

In excitation–contraction coupling, voltage-sensing modules (VSMs) of CaV1.1 Ca2+ channels simultaneously gate the associated pore and Ca2+ release channels in the sarcoplasmic reticulum. Ferreira Gregorio et al. find that VSMs adopt two inactivated states, and the degree of inactivation is dependent on external Ca2+ and the mouse strain used.
Abstract
In skeletal muscle, the four-helix voltage-sensing modules (VSMs) of CaV1.1 calcium channels simultaneously gate two Ca2+ pathways: the CaV1.1 pore itself and the RyR1 calcium release channel in the sarcoplasmic reticulum. Here, to gain insight into the mechanism by which VSMs gate RyR1, we quantify intramembrane charge movement associated with VSM activation (sensing current) and gated Ca2+ release flux in single muscle cells of mice and rats. As found for most four-helix VSMs, upon sustained depolarization, rodent VSMs lose the ability to activate Ca2+ release channels opening; their properties change from a functionally capable mode, in which the mobile sensor charge is called charge 1, to an inactivated mode, charge 2, with a voltage dependence shifted toward more negative voltages. We find that charge 2 is promoted and Ca2+ release inactivated when resting, well-polarized muscle cells are exposed to low extracellular [Ca2+] and that the opposite occurs in high [Ca2+]. It follows that murine VSMs are partly inactivated at rest, which establishes the reduced availability of voltage sensing as a pathogenic mechanism in disorders of calcemia. We additionally find that the degree of resting inactivation is significantly different in two mouse strains, which underscores the variability of voltage sensor properties and their vulnerability to environmental conditions. Our studies reveal that the resting and activated states of VSMs are equally favored by extracellular Ca2+. Promotion by an extracellular species of two states of the VSM that differ in the conformation of the activation gate requires the existence of a second gate, inactivation, topologically extracellular and therefore accessible from outside regardless of the activation state.
Introduction
Electrical signaling is used for multiple purposes in animal cells. In most cases, it is mediated by the opening and closing of ion channels by means of “gates” that may in turn be controlled electrically. In voltage-sensitive channels, electrical control is the task of a molecular motif expressed with high sequence identity across channels in different tissues and taxa. An indication of their ubiquity is that after many years of their characterization in channels of excitable tissues, similar voltage-sensing modules (VSMs) were identified in voltage-activated phosphatases (reviewed by Villalba-Galea, 2012). Another indication of the ubiquity of VSMs is the finding of a similar structure in Ca2+ channels of intracellular storage organelles (Efremov et al., 2015; Yan et al., 2015; Zalk et al., 2015), where the motif is called VSL, for voltage-sensitive-like.
A hint of the widespread biological applicability of VSMs appeared decades ago. Indeed, the first quantitative manifestation of VSM function, intramembranous charge movement (or sensing current), was measured in frog muscles (Schneider and Chandler, 1973), in which it is not associated with a standard gating function. Surprisingly, that charge movement was shown to occur within a membrane protein, the voltage sensor of excitation–contraction (EC) coupling, which controls an ion pathway not located in the membrane where the sensors move. It was only with the subsequent measurement of “gating” currents in squid giant axons (Armstrong and Bezanilla, 1973) that a VSM was functionally associated with gating within the same channel protein. The skeletal muscle VSM remains unique in that it controls gates in two ion pathways traversing separate membranes: CaV1.1, the L-type channel in transverse tubules, and RyR1, the Ca2+ release channel in the SR (reviewed by Rebbeck et al., 2014).
A salient property of VSMs, equally valid for EC coupling, channels and voltage-sensitive phosphatases (VSPs), is their tendency to enter a functionally disabled state upon sustained increase of the membrane potential to zero and beyond. This transition is called “relaxation,” to separate it from the closure of the pathway that most often associates with the VSM transition in ion channels. This form of channel closure, termed voltage-dependent inactivation (VDI), was found to be accompanied by major changes in the VSM sensing currents (Armstrong and Bezanilla, 1977; Bezanilla and Armstrong, 1977). VDI occurs via a variety of processes; one classification, never fully settled, distinguishes two types of VDI: N-type, whereby the N-terminal portion blocks the open channel, and C-type, a slower phenomenon, the effects of which accumulate during repetitive activation (Hoshi et al., 1990, 1991).
The effects of C-type inactivation on gating currents were first separated from the changes associated with faster N-type inactivation in squid axon Na+ channels (Bezanilla et al., 1982). A set of changes similar to C-type inactivation was described later for the EC coupling voltage sensor of frog muscle (Brum and Rios, 1987; Brum et al., 1988a,b), which at the time had been identified with the L-type Ca2+ channel CaV1.1 (Rios and Brum, 1987; Tanabe et al., 1988). Brum and Rios (1987) quantified the amount of intramembranous charge mobile in muscle cells inactivated by long-term depolarization, a component termed “charge 2” by Adrian and Almers (1976), and found that it was the same as that of “charge 1” that became available as cells were held well polarized (at holding potential, Vh, of −80 or −90 mV). The equality of charges 1 and 2, together with the simultaneity and reciprocity of their changes, was taken as evidence that they reflect the same sensors in different conditions or modes (defined later). This proposal was later confirmed in frog muscle (reviewed by Melzer et al., 1995), mammalian cardiac L-type Ca channels (Field et al., 1988; Shirokov et al., 1992), various Na+ and K+ voltage-sensitive channels, and VSPs (Villalba-Galea et al., 2008; Villalba-Galea, 2012). The term “relaxation” was introduced for VSPs while recognizing it as common to all VSMs; the terms “charge 1” and “charge 2” have been used only in skeletal and cardiac muscle.
Additionally, Brum et al. (1988a,b) and Pizarro et al. (1989) demonstrated that the presence of Ca2+ ions in the extracellular medium opposes this relaxation in frog muscles and proposed that C-type inactivation or relaxation includes conformational changes akin to a collapse of the pore in the regions that establish ionic selectivity (reviewed by Melzer et al., 1995).
The EC coupling sensor charge of mammalian muscle has been studied in rat myofibers (Hollingworth and Marshall, 1981; Simon and Beam, 1983; Delbono et al., 1991; Shirokova et al., 1996; Szentesi et al., 1997), myotubes (García et al., 1994; Avila and Dirksen, 2000), human muscle biopsies (García et al., 1992), and isolated mouse muscle fibers (Wang et al., 1999; Collet et al., 2003; Prosser et al., 2009). All these studies focused on the charge that moves in well-polarized cells. There have been no direct examinations of charge 2 or attempts to separate it from charge 1 in mammals.
Here we describe, in mammalian muscle, dynamic properties of the EC coupling-related sensing current, its relationship with the Ca2+ flux that it gates in RyR1, and the properties of the relaxed mode, including its sensitivity to the presence of extracellular Ca2+. The study demonstrates a fundamental property: like most other VSMs (reviewed by Villalba-Galea et al., 2008), those in CaV1.1 transition among four stable states, two of which are functionally inactivated. Ca2+ shifts the transition equilibria in favor of the non-inactivated states and correspondingly favors mode 1 over mode 2 of sensor charge movement. Analysis of the results constrains possible models of gating by CaV1.1 VSMs. Our results also indicate that changes in blood [Ca2+] within the range found in the human clinic can substantially alter the signaling ability of muscle VSMs and thereby modulate contractile activation, potentially contributing to the “disease phenotypes” of hypo- and hypercalcemia.
Materials and methods
Single-fiber preparation
Experiments were performed in single muscle fibers isolated from the flexor digitorum brevis (FDB) muscle of the mouse. Animals were obtained from the animal facility of the School of Medicine and killed by cervical dislocation in accordance with the protocol approved by Comisión Honoraria de Experimentación Animal of Universidad de la República. FDB muscles were dissected in Tyrode’s solution containing 145 mM NaCl, 4 mM KCl, 10 mM Hepes, 10 mM NaHPO4, 2 mM CaCl2, and 2 mM MgCl2 and then incubated for ∼1 h at 37°C in Tyrode’s solution to which 4% FBS and 2 mg ml−1 collagenase type Ia (Sigma-Aldrich) were added. After digestion, the solution was replaced by Tyrode’s solution containing 4% FBS and left for 1 h at 4°C or RT. Before the experiments, the fibers were gently dispersed using glass pipettes of decreasing size and transferred to the experimental chamber on an inverted microscope stage (Nikon Diaphot or Leica SP5 DM6000 for confocal recording). Fibers were obtained from 8–12-wk-old BALB/c and B6D2F1 mice. Additional measurements were performed in singly dissected rat muscle fibers. Rats were sacrificed by CO2 inhalation and extensor digitorum longus muscles removed. Fibers were dissected in a relaxing solution that contained 145 mM K-glutamate, 10 mM Hepes, 1 mM MgCl2, and 0.1 mM EGTA. Individual fibers were mounted in a 2-Vaseline gap chamber following the method of Shirokova et al. (1996). pH was adjusted to 7.4 and osmolarity to 310 mOsm/liter in all cases.
