SULFOTAG (MSD; Cat #R91AO-1) at a conjugation ratio of 1:15 (2G12:SULFOTAG)), which was diluted in assay diluent at 2 ug/mL and added to the plates (25 μL/well), then incubated for 1 hr on the vibrational shaker at 650 rpm. Plates were washed and read using the 1X read buffer (MSD Read Buffer T (4x); Cat# R92TC-1) on MSD Sector Imager 2400. Results shown are representative of those obtained in three experiments.
Flow cytometry
HIV-1 JR-FLΔCT Env plasmid was transfected with Effectene transfection reagent (Qiagen) into 293T cells, following manufacturer’s instructions. 48–72 hours later, cells were detached with 5 mM EDTA/PBS. 0.5–1 million cells were incubated briefly with 1 or 100 μM of BMS-378806, in some cases followed by sCD4 (20 μg/ml except in Figure 5c, where 100 μg/ml sCD4 was used) and thenC34-Ig (final concentration 20 μg/mL). After 30-minute incubation, the cells were washed twice. Allophycocyanin-conjugated F(ab’)2 fragment donkey anti-human IgG antibody (1:100 dilution; catalog no. 709-136-149; Jackson ImmunoResearch Laboratories) and Fluorescein isothiocyanate-conjugated anti-CD4 antibody (1:33 dilution, E-biosciences) was then added to the cells for 15 minutes. Cells were washed twice and analyzed with a BD FACSCanto II flow cytometer (BD Biosciences). APC, allophycocyanin; FSC, forward scatter; FITC, fluorescein isothiocyanate, MFI, mean fluorescence intensity. Results shown are representative of those obtained in two experiments.
Data Availability Statement
Coordinates and structure factors for the crystal structures of BMS-378806 and BMS-626529 have been deposited in the protein databank (PDB) under accessions numbers: 5U7M and 5U7O, respectively. All other data generated or analyzed during this study are included in this published article (and its supplementary information files) or are available from the corresponding author on reasonable request.
Supplementary Material
Acknowledgments
We thank J. Stuckey for assistance with figures, members of Structural Biology and Structural Bioinformatics Core Sections, Vaccine Research Center for discussions and comments on the manuscript. Funding was provided by the Intramural Research Program of the Vaccine Research Center, National Institute of Allergy and Infectious Diseases, the Intramural AIDS Targeted Antiretroviral Program, the National Institute of General Medical Sciences, National Institutes of Health (J.S.), the National Science Foundation MCB-1157506 (E.F.) and the National Institutes of Health GM56550 (E.F.). Use of sector 22 (Southeast Region Collaborative Access team) at the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science, under contract no. W-31-109-Eng-38.
Footnotes
Accession codes
Coordinates and structure factors for the crystal structures of BMS-378806 and BMS-626529 have been deposited in the protein data bank under accessions numbers: 5U7M and 5U7O respectively.
Author Contributions
M.P. determined the structures with assistance from Y-T.L., G-Y.C., D.I.S, D.R.L and P.D.K. T.B. and G-Y.C. assisted in resistance analysis. S.N. and A. McD. performed antigenic analyses. S.O’D., R.T.B., M. K. L. and J.R.M performed neutralization experiments. A.S. and E.F. performed ITC experiments. A.D. expressed proteins and H.G. and D.I.S purified proteins. R.R. performed sequence entropy analysis. A.F., A.H., N.M. and J.S. contributed mutagenesis analyses and competition analysis with CD4. M.P. and P.D.K. analyzed the data and wrote the paper, with contributions from J.S. and D.R.L.
Competing financial interests
The authors declare no competing financial interests. D.R.L owns stock in Bristol-Myers Squibb.
