

Abstract
Aflatoxins produced by several species in Aspergillus section Flavi are a significant problem in agriculture and a continuous threat to human health. To provide insights into the biology and global population structure of species in section Flavi, a total of 1,304 isolates were sampled across six species (A. flavus, A. parasiticus, A. nomius, A. caelatus, A. tamarii, and A. alliaceus) from single fields in major peanut‐growing regions in Georgia (USA), Australia, Argentina, India, and Benin (Africa). We inferred maximum‐likelihood phylogenies for six loci, both combined and separately, including two aflatoxin cluster regions (aflM/alfN and aflW/aflX) and four noncluster regions (amdS, trpC, mfs and MAT), to examine population structure and history. We also employed principal component and STRUCTURE analysis to identify genetic clusters and their associations with six different categories (geography, species, precipitation, temperature, aflatoxin chemotype profile, and mating type). Overall, seven distinct genetic clusters were inferred, some of which were more strongly structured by G chemotype diversity than geography. Populations of A. flavus S in Benin were genetically distinct from all other section Flavi species for the loci examined, which suggests genetic isolation. Evidence of trans‐speciation within two noncluster regions, whereby A. flavus SBG strains from Australia share haplotypes with either A. flavus or A. parasiticus, was observed. Finally, while clay soil and precipitation may influence species richness in Aspergillus section Flavi, other region‐specific environmental and genetic parameters must also be considered.
Keywords: Aspergillus, balancing selection, maximum likelihood, multilocus sequence typing, principal component analysis
1. INTRODUCTION
Aspergillus section Flavi contains the most serious mycotoxin‐producing fungi in this genus, being named for one of its well publicized toxigenic species, Aspergillus flavus Link (Horn, 2003; Raper & Fennell, 1965). Being a prolific colonizer of many taxonomic groups (e.g., plants, animals, and humans), A. flavus is distributed worldwide and occupies different ecological niches (Cleveland et al., 2009; Klich, 2007). A. flavus, as a species, encompasses two morphotypes and various chemotypes. Some strains produce large sclerotia (L‐strain) while others produce numerous small sclerotia (S‐strain) (Cotty, 1989). A. flavus L‐strains can exhibit either a nonaflatoxigenic phenotype or they will produce only one type (B) of aflatoxins, and the S‐strain morphotype will produce only B aflatoxins (SB), or they can produce B and G aflatoxins (SBG) (Cotty & Cardwell, 1999). Production of G aflatoxins is rarely observed in A. flavus L‐strains (Ehrlich et al., 2004). Most of the other species in section Flavi are soil‐inhabiting saprobes such as A. parasiticus, another producer of mycotoxins (Abbas et al., 2009; Horn, 2003). Aflatoxins are the most studied of the mycotoxins in this genus (Bennett, 2010; Horn, 2003); in fact the term “mycotoxin” was coined to describe aflatoxins after the Turkey X disease outbreak in the 1960s (Bennett & Klich, 2003). In developing countries, the risk to humans by carcinogenic aflatoxins is very high, and chronic exposure to aflatoxin is estimated to impact 4.5 billion people (CDC 2016). Strict regulations are being set worldwide, which means the need for effective control of aflatoxin‐producing fungi is crucial (Klich, 2007). Recent discoveries of the sexual states in A. flavus, A. nomius and A. parasiticus suggest that barriers to genetic recombination, such as heterokaryon incompatibility, are not impassable (Horn, Gell, Singh, Sorensen, & Carbone, 2016; Horn, Moore, & Carbone, 2011; Horn, Ramirez‐Prado, and Carbone, 2009a; Horn, Ramirez‐Prado, and Carbone, 2009b; Horn, Moore et al., 2009; Micali & Smith, 2005; Ramirez‐Prado et al., 2008), and genetic exchange and recombination could potentially create new strains or species that are better adapted to particular niches (Milgroom, 1996). Geiser and coworkers reported two reproductively isolated clades (identified as Groups I and II) within A. flavus for an Australian population, and they suggested that recombination could be occurring among strains within the species (Geiser, Pitt, & Taylor, 1998). Geiser's Group I included A. flavus L and SB strains, and Group II included A. flavus SB and SBG strains (Geiser et al., 1998). In the case of the two approved A. flavus biocontrol strains that are both nonaflatoxigenic, genetic recombination may restore toxigenicity (Geiser et al., 1998; Horn et al., 2016; Moore et al., 2009). In an experimentally recombining population of A. flavus, a single crossover recombination was shown to result in the gain of cluster regions that were able to restore the aflatoxigenic phenotype (Olarte et al., 2011); field experiments further showed that A. flavus sclerotia can be fertilized by native strains and yield recombinant progeny (Horn et al., 2016). Moreover, genetic exchange between species may result in hybrids that have novel chemotype profiles, are mycotoxin super‐producers, are better pathogens, and/or are more fit under adverse and changing environments; recent evidence shows hybridization is experimentally possible between A. flavus and A. parasiticus (Olarte et al., 2015).
Balancing selection acts to maintain genetic polymorphisms in a population and results in more genetic variation between alleles or haplotypes within a species than would be predicted by genetic drift alone (Hedrick, 2007; Schierup, Mikkelsen, & Hein, 2001). This signature of balancing selection has been inferred for the mating‐type loci of various fungi (May et al., 1999; Ramirez‐Prado et al., 2008), transcending species boundaries and resulting in trans‐speciation, a phenomenon where the loci under balancing selection are more similar among species than within a single species. Other evolutionary mechanisms that may result in a signature of trans‐speciation include hybridization and introgression, or incomplete lineage sorting, at specific loci (Sun, Corcoran et al., 2012). Trans‐speciation has been observed for chemotype‐specific alleles in A. parasiticus (Carbone et al., 2007) and A. flavus (Moore et al., 2009). In the aflatoxin gene cluster, selection may act to maintain the nonaflatoxigenic phenotype in A. flavus, and G1 or B1 dominance in A. parasiticus (Carbone et al., 2007; Moore et al., 2009). Because balancing selection could be acting on chemotype or morphological differences (Moore et al., 2009; Nei, 2007), species delimitation using cluster regions alone is tenuous, and a more holistic approach is warranted (Samson & Varga, 2009). For phylogenetic inference of species trees, multiple single‐copy neutral loci such as amdS and trpC should also be targeted (Michielse, Ram, & van den Hondel, 2004; Yelton, Hamer, & Timberlake, 1984).
Climate change can result in fragmentations and bottlenecks, eliminating or reducing the population sizes of endemic species, and selecting for more fit taxa (Ali & Roossinck, 2008; Opdam & Wascher, 2004). Climate has been reported to influence the aflatoxin producing ability of A. flavus, alter the numbers of aflatoxigenic fungi in the environment, and change fungal population structure (Cotty & Jaime‐Garcia, 2007). Soil type (e.g., clay, sand) and quality (i.e., edaphic factors such as pH and mineral content) can impact the presence and distribution of Aspergillus in a field (Ahmad & Singh, 1994; Wassila, Houda, & Mohamed, 2015). For example, it has been reported that soils high in clay content may favor the prevalence of the A. flavus S morphotype (Jaime‐Garcia & Cotty, 2006). Given the diverse habitats and differences in mean annual temperature and precipitation in peanut‐growing regions worldwide, inferences of genetic isolation should be examined with the possibility of climatic and environmental conditions driving genetic differentiation and adaptation (Moore et al., 2013). In this study, we examine the evolutionary history of populations of Aspergillus section Flavi from geographically isolated regions using a bottom‐up microevolutionary approach. We build on previously obtained and published data (Moore et al., 2013), to test whether there are significant associations of haplotypes and clades with chemotype production and with different abiotic and edaphic factors (e.g., soil type, precipitation, temperature). We found that while diversity in aflatoxin chemotype profile is a good predictor of species genetic diversity, our sampled fields that have clay soils, as well as high precipitation, exhibited less species diversity. Further exploration of the role of climate and other environmental parameters in adaptive evolution in Aspergillus section Flavi will allow us to better understand, and possibly improve, our biocontrol strategies of these agriculturally important species in different parts of the world.