Voltage clamp of single myofibers
Single mouse fibers were voltage clamped following the procedure of Wang et al. (1999, 2000). Short fibers (300–500 µm) were selected for whole-cell patch clamping. External solution, designed to suppress ionic currents, contained 145 mM TEACH3SO3, 10 mM Hepes, 1.8 mM CaCl2, 3.5 mM MgCl2, 0.5 mM CdCl2, 0.1 mM LaCl3, and 1 mM 3,4-diaminopyridine and 9-anthracene carboxylic acid. In some experiments, CaCl2 was changed, either reduced to zero with an equal concentration of MgCl2 added or increased to 5 mM (removing an equal concentration of MgCl2) or 10 mM, with MgCl2 reduced to zero. To avoid movement, 50 µM BTS (n-benzyl-p-toluene sulphonamide; Sigma-Aldrich) was added. Pipettes were made by conventional methods (P-1000 puller; Sutter Instruments; Corning 7056 glass; Warner Instruments) and polished on a microforge. Tip diameter was 10–15 µm, and pipette resistance filled with the internal solution ranged from 0.4 to 1 MΩ. The internal solution contained 145 mM Cs-glutamate or N-methyl-glucamine-glutamate, 10 mM Hepes, 5 mM ATP, and 10 mM EGTA. Mg2+ and Ca2+ were added to set Mg2+ to 0.9 mM and Ca2+ to 100 nM. Solution composition was calculated with Maxchelator (http://maxchelator.stanford.edu). pH was adjusted to 7.4 with CsOH, and osmolarity was set to 310 mOsm in all solutions. Fibers were patch-clamped with an Axon 200B patch amplifier (Molecular Devices). Whole-cell access resistance was 1.55 ± 0.06 MΩ for BALB/c mice (n = 158) and 1.06 ± 0.07 MΩ for B6D2F1 mice (n = 111). Membrane capacitance was 3.53 ± 0.09 nF for BALB/c mice and 2.88 ± 0.12 nF for B6D2F1 mice. Holding current mean was −5.33 ± 0.72 nA. When the current was >3 A/F, the experiment was discarded. For charge movement measurements, series resistance was compensated analogically before recording, and the setting was slightly adjusted during the experiment to maintain good voltage control. Membrane capacitance (Cm) was determined at regular intervals from the time integral of the current, during +10-mV pulses applied from a “subtracting holding potential” of +20 mV for fibers held depolarized (at Vh = 0 mV) or −10-mV pulses applied from a subtracting holding potential of −140 mV for polarized fibers (Vh = −80 or −90 mV), although a less negative subtracting holding potential was used in some experiments.
Rat fibers were voltage-clamped in a 2-Vaseline gap device, using the homemade clamp amplifier described by Brum et al. (2003). Solutions and recording procedures in rat were as described for mice. Mouse experiments were carried at 22°C and rat experiments between 10°C and 15°C.
Measurement and nomenclature of sensor charge
Nonlinear capacitive currents, obtained by conventional subtraction of test and scaled control currents, were corrected by sloping baselines fitted starting 50 ms into the transient, to yield charge movement currents (IQ). Charges displaced during pulses and after pulse termination (QON and QOFF) obtained by time integration of IQ were normalized to membrane capacitance for quantification in dimensions of voltage and units of nC/μF. The dependence of QON or the mean of QON and QOFF on test potential Vm, or V when meaning is clear, was usually described by a “Boltzmann” function:
| (1) |
where Qmax is the maximum charge displaced, the central voltage of the distribution, and κ a steepness factor.
When needed, a distinction is made between two modes of charge movement, charge 1 and charge 2, defined in Scheme 1 in Results. The amount of displaced charge in these modes is represented as Qi (where i = 1 or 2), and the corresponding parameters in fits with Eq. 1 are identified as and κi. Model entity Q1 is usually approximated by the charge mobilized from a well-polarized holding potential (Vh = −80 or −90 mV) and Q2 by that measured when the myofiber is held depolarized (Vh = 0 mV). The estimates in these conditions will be represented as QP (for “polarized”) and QD (for “depolarized”); the corresponding Boltzmann parameters will be identified with the same subindexes.
Scheme 1.
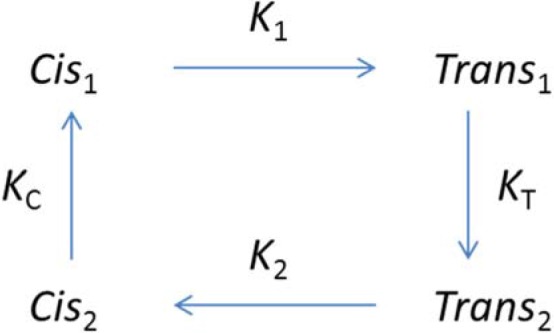
State model of the VSM. Horizontal transitions are V dependent and fast. Those between states 1 cause movement of charge 1. Charge 2 moves between states 2. Vertical transitions are slow and V independent. The pair of states with index i (1 or 2) will be referred to as mode i. Qi(V), the amount of charge i displaced across the membrane field upon application of voltage V, is proportional to the occupancy of Transi. The availability of charge i is proportional to the sum of occupancies of states i.
In modes 1 and 2, the displacement of sensing charge occurs over very different ranges of V. Therefore, in conditions in which the sensors move in a different mode, the control currents for determination of linear capacitance may be measured at very different values of V, that is, positive to 0 mV for determination of charge movement in depolarized myofibers and negative to −120 mV when fibers are well polarized. Finding a suitable voltage range for controls becomes especially difficult in cases studied herein, in which the fiber is well polarized but QP is thought to have contributions from both modes (therefore producing nonlinear charge movements at both extremes of the voltage range). In some of those cases, suitable controls for QP were obtained later at positive potentials, after sustained depolarization, when all or most of the sensor charge moves as charge 2 and pulses in the positive voltage range elicit only linear capacitive current.
Measurement of Ca2+ transients
Rhod-2 (Gentaur) was added to the internal solution in the patch pipette at a concentration of 50–100 µM. To allow for dye diffusion into the cell and internal solution equilibration, experiments were started 15–20 min after membrane breach. A confocal microscope (SP5; Leica Microsystems) in line-scan mode was used to record cytosolic Ca2+-monitoring signals. Fluorescence was excited with the 543-nm line of a solid-state laser. Emitted light was collected in the range 550–650 nm, at 1,400 or 2,800 512-pixel lines per second, to form images of 1,024 or 4,096 lines. Fluorescence intensity F(t) was normalized to its initial value, F0 ≡ F(0), and [Ca2+] calculated as described by Royer et al. (2010):
k−dye was set to 100 s−1 (Royer et al., 2008).
Ca2+ release flux (a magnitude with dimensions of concentration rate of change and units of mM/s, proportional to Ca2+ current through RyRs) was derived as described by Melzer et al. (1984) and Schuhmeier and Melzer (2004). A “removal” model with four components (a fast buffer, slow binding sites largely representing the added EGTA, the monitor dye, and a linear uptake term) was fitted to the decay of the Ca2+ transient after the pulse. Release flux was calculated to account for the transient during the pulse, assuming removal consistent with the fitted four-component model. Concentrations of EGTA and the dye in the cell were fitted or estimated to be 0.7 of the concentration in the pipette (Royer et al., 2008). Image analysis software was written in IDL (Harris Geospatial); other processing was done with ImageJ (National Institutes of Health).
Electrophysiological data collection was standard (PC, analogue-to-digital converter, Digidata [Molecular Devices], and pCLAMP v.10 software [Molecular Devices]). Signals were filtered at 2 kHz. Output signals generated under pCLAMP were used to synchronize voltage pulses with image acquisition.
Simulations
A kinetic version of the four-state model in Scheme 1 was implemented in the IDL environment. The voltage-dependent rate constants for the forward and backward transitions between Cisi and Transi were computed as
| (2) |
where i is 1 or 2, and is a voltage-independent factor fitted for all transients at 0.03 ms−1. Note that mobile charge with these kinetic characteristics will equilibrate with the distribution Q(V) of displaced charge described by Eq. 1; is the central voltage of the charge distribution and κi the steepness factor. The vertical transitions are voltage independent. Their kinetic constants (direction T1 to T2), , and were adjusted to fit the experimental charge values measured in response to test pulses applied after changing the holding potential from a polarized to a depolarized condition (see Fig. 3). was determined assuming microscopic reversibility. The differential equations of the model were solved using the Euler method.
Figure 3.

Interconversion of charge upon sustained membrane depolarization. (A) Pulse protocol. Fibers were held at −90 mV and depolarized to +20 mV for increasing time intervals. Sensor current was induced from −50 mV by a pulse to +10 mV (charge displacement represented in blue) or a negative-going pulse to −120 mV (displacement in red). (B) Charge displacement plotted versus depolarization time. In black are sums of the displacement in both pulses at the same conditioning duration. Note approximate constancy of this sum. Continuous curves plot the kinetic simulation described in Materials and methods, with = 5 × 10−5, = 4.5 × 10−3, = 5 × 10−4, = −25 mV, = −78 mV, κ1 = 12 mV, and κ2 =19 mV. Data are mean ± SEM from five BALB/c fibers.
Statistics
Values in text and figures correspond to mean ± SEM. Two-tailed Student’s t test was performed, and differences were considered significant at P < 0.05.
Symbols and glossary
V, Vh: transmembrane potential, holding transmembrane potential.
IQ: voltage sensor current (also known as intramembranous charge movement).
Q(V): charge displaced, obtained by time integration of IQ.
Q1, Q2: charges displaced in modes 1 and 2.
QP, QD: charges displaced at polarized and depolarized holding potentials.
Qmax: maxima of displaced charge, reached at saturating V, usually evaluated with Eq. 1.
,: central voltage in a fit of Q1 or Q2 with Eq. 1.
,: central voltage in a fit of QP or QD with Eq. 1.
κi: steepness factor in Eq. 1 fits; i can be 1, 2, P, D, or h.
Ci, Ti: occupancies of states Cisi and Transi (Scheme 1); i can be 1 or 2.
KC, KT: equilibrium ratios between states Cis and Trans in Scheme 1.
τi: time constant in exponential fit to IQ(t); i can be 1, 2, P, or D.
, b: Parameters in fit of τi(V) by Eq. 7.
kB, e,: Boltzmann constant, elemental charge, and effective valence.
, : V-independent transition rate constants between Trans states.
, : V-independent rate constants between Cis states.
, : V-dependent rate constants between states Cisi and Transi (Eq. 2).
,: Ca2+ release flux, with dimensions of d[Ca]/dt (a function of t and V).
: peak value of during a pulse at voltage V.
: rate of change of Ca2+ indicator fluorescence; approximates .
Results
This section consists of three parts. The first one presents the properties of intramembranous mobile (sensing) charge in myofibers of mouse FDB. These properties are both steady state (which include activation Q(V) and availability, a function of the holding potential Vh that characterizes VDI or relaxation of the voltage sensor) and kinetic (time course of charge movement currents and time dependence of the slow changes that characterize relaxation). The second section includes a description of activation and inactivation of one of the functional effectors of the voltage sensor, Ca2+ release from the SR quantified by flux . Finally, we probe the dependence of the inactivation process on extracellular Ca2+, found to play a major role in earlier work on frog muscle.