References
- 1.Lima VD, et al. Continued improvement in survival among HIV-infected individuals with newer forms of highly active antiretroviral therapy. Aids. 2007;21:685–692. doi: 10.1097/QAD.0b013e32802ef30c. [DOI] [PubMed] [Google Scholar]
- 2.Marcus JL, et al. Narrowing the Gap in Life Expectancy Between HIV-Infected and HIV-Uninfected Individuals With Access to Care. Jaids-Journal of Acquired Immune Deficiency Syndromes. 2016;73:39–46. doi: 10.1097/QAI.0000000000001014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vittinghoff E, et al. Combination antiretroviral therapy and recent declines in AIDS incidence and mortality. Journal of Infectious Diseases. 1999;179:717–720. doi: 10.1086/314623. [DOI] [PubMed] [Google Scholar]
- 4.Kwon YD, et al. Crystal structure, conformational fixation and entry-related interactions of mature ligand-free HIV-1 Env. Nat Struct Mol Biol. 2015;22:522–31. doi: 10.1038/nsmb.3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wyatt R, Sodroski J. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science. 1998;280:1884–8. doi: 10.1126/science.280.5371.1884. [DOI] [PubMed] [Google Scholar]
- 6.Munro JB, et al. Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science. 2014;346:759–63. doi: 10.1126/science.1254426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wild C, Greenwell T, Matthews T. A Synthetic Peptide from Hiv-1 Gp41 Is a Potent Inhibitor of Virus-Mediated Cell-Cell Fusion. Aids Research and Human Retroviruses. 1993;9:1051–1053. doi: 10.1089/aid.1993.9.1051. [DOI] [PubMed] [Google Scholar]
- 8.Oldfield V, Keating GM, Plosker G. Enfuvirtide - A review of its use in the management of HIV infection. Drugs. 2005;65:1139–1160. doi: 10.2165/00003495-200565080-00007. [DOI] [PubMed] [Google Scholar]
- 9.Dorr P, et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrobial Agents and Chemotherapy. 2005;49:4721–4732. doi: 10.1128/AAC.49.11.4721-4732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu X, et al. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science. 2010;329:856–61. doi: 10.1126/science.1187659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang J, et al. Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature. 2012;491:406–12. doi: 10.1038/nature11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muster T, et al. Cross-neutralizing activity against divergent human immunodeficiency virus type 1 isolates induced by the gp41 sequence ELDKWAS. J Virol. 1994;68:4031–4. doi: 10.1128/jvi.68.6.4031-4034.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang J, et al. Identification of a CD4-Binding-Site Antibody to HIV that Evolved Near-Pan Neutralization Breadth. Immunity. 2016;45:1108–1121. doi: 10.1016/j.immuni.2016.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwong PD, et al. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–59. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao Q, et al. Identification of N-phenyl-N′-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology. 2005;339:213–25. doi: 10.1016/j.virol.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 16.LaLonde JM, et al. Structure-based design, synthesis, and characterization of dual hotspot small-molecule HIV-1 entry inhibitors. J Med Chem. 2012;55:4382–96. doi: 10.1021/jm300265j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Madani N, et al. Small-molecule CD4 mimics interact with a highly conserved pocket on HIV-1 gp120. Structure. 2008;16:1689–701. doi: 10.1016/j.str.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang CC, et al. Scorpion-toxin mimics of CD4 in complex with human immunodeficiency virus gp120 crystal structures, molecular mimicry, and neutralization breadth. Structure (Camb) 2005;13:755–68. doi: 10.1016/j.str.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 19.Wang T, et al. Discovery of 4-benzoyl-1-[(4-methoxy-1H- pyrrolo[2,3-b]pyridin-3-yl)oxoacetyl]-2- (R)-methylpiperazine (BMS-378806): a novel HIV-1 attachment inhibitor that interferes with CD4-gp120 interactions. J Med Chem. 2003;46:4236–9. doi: 10.1021/jm034082o. [DOI] [PubMed] [Google Scholar]