2. MATERIALS AND METHODS
2.1. Sampling, DNA isolation, and target loci
Species in Aspergillus section Flavi were sampled from geographically isolated peanut fields spanning five continents (Tables 1 and S1). We focused our attention on major peanut‐growing regions that were considered hotspots for aflatoxin contamination in 2005. As our goal was to maximize our sampling of genetic diversity in Aspergillus section Flavi, we sampled species diversity at southern latitudes, which is typically higher than in northern latitudes (Horn, 2007). For this reason, the European continent is not represented in our sampling. Aspergillus oryzae isolates from Japan were included in this global study of section Flavi, although they were not sampled in peanut fields. A total of 1,304 isolates were examined for this study. DNA was extracted from freeze‐dried mycelia, and target loci for phylogenetic inference were amplified and sequenced, as described previously (Moore et al., 2009). DNA sequence variation was assayed in six genomic regions: aflM/aflN and aflW/aflX intergenic regions within the aflatoxin gene cluster, and a major facilitator superfamily (mfs) gene adjacent to the cluster on chromosome 3; acetamidase (amdS) and mating‐type (MAT) genes on chromosome 6; and the tryptophan synthase (trpC) gene on chromosome 4. The chromosomal locations associated with these loci have been reported for A. flavus (Moore et al., 2013; Ramirez‐Prado et al., 2008; Smith, Woloshuk, Robertson, & Payne, 2007) but have not been confirmed for all species examined in this study. For each locus, the sequence of at least one representative isolate from each putative species was subjected to BLASTn searches against the NCBI nonredundant (nr) database to confirm species identity. Table 2 lists the numbers of individuals in each species examined per locus. Mating‐type distributions and chemotype profiles for the global populations of A. flavus and A. parasiticus included in this study have been previously examined as well as field data relating to soil type, temperature and precipitation (Moore et al., 2013).
Table 1.
Species and total individual counts for each geographic region
| Argentina | Australia | Benin | India | Japan | USA | |
|---|---|---|---|---|---|---|
| A. alliaceus | 0 | 9 | 0 | 0 | 0 | 0 |
| A. caelatus | 80 | 0 | 0 | 0 | 0 | 31 |
| A. flavus L | 80 | 80 | 80 | 80 | 0 | 104 |
| A. flavus S | 4 | 80 | 44 | 0 | 0 | 26 |
| A. nomius | 0 | 0 | 0 | 0 | 0 | 33 |
| A. oryzae | 0 | 0 | 0 | 0 | 51 | 1 |
| A. parasiticus | 80 | 80 | 1 | 0 | 0 | 182 |
| A. sojae | 0 | 0 | 0 | 0 | 1 | 0 |
| A. tamarii | 0 | 6 | 80 | 56 | 0 | 35 |
| Total | 244 | 255 | 205 | 136 | 52 | 412 |
Table 2.
Species and total isolate and haplotype counts for each genomic region
| aflM/aflN | aflW/aflX | amdS | trpC | mfs | MAT1‐1 | MAT1‐2 | |
|---|---|---|---|---|---|---|---|
| A. alliaceus | 4 (1) | 5 (3) | 1 (1) | 1 (1) | 1 (1) | 2 (2) | 2 (2) |
| A. caelatus | n/a | 42 (12) | 40 (8) | 55 (9) | 46 (8) | 56 (5) | 19 (3) |
| A. flavus L | 346 (29) | 357 (17) | 352 (15) | 352 (13) | 351 (11) | 192 (2) | 178 (4) |
| A. flavus S | 90 (28) | 90 (15) | 89 (13) | 89 (9) | 89 (11) | 39 (4) | 48 (5) |
| A. nomius | 1 (1) | 19 (2) | 7 (4) | 9 (4) | 0 | 15 (9) | 24 (15) |
| A. oryzae | 34 (5) | 49 (12) | 34 (2) | 34 (3) | 34 (2) | 32 (3) | 7 (1) |
| A. parasiticus | 243 (24) | 245 (10) | 245 (9) | 255 (4) | 245 (16) | 208 (5) | 68 (1) |
| A. sojae | 1 (1) | 1 (1) | 1 (1) | 1 (1) | 1 (1) | 0 | 0 |
| A. tamarii | n/a | 35 (6) | 84 (11) | 66 (3) | 1 (1) | 36 (6) | 68 (6) |
| Total | 719 (89) | 844 (78) | 854 (64) | 862 (47) | 768 (51) | 580 (36) | 408 (37) |
The number of haplotypes is shown in parentheses.
2.2. Phylogenetic inference, principal component, and structure analysis
The GenBank accessions for sequences from each locus are listed by species in Tables [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link]. Mutliple DNA sequence alignment was performed using Sequencher® version 5.0 software (Gene Codes Corporation, Ann Arobor, MI, USA), and each alignment was refined by eye. The alignments were then exported in NEXUS format for use in SNAP Workbench (Monacell & Carbone, 2014; Price & Carbone, 2005). Multiple sequence alignments for each locus were collapsed into haplotypes using SNAP Map (Aylor, Price, & Carbone, 2006). Collapsing was performed with the options of recoding indels and excluding infinite sites violations. Indels were recoded as single unique events and weighted equally in phylogenetic analyses. Recoded indels that were not binary violated infinites sites and were excluded. Because our variation spans the population–species interface, collapsing in this fashion allowed us to take full advantage of indels arising within species and exclude only those indels that violate position homology between species. All but one of the alignments were rooted with A. nomius type strain NRRL 13137 as an outgroup species, an appropriate outgroup according to Peterson et al. (2008). Since NRRL 13137 was found to contain only the MAT1‐1 idiomorph based on genomic sequencing (Moore, Mack, & Beltz, 2015), a MAT1‐2 A. nomius isolate (IC1493) was used for the respective phylogeny. We performed heuristic parsimony searches in PAUP* 4.0 (Swofford, 1998) to infer phylogenies for all section Flavi species sampled, and if parsimony searches inferred more than one equally parsimonious tree, bootstrap values were calculated for branch length support. Bootstrap consensus trees were based on 1,000 replicates. Statistical support for specific clades in the tree was based on branches with bootstrap percentages greater than 70 (Hillis & Bull, 1993). For the amdS and trpC loci, a haplotype comparison was made through separate alignments (data not shown) between our sampled global species and Geiser's Groups I and II (Geiser et al., 1998). Briefly, Geiser and coworkers examined 11 genomic loci for an Australian A. flavus population and reported the existence of two reproductively isolated clades or groups that could not be delineated by geography or morphology.
Additional single‐locus studies, for the sampled global species in section Flavi, involved inference of maximum‐likelihood (ML) phylogenies using the program Randomized Axelerated Maximum Likelihood or RAxML (Stamatakis, 2014), and principal component analysis (PCA). These analyses were performed with the exclusion of indels and infinite sites violations from a DNA sequence alignment. PCA uses a variance–covariance matrix to reduce a sample into groupings of uncorrelated variables called principal components (Abdi & Williams, 2010). The components are known as eigenvectors and refer to different levels of variation. The first eigenvector considers the most variation possible with subsequent eigenvectors having less variation. Principal components were normalized to sum to 1, and the number of significant axes of variation (eigenvectors) was determined using the Tracy–Widom statistic (Tracy & Widom, 1994). The number of k clusters was estimated using the Gap Statistic (Tibshirani, Walther, & Hastie, 2001), which is an unbiased estimate of the number of distinct clusters in the population sample. Significant principal components and PCA clusters were displayed graphically using the SCATTERPLOT3D package in R (Ligges & Mächler, 2003). In a previous study, we showed evidence of balancing selecting in the aflW/aflX cluster region, which delimited two evolutionary distinct lineages: IB and IC. Lineage IB included only nonaflatoxigenic isolates while IC included both toxigenic and atoxigenic strains (Moore et al., 2009). To explore this partitioning of variation into lineages IB and IC on a global scale, and across multiple species in section Flavi, single‐locus phylogenies were inferred for the aflW/aflX region, as well as noncluster loci (amdS, trpC and MAT).
Multilocus analyses were performed for the A. flavus and A. parasiticus populations. The A. flavus isolates included both large sclerotium (L) and small sclerotium (S) morphotypes (Cotty, 1989). The six genomic loci were first concatenated using SNAP Combine (Aylor et al., 2006), and collapsed with SNAP Map having recoded indels and excluded infinite sites violations. Each geographic location was examined separately with focus on A. flavus and A. parasiticus isolates and their associated chemotype profiles. Phylogenies were displayed in circle tree format using FigTree v1.3.1 (http://tree.bio.ed.ac.uk/software/figtree/) or T‐BAS v2.0 (Carbone, unpublished), an extension of the T‐BAS v1.0 toolkit (Carbone et al., 2016). Clade groupings were labeled along the tree perimeter based on species or morphotype (e.g., A. flavus L, A. flavus S, A. parasiticus). The PCA scatter plots considered up to three eigenvectors (axes of variation). Additional PCA scatter plots were inferred for the global combined multilocus dataset to investigate possible associations of temperature, precipitation, or soil type with inferred clusters.