Steady properties of the sensing charge; charge 1 and charge 2
Fig. 1 A represents currents obtained in the same mouse fiber, at different Vh. Shown are nonlinear capacitive (“charge movement”) currents IQ. With the fiber at Vh = −80 mV (blue traces), pulses are applied in the positive direction; this is reversed when the fiber is held at Vh = 0 mV (green). These currents satisfy the classic criteria of sensing current, namely, saturation at the extremes of V and near equality of ON and OFF charge displacement.
Figure 1.

Nonlinear capacitive currents in polarized and depolarized condition. (A) Sensor current IQ recorded at holding potential (HP) −80 mV (blue) and 0 mV (green). Test pulse voltage listed in mV. (B) Mean charge distribution at the two HPs: QD, at Vh = 0 mV (green), and QP, at Vh = −80 mV (blue) (mean ± SEM). In terms of the model of Scheme 1, QD(V) approximates the properties of charge 2, and QP(V) approximates those of charge 1, with limitations demonstrated later. Best fit parameters of Eq. 1, in myofibers held depolarized: Qmax = 53 ± 2.1 nC/µF, = −78 ± 3.0 mV, and κ = 21 ± 2.2 mV (n = 5); for polarized fibers: Qmax = 50 ± 1.3 nC/µF, = −25 ± 1.5 mV, and κ = 16 ± 1.1 mV (n = 9). (C) Time constant of decay of IQ(t) in the two conditions of polarization. Points plot mean and SEM for eight (depolarized) or four (polarized) fibers. Curves plot best fit with Eq. 7: = 6.0 ± 0.15 ms, = −82 ± 3.3 mV, κD = 21 mV, and bD = 0.22 ± 0.02; = 4.0 ± 0.5 ms, = −36 ± 7.7 mV, κP = 16 mV, and bP = 0.21 ± 0.02. Inset: superimposed ON transients in response to −100, −80, −40, and −20 mV test pulses from HP 0 mV. In red, exponential function fitted to the individual records. All data were obtained in myofibers of BALB/c mice. For cells of the B6D2F1 strain the values were similar in the depolarized condition: = 5.4 ± 1.5 ms, = −91 ± 4.7 mV, κD = 20 mV, and bD = 0.2 ± 0.03; n = 10. Data in polarized B6D2F1 are shown in Fig. 5.
The charge displacement Q(V), time integral of the ON and OFF transients, averaged in 10 fibers, is plotted in Fig. 1 B for the two conditions of Vh. The subindex P (for polarized) identifies variables and parameters pertaining to fibers held at Vh = −80 mV, and D marks corresponding values, measured in fibers (usually the same) held at Vh = 0 mV. The dependence on pulse voltage of the charge displaced was parameterized by Boltzmann fits to the averages (Eq. 1, smooth curves). The maximum charge displaced (Qmax) was approximately the same in both cases; the central voltage was −20 mV, and , pertaining to the depolarized condition, was −80 mV.
The aforementioned results were obtained in BALB/c mice; to our surprise, in murine strain B6D2F1 (used when BALB/c mice temporarily became unavailable; see Fig. 5 and Tables 1 and 2 for a detailed presentation of these data) was about the same as in BALB/c mice, but was almost 30 mV more negative. The implications of the strikingly unequal shift are considered in the Discussion.
Figure 5.

Dependence of charge distribution on extracellular Ca2+. (A) Data obtained in the BALB/c strain. QP(V) in three solutions with different [Ca2+], as indicated. The fit to QD(V) in reference [Ca2+]e (green) is replotted from Fig. 1. Curves: Eq. 1 fitted to averages. In 1.8 mM Ca2+, Qmax = 36 nC/µF, = −24 mV, and κ = 15 mV (n = 5 myofibers). In 0 mM, Qmax = 39 nC/µF, = −41 mV, and κ = 12 mV (n = 6). Other parameters are in Table 2. Inset: comparison of QD(V) in 1.8 mM (black) and 0 mM (blue) [Ca2+]e. Same fit for both conditions, = −94 mV, κ = 28 mV (n = 3). (B) Same variables in B6D2F1 mice. Fit to QD(V): κ = 20 mV, = −79 mV (n = 21). Fits to QP(V); in 0 mM [Ca2+]e: κ = 14 mV, = −58 mV (n = 12). In 1.8 mM, κ = 20 mV, = −45 mV (n = 16). In 10 mM, κ = 14 mV, = −27 mV (n = 37). In both panels, data points correspond to mean ± SEM.
Table 1. Charge distribution parameters.
| Species or strain | Polarized or depolarized condition | Qmax | κ | N cells | |
|---|---|---|---|---|---|
| nC/µF | mV | mV | |||
| BALB/c | QP | 38.9 (4.0) | 13.6 (0.9) | −21.6 (2.4) | 18 |
| QD | 40.4 (4.3) | 20.7 (1.0) | −79.3 (3.7) | 24 | |
| B6D2F1 | QP | 38.9 (4.0) | 13.6 (0.9) | −46.1 (2.4) | 16 |
| QD | 41.4 (4.8) | 19.1 (0.9) | −81.1 (2.1) | 21 | |
| Rat | QP | 47.4 (4.4) | 10.3 (0.5) | −22.7 (2.4) | 2 |
| QD | 46.0 (24.9) | 23.8 (11.5) | −81.0 (2.1) | 2 |
Data were fitted by Eq. 1, and fitted parameters were averaged. Differences in Qmax are not statistically significant, whereas κ and differ significantly in the polarized and depolarized conditions (P < 0.001). Values listed are mean (SEM) except for rat where range is indicated.
Table 2. Effects of variation of external [Ca2+] on charge distribution.
| Strain | Condition | Qmax | κ | N cells | |
|---|---|---|---|---|---|
| nC/µF | mV | mV | |||
| BALB/c | 0 Ca2+, 5.3 Mg2+ | 29 (2.5) | 6.8 (0.7) | −40 (1.9) | 9 |
| 1.8 Ca2+, 3.5 Mg2+ | 39 (3.9) | 14 (0.9) | −22 (2.4) | 18 | |
| 10 Ca2+, 0 Mg2+ | 38 (3.0) | 12 (2.3) | −5 (2.9) | 4 | |
| B6D2F1 | 0 Ca2+, 5.3 Mg2+ | 33 (5.0) | 10 (1.3) | −55 (2.8) | 14 |
| 1.8 Ca2+, 3.5 Mg2+ | 39 (4.0) | 14 (0.9) | −46 (2.4) | 16 | |
| 10 Ca2+, 0 Mg2+ | 30 (12.3) | 12.3 (0.6) | −23 (2.6) | 37 |
Charge distributions were obtained from BALB/c and B6D2F1 fibers. Values listed are mean (SEM) of best parameters in fits with Eq. 1. Free Ca2+ and Mg2+ concentrations are in mM. Data for 1.8 mM Ca2+ are the same as in Table 1; they are included here to facilitate comparison. Qmax in both strains did not differ significantly in any of the conditions tested.
Mean parameters are listed in Table 1. The slope factor κ was different at the two values of Vh; whereas at −80 mV it was 12 or 13 mV, it was ∼20 mV in the cells held depolarized. The difference was significant and similar for both murine strains.
Based largely on the equality of Qmax in both conditions of polarization found for sensors of NaV channels of the squid giant axon, Bezanilla et al. (1982) proposed that the currents correspond to transitions between separate pairs of conformations of the same voltage sensor (later identified with charge 1 and charge 2 modes in skeletal muscle). A shift of Q(V) toward negative voltages upon sustained depolarization turned out to be shared by all known four-helix VSMs (reviewed by Villalba-Galea, 2012, 2016).
As proposed by Brum and Rios (1987), a minimum of four conformations, illustrated in Scheme 1, is necessary to explain the observations.
The model, which has been applied with varied nomenclature, consists of two functional states here identified with the index 1 (Cis1 is resting but available to convert to Trans1, which is activating for the ion pathways) and two nonfunctional states, of index 2, which are generally unable to activate the release channels (and were termed relaxed by Villalba-Galea et al., 2008). Horizontal transitions are voltage dependent and produce intramembranous charge movement; namely, transitions between states 1 result in movement of charge 1 (the displaced charge is represented as Q1(V)), and those between states 2 result in displacement of charge 2 (Q2(V)). Cis names states of sensors located on the intracellular side of the membrane electric field and Trans states in which the charge has crossed the field. As shown in Fig. 1, the characteristic times of charge movement are 20 ms or less. Transitions between states 1 and 2 (Cis or Trans), which are accompanied by changes in the functional availability of the effectors, take seconds or longer (see Fig. 3) and are voltage independent. Occupancies are represented as Ci, Ti, where i is 1 or 2. Qi and Ti are trivially proportional. The charge distribution QD(V), measured at Vh = 0 mV or greater, approximates Q2, and QP(V) approximates Q1. The correspondence between Q1 and QP breaks down in conditions described later; therefore it is necessary to use different nomenclature for experimental and model entities.
With one exception, the four-state model describes well the observations on “steady-state” or “quasi-equilibrium” properties of the sensor charge. As shown by the fits in Fig. 1 B, the Boltzmann function (Eq. 1), which applies to equilibria between two states in a canonical ensemble, describes well the dependence Q(V), separately for QP and QD, at times short compared with the time constants of the slow transitions. In terms of the four-state model, the canonical distribution prescribes for the equilibrium constants
where is the effective valence of the moving particle. This is consistent with a Boltzmann distribution for Q1 and Q2 provided that the slope factor κi is . A salient failure in the model description is that conservation of charge requires equality of z′1 and z′2, but the slope factors are different for the measured QP(V) and QD(V). A simple change in the model (consisting of the addition of an intermediate state in the transition between Cis2 and Trans2) accounts for this difference (Villalba-Galea et al., 2008).
The equilibrium occupancies of the four states, as functions of Vh, are given by Brum et al. (1988a) as
| (3) |
| (4) |
Availability of sensing charge
Charge 1 is progressively reduced at increasing values of Vh. This is illustrated in Fig. 2, in which the charge moved by a pulse from −70 to 0 mV (an experimental measure of the available charge 1) is plotted as a function of Vh. Charge displaced in this range follows a decreasing sigmoidal dependence with Vh, well fitted as
Figure 2.

