

Abstract
Crystallinity is seldomly utilized as part of the microphase segregation process in ultralow-molecular-weight block copolymers. Here, we show the preparation of two types of discrete, semicrystalline block co-oligomers, comprising an amorphous oligodimethylsiloxane block and a crystalline oligo-l-lactic acid or oligomethylene block. The self-assembly of these discrete materials results in lamellar structures with unforeseen uniformity in the domain spacing. A systematic introduction of dispersity reveals the extreme sensitivity of the microphase segregation process toward chain length dispersity in the crystalline block.
Self-organization in (di)block copolymers (BCPs) has proven itself, both theoretically and experimentally, as a robust and well-established strategy to generate organized structures at nano- and mesoscopic length scales reaching beyond that of small, organic molecules.1−3 Yet, the gap between small, perfectly defined molecules and traditional polymers is slowly fading.4,5 In earlier work, we illustrated our efforts to amalgamate full synthetic control and BCP self-assembly.6 Low-molecular-weight (MW) block co-oligomers (BCOs) composed of discrete oligodimethylsiloxane (oDMS) and discrete, atactic oligolactic acid (oLA) exhibited self-assembly behavior that is qualitatively analogous to that of traditional, disperse block copolymers but allowed the exploration of a far lower MW regime.7 In line with this work, Hawker and co-workers simultaneously reported similar observations.8
To evolve nano- and mesoscale organization of BCOs into a higher level of perfection, the introduction of additional interactions that further guide self-assembly and increase the effective interaction parameter χ of the incompatible blocks is required. Such interactions can be diverse in nature—van der Waals, dipole, π–π, and others—and have already been exploited in, for example, the molecular designs of liquid crystalline materials.9 Also the use of one or two (semi)crystalline blocks in (diblock) BCPs has received a great deal of attention.10−12 Furthermore, numerous examples highlight the importance of molecular perfection (i.e., sequence control) in other classes of self-assembling oligomeric sequences.13,14
In the context of a “simple” diblock copolymer design, understanding and exploiting the optimal blend of (directional) molecular interactions and full synthetic control is still in its infancy. Even though the development of “high χ–low N” BCPs has led to ever smaller domain spacings, the general trend is to avoid the use of crystalline blocks, and the extent of long-range order in the resulting systems is often limited.6,8,15−18 A notable exception here is the work of Booth and co-workers on poly(ethylene oxide-block-propylene oxide),19−21 poly(ethylene oxide-block-butylene oxide),22−24 and poly(ethylene oxide-block-methylene)25−29 in di- and triblock configurations.
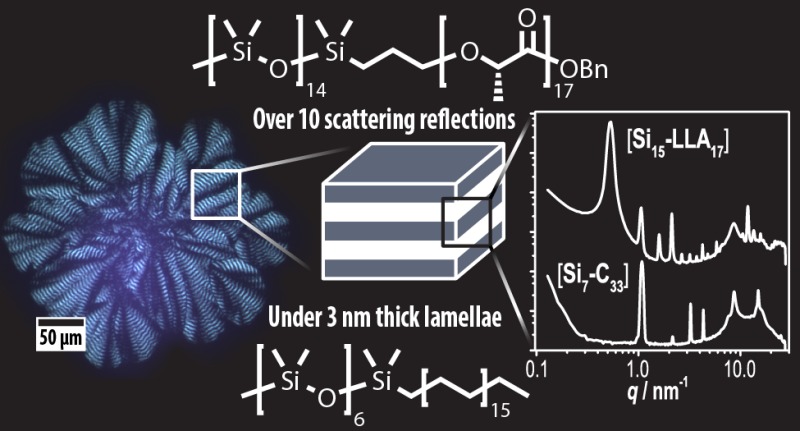
We prepared two types of discrete BCOs that consist of amorphous oDMS linked to a crystalline block. The first is related to oDMS-oLA, and the second contains oDMS covalently bound to an alkane of intermediate length (Figure 1A,B). As shown before, discrete, isotactic oligo-l-lactic acid (oLLA) is crystalline, forming well-defined lamellea.30 Likewise, poly- and oligomethylene are semicrystalline materials, also organizing into lamellar domains.31 The oDMS-oLLA BCO studied in this work consists of 15 siloxane and 17 l-lactic acid repeat units, abbreviated as [Si15-LLA17]. This BCO design permits a systematic study of discrete and (partially) disperse semicrystalline oDMS-oLLA and a direct comparison with our previous study on the fully amorphous sister compounds (vide infra).7 Additionally, an oligodimethylsiloxane–oligomethylene (oDMS-oM) BCO was chosen, comprising 7 siloxane repeat units and a C33 alkane (i.e., [Si7-M33]). Similar to [Si15-LLA17], the volume fraction of the crystalline block is close to 0.50.