The optimal number of k clusters was also determined using STRUCTURE version 2.3.1 (Falush, Stephens, & Pritchard, 2003; Pritchard, Stephens, & Donnelly, 2000), which assigns individuals to clusters assuming a model of admixture and correlated allele frequencies. To achieve good mixing and convergence, we used an MCMC sampling strategy of 20,000 iterations after a burn‐in period of 20,000. Three simulations were performed for k ranging from 1 to 10 to assess convergence of log‐likelihood values using the “full search” method in CLUMPP v1.1.2 (Jakobsson & Rosenberg, 2007). Two methods, LnP(D) and delta K (Evanno, Regnault, & Goudet, 2005) implemented in Structure Harvester v0.6.93 (Dent & vonHoldt, 2012), were used to estimate the optimal number of k clusters. The results were visualized in T‐BAS v2.0 as outer rings of a multilocus global ML phylogeny. This holistic analysis combined results from PCA, STRUCTURE, and phylogenetic inference. The ML analysis was performed using RAxML version 8, which is accessible through the CIPRES RESTful application (CRA) programmer interface (Miller et al., 2015). Specifically, the multilocus sequence alignment file was subjected to a ML search strategy under the GTRGAMMA model of rate heterogeneity, and branch support values were based on rapid bootstrapping using 500 replicates; A. nomius (NRRL 13137) was specified as the outgroup. The best ML tree and associated locality and chemotype metadata were examined using T‐BAS v2.0 to look for evidence of population structuring based on geography and among closely related species (A. nomius, A. oryzae and A. sojae). We performed statistical analyses to determine how well individual cluster membership can predict chemotype using multiple linear regressions in R (R Development Core Team 2014) on the matrix of individual membership coefficients and different toxin chemotype concentrations, normalized between 0 and 1. Population structure was investigated by hierarchical analysis of molecular variance (AMOVA) and quantifying total variance within and between localities by F ST using Arlequin v.3.5.1.2 (Excoffier & Lischer, 2010). Significance of F ST estimates was determined using 1,000 permutations.
3. RESULTS
On a global scale, all geographic localities sampled showed some degree of genetic cohesion indicative of shared evolutionary history. This was evident by the sharing of haplotypes among strains from distant localities across six combined genomic loci (Figure 1). There was little genetic differentiation based on geography (fourth ring; Figure 1), except for a single clade of SBG from Benin (Figures 1 and 2). Lineage IB was represented in this analysis, but on a global scale it also included toxigenic A. flavus strains such as IC445 and IC457 from Argentina, as well as IC640, IC642, and IC672 from Australia. Also, several haplotypes and clades (Figure 1) were associated with specific chemotype profiles for A. parasiticus (e.g., B‐ or G‐dominance, OMST production). Structure analysis, using the Evanno method (first innermost ring; Figure 1) and the gap statistic (third ring), inferred two major genetic clusters that corresponded with A. parasiticus (blue‐colored branches) and A. flavus sensu lato (predominantly green‐colored branches); species boundaries were well defined, with only a small percentage (~5%) of isolates showing admixture (Table 3). Seven distinct clusters were inferred using the STRUCTURE LnP method (second innermost ring) and separated isolates belonging to different species/lineages and showing differences in mycotoxin production. The three outermost rings correspond to production of B aflatoxins (fifth ring), G aflatoxins (sixth ring), and OMST (seventh ring), respectively. Each individual was assigned a membership coefficient in the cluster, with coefficients summing to 1 across the seven clusters. Of the three toxin chemotype categories, G aflatoxin production was more strongly correlated (r 2 = .7220; p < .001; residual standard error: 0.2471) with the seven clusters than was B aflatoxin (r 2 = .2685; p < .001; residual standard error: 0.3801) or OMST (r 2 = .3363; p < .001; residual standard error: 0.221). About 72% of the variance found in G aflatoxin production can be explained by a strain's membership into one of the seven clusters, whereas only 33% of the variation (F ST = 0.3335, p < .001) was explained by variation between localities. This was also supported in PCA scatter plots in Figure 2; with the exception of a subset of isolates sampled in Benin, there was tight clustering of isolates from different localities.
Figure 1.
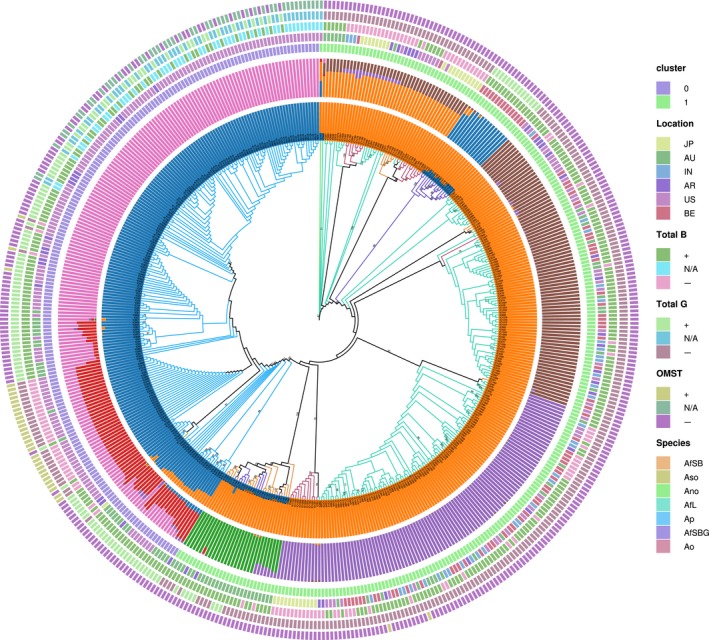
Graphical representation of the results from Structure analysis using the Evanno and Structure (LnP) methods, maximum‐likelihood phylogenetic analysis and principal component analysis (PCA). The circle tree represents the global phylogeny for six combined genomic regions (aflM/aflN, aflW/aflX, amdS, trpC, mfs, and MAT) for A. flavus (AfL, AfSB, AfSBG), A. nomius (Ano), A. oryzae (Ao), A. parasiticus (Ap), and A. sojae (Aso) populations. The innermost rings show the inferred clusters, using the Evanno method (k = 2; first ring) followed by the LnP method (k = 7; second ring), and the gap statistic (k = 2; third ring). The six geographical localities (JP, AU, IN, AR, US, and BE) are shown in the fourth ring. The three outermost rings show whether isolates are producing (+) or not producing (−) B aflatoxin (fifth ring), G aflatoxin (sixth ring) and OMST (seventh ring); N/A is for data not available
Figure 2.
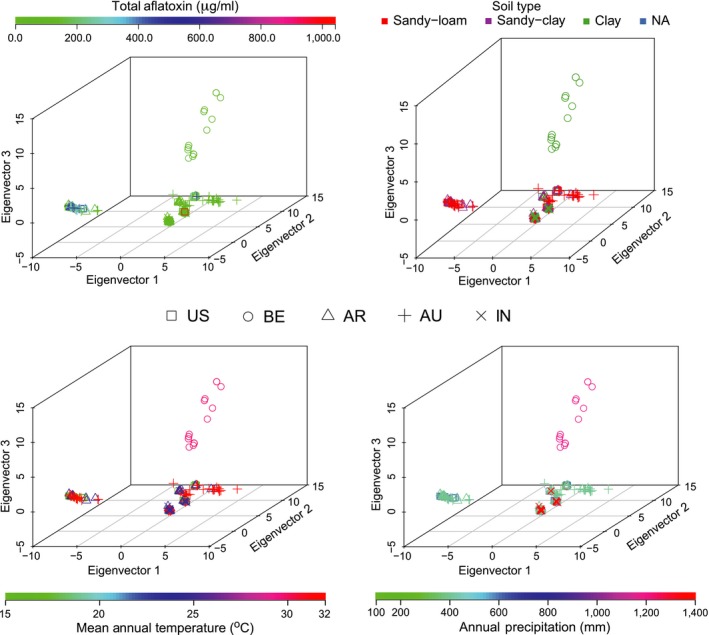
Principal component analysis (PCA) scatter plots for the global combined populations of A. flavus and A. parasiticus that represent associations based on four attributes: total aflatoxins, soil type, annual temperature, and annual precipitation. Each scatter plot's color scheme is unique to the particular attribute examined, and the different shapes relate to the geographic origin of each isolate
Table 3.