Charge availability as a function of holding potential. (A) Pulse protocol. Fibers were held for 2 min at each holding potential (HP) before the test pulse. (B) Charge displaced (mean ± SEM) between −70 and 0 mV (QP) versus Vh. The curve corresponds to a fit by Eq. 5. Parameters: = −57.4 mV, κh = 7.48 mV, and Q∞= 0.26. Data from five BALB/c fibers.
| (5) |
As shown by Brum et al. (1988a), this dependence is approximately predicted by the equilibrium equations of the four-state model. Indeed, at any value of holding voltage, the availability of charge 1 is equal to the sum of occupancies of states Cis1 and Trans1. From Eqs. 3 and 4, and assuming that κ1 = κ2 = κ, this sum can be shown to have the same mathematical form as Eq. 5, with κh = κ. Moreover, the half-inactivation voltage satisfies
| (6) |
Therefore, is the logarithmic mean of and with weights 1 and KT. When the inactivation tendency is large, the half-inactivation voltage is close to .
In the present case, obtained fitting measurements in the BALB/c strain with Eq. 5 is −57.4 mV. Using instead Eq. 6, with parameters derived under the assumption that Q1(V) and Q2(V) are approximated by QP and QD (Table 1) and κh = (κ1 + κ2)/2, the calculated value is the same provided that KT = 90. That the same value can be derived from the fit to a direct measurement of available charge (Fig. 2) or from model-based operations on the voltage dependence of charge displacement (Eq. 6) establishes the four-state model as an adequate first approximation.
Note in Eq. 5 the additive term Q∞, representing the amount of charge measured at the highest values of Vh. This is not an actual remnant of charge 1 (i.e., it does not reflect transitions between states Cis1 and Trans1) but is instead charge 2 displaced by the test pulse as states 2 are populated because of the steady depolarization. On the basis of limited measurements of the charge displaced with pulses in a highly depolarized voltage range (unpublished data), we estimate that the remnant of charge 1 at Vh beyond 0 mV corresponds to a joint occupancy C1 + T1 of 0.01 or less.
Kinetics of charge movement: fast transitions
We characterized the voltage-dependent kinetics of sensing currents by fitting to the IQ(t) transients recorded at both values of Vh an exponential term (of time constant correspondingly represented as τP or τD) plus a constant (Fig. 1 C, inset). The dependence of the time constants on test potential V was fitted with Eq. 7, derived for a second-order, single-barrier Eyring-rate model (Tsien and Noble, 1969) as implemented by Simon and Beam (1985):
| (7) |
Here i is P or D, represents the time constant at a voltage where τ is maximum, b is the weight of the second-order term in a polynomial description of the dependence of free energy on V, and. The fairly good fits, with similar parameter values for the voltage dependence of charge distribution and kinetic rates, show that the two-state Eyring-rate model captures well the basic kinetic and steady-state properties of sensor charge in both modes.
Kinetics of slow transitions: Charge interconversion
Fig. 3 illustrates experiments designed to monitor the inactivation transition, which is associated with loss of function of the voltage sensor and is depicted vertically in Scheme 1 (Trans1 to Trans2). The pulse protocol is in Fig. 3 A: starting from a well-polarized fiber (Vh = −90 mV), the holding potential is changed to 0 mV for a variable time, then charge displacement is measured in two ranges that separately probe the existing amounts of charge 1 or 2. In Fig. 3 B are averages of charge displaced in these ranges versus time elapsed at Vh = +20 mV. The curve in blue, labeled Q1, traces the charge displaced by the positive-going pulse (a measure of dwindling charge 1). Q2 represents the charge displaced by the negative pulse, a measure of available charge 2. The changes are reciprocal and of similar magnitude. The kinetics of both changes is roughly exponential with τ ∼200 ms. The conservation of the sum, consistent with the simple four-state model, justifies describing the phenomenon as “charge interconversion” (Brum and Rios, 1987) to mark the contrast with the “charge immobilization” that characterizes N-type inactivation of ion channels (Armstrong and Bezanilla, 1977). The continuous curves in Fig. 3 are generated with a kinetic version of the four-state model, using the parameter values experimentally determined above. (According to the kinetics revealed in Fig. 3, there is some conversion of charge 1 to charge 2 during the pulses to high V in the activation experiments illustrated in Fig. 1. Specifically, in a 100-ms pulse to +20 mV, 34% of the available Q1 converts to Q2. From the measured distribution of Q1 and Q2 it can be further calculated that the charge displaced in the OFF transient will be ∼10% less than the amount in the ON. This error would have been reduced substantially by monitoring QP with briefer pulses at the higher V, which was not done because we were not aware at the time of the rapid inactivation in these rodents.)
The conclusion from this section is that the movements of sensor charge are well described as rapid voltage-dependent transitions inside pairs of states (modes) and that these pairs are connected by voltage-independent transitions, which are ∼100 times slower.
Activation of Ca2+ release by the voltage sensors
In mouse muscle the bulk of charge movement controls activation of Ca2+ release from the SR (Wang et al., 1999; Collet et al., 2004; Prosser et al., 2009). Fig. 4 A shows IQ(t) and Ca2+ transients recorded in the same myofiber at Vh= −80 mV. Averages in BALB/c myofibers of charge displacement (red symbols) and peak release flux derived from the Ca2+ transients are in Fig. 4 B. Fig. 4 C plots the “transfer function” , which summarizes the relationship between response and sensing charge (Ríos et al., 1993). As reported before for frog muscle (Ríos et al., 1993), this function is concave upwards at low values of Q, suggesting a cooperative action of activating sensors. Given that four CaV1.1 complexes face every other RyR channel in the triadic junction (Block et al., 1988), cooperativity or collective action of some sort is expected. The dependency saturates at high values of Q, suggesting the presence of other voltage-dependent movements, which may be redundant, be functionally silent, or occur at unrelated VSMs (Sigg and Bezanilla, 1997). Some of this redundant charge might arise for example from CaVs in excess of the minimum required for activating the underlying RyR.
Figure 4.

Gating of Ca2+ release. (A) Sensor currents and Ca2+ transients recorded in a BALB/c myofiber held at −80 mV. Test pulse voltages are listed. (B) Ca2+ release flux () and charge displacement as a function of test voltage. Data (mean ± SEM) were normalized to individual maxima in five fibers and averaged. Parameters for Q/Qmax adjusted with Eq. 1: = −19 mV, κ = 15 mV; for , = −30 mV, κ = 8.9 mV. (C) Transfer function: release flux versus charge displacement (mean ± SEM), plotted parametrically from data (and fits) in B.
VSMs require extracellular Ca2+ for their gating functions
Work on frog muscle by several laboratories has shown effects of many pharmacological agents and ions on the functional availability of Ca2+ release, via a change in the balance of charge 1 and charge 2 modes. Specifically “Ca antagonist” drugs (dihydropyridines, phenylalkylamines, diphenyl-butylpiperidines, and benzolactams) promote sensor relaxation (reviewed by Romey et al., 1988; Ríos and Pizarro, 1991), whereas metal cations, chiefly Ca2+, antagonize it. The key observations are that Ca2+ release is disabled upon removal of extracellular Ca2+ and that the change is accompanied by and due to full transition of VSMs to charge 2 mode (Brum et al., 1988b; reviewed by Melzer et al., 1995).
To assess whether extracellular Ca2+ has a similar role in mammalian muscle, we explored the effects of changing its concentration [Ca2+]e in fibers held at Vh = −80 mV, while keeping total divalent cation concentration constant (replacing Ca2+ by an equal concentration of Mg2+) to minimize changes in electrostatic shielding. Anticipating the presence of charge 2, controls for calculation of asymmetric current were obtained at highly negative potentials, as far as possible from the range where most of charge 2 movement occurs.
The mean QP(V) in 0 mM Ca2+ is shown in Fig. 5 A. Although the total mobile charge did not change from that in normal [Ca2+]e, the mid-voltage had a large negative shift (from −22 to −40 mV). In the same graph, the distribution QD(V) of mobile charge (in fibers held at Vh = 0 mV, replotted from Fig. 1) is seen to be centered at = −79 mV. The inset in the figure confirms that QD(V) has the same distribution in normal and 0 mM [Ca2+]e. In agreement with the results in frog muscle, the effect of removing external Ca2+ can be attributed to partial inactivation of the voltage sensor, with partial conversion of charge 1 to charge 2.
That switching to zero Ca2+ led to partial inactivation raised the unexpected possibility that the VSMs may not be fully available in the physiological condition (Vh = −80 mV, [Ca2+] = 1.8 mM). If this were the case, increasing [Ca2+] in the conventional saline solution would increase activation at normal Vh (after an increase in the availability of sensors). This was indeed the case, as shown with QP(V) in 10 mM [Ca2+]e (in red in Fig. 5 A). The distribution shifted positive to a midvoltage of −5 mV. The shift, by nearly +30 mV, is much greater than what can be attributed to electrostatic shielding by the extra calcium (10 mV at most, as established by Brum and Rios, 1987). From these effects and effects on release flux presented later, we conclude that the voltage sensor is indeed partially inactivated in the resting condition. Increasing [Ca2+]e then reduces inactivation, whereas reducing it to zero increases inactivation, bringing the sensor closer to the fully inactivated condition or charge 2 mode.
The study was repeated in mouse strain B6D2F1, with qualitatively similar but quantitatively different results. Q(V) averages are in Fig. 5 B; Boltzmann fit parameters at various holding voltages and in solutions with different [Ca2+] are listed in Table 2. The distribution in depolarized fibers (QD, green), centered at −81 mV, is similar to that in BALB/c fibers. QP(V), however, was shifted negative to that in BALB/c. The midvoltage in standard Tyrode’s solution was 25 mV more negative (−46 mV), and the change to [Ca2+]e = 0 brought the distribution much closer to QD(V) ( = −55 mV).
The experiments show that the function QP(V) shifts negative as extracellular Ca2+ is reduced. This may reflect either a shift in Q1(V) (reflecting a change in the properties of the sensors that dwell in mode 1, i.e., the pair of states Cis1 and Trans1) or a greater inactivation of VSMs (so that Q2 increases as a fraction of QP and therefore QP(V) approaches the negatively shifted distribution Q2(V)). The second alternative predicts a reduction in maximum as [Ca2+]e is decreased.