- 20.Guo Q, et al. Biochemical and genetic characterizations of a novel human immunodeficiency virus type 1 inhibitor that blocks gp120-CD4 interactions. J Virol. 2003;77:10528–36. doi: 10.1128/JVI.77.19.10528-10536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ho HT, et al. Envelope conformational changes induced by human immunodeficiency virus type 1 attachment inhibitors prevent CD4 binding and downstream entry events. J Virol. 2006;80:4017–25. doi: 10.1128/JVI.80.8.4017-4025.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin PF, et al. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc Natl Acad Sci U S A. 2003;100:11013–8. doi: 10.1073/pnas.1832214100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang T, et al. Inhibitors of human immunodeficiency virus type 1 (HIV-1) attachment. 5. An evolution from indole to azaindoles leading to the discovery of 1-(4-benzoylpiperazin-1-yl)-2-(4,7-dimethoxy-1H-pyrrolo[2,3-c]pyridin-3-yl)ethane -1,2-dione (BMS-488043), a drug candidate that demonstrates antiviral activity in HIV-1-infected subjects. J Med Chem. 2009;52:7778–87. doi: 10.1021/jm900843g. [DOI] [PubMed] [Google Scholar]
- 24.Kadow J, Wang HG, Lin PF. Small-molecule HIV-1 gp120 inhibitors to prevent HIV-1 entry: an emerging opportunity for drug development. Curr Opin Investig Drugs. 2006;7:721–6. [PubMed] [Google Scholar]
- 25.Nowicka-Sans B, et al. In vitro antiviral characteristics of HIV-1 attachment inhibitor BMS-626529, the active component of the prodrug BMS-663068. Antimicrob Agents Chemother. 2012;56:3498–507. doi: 10.1128/AAC.00426-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown J, et al. Compartmental absorption modeling and site of absorption studies to determine feasibility of an extended-release formulation of an HIV-1 attachment inhibitor phosphate ester prodrug. J Pharm Sci. 2013;102:1742–51. doi: 10.1002/jps.23476. [DOI] [PubMed] [Google Scholar]
- 27.Lalezari JP, et al. Safety and efficacy of the HIV-1 attachment inhibitor prodrug BMS-663068 in treatment-experienced individuals: 24 week results of AI438011, a phase 2b, randomised controlled trial. Lancet HIV. 2015;2:e427–37. doi: 10.1016/S2352-3018(15)00177-0. [DOI] [PubMed] [Google Scholar]
- 28.Nettles RE, et al. Pharmacodynamics, safety, and pharmacokinetics of BMS-663068, an oral HIV-1 attachment inhibitor in HIV-1-infected subjects. J Infect Dis. 2012;206:1002–11. doi: 10.1093/infdis/jis432. [DOI] [PubMed] [Google Scholar]
- 29.Ray N, et al. Prediction of virological response and assessment of resistance emergence to the HIV-1 attachment inhibitor BMS-626529 during 8-day monotherapy with its prodrug BMS-663068. J Acquir Immune Defic Syndr. 2013;64:7–15. doi: 10.1097/QAI.0b013e31829726f3. [DOI] [PubMed] [Google Scholar]
- 30.Zhou N, et al. Genotypic correlates of susceptibility to HIV-1 attachment inhibitor BMS-626529, the active agent of the prodrug BMS-663068. J Antimicrob Chemother. 2014;69:573–81. doi: 10.1093/jac/dkt412. [DOI] [PubMed] [Google Scholar]
- 31.Pancera M, et al. Structure and immune recognition of trimeric pre-fusion HIV-1 Env. Nature. 2014;514:455–61. doi: 10.1038/nature13808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanders RW, et al. A next-generation cleaved, soluble HIV-1 Env Trimer, BG505 SOSIP.664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog. 2013;9:e1003618. doi: 10.1371/journal.ppat.1003618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Si Z, et al. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc Natl Acad Sci U S A. 2004;101:5036–41. doi: 10.1073/pnas.0307953101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walker LM, et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature. 2011;477:466–70. doi: 10.1038/nature10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang J, et al. Broad and potent HIV-1 neutralization by a human antibody that binds the gp41-gp120 interface. Nature. 2014;515:138–42. doi: 10.1038/nature13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strong M, et al. Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2006;103:8060–5. doi: 10.1073/pnas.0602606103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Madani N, et al. Localized changes in the gp120 envelope glycoprotein confer resistance to human immunodeficiency virus entry inhibitors BMS-806 and #155. J Virol. 2004;78:3742–52. doi: 10.1128/JVI.78.7.3742-3752.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou N, et al. In vivo patterns of resistance to the HIV attachment inhibitor BMS-488043. Antimicrob Agents Chemother. 