Isolates showing inferred admixture between genetic clusters of A. parasiticus and A. flavus, based on Structure analysis using the Evanno method in Figure 1
| Isolate | Species | Locality | Total B (μg/ml) | Total G (μg/ml) |
|---|---|---|---|---|
| IC73 | A. parasiticus | USA | 76 | 399.7 |
| IC157a | A. nomius | USA | + | + |
| IC328 | A. parasiticus | USA | 9 | 93.2 |
| IC329 | A. parasiticus | USA | 23.2 | 245 |
| IC330 | A. parasiticus | USA | 20.4 | 230.2 |
| IC331 | A. parasiticus | USA | 31 | 314 |
| IC490 | A. parasiticus | Argentina | 23.4 | 45.5 |
| IC517 | A. parasiticus | Argentina | 37.8 | 160 |
| IC642b | A. flavus L | Australia | 165.1 | 0 |
| IC477 | A. flavus SBG | Argentina | 2.3 | 6.2 |
| IC478 | A. flavus SBG | Argentina | 2 | 7.2 |
| IC494 | A. parasiticus | Argentina | 68.4 | 221.2 |
| IC526 | A. parasiticus | Argentina | 0.2 | 1.1 |
| IC720 | A. flavus SBG | Australia | 4.6 | 11.3 |
| IC723 | A. flavus SB | Australia | 77.1 | 0 |
| IC731 | A. flavus SBG | Australia | 1 | 1.3 |
| IC733 | A. flavus SBG | Australia | 1.3 | 2 |
| IC735 | A. flavus SBG | Australia | 1 | 1.6 |
| IC741 | A. flavus SB | Australia | 68.6 | 0 |
| IC742 | A. flavus SBG | Australia | 3.2 | 5.9 |
| IC743 | A. flavus SB | Australia | 23.9 | 0 |
| IC744 | A. flavus SBG | Australia | 8 | 17.6 |
| IC751 | A. flavus SB | Australia | 15 | 0 |
| IC753 | A. flavus SB | Australia | 34.4 | 0 |
| IC755 | A. flavus SB | Australia | 10 | 0 |
| IC758 | A. flavus SB | Australia | 6.9 | 0 |
| IC760 | A. flavus SB | Australia | 79 | 0 |
| IC768 | A. flavus SB | Australia | 14 | 0 |
| IC770 | A. flavus SB | Australia | 58.1 | 0 |
| IC777 | A. flavus SB | Australia | 12.9 | 0 |
| IC780 | A. flavus SB | Australia | 77.5 | 0 |
| IC785 | A. flavus SB | Australia | 34.9 | 0 |
| IC786 | A. flavus SB | Australia | 32 | 0 |
| IC787 | A. flavus SB | Australia | 27.7 | 0 |
| IC788 | A. flavus SBG | Australia | 0.4 | 0.3 |
| IC790 | A. flavus SB | Australia | 16.7 | 0 |
| IC791 | A. flavus SB | Australia | 13.9 | 0 |
| IC792 | A. flavus SB | Australia | 73.7 | 0 |
| IC793 | A. flavus SB | Australia | 0 | 0 |
| IC798 | A. flavus SB | Australia | 14.3 | 0 |
| IC806 | A. parasiticus | Australia | 53.5 | 278.1 |
| IC811 | A. parasiticus | Australia | 21.2 | 65.5 |
| IC813 | A. parasiticus | Australia | 19.1 | 52.2 |
| IC832 | A. parasiticus | Australia | 383.2 | 8.6 |
| IC836 | A. parasiticus | Australia | 22.1 | 60.5 |
| IC837 | A. parasiticus | Australia | 45.8 | 127.7 |
| IC839 | A. parasiticus | Australia | 107.4 | 223.9 |
| IC851 | A. parasiticus | Australia | 135.1 | 215.9 |
| IC860 | A. parasiticus | Australia | 15.3 | 44.3 |
| IC863 | A. parasiticus | Australia | 49.7 | 127.7 |
| IC867 | A. parasiticus | Australia | 115.5 | 92.2 |
| IC875 | A. parasiticus | Australia | 230.7 | 202.7 |
| IC876 | A. parasiticus | Australia | 17.4 | 53.5 |
| IC906 | A. parasiticus | USA | 37 | 222.5 |
| IC907 | A. parasiticus | USA | 65.7 | 420.5 |
| IC911 | A. parasiticus | USA | 16.5 | 172.8 |
| IC920 | A. parasiticus | USA | 84.7 | 361 |
| IC1112 | A. flavus SBG | Benin | 35.4 | 312.6 |
| IC1113 | A. flavus SBG | Benin | 11 | 13.7 |
| IC1117 | A. flavus SBG | Benin | 13.2 | 26.1 |
| IC1118 | A. flavus SBG | Benin | 14.4 | 18 |
| IC1119 | A. flavus SBG | Benin | 13.4 | 18.8 |
| IC1133 | A. flavus SBG | Benin | 11.8 | 14.6 |
| IC1134 | A. flavus SBG | Benin | 12.6 | 15.8 |
| IC1140 | A. flavus SBG | Benin | 4.8 | 18 |
| IC1141 | A. flavus SBG | Benin | 12.9 | 16.8 |
| IC1142 | A. flavus SBG | Benin | 47.1 | 117.7 |
| IC1144 | A. flavus SBG | Benin | 15.1 | 30.8 |
| IC1145 | A. flavus SBG | Benin | 15.8 | 12.5 |
| IC1147 | A. flavus SBG | Benin | 14.1 | 9.2 |
| IC1149 | A. flavus SBG | Benin | 18.9 | 40.6 |
| IC1150 | A. flavus SBG | Benin | 21.6 | 48.1 |
| IC1215a | A. sojae | Japan | – | – |
| IC1258 | A. flavus L | India | 10.7 | 0 |
No aflatoxin quantification data acquired. Strain is designated aflatoxin positive (+) or negative (−).
Isolate produces G1 < 0.5 μg/ml.
A single most parsimonious tree for each locus is illustrated in Figs [Link], [Link], [Link], [Link]. Haplotype designations for each phylogeny are found in supplemental Tables [Link], [Link], [Link], [Link], [Link], [Link], [Link]. Phylogenies inferred for noncluster loci amdS and trpC show well‐supported clades that are associated with species or morphotype. Shared haplotypes between species from different geographic regions suggest recent descent from a common ancestor prior to geographic isolation, or alternatively, extensive migration between localities. For amdS (Fig. S1; S10), there is strong bootstrap support (>75%) for many nodes; however, clades are not monophyletic and not associated with a single geographic region. For example, haplotype H12 includes a mix of A. alliaceus, A. caelatus, A. flavus L and S, A. oryzae and A. tamarii isolates from different localities. Although there was no evidence of geographic structure, some haplotypes based on species or morphotype, such as A. flavus S haplotypes H48‐H53, were associated only with Argentina and Australia and included a mix of AFB or AFB+G producers. With the exception of an Australian A. flavus SBG isolate (H5) and an Australian A. flavus L isolate (H42), global A. parasiticus is monophyletic.
Isolates associated with Geiser's Groups I and II are dispersed throughout the amdS phylogeny in haplotypes H12, H20, and H25, and haplotypes H9, H49, and H50, respectively. Haplotype H12 includes multiple species, while H20 and H25 include only A. flavus L strains. Haplotype H20 includes lineage IC isolates such as the AF36 biocontrol strain and NRRL 3357, while H12 encompasses isolates from both IC and IB lineages. Multiple geographic origins are represented in each haplotype. Although all of our Geiser's Group II isolates were of the S morphotype, their chemotype profiles were a mixture of AFB and AFB+G. All but one of the isolates, IC477, represented in Group II haplotypes are from Australia. Isolate IC477 (H49) is an AFB+G strain from Argentina, whereas haplotype H9 is solely comprised of SB stains.
The trpC phylogeny (Fig.S1; Table S11) supports the monophyly of species in Aspergillus section Flavi (Peterson et al., 2008). Four main clades were observed: A. flavus, A. parasiticus, A. caelatus with A. tamarii, and A. nomius. Though there is evidence of shared haplotypes within the A. flavus and A. parasiticus clades (haplotypes H9 and H30, respectively); overall, species groupings are upheld. The trpC Geiser Group I haplotypes are spread throughout the A. flavus clade and includes isolates of A. flavus L and S, A. oryzae and A. caelatus. Isolates associated with Geiser's Group II were found in only a single haplotype (H15), and are comprised of A. flavus S‐strains from Australia and Argentina, with AFB and AFB+G chemotypes.