To test this prediction, Ca2+ transients and sensor currents were recorded simultaneously in an external solution with 1.8 mM Ca2+ and immediately after changing to 0 mM [Ca2+]e. The change had the predicted effect; as shown in Fig. 6, was reduced to almost zero, and the effect was reversible immediately upon return to normal [Ca2+]e. On average in 10 BALB/c myofibers, peak was reduced by 62% (SEM = 8.7), the quasi-steady value reached by after the peak, by 64% (SEM = 5.6), and the available sensor charge Q1, evaluated in a pulse from −90 to 0 mV, was reduced by 36% (SEM = 5.7).
Figure 6.

Ca2+ release in low extracellular Ca2+. Top: Ca2+ transients in response to test pulses from −80 to 0 mV in 1.8 mM [Ca2+]e (left), after changing to 0 mM (center; 2 min and 6 s later), and after returning to reference solution (right). Middle: derived from the Ca2+ transients shown. Bottom: asymmetric (sensor) current, recorded simultaneously. (While the sensor current is indeed reduced in the 0 Ca2+ condition, the reduction is exaggerated in the example. Indeed, the sensor current records were obtained by subtraction from the current elicited by the positive-going pulses, the scaled “control” currents elicited by pulses between −90 and −110 mV. These pulses displaced some charge 2 in the 0 Ca2+ condition, which when scaled and subtracted from the test current, resulted in an erroneous further reduction of the difference.) BALB/c myofiber.
A trivial explanation for this effect is that Ca2+ transients are reduced in zero Ca2+ because they rely on Ca2+ influx via ICa, which could contribute directly and has a demonstrated role in SR loading (Robin and Allard, 2015). The direct contribution appears to be negligible in frog muscle (e.g., Armstrong et al., 1972; Brum et al., 1987); besides, it was minimized here by measuring at V close to the reversal potential of ICa. Changes in SR Ca2+ content under the present conditions can be ruled out because the interval between measurements in different [Ca2+]e was 2 min, and >10 min is required for releasable Ca2+ to decay by 50% in mouse myofibers exposed to zero [Ca2+]e (Royer et al., 2010). Note also that the transfer function (Fig. 4) predicts that a reduction in Q1 will cause a greater than proportional reduction in , as observed. In sum, the simultaneous reduction in and Q1, commensurate with , indicates that the functional deficit is due to a primary impairment of the voltage sensors. (It should also be noted, in this regard, that the record of IQ(t) in [Ca2+] = 0 in Fig. 6 provides an exaggerated version of the reduction in Q1, as in this case the subtracted “control” current is likely in error, increased by the contribution by Q2 in the voltage range where control currents are collected.)
Our inference that VSMs are partially inactivated in normal Tyrode’s solution requires that raising extracellular [Ca2+] above normal values increase maximal and that the increase be more marked in B6D2F1 mice (because the rested-state inactivation of VSMs in these was greater than in the BALB/c strain; Fig. 5). These expectations were tested by comparing maximally activated immediately before and after transfer between [Ca2+]e = 1.8 and 5.3 mM. Results are summarized in Table 3. To reduce the possible changes in SR calcium load, we compared , the maximum rate of change in [Ca2+]cyto, rather than (the calculation of which, on the basis of fits to multiple Ca2+ transients, requires application of many pulses, some of which must be long-lasting; González and Ríos, 1993; Schuhmeier and Melzer, 2004). In B6D2F1 mouse fibers, the fractional increase in associated with the change was 0.77. In BALB/c it was 0.28. The difference between strains was statistically significant. We conclude therefore that VSMs are partially inactivated in normal [Ca2+]e, to a variable degree in different mouse strains.
Table 3. Effects of variation of extracellular [Ca2+] on Ca2+ transients.
| Strain | Myofiber ID | Vtest | 1.8 mM Ca2+ | 5.3 mM Ca2+ | Ratio | |
|---|---|---|---|---|---|---|
| Peak | Peak | |||||
| mV | u.a./ms | u.a./ms | ||||
| BALB/c | 20160907a | 20 | 22.29 | 28.06 | 1.26 | |
| 20160908a | 20 | 12.00 | 15.34 | 1.28 | ||
| 20160908e | 40 | 16.30 | 17.20 | 1.05 | ||
| 20160908f | 20 | 19.47 | 24.29 | 1.25 | ||
| Mean | 1.21 | |||||
| SEM | 0.05 | |||||
| B6D2F1 | 20160504d | 20 | 40.24 | 48.43 | 1.20 | |
| 20160520a | 40 | 5.45 | 4.64 | 0.85 | ||
| 20160506d | 40 | 4.24 | 9.12 | 2.15 | ||
| 20160506a | 40 | 7.09 | 15.64 | 2.21 | ||
| 20160506c | 20 | 7.25 | 14.22 | 1.96 | ||
| 20160504e | 40 | 65.17 | 94.35 | 1.45 | ||
| 20160511c | 40 | 22.89 | 59.42 | 2.59 | ||
| Mean | 1.8 | |||||
| SEM | 0.24 |
Fluorescence signals were recorded in response to large depolarizing pulses in reference 1.8 mM external [Ca2+] and after changing external [Ca2+] to 5.3 mM to keep total divalent cation concentration constant. [Mg2+] was 3.5 mM in the reference solution and 0 mM in the high-[Ca2+] solution. In both strains, the differences in peak amplitude in the two conditions were significantly different (P < 0.05).
Charge 2 is present in rat fibers
To confirm and generalize the observations, we recorded sensor charge movement in a different mammal using a different method. Fibers from rat extensor digitorum longus were studied by the 2-Vaseline gap technique, which allows for clamping greater areas of junctional membrane and repeating measurements on the same preparation for longer times, thus providing more reliable measurements. Fig. 7 summarizes the results. Currents at different potentials, applied from Vh = 0 mV, are plotted in Fig. 7 A; QP(V) and QD(V) are in Fig. 7 B. The parameters of QD(V) are similar to those in either mouse strain (Table 1). QP(V) is centered near −20 mV, as previously reported (Hollingworth and Marshall, 1981; Delbono et al., 1991; Szentesi et al., 1997). In both conditions of holding potential and in physiological [Ca2+], the distributions of mobile charge are remarkably similar to those of the BALB/c mouse strain.
Figure 7.

Sensor current and charge distribution in rat fibers. (A) IQ records in response to negative pulses from Vh = 0 mV in −10-mV steps from −20 to −100 mV and in −20-mV steps up to −140 mV. (B) Mean QP (V) and QD (V) in three fibers. = −21 mV, κP = 13 mV; = −77 mV, κD = 21 mV. Qmax was 36 ± 10 nC/µF. Data points represent mean ± SEM.
Discussion
The present study revealed multiple properties of the EC coupling voltage sensors of skeletal muscle. We characterized for the first time in mammalian skeletal muscle the phenomenon of VSM inactivation or relaxation, which occurs upon holding the membrane potential at 0 mV (or higher) and results in closure with loss of gating function of both VSM-gated ion pathways; we found that the process is similar qualitatively but has substantial quantitative differences with the well-studied relaxation of the corresponding VSMs of frog skeletal muscle. We also demonstrated that extracellular Ca2+ antagonizes the relaxation process and conversely that its absence promotes VDI. This effect is quantitatively different than its counterpart in frog muscle, with the consequence that murine VSMs appear to be partially relaxed in the muscles at resting potential in physiological extracellular Ca2+ concentration. In this discussion, we focus on the consequences of these observations for physiology and mechanism of control of Ca2+ release in EC coupling.
The EC coupling VSMs have four major conformations and two modes
The properties of the VSMs in the rodents studied here and in amphibians uphold the suitability of a simple four-state biophysical model to describe approximately the observed Q(V) and its steady-state inactivation properties, as well as the kinetics of its displacements.
The most salient steady-state property of sensor charge is the adoption of two distributions Q(V) at extremes of holding potential. These distributions reflect the voltage sensitivity of the VSMs in two sets of states (modes), which in muscle are named charge 1 and charge 2. A similar two-mode scheme has been found to apply to other four-helix VSMs. The discovery sequence started with NaV channels of squid axon (Bezanilla et al., 1982), continued with EC coupling of frog skeletal muscle (Brum and Rios, 1987; reviewed by Melzer et al., 1995), NaV of crayfish axon (Rayner and Starkus, 1989), and CaV1.2 of mammalian cardiac muscle (Field et al., 1988; Bean and Rios, 1989; Shirokov et al., 1992). Similar modes were found later for Shaker and HERG K (Olcese et al., 1997; Piper et al., 2003), the bacterial Na channel (Kuzmenkin et al., 2004), and hyperpolarization-activated HCN channels (Bruening-Wright and Larsson, 2007). In most channels, the transitions to mode 2 are accompanied by loss of permeation (reviewed by Villalba-Galea et al., 2008). The existence of four main conformations of the sensor has been directly demonstrated via changes in the fluorescence of tetramethylrhodamine, methyl ester, placed as label on the VSM of the phosphatase Ci-VSP (Villalba-Galea et al., 2008). The description of VSM states by Scheme 1 is therefore approximately valid for every four-helix VSM in which charge movements have been measured. VSM relaxation was reviewed recently within the wider set of hysteresis phenomena (Villalba-Galea, 2016).
Variation across taxa
Within a general picture of similarity, there are salient quantitative differences between sensors of rodents and frogs. The properties measured here for charge movement in normally polarized fibers (when most of the sensors operate in charge 1 mode) are as previously described in mouse (Wang et al., 1999; Prosser et al., 2009) and rat muscle (Hollingworth and Marshall, 1981; Delbono et al., 1991; Szentesi et al., 1997). The steepness factor values found here for rodents are similar in frogs. Large differences, however, were found in the central voltage of QP(V), the charge mobile in polarized conditions. The variation resulted in different shifts ; whereas in the frog the shift is approximately −90 mV, in mice it ranged from −60 mV in BALB/c to −32 mV in the B6D2F1 strain. The shift in rats was similar to that in BALB/c mice.
and estimate the central voltages and in the four-state model. In the context of the model, is a critical measure of steady-state properties. As shown by Brum et al. (1988a), the equality applies (see Scheme 1 for definitions), which implies that the shift measures the thermodynamic separation of the modes. Indeed, KCKT is a joint measure of the tendency of the system to inactivate at a depolarized potential (KT) and be available in the well-polarized cell (KC). That is so much greater in frogs than mice might reflect actual differences in KC and/or KT. Alternatively, , the central voltage in the distribution of charge measured from a polarized holding potential, may not be equivalent to , the central voltage of the Cis1 → Trans1 transition. In other words, QP, the charge displaced in well-polarized cells, which in frog muscle is about equal to Q1, may have a major component of Q2 in mice.