Figure 1.

Molecular structures of [Si15-LLA17] (A) and [Si7-M33] (B). (C) DSC traces of [Si15-LLA17] (black) and [Si7-M33] (red). The data are shifted vertically for clarity. The order–disorder transition unrelated to the crystallization process is indicated with an asterisk. (D) Room-temperature SAXS data for discrete [Si15-LLA17] (top) and [Si7-C33] (bottom). The data are shifted vertically for clarity. A selection of higher order reflections is indicated.
oDMS-oLLA was synthesized according to the same protocol employed for the synthesis of atactic oDMS-oLA, with the sole difference that optically pure l-lactide was used as the starting material instead of the racemic dl-lactide (see Schemes S1–S3). The synthetic strategy toward discrete [Si7-M33] (Scheme S4) employed the efficient and selective Karstedt hydrosilylation as a key reaction step.33 This enabled the use of synthetically accessible oDMS7 hydride and a C33 α-olefin. The high purity of both BCOs and absence of oligomers with a different number of repeat units were confirmed with NMR and mass spectrometry (MALDI-TOF) (Figures S1–S5).
The thermal signatures of [Si15-LLA17] and [Si7-M33] were captured with differential scanning calorimetry (DSC; Figure 1C and Table 1, entries 1 and 2). Each compound revealed strong and narrow endothermic transitions. The transition of [Si15-LLA17] started close to the known melting point of pure oligo-l-lactic acid LLA16 (Tm = 90 °C, ΔHfus = 71 kJ mol–1) and showed a similar transition enthalpy. As a result of the 7 °C hysteresis upon cooling, a new, low-intensity exotherm appeared at 79 °C, preceding the primary peak (indicated in Figure 1C with an asterisk). BCO [Si7-M33] shows two transitions, at 36 and 44 °C. Interestingly, no further transitions were observed at higher temperatures, although pure tritriacontane (C33H68) has a melting point at 72 °C.34 The enthalpy associated with the transition at 44 °C (71 kJ (mol BCO)−1) has a value similar to that of the enthalpy of fusion of tritriacontane (79.5 kJ mol–1).34
Table 1. Molecular and Morphologic Characterization Data.
| entry | compounda | no. Sib | no. M or LLAb | Mn,calcdc [Da] | Mn,SEC [Da] | Đ | Nd | fcre | Tm [°C] | ΔHfus [kJ mol–1]f | TODT [°C] | d* [nm] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | [Si7-M33] | 7 | 33 | 982 | 2114 | <1.00001h | 15.6 | 0.51 | 43.8 | 71 | n.o. | 5.77 |
| 2 | [Si15-LLA17] | 15 | 17 | 2486 | 4380 | <1.00001h | 32.0 | 0.47 | 85.1 | 78 | 79.1 | 11.7 |
| 3 | [Si∼15-LLA17] | 15.8g | 17 | 2587 | 4444 | 1.04 | 33.0 | 0.45 | 87.9 | 80 | 83.5 | 12.4 |
| 4 | [Si15-LLA∼17] | 15 | 15.2g | 2356 | 4482 | 1.13 | 30.5 | 0.44 | 87–125i | 42 | n.o. | 12.7 |
| 5 | [Si∼15-LLA∼17] | 15g | 16.2g | 2470 | 4463 | 1.13 | 31.3 | 0.45 | 90–129i | 25 | n.o. | 18.3 |
Disperse blocks are indicated with a tilde (∼) character preceding the (desired) average block lengths.
Number of siloxane, methylene, or l-lactic acid repeat units.
Calculation based on the total number of repeat units.
Number of segments based on a 110 Å3 reference volume.
Volume fraction of the crystalline block, calculated using bulk densities for PDMS (0.95 g mL–1),6,7 PLLA (1.29 g mL–1),32 and tritriacontane (0.82 g mL–1).
Heat effect per mole of BCO.
Average value.
Calculated from relative peak intensities in the MALDI-TOF spectra.