Although highly divergent when compared to each other, the MAT1‐1 and MAT1‐2 idiomorphs are highly conserved on the DNA and amino acid levels when examined separately, both within and between species (Ramirez‐Prado et al., 2008). This was not the case for the MAT loci in A. nomius (Fig. S2; Tables S12 and S13), which showed elevated haplotype diversity; there were five haplotypes inferred for 15 MAT1‐1 isolates and fourteen haplotypes for 25 MAT1‐2 isolates. Moreover, A. nomius harbors both MAT idiomorphs within a single genome for seven of the 32 (22%) sampled isolates. Nine of the 22 haplotypes (41%) for the MAT1‐1 phylogeny, and 15 of the 30 haplotypes (50%) for the MAT1‐2 phylogeny include A. nomius isolates. Both mating‐type phylogenies show evidence of trans‐speciation, and there is little evidence of clades that are structured geographically. An Australian A. alliaceus isolate (IC892) appears most divergent from other sampled species for both MAT1‐1 (H4) and MAT1‐2 (H17). This isolate may be homothallic since it has both MAT loci, whereas Australian A. allicaeus isolates IC888 (H7; MAT1‐1) and IC894 (H21; MAT1‐2) appear to have only one MAT locus present in their genomes and are likely heterothallic.
The mfs phylogeny (Fig. S2; Table S14) shows three major clades with strong bootstrap support: A. parasiticus (top), A. caelatus (middle), and A. flavus (bottom). This is concordant with the species tree inferred from the trpC locus. Although clades show extensive geographic mixing, haplotypes H43‐H46 representing a subset of Benin A. flavus SBG strains are well differentiated from the rest of the global sample. The only two SB isolates sampled in Benin share haplotypes (H15 and H31) with A. flavus L strains. A second well‐supported group of A. flavus S isolates includes haplotypes H23, H24, and H26, which are a mixture of SB and SBG from more than one locality. Haplotype H2 includes an Argentinian A. caelatus isolate among global A. parasiticus individuals. This same A. caelatus isolate (IC568) grouped with a haplotype that includes mostly A. flavus for amdS (H12 in Fig. S1; Table S10). There was some evidence of chemotype/morphotype admixture with an Australian SBG isolate sharing haplotype H15 with global L and SB strains. Other mfs haplotypes such as H2, H17, and H36 may harbor chemotype‐specific differences that are maintained via trans‐speciation.
Phylogenetic inference for the cluster locus, aflM/aflN (Fig. S3; Table S15), indicates that species are not as monophyletic as observed in some of the noncluster loci. Haplotype groupings do exist that show geographic and species differentiation such as A. flavus SBG from Benin (H55, H58‐H61). Although not well supported, this clade of SBG from Benin shares a recent common ancestor with another group of predominantly SB isolates from different localities (H2‐H8) and includes isolates that associate with Geiser's Group II. Isolate IC793 (H6) is nonaflatoxigenic which is considered rare for A. flavus S‐strains (Horn & Dorner, 1999). Other haplotypes contain isolates that associate with Geiser's Group I (H10 and H39) and Group II (H78). There is also evidence of trans‐speciation in aflM/aflN, which has been reported previously (Carbone et al., 2007). For example, in haplotype H39, an A. parasiticus isolate from Argentina groups with global A. flavus isolates. Interestingly, haplotype H47 includes the AF36 biocontrol strain, and despite being sampled from different geographic regions (Arizona, USA; Texas, USA; Karnataka, India), all four of the nonaflatoxigenic strains in this haplotype have the same nonsense mutation in aflC (data not shown), which suggests dispersal that transcends geographic boundaries as reported in other studies (Grubisha & Cotty, 2015; Ortega‐Beltran, Grubisha, Callicott, & Cotty, 2016). Moreover, many nonaflatoxigenic A. flavus L isolates, from various localities, group with A. oryzae. More than 90% of these nonaflatoxigenic isolates belong to Lineage IB (Moore et al., 2009). Nestled among these haplotypes was a group of B‐ and G‐dominant A. parasiticus isolates (H31‐H36) from various localities.
The aflW/aflX locus (Fig. S4; Table S16) appears more concordant with the noncluster loci in that there is evidence of grouping based on species and/or geography. There are clades that are predominantly A. flavus L, A. parasiticus, A. flavus S, and a backbone of A. oryzae that separates most of the sample from a second A. flavus clade, which includes Lineage IB isolates. Many of the haplotypes for this region of the aflatoxin cluster show evidence of trans‐speciation (Table S16) such that some isolates of nonaflatoxigenic A. caelatus, A. tamarii, A. oryzae and A. alliaceus are distributed across haplotypes shared with A. flavus (H20, H29, H34, H36, H40, H41, H48, H50) or A. parasiticus (H5, H8). Eleven of the 55 haplotypes harbor trans‐specific polymorphisms. Haplotypes encompassing isolates that associate with Geiser's Group I are H40, H50 and H53, while haplotypes H19‐H22 include isolates that associate with Group II. The single‐locus RAxML analysis of the aflW/aflX region (Fig. S5) shows that clades are predominantly lineage‐specific rather than species‐ or geography‐specific. This is illustrated through the mixed branch colors in the tree. The branches that include Lineage IB isolates are labeled along the tree's perimeter and include species other than A. flavus. For example, A. caelatus, A. nomius and A. oryzae are each split into two separate lineages. PCA for the aflW/aflX region (Fig. S5) for the global sample suggests seven distinct clusters that do not align with species or geography designations. The Lineage IB cluster is encircled and is not entirely comprised of nonaflatoxigenic isolates. For the amdS and trpC loci (Fig. S5), the RAxML phylogenies are concordant with the species tree. These species are not as clearly delimited in the PCA scatter plots of amdS and trpC for which k‐values indicate the presence of six and seven distinct clusters, respectively. There was limited resolution of species boundaries in the MAT1‐1 mating‐type gene in both the maximum‐likelihood phylogeny and PCA scatter plots (Fig. S6). The k‐value of four includes one cluster of A. alliaceus from Australia, a disperse cluster containing A. nomius and A. tamarii from the USA and Benin, a cluster containing A. caelatus from the USA and Argentina, and a fourth cluster containing a mixture of species from various geographic localities. Similarly for the MAT1‐2 mating gene (Fig. S7), the maximum‐likelihood tree and PCA scatter plots show limited resolution of species. The PCA results only reveal two unique clusters, grouping a genetically distinct strain of A. alliaceus from Australia with an outlier strain of A. tamarii from India; the remaining isolates comprise the second cluster.
Multilocus maximum‐likelihood tree and PCA scatter plot comparisons of A. flavus and A. parasiticus populations within each geographic region can be seen in Figs [Link], [Link], [Link], [Link], [Link]. For the US population (Fig. S8), some clustering is observed based on species/morphotype and chemotype profile. In each PCA scatter plot, a k‐value of seven indicates there is an additional cluster being masked by one of the six observed clusters. Some of these clusters are species‐specific, but not necessarily field‐specific (top scatter plot) or chemotype‐specific (bottom scatter plot). Figure S9 illustrates the maximum‐likelihood tree and PCA scatter plot results for the Argentina populations. Species/morphotype delimitation is more easily discernible than chemotype grouping in the maximum‐likelihood phylogeny as well as in the PCA scatter plots that have a k‐value of three. The first principal component (eigenvector 1) indicated that the A. flavus L and SB isolates share a cluster, with SBG and A. parasiticus isolates comprising the second and third clusters, respectively. Based on the second principal component (eigenvector 2), three possible clusters are discernible, except the A. flavus L cluster is subdivided and a portion of them are loosely clustered with the SB, SBG and A. parasiticus strains. In Fig. S10, there is evidence of strong species/morphotype structure within the Australian maximum‐likelihood phylogeny, although there is also evidence of admixture of different chemotypes in each clade. The PCA for the Australian populations of A. flavus and A. parasiticus in this figure suggests a k‐value of two and indicates a degree of similarity among A. flavus morphotypes (L‐ and S‐strains) that loosely groups the two morphotypes while separating them from A. parasiticus. The second principal component (eigenvector 2) for this group of isolates indicates subdivision in A. flavus L, in which some isolates group more closely with SB, SBG and A. parasiticus. The Benin ML tree mainly shows clades associated with chemotype profile (Fig. S11). This is supported by PCA and a k‐value of six despite all of the isolates belonging to A. flavus. Most apparent is the subdivision of A. flavus SBG isolates into multiple groupings. Two of the clusters are each a mixture of A. flavus L and SB isolates; the other four clusters are comprised of one or more A. flavus SBG isolates. The ML tree for the India A. flavus population (Fig. S12) is only represented by a single morphotype (L‐strains), and clades are associated with chemotype profiles being either aflatoxin positive or negative. PCA scatter plots indicate many distinct groupings of isolates, but there is not enough variation for reliable estimation of k value.