In the framework of the four-state model (Scheme 1), if QP consists of Q1 and Q2 components, its dependence QP(V) should equal a linear combination of Q1(V) and Q2(V). While this fit worked in some cases (not depicted), in others QP(V) did not feature two clearly separated components. As shown by Shirokov et al. (1998), a continuum model that describes activation without recourse to discrete barriers (Läuger, 1988; Levitt, 1989) fits better the CaV1.2 sensor in situations of partial inactivation, without introducing more parameters. Such models have the additional advantage of accounting for “preopening” transitions of the sensor, which are obviously ignored by the four-state model and become prominent in CaV1.2 (Olcese et al., 1997). More simply, the Q2 component may be underestimated because of partial cancellation with sensor charge contributing to the “linear capacitive current” displaced by control pulses.
Measurement of in high extracellular [Ca2+] (which evidenced a large potentiation attributable to a reactivated voltage sensor rather than an increase in SR Ca2+ load) showed this indeed to be the case. Voltage sensors are partially inactivated in resting physiological conditions in these mice.
Sensor availability and Ca2+ flux depend on extracellular Ca2+
The present experiments extend to mammalian muscle the observation of antagonism between extracellular cations and VDI of the VSM, first made by Brum et al. (1988b) in frog muscle. Pizarro et al. (1989) showed that other cations substitute for Ca2+ with efficiency scalable to their CaV1.1 permeability; on the basis of this correspondence, they proposed that C-type inactivation or relaxation involves a collapse of the pore at the selectivity filter, a collapse opposed by permeant ions within the pore. Similar effects of permeant ions have been found in most channels that undergo C-type inactivation (Grissmer and Cahalan, 1989; Hoshi et al., 1991; Demo and Yellen, 1992; López-Barneo et al., 1993).
The reduction or removal of extracellular Ca2+ has effects on sensor charge and associated Ca2+ currents similar to those of sustained depolarization, dihydropyridines, and other inactivation-promoting drugs in both frog CaV1.1 (Ríos and Pizarro, 1991; Schnier et al., 1993; Melzer et al., 1995) and mammalian CaV1.2 (Field et al., 1988; Shirokov et al., 1993). In agreement with the early findings, we now demonstrate that the Q(V) distribution present in depolarized cells does not change in 0 [Ca2+]e (Fig. 5 A, inset). Collectively, these observations indicate that the inactivated mode reached by three means (depolarization, removal of permeant ions, and drugs) is the same and is shared by all CaV1 channel isoforms.
Large quantitative differences in the effects of [Ca2+]e were found between the two mouse strains. As listed in Table 1, is similar in both mouse strains and in the rat, at about −80 mV. This agreement indicates that in depolarized cells all sensors adopt the charge 2 mode, and therefore is equal to . In contrast, is 37 mV more negative in B6D2F1 than in BALB/c. The variability of suggests that the charge mobile in well-polarized mouse myofibers in solutions of physiological [Ca2+] is contributed by sensors in charge 1 and charge 2 modes, with greater contribution of charge 2 in the B6D2F1 strain. This interpretation justifies the greater shift in observed in this strain upon exposure to 10 mM [Ca2+]e as the consequence of the greater starting proportion of charge 2. Also in agreement, the distribution QP(V) in 0 mM [Ca2+]e is left-shifted in B6D2F1 relative to that in BALB/c (Fig. 5 B). The conclusions are that the mouse VSMs are more prone to inactivation than the frog’s—especially in B6D2F1—and that changes in extracellular [Ca2+] near its physiological value alter the availability of sensors.
A potential clinical implication
Extrapolation of the present results to humans has implications for pathophysiology. That is nearly zero when [Ca2+]e is absent (Fig. 6) and increases by 28% or 77% when [Ca2+]e is raised from 1.8 to 5 mM (Table 3) implies that the protective (inactivation-antagonizing) effect of the ion is far from saturated at normal [Ca2+]e. Ionic [Ca2+] in human plasma is ∼1.25 mM, but in hypocalcemia it may reach <0.7 mM (e.g., Hästbacka and Pettilä, 2003; Cecchi et al., 2015). A change of such magnitude in [Ca2+]e would diminish Ca2+ release flux substantially just by reducing VSM availability (to 0.7/1.25 of the normal value; proportionality is a suitable first approximation here). Conversely, hypercalcemia would increase VSM availability and Ca2+ release flux. Of course, the effects will not stop there; long-term changes in calcemia will eventually change SR calcium content and other variables that affect EC coupling. The present results simply reveal that VSM availability, a variable of physiological impact, will be directly and significantly affected by changes in calcemia within the clinical range.
Models of coupling
That a permeating ion (Ca2+ in this case) may affect a channel’s VSMs from an extracellular location has mechanistic implications. Here we explore these and conclude that these effects rule out the class of models of electromechanical coupling that achieve activation and inactivation with only one physical gate. The diagram in Fig. 8 A represents one such model for the CaV1.1 channel (the same argument can be made for the RyR, but the presence of two membranes makes the graphics more complex in that case). In correspondence with Scheme 1, the model has four states; in two of them (those of index 1) the mobile charge is engaged with the gate, which favors the closed state at the resting potential. The bias is represented by a spring that keeps the gate closed and requires energy to allow its opening. In the engaged states, the energy is provided by the field acting on the mobile charge, which can be in position Cis (favored at negative potentials) or Trans. In the two other states, of index 2, the sensor is disengaged from the gate; disengagement is the simplest possible form of inactivation, it allows the model to work without a dedicated inactivation gate. An alternative representation of the model, as four free energy local minima separated by barriers of arbitrary height along a single reaction coordinate, is in Fig. 8 B. The electric field changes the energy linearly between Cis and Trans locations.
Figure 8.

Two models of electromechanical coupling. (A) Hypothetical single-gate model of the CaV1.1 channel. A conventional activation gate can be opened by the electric field acting on an “engaged” charged sensor. Inactivation is represented by disengagement of sensor and gate (states 2). (B) Representation of the model in terms of free energy along a reaction coordinate. Negative polarization (black trace) lowers the energy of states Cis. The spontaneous tendency to inactivation is reflected in the voltage-independent difference between the energies of Trans states. (C) Right shift in the Q (V) curves reflects the additional work required to displace the sensor when it is engaged with the gate. (D) A two-gate model; the inactivation “gate” is represented by narrowing of the pore at locations external to the activation gate (contours in red). Whereas in the single-gate model (A), states Cis1, Trans2, and Cis2 present an identical aspect to extracellular Ca2+, in D, the presence of an inactivation gate, closed in states Cis2 and Trans2, defines a site that presents the same properties to extracellular ions in both states, thus justifying the differential promotion of states 1 by extracellular Ca2+.
This single-gate model is good in first analysis, as it justifies the main change induced by sustained depolarization. Indeed, the work done by the electric field on the system is proportional to the area comprised between the Q(V) curve and the vertical axis (Chowdhury and Chanda, 2012). When the sensing charge is engaged (i.e., in states 1), the Cis to Trans transition requires additional work to open the gate, compared with the disengaged situation 2, in which the sensor moves without the load imposed by the gating interaction. This is reflected by a right shift of Q(V) and growth in the area of work in states 1 (the difference is in dark gray in Fig. 8 C).
This simple model, however, fails to account for the effect of [Ca2+]e. In the single-gate hypothesis, extracellular Ca2+ can only promote activation by stabilizing the open state (Trans1), as all the other states are closed to extracellular agents. Such stabilization would ease the opening (lower the elastic constant or shorten the excursion of the spring in the cartoon representation). Therefore the work of opening would be reduced, and the Q(V) curve would shift to the left on the V axis, contrary to the observation of right shifts upon increase in [Ca2+]e. In more intuitive terms, because the extracellular ion modifies equally (the free energy of) two states with the activation gate in different position (closed or open), its action must be independent of this gate.
The alternative is to assume a second process that closes the channel independently, an “inactivation gate,” as represented in Fig. 8 D. The assumption and cartoon match the current structural picture of the CaV1.1 channel complex, which, similar to that of other V-sensitive channels, includes a selectivity filter and a separate “helix crossing” stricture, located more deeply along the permeation pathway (Wu et al., 2015). In Fig. 8 D, inactivation includes narrowing of the pore at a location near the outside of the cell, presumably corresponding to the selectivity filter, while the activation gate is placed closer to the interior of the cell, in correspondence with the helix crossing. Under this assumption, high [Ca2+]e favors Cis1 and Trans1, the states that have the inactivation gate open (represented by no narrowing in this cartoon), increasing their occupancy at any voltage without favoring one over the other. The effect may be justified by the steric “foot in the door” mechanism envisioned for other channels. Under this assumption, the shift of Q(V) in the positive direction at high [Ca2+]e simply reflects a greater occupancy of noninactivated states, rather than changes in voltage dependence of the activating transition. Unlike the single-gate model, this scheme does not require a change in voltage dependence of activation of release flux, consistent with the observed properties of in high [Ca2+]e.
In the single-gate model, the sensor has only two states, linked by a voltage-dependent transition; the changes observed in the sensor upon relaxation are entirely due to its linkage to the effector. In the two-gate model, the sensor remains connected to the effector when the system is inactivated. Therefore, in this model, inactivation/relaxation is an intrinsic property of the sensor. In other words, the sensor has four states. Evidence that relaxation is intrinsic to the VSM was first obtained by Villalba-Galea et al. (2008), who showed that the VSM of Ci-VSP may adopt four states even when separated from its effector, the intracellular stretch with enzymatic activity.