2011;55:729–37. doi: 10.1128/AAC.01173-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schader SM, et al. HIV gp120 H375 is unique to HIV-1 subtype CRF01_AE and confers strong resistance to the entry inhibitor BMS-599793, a candidate microbicide drug. Antimicrob Agents Chemother. 2012;56:4257–67. doi: 10.1128/AAC.00639-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herschhorn A, et al. A broad HIV-1 inhibitor blocks envelope glycoprotein transitions critical for entry. Nat Chem Biol. 2014;10:845–52. doi: 10.1038/nchembio.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herschhorn A, et al. Release of gp120 Restraints Leads to an Entry-Competent Intermediate State of the HIV-1 Envelope Glycoproteins. MBio. 2016;7 doi: 10.1128/mBio.01598-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pancera M, et al. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc Natl Acad Sci U S A. 2010;107:1166–71. doi: 10.1073/pnas.0911004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Langley DR, et al. Homology models of the HIV-1 attachment inhibitor BMS-626529 bound to gp120 suggest a unique mechanism of action. Proteins. 2015;83:331–50. doi: 10.1002/prot.24726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kong R, Tan JJ, Ma XH, Chen WZ, Wang CX. Prediction of the binding mode between BMS-378806 and HIV-1 gp120 by docking and molecular dynamics simulation. Biochim Biophys Acta. 2006;1764:766–72. doi: 10.1016/j.bbapap.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 45.Teixeira C, Serradji N, Maurel F, Barbault F. Docking and 3D-QSAR studies of BMS-806 analogs as HIV-1 gp120 entry inhibitors. Eur J Med Chem. 2009;44:3524–32. doi: 10.1016/j.ejmech.2009.03.028. [DOI] [PubMed] [Google Scholar]
- 46.Li L, Chen H, Zhao RN, Han JG. The investigations on HIV-1 gp120 bound with BMS-488043 by using docking and molecular dynamics simulations. J Mol Model. 2013;19:905–17. doi: 10.1007/s00894-012-1619-5. [DOI] [PubMed] [Google Scholar]
- 47.Liu Q, et al. Quaternary contact in the initial interaction of CD4 with the HIV-1 envelope trimer. Nat Struct Mol Biol. 2017;24:370–378. doi: 10.1038/nsmb.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guttman M, et al. CD4-induced activation in a soluble HIV-1 Env trimer. Structure. 2014;22:974–84. doi: 10.1016/j.str.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Da LT, Quan JM, Wu YD. Understanding the binding mode and function of BMS-488043 against HIV-1 viral entry. Proteins. 2011;79:1810–9. doi: 10.1002/prot.23005. [DOI] [PubMed] [Google Scholar]
- 50.Shrivastava IH, Wendel K, LaLonde JM. Spontaneous rearrangement of the beta20/beta21 strands in simulations of unliganded HIV-1 glycoprotein, gp120. Biochemistry. 2012;51:7783–93. doi: 10.1021/bi300878d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li M, et al. Human Immunodeficiency Virus Type 1 env Clones from Acute and Early Subtype B Infections for Standardized Assessments of Vaccine-Elicited Neutralizing Antibodies. J Virol. 2005;79:10108–25. doi: 10.1128/JVI.79.16.10108-10125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seaman MS, et al. Tiered categorization of a diverse panel of HIV-1 Env pseudoviruses for assessment of neutralizing antibodies. J Virol. 2010;84:1439–52. doi: 10.1128/JVI.02108-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 54.Weiss MS. Global indicators of X-ray data quality. Journal of Applied Crystallography. 2001;34:130–135. [Google Scholar]
- 55.Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–3. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Collaborative Computational Project, N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 57.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 58.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Davis IW, Murray LW, Richardson JS, Richardson DC. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32:W615–9. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and structure factors for the crystal structures of BMS-378806 and BMS-626529 have been deposited in the protein databank (PDB) under accessions numbers: 5U7M and 5U7O, respectively. All other data generated or analyzed during this study are included in this published article (and its supplementary information files) or are available from the corresponding author on reasonable request.