4. DISCUSSION
This study focused on species in Aspergillus section Flavi that were present in peanut fields at the time of sampling (2004–2006). Although other species have since been described (e.g., A. minisclerotigenes, A. arachidicola, A. mottae; see Pildain et al., 2008; Soares, Rodrigues, Peterson, & Venâncio, 2012), these were not sampled from the peanut fields examined in this study, nor did comprehensive BLASTn searches of NCBI databases reveal close matches to these other species. Our inferences were therefore limited to species that were sampled in peanut fields. The results from this expanded global sample of species in Aspergillus section Flavi confirms and strengthens inferences of population processes in the aflatoxin gene cluster of these agriculturally important species. First, we see widespread evidence of balancing selection acting on G1‐ and B1‐dominant A. parasiticus chemotypes in both the aflM/aflN and aflW/aflX regions. Previously this was detected only in the aflM/aflN (hypE) locus for the Georgia A. parasiticus population (Carbone et al., 2007). Similarly, there is global evidence for Lineage IB (predominantly nonaflatoxigenic) in A. flavus that was first inferred from this same Georgia field (Horn & Greene, 1995; Moore et al., 2009). It appears that these patterns of trans‐speciation for specific chemotype profiles transcend not only species but also geographic boundaries. Seven distinct genetic clusters were more strongly structured by G chemotype diversity than geography. Although there can be other factors that underlie these seven clusters, chemotype‐specific evolutionary lineages were observed in previous studies (Carbone et al., 2007; Moore et al., 2009). Whether these chemotype‐specific lineages have increased pathogenicity or enhanced fitness for some other trait or ecological condition is unknown and warrants further study.
Climate has been reported to influence toxin production and population size of A. flavus S in the USA (Bock, Mackey, & Cotty, 2004; Cotty & Jaime‐Garcia, 2007). Although reports of A. flavus S‐strains in the USA producing G aflatoxins (SBG) are rare, they are common elsewhere (Cotty & Cardwell, 1999). For example, the Benin population we examined included SBG isolates; however, no A. parasiticus isolates were sampled in our Benin field. With regard to populations of aflatoxin‐producing fungi, species that produce both B and G aflatoxins such as A. parasiticus and A. nomius are seldom found in certain localities (Cotty & Cardwell, 1999). Given that recombination in fungi can occur during times of high stress (Meng et al., 2015), one possibility is that A. parasiticus found its way out of an inhospitable soil environment, through hybridization with more adaptable A. flavus. However, attempting to prove this hypothesis would require more extensive sampling and analyses. The observation that A. flavus SBG isolates from Benin, which may or may not be related to the unnamed taxon SBG isolated from Nigerian groundnut in the 1960s (Probst, Callicott, & Cotty, 2012), are genetically distinct from other A. flavus populations sampled could indicate that this subpopulation is under strong selection and possibly evolving into its own distinct lineage. PCA analysis suggests that there could be further splitting of the Benin A. flavus SBG isolates into multiple groups (Figure 2), each associated with a specific chemotype profile; this is unknown and warrants further investigation. Egel, Cotty, and Elias (1994) reported that A. flavus SB are closely related to the A. flavus L‐type strain based on molecular variation, suggesting a possible common ancestor between SB and A. flavus L. The high sequence diversity observed within the MAT locus among A. nomius isolates sampled in the USA could relate to multiple A. nomius lineages as reported by Ehrlich et al. (2007) among isolates from Thailand.
Collectively, molecular sequence data in cluster and noncluster loci indicate a potential hybrid origin for some strains. For example, analysis of the amdS locus showed Australian SBG isolate IC721 sharing a haplotype with A. flavus L and SB isolates, while another Australian SBG isolate, IC744, shared a haplotype with A. parasiticus isolates based on the mfs locus. As the other loci examined were distinct in these SBG strains it is plausible that the mfs locus was inherited from one or the other parent species during an interspecific recombination event. Alternatively, the similarity between A. flavus and A. parasiticus could be the result of balancing selection, which has been reported in the aflatoxin gene cluster (Carbone et al., 2007). Although mfs is adjacent to the aflatoxin cluster, it does not show the level of variation observed in cluster genes (Moore et al., 2009). The apparent similarity between these two species at the mfs locus may be the result of balancing selection acting on cluster genes that sweeps variation out of adjacent linked regions (Charlesworth, 2006).
Considering the chemotype profiles of the sampled Australian SBG strains, 12 of the 26 examined strains also produce measurable quantities of O‐methylsterigmatocystin (OMST). This toxic secondary metabolite is an intermediate in aflatoxin synthesis, that is, often secreted by nonaflatoxigenic A. parasiticus strains (Bhatnagar et al., 1987) and further hints at possible interspecific hybridization among the A. flavus and A. parasiticus populations in Australia. The accumulation of OMST and its precursor sterigmatocystin (ST) has been reported in Aspergillus and allied fungi (Rank et al., 2011). While these and other secondary metabolites have been useful for species delimitation (Frisvad, Andersen, & Thrane, 2008) the mechanisms generating new chemotypes are less clear. Olarte et al. (2015) showed that A. flavus and A. parasiticus, when crossed in the laboratory, yielded offspring that exhibited chemotypes representative of both parent species, such as aflatoxin (A. flavus parent) and OMST production (A. parasiticus parent). The PCA scatter plots for Argentina (Fig. S9) and Australia (Fig. S10) show some proximity of SBG isolates to a subset of the A. flavus L, SB and A. parasiticus populations along the second principal component (eigenvector 2), which could be the result of interspecific recombination, a hypothesis which warrants further investigation. The discrepancy in the number of sampled SBG strains between these two localities relates to differences in population sizes as seen in their mating‐type distributions. The Argentina populations are disproportionately clonal and dominated by mating‐type MAT1‐1, while the Australian populations are predominantly sexual and have an approximately 1:1 distribution of MAT1‐1 and MAT1‐2 (Moore et al., 2013).
Our results indicate the existence of genetic exchange and recombination between distinct morphotypes (A. flavus L, A. flavus SB, A. flavus SBG), evolutionary lineages (IB and IC) and species (e.g., A. flavus and A. parasiticus). For example, it has been suggested that the AF36 biocontrol strain is a recombinant that resulted from the hybridization of parents representing both A. flavus L and S morphotypes (Chang et al., 2012). We observed multiple isolates, from different localities, that share the same nonsense mutation found in the aflC locus of AF36; however, we have not determined whether they are recombinants in their aflatoxin clusters. Multilocus genotyping revealed toxigenic strains that group with Lineage IB strains. These strains could be recombinant offspring between Lineages IB and IC that have genomic signatures of IB, and have inherited aflatoxigenicity. Further sampling and examining more genomic regions are necessary to understand the origins of toxigenic stains in Lineage IB. Some of the aflatoxigenic isolates that share the signature of Lineage IB in their aflW locus produce high concentrations of aflatoxin. This “super‐producer” toxin phenotype might result when the nonaflatoxigenic phenotype is repaired via crossover recombination or independent assortment. One of these high‐producing strains, IC642 from Australia, is an A. flavus L‐strain that shares a haplotype with A. parasiticus isolates. This isolate, as well as other aflatoxigenic strains from Australia and Argentina, may have resulted from interspecific hybridization between A. flavus and A. parasiticus. The possibility of intra‐ and inter‐specific genetic exchange as driving forces for adaptive evolution and speciation in Aspergillus section Flavi merits further investigation.
5. CONCLUSIONS
There is widespread evidence of balancing selection acting on the presence or absence of aflatoxin‐specific chemotypes, which can be useful in delimiting species and population boundaries in Aspergillus section Flavi. For example, A. flavus SBG strains in Benin are genetically and chemotypically distinct from A. flavus SB strains in the US. A signature of trans‐speciation was observed in A. flavus L‐strains with the maintenance of nontoxigenic and toxigenic strains that belong to Lineages IB and IC, respectively. The predominance of A. oryzae in lineage IB supports the distinctiveness of these two evolutionary lineages. Previously we provided evidence of balancing selection acting on G1‐dominant strains in A. parasiticus (Carbone et al., 2007). We now show that balancing selection on G production is a major driver of species diversification in section Flavi. While recombination could explain differences in chemotype profiles, clonality and environmental conditions could also be important in maintaining diversity. A better understanding of this genotype by chemotype by environment interactions has implications not only in species delimitation but also in biological control using nonaflatoxigenic strains of A. flavus.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
GGM performed laboratory bench work, PCR and sequencing of genomic loci, clean‐up and preparation of genomic sequences and performed population analyses, drafted the manuscript, and created tables and figures for publication. RAO assisted with population analyses, assisted with manuscript drafting, and addressing reviewer concerns. BWH acquired, verified the species identities of, and shared fungal isolates, and served as a proof‐reader for the manuscript. JLE assisted with the laboratory bench work, as well as PCR and sequencing of genomic loci. RS assisted with the laboratory bench work, PCR and sequencing of genomic loci. CJO assisted with the laboratory bench work, PCR and sequencing of genomic loci. IC helped develop the population analysis software, assisted with population analyses, helped draft the manuscript and address reviewer concerns.