The highest resolution structure of CaV1.1 available to date (Wu et al., 2016), derived from cryoelectron microscopic images of protein particles in 10 mM Ca, tentatively assigns two Ca2+ ions to pore spaces at the selectivity filter. The channels are closed at the helix crossing and the voltage sensing helices are “UP,” with most charged residues on the extracellular side of the charge transfer center, in what we call a Trans position. The authors tentatively assign this structure to an inactivated state, a view according to which the voltage sensors in an activating, Trans location coexist with a closed activation gate—in essence a one-gate model in which inactivation amounts to disengagement of sensor and gate. Our results and the observation in the same paper (Wu et al., 2016, Extended Data Fig. S7) of structural differences induced by extracellular Ca2+ are instead consistent with the operation of two gates. Therefore, the conclusions from structural and “biophysical” approaches are yet to be reconciled.
Conclusions
We evaluated quantitatively in skeletal muscle of rodents the relationship between the sensing current in VSMs of CaV1.1 and the gating of the associated Ca2+ release pathway. The sensor’s transitions are reproduced approximately by a four-state model with two functionally inactivated states, common to every four-helix VSM in which these transitions have been studied. The energy differences that favor inactivation at depolarized membrane potentials and recovery at the resting potential are lesser and more variable in these rodents than in frog muscle, with the unexpected consequence that the voltage sensors appear to be partly inactivated in muscles at rest. As found previously with frog muscle, extracellular [Ca2+] antagonizes inactivation. Combined with the presence of inactivated sensors in resting muscle, the observation predicts that the fraction of inactivated sensors in working muscles, and hence their performance, will vary steeply with changes in calcemia within the clinical range. The inactivation-opposing effect of extracellular Ca2+ favors a class of biophysical models in which inactivation is intrinsic to VSMs rather than a consequence of their disengagement from effector gates.
Acknowledgments
We are grateful to Dirk Gillespie (Rush University) for an insightful critique of the manuscript and to Mariana Di Doménico and Magdalena Guarino (Universidad de la República) for help with experiments in the final stages of this study.
This work was supported by a grant from Fondo Clemente Estable (Agencia Nacional de Investigación e Innovación, Uruguay), by a Physiological Society Senior Grant (UK), and by funds from Programa de Desarrollo de las Ciencias Básicas (PEDECIBA) and a Research Grant from Comisión Sectorial de Investigación Científica de la Universidad de la República (CSIC) to G. Brum and a Research Starting Grant from CSIC to J. Ferreira Gregorio. E. Ríos was supported by National Institute of General Medical Sciences grant R01GM111254 and National Institute of Arthritis and Musculoskeletal and Skin Diseases R01AR071381.
The authors declare no competing financial interests.
Mark Nelson served as guest editor.
Footnotes
Abbreviations used:
- EC
- excitation–contraction
- FDB
- flexor digitorum brevis
- VDI
- voltage-dependent inactivation
- VSM
- voltage-sensing module
- VSP
- voltage-sensitive phosphatase
References
- Adrian R.H., and Almers W.. 1976. Charge movement in the membrane of striated muscle. J. Physiol. 254:339–360. 10.1113/jphysiol.1976.sp011235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong C.M., and Bezanilla F.. 1973. Currents related to movement of the gating particles of the sodium channels. Nature. 242:459–461. 10.1038/242459a0 [DOI] [PubMed] [Google Scholar]
- Armstrong C.M., and Bezanilla F.. 1977. Inactivation of the sodium channel. II. Gating current experiments. J. Gen. Physiol. 70:567–590. 10.1085/jgp.70.5.567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong C.M., Bezanilla F.M., and Horowicz P.. 1972. Twitches in the presence of ethylene glycol bis(β-aminoethyl ether)-N,N′-tetracetic acid. Biochim. Biophys. Acta. 267:605–608. 10.1016/0005-2728(72)90194-6 [DOI] [PubMed] [Google Scholar]
- Avila G., and Dirksen R.T.. 2000. Functional impact of the ryanodine receptor on the skeletal muscle L-type Ca(2+) channel. J. Gen. Physiol. 115:467–480. 10.1085/jgp.115.4.467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean B.P., and Rios E.. 1989. Nonlinear charge movement in mammalian cardiac ventricular cells. Components from Na and Ca channel gating. J. Gen. Physiol. 94:65–93. 10.1085/jgp.94.1.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F., and Armstrong C.M.. 1977. Inactivation of the sodium channel. I. Sodium current experiments. J. Gen. Physiol. 70:549–566. 10.1085/jgp.70.5.549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F., Taylor R.E., and Fernández J.M.. 1982. Distribution and kinetics of membrane dielectric polarization. 1. Long-term inactivation of gating currents. J. Gen. Physiol. 79:21–40. 10.1085/jgp.79.1.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block B.A., Imagawa T., Campbell K.P., and Franzini-Armstrong C.. 1988. Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J. Cell Biol. 107:2587–2600. 10.1083/jcb.107.6.2587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruening-Wright A., and Larsson H.P.. 2007. Slow conformational changes of the voltage sensor during the mode shift in hyperpolarization-activated cyclic-nucleotide-gated channels. J. Neurosci. 27:270–278. 10.1523/JNEUROSCI.3801-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brum G., and Rios E.. 1987. Intramembrane charge movement in frog skeletal muscle fibres. Properties of charge 2. J. Physiol. 387:489–517. 10.1113/jphysiol.1987.sp016586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brum G., Stefani E., and Rios E.. 1987. Simultaneous measurements of Ca2+ currents and intracellular Ca2+ concentrations in single skeletal muscle fibers of the frog. Can. J. Physiol. Pharmacol. 65:681–685. 10.1139/y87-112 [DOI] [PubMed] [Google Scholar]
- Brum G., Fitts R., Pizarro G., and Ríos E.. 1988a Voltage sensors of the frog skeletal muscle membrane require calcium to function in excitation-contraction coupling. J. Physiol. 398:475–505. 10.1113/jphysiol.1988.sp017053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brum G., Ríos E., and Stéfani E.. 1988b Effects of extracellular calcium on calcium movements of excitation-contraction coupling in frog skeletal muscle fibres. J. Physiol. 398:441–473. 10.1113/jphysiol.1988.sp017052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brum G., Piriz N., DeArmas R., Rios E., Stern M., and Pizarro G.. 2003. Differential effects of voltage-dependent inactivation and local anesthetics on kinetic phases of Ca2+ release in frog skeletal muscle. Biophys. J. 85:245–254. 10.1016/S0006-3495(03)74470-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecchi E., Grossi F., Rossi M., Giglioli C., and De Feo M.L.. 2015. Severe hypocalcemia and life-threatening ventricular arrhytmias: case report and proposal of a diagnostic and therapeutic algorithm. Clin. Cases Miner. Bone Metab. 12:265–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury S., and Chanda B.. 2012. Estimating the voltage-dependent free energy change of ion channels using the median voltage for activation. J. Gen. Physiol. 139:3–17. 10.1085/jgp.201110722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collet C., Csernoch L., and Jacquemond V.. 2003. Intramembrane charge movement and L-type calcium current in skeletal muscle fibers isolated from control and mdx mice. Biophys. J. 84:251–265. 10.1016/S0006-3495(03)74846-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collet C., Pouvreau S., Csernoch L., Allard B., and Jacquemond V.. 2004. Calcium signaling in isolated skeletal muscle fibers investigated under “silicone voltage-clamp” conditions. Cell Biochem. Biophys. 40:225–236. 10.1385/CBB:40:2:225 [DOI] [PubMed] [Google Scholar]
- Delbono O., García J., Appel S.H., and Stefani E.. 1991. Calcium current and charge movement of mammalian muscle: action of amyotrophic lateral sclerosis immunoglobulins. J. Physiol. 444:723–742. 10.1113/jphysiol.1991.sp018903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demo S.D., and Yellen G.. 1992. Ion effects on gating of the Ca(2+)-activated K+ channel correlate with occupancy of the pore. Biophys. J. 61:639–648. 10.1016/S0006-3495(92)81869-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremov R.G., Leitner A., Aebersold R., and Raunser S.. 2015. Architecture and conformational switch mechanism of the ryanodine receptor. Nature. 517:39–43. 10.1038/nature13916 [DOI] [PubMed] [Google Scholar]
- Field A.C., Hill C., and Lamb G.D.. 1988. Asymmetric charge movement and calcium currents in ventricular myocytes of neonatal rat. J. Physiol. 406:277–297. 10.1113/jphysiol.1988.sp017380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García J., McKinley K., Appel S.H., and Stefani E.. 1992. Ca2+ current and charge movement in adult single human skeletal muscle fibres. J. Physiol. 454:183–196. 10.1113/jphysiol.1992.sp019259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García J., Tanabe T., and Beam K.G.. 1994. Relationship of calcium transients to calcium currents and charge movements in myotubes expressing skeletal and cardiac dihydropyridine receptors. J. Gen. Physiol. 103:125–147. 10.1085/jgp.103.1.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González A., and Ríos E.. 1993. Perchlorate enhances transmission in skeletal muscle excitation-contraction coupling. J. Gen. Physiol. 102:373–421. 10.1085/jgp.102.3.373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissmer S., and Cahalan M.. 1989. TEA prevents inactivation while blocking open K+ channels in human T lymphocytes. Biophys. J. 55:203–206. 10.1016/S0006-3495(89)82793-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hästbacka J., and Pettilä V.. 2003. Prevalence and predictive value of ionized hypocalcemia among critically ill patients. Acta Anaesthesiol. Scand. 47:1264–1269. 10.1046/j.1399-6576.2003.00236.x [DOI] [PubMed] [Google Scholar]