A broad melting range was observed. n.o. = not observed.
For both BCOs, we attribute the endothermic transitions at the highest temperatures to melting transitions of the crystalline blocks and the formation of an isotropic state. This was corroborated with polarized optical microscopy (Figure S6) and small-angle X-ray scattering (SAXS, Figure S7) experiments at temperatures above and below these transitions. Possibly, the observed reduction in Tm of the BCO [Si7-M33] is caused by alkyl chain folding, which is (partially) supported by X-ray scattering measurements (vide infra). Apparently, chain folding does not occur in BCO [Si15-LLA17]. The remaining transition in the cooling trace of [Si15-LLA17] is attributed to an order–disorder transition, which is also present in most of the atactic oDMS-oLA BCOs previously published.6 In contrast, we ascribe the second transition in BCO [Si7-M33] to a solid–solid phase transition involving a reorganization of the alkyl chains.34−37
The formation of an ordered crystalline state was further investigated with SAXS at room temperature in medium- (MAXS) and wide-angle X-ray scattering (WAXS) operating modes (see Figure 1D). Remarkably, both discrete BCOs reveal an unprecedentedly high number of equally spaced reflection peaks (q/q* up to √121 for [Si15-LLA17]), indicative for a high degree of long-range order. The distance between the reflections unambiguously points to the presence of a lamellar structure with a lamellar domain spacing dlam = 2π/q* = 11.7 nm for [Si15-LLA17] and dlam = 5.8 nm for [Si7-M33]. The latter is one of the smallest feature sizes hitherto observed.15 At higher q-values, additional scattering peaks reveal further interchain organization. For BCO [Si15-LLA17], a broad reflection at q = 8.6 nm–1 is attributed to the amorphous siloxane block. The sharp reflections at larger q originate from the crystalline oLLA block, which is in concordance with the scattering profile of the pure LLA16 (Figure S7A) and known profiles for PLLA.38 Interestingly, BCO [Si7-M33] gave a significantly narrower siloxane reflection around q = 8.7 nm–1, along with a sharp peak at 14.9 nm–1, a shoulder at q = 16.7 nm–1, and a weak reflection at 24.9 nm–1. The last three reflections suggest that the alkyl blocks crystallize in the same orthorhombic substructure as linear alkanes.39,40 In addition, sharpening of the siloxane reflection band is an indication that the variability of interchain distances within the oDMS block is smaller in [Si7-M33] than in [Si15-LLA17]. Presumably, this is the result of the combined effect of crystallinity in the oM block and the short stretched-chain length of the oDMS block in BCO [Si7-M33].
The nearly symmetric composition of the BCOs dictates that the thicknesses of the amorphous and crystalline blocks both should be around half dlam. For BCO [Si15-LLA17] (dlam = 11.7 nm)—taking into account the domain spacing of pure LLA16 (dlam = 5.2 nm, see Figure S7A)—we propose that the lamellar structure is composed of interdigitated, fully stretched oLLA chains, aligned perpendicularly to the domain boundary. Additionally, the short molecular length of the Si15 block causes the amorphous part to consist of an elongated oDMS coil, as depicted in Figure S8A. In the case of BCO [Si7-M33], we envision a similar structure. However, the estimated extended chain length in the crystalline C33 alkyl block (∼4.4 nm, see Figure S7B and literature values)34 is 150% larger than half of the domain spacing of the BCO. As a consequence, the oM long-axis might be tilted relative to the normal of the interdomain planes27 or folded once28 (Figure S8B,C). The proposed chain orientation of the oLLA and oM blocks is in correspondence with the melting transitions of the BCOs compared to those of the crystalline homoblocks.
The large number of observed reflection peaks is uncommon for BCPs, particularly in such low-MW materials.27,41,42 According to paracrystalline theory,43 the high number of observed reflections permits only a small variability in the lamellar thickness throughout the microphase-segregated sample. Although crystallinity undoubtedly enhances the uniformity of the lamellar domains, we wondered to what extent the discrete nature of the materials plays a role in obtaining such highly organized structures. We therefore synthesized three additional BCOs, [Si∼15-LLA17], [Si15-LLA∼17], and [Si∼15-LLA∼17], incorporating a disperse oDMS, a disperse oLLA, or two disperse blocks, respectively. This allows a systematic screening of the effect of a discrete versus a disperse block on the crystallization of the oLLA block and subsequent microphase separation. In the abbreviations, disperse blocks are indicated with a tilde (∼) character preceding the (desired) average block lengths. The synthesis of the materials is documented in the Supporting Information. As summarized in Table 1, entries 3–5, oDMS and oLLA block lengths and Mn values of the (partially) disperse materials were close to those of the discrete analogues. Values for the molar mass dispersity increased after introduction of one or two disperse blocks, although the maximum value of Đ = 1.13 (for both [Si15-LLA∼17] and [Si∼15-LLA∼17]) still can be considered as relatively low. Mass spectrometry nicely captures the striking differences between “no” and “low” dispersity (Figure S9).