DATA ACCESSIBILITY
DNA Sequences: GanBank accessions DQ390825‐DQ391160; FJ871513‐FJ878506; KX853136‐KX853999; HM353299‐HM355372; HM745598‐HM745901; HQ000095‐HM002855.
Supporting information
ACKNOWLEDGMENTS
Funding is from the North Carolina Cooperative State Research, Education and Extension Service, grant nos. 2008‐34500‐19396 and 2010‐34500‐21676 and the National Research Initiative of the USDA Cooperative State Research, Education and Extension Service, grant no. 2005‐35319‐16126. This project was also supported by the Agriculture and Food Research Initiative Competitive Grants Program grant no. 2013‐68004‐20359 from the USDA National Institute of Food and Agriculture (NIFA). We also thank the National Science Foundation's Dimensions of Biodiversity (DoB) Program for financial support, DEB‐1046167, to I. Carbone. Development of T‐BAS v2.0 and SNAP Workbench is supported by the National Science Foundation (NSF) Genealogy of Life (GoLife) Program to IC (DEB‐1541418). This work was also supported in part by the University of North Carolina General Administration under an award for High Performance Computing (HPC) and Computational Sciences. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Moore GG, Olarte RA, Horn BW, et al. Global population structure and adaptive evolution of aflatoxin‐producing fungi. Ecol Evol. 2017;7:9179–9191. https://doi.org/10.1002/ece3.3464
REFERENCES
- Abbas, H. K. , Wilkinson, J. R. , Zablotowicz, R. M. , Accinelli, C. , Abel, C. A. , Bruns, H. A. , … Weaver, M. A. (2009). Ecology of Aspergillus flavus, regulation of aflatoxin production, and management strategies to reduce aflatoxin contamination of corn. Toxin Reviews, 28, 142–153. [Google Scholar]
- Abdi, H. , & Williams, L. J. (2010). Principal component analysis. WIREs Computational Statistics, 2, 433–459. [Google Scholar]
- Ahmad, S. K. , & Singh, P. L. (1994). Distribution of Aspergillus flavus in soil and air of agricultural fields. Indian Phytopathology, 47, 81–86. [Google Scholar]
- Ali, A. , & Roossinck, M. J. (2008). Genetic bottlenecks In Roossinck M. J. (Ed.), Plant virus evolution (pp. 123–131). Berlin Heidelberg: Springer‐Verlag. [Google Scholar]
- Aylor, D. L. , Price, E. W. , & Carbone, I. (2006). SNAP: Combine and Map modules for multilocus population genetic analysis. Bioinformatics, 22, 1399–1401. [DOI] [PubMed] [Google Scholar]
- Bennett, J. W. (2010). An overview of the genus Aspergillus In Machida M., & Gomi K. (Eds.), Aspergillus: Molecular biology and genomics (pp. 1–17). Wymondham, UK: Caister Academic. [Google Scholar]
- Bennett, J. W. , & Klich, M. A. (2003). Mycotoxins. Clinical Microbiology Reviews, 16, 497–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar, D. , McCormick, S. P. , Lee, L. S. , & Hill, R. A. (1987). Identification of O‐methylsterigmatocystin as an aflatoxin B1 and G1 precursor in Aspergillus parasiticus . Applied and Environmental Microbiology, 53, 1028–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock, C. H. , Mackey, B. , & Cotty, P. J. (2004). Population dynamics of Aspergillus flavus in the air of an intensively cultivated region of south‐west Arizona. Plant Pathology, 53, 422–433. [Google Scholar]
- Carbone, I. , Jakobek, J. L. , Ramirez‐Prado, J. H. , & Horn, B. W. (2007). Recombination, balancing selection and adaptive evolution in the aflatoxin gene cluster of Aspergillus parasiticus . Molecular Ecology, 16, 4401–4417. [DOI] [PubMed] [Google Scholar]
- Carbone, I. , White, J. B. , Miadlikowska, J. , Arnold, A. E. , Miller, M. A. , Kauff, F. , … Lutzoni, F. (2016). T‐BAS: Tree‐Based Alignment Selector toolkit for phylogenetic‐based placement, alignment downloads, and metadata visualization: An example with the Pezizomycotina tree of life. Bioinformatics, 33, 1160–1168. [DOI] [PubMed] [Google Scholar]
- CDC (2016). Health Studies Branch. Understanding Chemical Exposures: Aflatoxin. Retrieved from http://www.cdc.gov/nceh/hsb/chemicals/aflatoxin.htm (accessed November 2, 2016).
- Chang, P.‐K. , Abbas, H. K. , Weaver, M. A. , Ehrlich, K. C. , Scharfenstein, L. L. , & Cotty, P. J. (2012). Identification of genetic defects in the atoxigenic biocontrol strain Aspergillus flavus K49 reveals the presence of a competitive recombinant group in field populations. International Journal of Food Microbiology, 154, 192–196. [DOI] [PubMed] [Google Scholar]
- Charlesworth, D. (2006). Balancing selection and its effects on sequences in nearby genome regions. PLoS Genetics, 2, e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland, T. E. , Yu, J. J. , Federova, N. , Bhatnagar, D. , Payne, G. A. , Nierman, W. C. , & Bennett, J. W. (2009). Potential of Aspergillus flavus genomics for applications in biotechnology. Trends in Biotechnology, 27, 151–157. [DOI] [PubMed] [Google Scholar]
- Cotty, P. J. (1989). Virulence and cultural characteristics of two Aspergillus flavus strains pathogenic on cotton. Phytopathology, 79, 808–814. [Google Scholar]
- Cotty, P. J. , & Cardwell, K. F. (1999). Divergence of West African and North American communities of Aspergillus section Flavi. Applied and Environmental Microbiology, 65, 2264–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotty, P. J. , & Jaime‐Garcia, R. (2007). Influences of climate on aflatoxin producing fungi and aflatoxin contamination. International Journal of Food Microbiology, 119, 109–115. [DOI] [PubMed] [Google Scholar]
- Dent, E. A. , & vonHoldt, B. M. (2012). STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genetics Resources, 4(2), 359–361. [Google Scholar]
- Egel, D. S. , Cotty, P. J. , & Elias, K. S. (1994). Relationships among isolates of Aspergillus sect. Flavi that vary in aflatoxin production. Phytopathology, 84, 906–912. [Google Scholar]
- Ehrlich, K. C. , Chang, P.‐K. , Yu, J. , & Cotty, P. J. (2004). Aflatoxin biosynthesis cluster gene cypA is required for G aflatoxin formation. Applied and Environmental Microbiology, 70, 6518–6524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich, K. C. , Kobbeman, K. , Montalbano, B. G. , & Cotty, P. J. (2007). Aflatoxin‐producing Aspergillus species from Thailand. International Journal of Food Microbiology, 114, 153–159. [DOI] [PubMed] [Google Scholar]
- Evanno, G. , Regnault, S. , & Goudet, J. (2005). Detecting the number of clusters of individuals using the software structure. A simulation study. Molecular Ecology, 14, 2611–2620. [DOI] [PubMed] [Google Scholar]
- Excoffier, L. , & Lischer, H. E. L. (2010). Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 10, 564–567. [DOI] [PubMed] [Google Scholar]
- Falush, D. , Stephens, M. , & Pritchard, J. K. (2003). Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics, 164, 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisvad, J. C. , Andersen, B. , & Thrane, U. (2008). The use of secondary metabolite profiling in chemotaxonomy of filamentous fungi. Mycological Research, 112, 231–240. [DOI] [PubMed] [Google Scholar]
- Geiser, D. M. , Pitt, J. I. , & Taylor, J. W. (1998). Cryptic speciation and recombination in the aflatoxin‐producing fungus Aspergillus flavus . Proceedings of the National Academy of Science USA, 95, 388–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubisha, L. C. , & Cotty, P. J. (2015). Genetic analysis of the Aspergillus flavus vegetative compatibility group to which a biological control agent that limits aflatoxin contamination in US crops belongs. Applied and Environmental Microbiology, 81, 5889–5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick, P. W. (2007). Balancing selection. Current Biology, 17, R230–R231. [DOI] [PubMed] [Google Scholar]