- Hollingworth S., and Marshall M.W.. 1981. A comparative study of charge movement in rat and frog skeletal muscle fibres. J. Physiol. 321:583–602. 10.1113/jphysiol.1981.sp014004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T., Zagotta W.N., and Aldrich R.W.. 1990. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 250:533–538. 10.1126/science.2122519 [DOI] [PubMed] [Google Scholar]
- Hoshi T., Zagotta W.N., and Aldrich R.W.. 1991. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 7:547–556. 10.1016/0896-6273(91)90367-9 [DOI] [PubMed] [Google Scholar]
- Kuzmenkin A., Bezanilla F., and Correa A.M.. 2004. Gating of the bacterial sodium channel, NaChBac: voltage-dependent charge movement and gating currents. J. Gen. Physiol. 124:349–356. 10.1085/jgp.200409139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Läuger P. 1988. Internal motions in proteins and gating kinetics of ionic channels. Biophys. J. 53:877–884. 10.1016/S0006-3495(88)83168-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt D.G. 1989. Continuum model of voltage-dependent gating. Macroscopic conductance, gating current, and single-channel behavior. Biophys. J. 55:489–498. 10.1016/S0006-3495(89)82842-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Barneo J., Hoshi T., Heinemann S.H., and Aldrich R.W.. 1993. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1:61–71. [PubMed] [Google Scholar]
- Melzer W., Rios E., and Schneider M.F.. 1984. Time course of calcium release and removal in skeletal muscle fibers. Biophys. J. 45:637–641. 10.1016/S0006-3495(84)84203-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzer W., Herrmann-Frank A., and Lüttgau H.C.. 1995. The role of Ca2+ ions in excitation-contraction coupling of skeletal muscle fibres. Biochim. Biophys. Acta. 1241:59–116. 10.1016/0304-4157(94)00014-5 [DOI] [PubMed] [Google Scholar]
- Olcese R., Latorre R., Toro L., Bezanilla F., and Stefani E.. 1997. Correlation between charge movement and ionic current during slow inactivation in Shaker K+ channels. J. Gen. Physiol. 110:579–589. 10.1085/jgp.110.5.579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper D.R., Varghese A., Sanguinetti M.C., and Tristani-Firouzi M.. 2003. Gating currents associated with intramembrane charge displacement in HERG potassium channels. Proc. Natl. Acad. Sci. USA. 100:10534–10539. 10.1073/pnas.1832721100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro G., Fitts R., Uribe I., and Ríos E.. 1989. The voltage sensor of excitation-contraction coupling in skeletal muscle. Ion dependence and selectivity. J. Gen. Physiol. 94:405–428. 10.1085/jgp.94.3.405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser B.L., Hernández-Ochoa E.O., Zimmer D.B., and Schneider M.F.. 2009. Simultaneous recording of intramembrane charge movement components and calcium release in wild-type and S100A1-/- muscle fibres. J. Physiol. 587:4543–4559. 10.1113/jphysiol.2009.177246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayner M.D., and Starkus J.G.. 1989. The steady-state distribution of gating charge in crayfish giant axons. Biophys. J. 55:1–19. 10.1016/S0006-3495(89)82775-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbeck R.T., Karunasekara Y., Board P.G., Beard N.A., Casarotto M.G., and Dulhunty A.F.. 2014. Skeletal muscle excitation-contraction coupling: who are the dancing partners? Int. J. Biochem. Cell Biol. 48:28–38. 10.1016/j.biocel.2013.12.001 [DOI] [PubMed] [Google Scholar]
- Rios E., and Brum G.. 1987. Involvement of dihydropyridine receptors in excitation-contraction coupling in skeletal muscle. Nature. 325:717–720. 10.1038/325717a0 [DOI] [PubMed] [Google Scholar]
- Ríos E., and Pizarro G.. 1991. Voltage sensor of excitation-contraction coupling in skeletal muscle. Physiol. Rev. 71:849–908. [DOI] [PubMed] [Google Scholar]
- Ríos E., Karhanek M., Ma J., and González A.. 1993. An allosteric model of the molecular interactions of excitation-contraction coupling in skeletal muscle. J. Gen. Physiol. 102:449–481. 10.1085/jgp.102.3.449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin G., and Allard B.. 2015. Voltage-gated Ca(2+) influx through L-type channels contributes to sarcoplasmic reticulum Ca(2+) loading in skeletal muscle. J. Physiol. 593:4781–4797. 10.1113/JP270252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romey G., Garcia L., Rieger F., and Lazdunski M.. 1988. Targets for calcium channel blockers in mammalian skeletal muscle and their respective functions in excitation-contraction coupling. Biochem. Biophys. Res. Commun. 156:1324–1332. 10.1016/S0006-291X(88)80777-0 [DOI] [PubMed] [Google Scholar]
- Royer L., Pouvreau S., and Ríos E.. 2008. Evolution and modulation of intracellular calcium release during long-lasting, depleting depolarization in mouse muscle. J. Physiol. 586:4609–4629. 10.1113/jphysiol.2008.157990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer L., Sztretye M., Manno C., Pouvreau S., Zhou J., Knollmann B.C., Protasi F., Allen P.D., and Ríos E.. 2010. Paradoxical buffering of calcium by calsequestrin demonstrated for the calcium store of skeletal muscle. J. Gen. Physiol. 136:325–338. 10.1085/jgp.201010454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider M.F., and Chandler W.K.. 1973. Voltage dependent charge movement of skeletal muscle: a possible step in excitation-contraction coupling. Nature. 242:244–246. 10.1038/242244a0 [DOI] [PubMed] [Google Scholar]
- Schnier A., Lüttgau H.C., and Melzer W.. 1993. Role of extracellular metal cations in the potential dependence of force inactivation in skeletal muscle fibres. J. Muscle Res. Cell Motil. 14:565–572. 10.1007/BF00141553 [DOI] [PubMed] [Google Scholar]
- Schuhmeier R.P., and Melzer W.. 2004. Voltage-dependent Ca2+ fluxes in skeletal myotubes determined using a removal model analysis. J. Gen. Physiol. 123:33–51. 10.1085/jgp.200308908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirokov R., Levis R., Shirokova N., and Ríos E.. 1992. Two classes of gating current from L-type Ca channels in guinea pig ventricular myocytes. J. Gen. Physiol. 99:863–895. 10.1085/jgp.99.6.863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirokov R., Levis R., Shirokova N., and Ríos E.. 1993. Ca(2+)-dependent inactivation of cardiac L-type Ca2+ channels does not affect their voltage sensor. J. Gen. Physiol. 102:1005–1030. 10.1085/jgp.102.6.1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirokov R., Ferreira G., Yi J., and Ríos E.. 1998. Inactivation of gating currents of L-type calcium channels. Specific role of the α2δ subunit. J. Gen. Physiol. 111:807–823. 10.1085/jgp.111.6.807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirokova N., García J., Pizarro G., and Ríos E.. 1996. Ca2+ release from the sarcoplasmic reticulum compared in amphibian and mammalian skeletal muscle. J. Gen. Physiol. 107:1–18. 10.1085/jgp.107.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigg D., and Bezanilla F.. 1997. Total charge movement per channel. The relation between gating charge displacement and the voltage sensitivity of activation. J. Gen. Physiol. 109:27–39. 10.1085/jgp.109.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon B.J., and Beam K.G.. 1983. Charge movement in a fast twitch skeletal muscle from rat. Biophys. J. 41:223–226. 10.1016/S0006-3495(83)84423-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon B.J., and Beam K.G.. 1985. The influence of transverse tubular delays on the kinetics of charge movement in mammalian skeletal muscle. J. Gen. Physiol. 85:21–42. 10.1085/jgp.85.1.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szentesi P., Jacquemond V., Kovács L., and Csernoch L.. 1997. Intramembrane charge movement and sarcoplasmic calcium release in enzymatically isolated mammalian skeletal muscle fibres. J. Physiol. 505:371–384. 10.1111/j.1469-7793.1997.371bb.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe T., Beam K.G., Powell J.A., and Numa S.. 1988. Restoration of excitation-contraction coupling and slow calcium current in dysgenic muscle by dihydropyridine receptor complementary DNA. Nature. 336:134–139. 10.1038/336134a0 [DOI] [PubMed] [Google Scholar]
- Tsien R.W., and Noble D.. 1969. A transition state theory approach to the kinetics of conductance changes in excitable membranes. J. Membr. Biol. 1:248–273. 10.1007/BF01869785 [DOI] [PubMed] [Google Scholar]
- Villalba-Galea C.A. 2012. Voltage-controlled enzymes: the new JanusBifrons. Front. Pharmacol. 3:161 10.3389/fphar.2012.00161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba-Galea C.A. 2016. Hysteresis in voltage-gated channels. Channels (Austin). 11:140–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba-Galea C.A., Sandtner W., Starace D.M., and Bezanilla F.. 2008. S4-based voltage sensors have three major conformations. Proc. Natl. Acad. Sci. USA. 105:17600–17607. 10.1073/pnas.0807387105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.M., Messi M.L., and Delbono O.. 1999. Patch-clamp recording of charge movement, Ca2+ current, and Ca2+ transients in adult skeletal muscle fibers. Biophys. J. 77:2709–2716. 10.1016/S0006-3495(99)77104-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.M., Messi M.L., and Delbono O.. 2000. L-Type Ca(2+) channel charge movement and intracellular Ca(2+) in skeletal muscle fibers from aging mice. Biophys. J. 78:1947–1954. 10.1016/S0006-3495(00)76742-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Yan Z., Li Z., Yan C., Lu S., Dong M., and Yan N.. 2015. Structure of the voltage-gated calcium channel Cav1.1 complex. Science. 350:aad2395 10.1126/science.aad2395 [DOI] [PubMed] [Google Scholar]
- Wu J., Yan Z., Li Z., Qian X., Lu S., Dong M., Zhou Q., and Yan N.. 2016. Structure of the voltage-gated calcium channel Ca(v)1.1 at 3.6 Å resolution. Nature. 537:191–196. 10.1038/nature19321 [DOI] [PubMed] [Google Scholar]
- Yan Z., Bai X., Yan C., Wu J., Li Z., Xie T., Peng W., Yin C., Li X., Scheres S.H.W., et al. . 2015. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature. 517:50–55. 10.1038/nature14063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalk R., Clarke O.B., des Georges A., Grassucci R.A., Reiken S., Mancia F., Hendrickson W.A., Frank J., and Marks A.R.. 2015. Structure of a mammalian ryanodine receptor. Nature. 517:44–49. 10.1038/nature13950 [DOI] [PMC free article] [PubMed] [Google Scholar]