The thermal behavior of [Si∼15-LLA17] is very similar to that of its discrete analogue, albeit with a slightly higher Tm of the oLLA block. Currently, we do not have an explanation for this observation. The upward shift of TODT with increasing dispersity (see the transitions marked with an asterisk in Figure S10) is similar to the trend observed in the atactic BCOs.7 Interestingly, we find significant changes in the thermal behavior if a disperse oLLA block is incorporated. Instead of a sharp melting transition, melting occurs over a broad temperature range. Moreover, ΔHfus decreases by 45–70%, albeit that the determination of ΔHfus became less accurate as a result of the peak broadening.
The phase behavior of the disperse polymers at room temperature was studied with SAXS (Figure 2). Discrete [Si15-LLA17] and the BCO with a disperse Si block [Si∼15-LLA17] reveal very similar scattering patterns, both indicative for a highly ordered lamellar structure. Introduction of a disperse oDMS block causes a 6% increase of the domain spacing (dlam = 12.4 nm). This is accompanied by a broadening of the principal scattering peak, indicated by a 35% increase of the full width at half-maximum value (see Figure 2). Remarkably, the degree of ordering decreases dramatically in the two BCOs comprising a disperse oLLA block. Here, the principal scattering peak at low q-values becomes very broad, and only a single, very weak second reflection is visible (indicated with the black triangles in Figure 2). In contrast, all scattering patterns look similar at large scattering angles. This suggests a very similar orthorhombic subunit cell in the crystalline oLLA domain of all four BCOs, albeit the DSC data show that a fraction of the disperse oLLA blocks resides in an amorphous state.
Figure 2.

Room-temperature SAXS data for discrete and (partially) disperse oDMS-oLLA BCOs. The data are shifted vertically for clarity. Select higher order reflections are indicated as q/q* values or with black triangles. fwhm = full width at half-maximum. The broad peak at q = 4 nm–1 results from background scattering (Kapton tape).
We propose that the crystalline nature of the oLLA precisely aligns the chains in a parallel manner, unaffected by dispersity in the block. In case of the BCOs with discrete oLLA chains, this results in near-perfect localization of the chain ends and interblock connections in three-dimensional space, greatly reducing any variations and fluctuations in domain spacing that generally exist in completely amorphous systems. In contrast, dispersity in the crystalline block (primarily) forces the ordering of the block links along the domain boundary out of alignment. This results in an increased variation of the lamellar thickness, at the cost of (long-range) order. Likely, this effect is amplified by the absence of chain folds in this low-MW system that can act as a buffer for variations in chain length during the crystallization process.
In summary, we studied the self-assembly behavior of oDMS-oLLA and oDMS-oM, incorporating additional crystalline interactions in the oligolactic acid and oligomethylene blocks. Our results show that the introduction of crystallinity indeed improves the self-assembly in BCOs, provided that the crystalline block is discrete. This leads to exceptionally uniform microphase-segregated domains and new avenues to further decrease feature sizes. However, dispersity plays a crucial role in the formation of ordered structures, causing a nearly complete loss of ordering if the crystalline block has a nonuniform length. Although this is easily explained with a simple, intuition-based molecular picture, no comparative experimental studies existed to date. Currently, we further elucidate the exact molecular organization in semicrystalline BCOs, and their intriguing temperature-dependent behavior, by using other block (length) combinations and architectures.
Acknowledgments
This work is financed by the Royal Netherlands Academy of Arts and Sciences and the Dutch Ministry of Education, Culture and Science (Gravity program 024.001.035).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b08627.
Experimental procedures and characterization data for all compounds, including Schemes S1–S4 and Figures S1–S10 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Leibler L. Macromolecules 1980, 13, 1602. 10.1021/ma60078a047. [DOI] [Google Scholar]
- Bates C. M.; Bates F. S. Macromolecules 2017, 50, 3. 10.1021/acs.macromol.6b02355. [DOI] [Google Scholar]
- Schacher F. H.; Rupar P. a; Manners I. Angew. Chem., Int. Ed. 2012, 51, 7898. 10.1002/anie.201200310. [DOI] [PubMed] [Google Scholar]
- Sinturel C.; Bates F. S.; Hillmyer M. A. ACS Macro Lett. 2015, 4, 1044. 10.1021/acsmacrolett.5b00472. [DOI] [PubMed] [Google Scholar]
- Hawker C. J. Science 2005, 309, 1200. 10.1126/science.1109778. [DOI] [PubMed] [Google Scholar]
- van Genabeek B.; de Waal B. F. M.; Gosens M. M. J.; Pitet L. M.; Palmans A. R. A.; Meijer E. W. J. Am. Chem. Soc. 2016, 138, 4210. 10.1021/jacs.6b00629. [DOI] [PubMed] [Google Scholar]
- van Genabeek B.; de Waal B. F. M.; Ligt B.; Palmans A. R. A.; Meijer E. W. ACS Macro Lett. 2017, 6, 674. 10.1021/acsmacrolett.7b00266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oschmann B.; Lawrence J.; Schulze M. W.; Ren J. M.; Anastasaki A.; Luo Y.; Nothling M. D.; Pester C. W.; Delaney K. T.; Connal L. A.; McGrath A. J.; Clark P. G.; Bates C. M.; Hawker C. J. ACS Macro Lett. 2017, 6, 668. 10.1021/acsmacrolett.7b00262. [DOI] [PubMed] [Google Scholar]
- Handbook of Liquid Crystals; Goodby J. W., Tschierske C., Raynes P., Gleeson H., Kato T., Collings P. J., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2014. [Google Scholar]
- He W.-N.; Xu J.-T. Prog. Polym. Sci. 2012, 37, 1350. 10.1016/j.progpolymsci.2012.05.002. [DOI] [Google Scholar]
- Nandan B.; Hsu J.-Y.; Chen H.-L. J. Macromol. Sci., Polym. Rev. 2006, 46, 143. 10.1080/15321790600646802. [DOI] [Google Scholar]
- Hamley I. W.Interfaces Crystallization Viscoelasticity; Springer: Berlin/Heidelberg, 1999; pp 113–137. [Google Scholar]
- Sun J.; Zuckermann R. N. ACS Nano 2013, 7, 4715. 10.1021/nn4015714. [DOI] [PubMed] [Google Scholar]
- Badi N.; Chan-Seng D.; Lutz J.-F. Macromol. Chem. Phys. 2013, 214, 135. 10.1002/macp.201200475. [DOI] [Google Scholar]
- Jeong G.; Yu D. M.; Mapas J. K. D.; Sun Z.; Rzayev J.; Russell T. P. Macromolecules 2017, 50, 7148. 10.1021/acs.macromol.7b01443. [DOI] [Google Scholar]
- Kwak J.; Mishra A. K.; Lee J.; Lee K. S.; Choi C.; Maiti S.; Kim M.; Kim J. K. Macromolecules 2017, 50, 6813. 10.1021/acs.macromol.7b00945. [DOI] [Google Scholar]
- Nowak S. R.; Hwang W.; Sita L. R. J. Am. Chem. Soc. 2017, 139, 5281. 10.1021/jacs.6b13285. [DOI] [PubMed] [Google Scholar]
- Carter M. C. D.; Jennings J.; Speetjens F. W.; Lynn D. M.; Mahanthappa M. K. Macromolecules 2016, 49, 6268. 10.1021/acs.macromol.6b01268. [DOI] [Google Scholar]
- Booth C.; Pickles C. J. J. Polym. Sci. Part A-2 Polym. Phys. 1973, 11, 249. 10.1002/pol.1973.180110206. [DOI] [Google Scholar]
- Viras F.; Luo Y.-Z.; Viras K.; Mobbs R. H.; King T. A.; Booth C. Makromol. Chem. 1988, 189, 459. 10.1002/macp.1988.021890219. [DOI] [Google Scholar]
- Šimek L.; Petřík S.; Hadobaš F.; Bohdanecký M. Eur. Polym. J. 1990, 26, 371. 10.1016/0014-3057(90)90256-4. [DOI] [Google Scholar]
- Yang Y.-W.; Tanodekaew S.; Mai S.-M.; Booth C.; Ryan A. J.; Bras W.; Viras K. Macromolecules 1995, 28, 6029. 10.1021/ma00122a008. [DOI] [Google Scholar]
- Mai S.-M.; Fairclough J. P. A.; Viras K.; Gorry P. A.; Hamley I. W.; Ryan A. J.; Booth C. Macromolecules 1997, 30, 8392. 10.1021/ma971158f. [DOI] [Google Scholar]
- Ryan A. J.; Fairclough J. P. A.; Hamley I. W.; Mai S.-M.; Booth C. Macromolecules 1997, 30, 1723. 10.1021/ma960943+. [DOI] [Google Scholar]
- Cooper D. R.; Leung Y.-K.; Heatley F.; Booth C. Polymer 1978, 19, 309. 10.1016/0032-3861(78)90224-0. [DOI] [Google Scholar]
- Domszy R. C.; Mobbs R. H.; Leung Y.-K.; Heatley F.; Booth C. Polymer 1979, 20, 1204. 10.1016/0032-3861(79)90144-7. [DOI] [Google Scholar]
- Swales T. G. E.; Domszy R. C.; Beddoes R. L.; Price C.; Booth C. J. Polym. Sci., Polym. Phys. Ed. 1985, 23, 1585. 10.1002/pol.1985.180230811. [DOI] [Google Scholar]
- Yeates S. G.; Booth C. Eur. Polym. J. 1985, 21, 217. 10.1016/0014-3057(85)90222-8. [DOI] [Google Scholar]
- Campbell C.; Viras K.; Richardson M. J.; Masters A. J.; Booth C. Makromol. Chem. 1993, 194, 799. 10.1002/macp.1993.021940305. [DOI] [Google Scholar]
- Takizawa K.; Nulwala H.; Hu J.; Yoshinaga K.; Hawker C. J. J. Polym. Sci., Part A: Polym. Chem. 2008, 46, 5977. 10.1002/pola.22944. [DOI] [Google Scholar]
- Mnyukh Y. V. J. Struct. Chem. 1960, 1, 346. 10.1007/BF00748864. [DOI] [Google Scholar]
- Okihara T.; Tsuji M.; Kawaguchi A.; Katayama K.-I.; Tsuji H.; Hyon S.-H.; Ikada Y. J. Macromol. Sci., Part B: Phys. 1991, 30, 119. 10.1080/00222349108245788. [DOI] [Google Scholar]
- Karstedt B. D.Platinum complexes of unsaturated siloxanes and platinum containing organopolysiloxanes. U.S. Patent US3775452, 1973.
- Strobl G.; Ewen B.; Fischer E. W.; Piesczek W. J. Chem. Phys. 1974, 61, 5257. 10.1063/1.1681871. [DOI] [Google Scholar]
- Ungar G. J. Phys. Chem. 1983, 87, 689. 10.1021/j100227a032. [DOI] [Google Scholar]
- Sirota E.; Singer D. J. Chem. Phys. 1994, 101, 10873. 10.1063/1.467837. [DOI] [Google Scholar]
- Wentzel N.; Milner S. T. J. Chem. Phys. 2010, 132, 044901. 10.1063/1.3276458. [DOI] [PubMed] [Google Scholar]
- Tsuji H. Macromol. Biosci. 2005, 5, 569. 10.1002/mabi.200500062. [DOI] [PubMed] [Google Scholar]
- Muller A. Proc. R. Soc. London, Ser. A 1928, 120, 437. 10.1098/rspa.1928.0158. [DOI] [Google Scholar]
- Smith A. E. J. Chem. Phys. 1953, 21, 2229. 10.1063/1.1698826. [DOI] [Google Scholar]
- Lynd N. A.; Hamilton B. D.; Hillmyer M. A. J. Polym. Sci., Part B: Polym. Phys. 2007, 45, 3386. 10.1002/polb.21321. [DOI] [Google Scholar]
- Hashimoto T.; Tanaka H.; Hasegawa H. Macromolecules 1985, 18, 1864. 10.1021/ma00152a013. [DOI] [Google Scholar]
- Hosemann R.; Bagchi S. N.. Direct Analysis of Diffraction by Matter; North-Holland Publishing: Amsterdam, 1962. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.