- Hillis, D. M. , & Bull, J. J. (1993). An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. Systematic Biology, 42, 182–192. [Google Scholar]
- Horn, B. W. (2003). Ecology and population biology of aflatoxigenic fungi in soil. Journal of Toxicology‐Toxin Reviews, 22, 351–379. [Google Scholar]
- Horn, B. W. (2007). Biodiversity of Aspergillus section Flavi in the United States: A review. Food Additives and Contaminants, 24, 1088–1101. [DOI] [PubMed] [Google Scholar]
- Horn, B. W. , & Dorner, J. W. (1999). Regional differences in production of aflatoxin B1 and cyclopiazonic acid by soil isolates of Aspergillus flavus along a transect within the United States. Applied and Environmental Microbiology, 65, 1444–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn, B. W. , Gell, R. M. , Singh, R. , Sorensen, R. B. , & Carbone, I. (2016). Sexual reproduction in Aspergillus flavus sclerotia: Acquisition of novel alleles from soil populations and uniparental mitochondrial inheritance. PLoS ONE, 11, e0146169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn, B. W. , & Greene, R. L. (1995). Vegetative compatibility within populations of Aspergillus flavus, A. parasiticus, and A. tamarii from a peanut field. Mycologia, 87, 324–332. [Google Scholar]
- Horn, B. W. , Moore, G. G. , & Carbone, I. (2011). Sexual reproduction in aflatoxin‐producing Aspergillus nomius . Mycologia, 103, 174–183. [DOI] [PubMed] [Google Scholar]
- Horn, B. W. , Moore, G. G. , & Carbone, I. (2009). Sexual reproduction in Aspergillus flavus . Mycologia, 101, 423–429. [DOI] [PubMed] [Google Scholar]
- Horn, B. W. , Ramirez‐Prado, J. H. , & Carbone, I. (2009a). Sexual reproduction and recombination in the aflatoxin‐producing fungus Aspergillus parasiticus . Fungal Genetics and Biology, 46, 169–175. [DOI] [PubMed] [Google Scholar]
- Horn, B. W. , Ramirez‐Prado, J. H. , & Carbone, I. (2009b). The sexual state of Aspergillus parasiticus . Mycologia, 101, 275–280. [DOI] [PubMed] [Google Scholar]
- Jaime‐Garcia, R. , & Cotty, P. J. (2006). Spatial relationships of soil texture and crop rotation to Aspergillus flavus community structure in South Texas. Phytopathology, 96, 599–607. [DOI] [PubMed] [Google Scholar]
- Jakobsson, M. , & Rosenberg, N. A. (2007). CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics, 23, 1801–1806. [DOI] [PubMed] [Google Scholar]
- Klich, M. A. (2007). Aspergillus flavus: The major producer of aflatoxin. Molecular Plant Pathology, 8, 713–722. [DOI] [PubMed] [Google Scholar]
- Ligges, U. , & Mächler, M. (2003). Scatter plot3d ‐ an R Package for Visualizing Multivariate Data. Journal of Statistical Software, 8, 1–20. [Google Scholar]
- May, G. , Shaw, F. , Badrane, H. , & Vekemans, X. (1999). The signature of balancing selection: Fungal mating compatibility gene evolution. Proceedings of the National Academy of Science USA, 96, 9172–9177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng, J.‐W. , Zhu, W. , He, M.‐H. , Wu, E.‐J. , Duan, G.‐H. , Xie, Y.‐K. , … Zhan, J. (2015). Population genetic analysis reveals cryptic sex in the phytopathogenic fungus Alternaria alternata . Scientific Reports, 5, 18250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micali, O. C. , & Smith, M. L. (2005). Biological concepts of vegetative self and nonself recognition in fungi In Xu J. (Ed.), Evolutionary genetics of fungi (pp. 63–86). Wymondham, UK: Horizon Bioscience. [Google Scholar]
- Michielse, C. B. , Ram, A. F. J. , & van den Hondel, C. A. (2004). The Aspergillus nidulans amdS gene as a marker for the identification of multicopy T‐DNA integration events in Agrobacterium‐mediated transformation of Aspergillus awamori . Current Genetics, 45, 399–403. [DOI] [PubMed] [Google Scholar]
- Milgroom, M. G. (1996). Recombination and the multilocus structure of fungal populations. Annual Review of Phytopathology, 34, 457–477. [DOI] [PubMed] [Google Scholar]
- Miller, M. A. , Schwartz, T. , Pickett, B. E. , Miller, M. A. , Schwartz, T. , Pickett, B. E. , He, S. , Klem, E. B. , Scheuermann, R. H. , … O'Leary, M. A. . (2015). A RESTful API for access to phylogenetic tools via the CIPRES science gateway. Evolutionary Bioinformatics, 11, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monacell, J. T. , & Carbone, I. (2014). Mobyle SNAP Workbench: A web‐based analysis portal for population genetics and evolutionary genomics. Bioinformatics, 30, 1488–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, G. G. , Elliott, J. L. , Singh, R. , Horn, B. W. , Dorner, J. W. , Stone, E. A. , … Carbone, I. (2013). Sexuality generates diversity in the aflatoxin gene cluster: Evidence on a global scale. PLoS Pathogens, 9, e1003574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, G. G. , Mack, B. M. , & Beltz, S. B. (2015). Genomic sequence of the aflatoxigenic filamentous fungus Aspergillus nomius . BMC Genomics, 16, 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, G. G. , Singh, R. , Horn, B. W. , & Carbone, I. (2009). Recombination and lineage‐specific gene loss in the aflatoxin gene cluster of Aspergillus flavus . Molecular Ecology, 18, 4870–4887. [DOI] [PubMed] [Google Scholar]
- Nei, M. (2007). The new mutation theory of phenotypic evolution. Proceedings of the National Academy of Science USA, 104, 12235–12242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olarte, R. A. , Horn, B. W. , Dorner, J. W. , Monacell, J. T. , Singh, R. , Stone, E. A. , … Carbone, I. (2011). Effect of sexual recombination on population diversity in aflatoxin production by Aspergillus flavus and evidence for cryptic heterokaryosis. Molecular Ecology, 21, 1453–1476. [DOI] [PubMed] [Google Scholar]
- Olarte, R. A. , Worthington, C. J. , Horn, B. W. , Moore, G. G. , Singh, R. , Monacell, J. T. , … Carbone, I. (2015). Enhanced diversity and aflatoxigenicity in interspecific hybrids of Aspergillus flavus and Aspergillus parasiticus . Molecular Ecology, 24, 1889–1909. [DOI] [PubMed] [Google Scholar]
- Opdam, P. , & Wascher, D. (2004). Climate change meets habitat fragmentation: Linking landscape and biogeographical scale level in research and conservation. Biological Conservation, 117, 285–297. [Google Scholar]
- Ortega‐Beltran, A. , Grubisha, L. C. , Callicott, K. A. , & Cotty, P. J. (2016). The vegetative compatibility group to which the US biocontrol agent Aspergillus flavus AF36 belongs is also endemic to Mexico. Journal of Applied Microbiology, 120, 986–998. [DOI] [PubMed] [Google Scholar]
- Peterson, S. W. , Varga, J. , Frisvad, J. C. , & Samson, R. A. (2008). Phylogeny and subgeneric taxonomy of Aspergillus In Varga J., & Samson R. A. (Eds.), Aspergillus in the Genomic Era (pp. 33–56). Wageningen, Netherlands: Wageningen Academic Publishers. [Google Scholar]
- Pildain, M. B. , Frisvad, J. C. , Vaamonde, G. , Cabral, D. , Varga, J. , & Samson, R. A. (2008). Two novel aflatoxin‐producing Aspergillus species from Argentinean peanuts. International Journal of Systematic and Evolutionary Microbiology, 58, 725–735. [DOI] [PubMed] [Google Scholar]
- Price, E. W. , & Carbone, I. (2005). SNAP: Workbench management tool for evolutionary population genetic analysis. Bioinformatics, 21, 402–404. [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K. , Stephens, M. , & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155, 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst, C. , Callicott, K. A. , & Cotty, P. J. (2012). Deadly strains of Kenyan Aspergillus are distinct from other aflatoxin producers. European Journal of Plant Pathology, 132, 419–429. [Google Scholar]
- R Development Core Team (2014). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Ramirez‐Prado, J. H. , Moore, G. G. , Horn, B. W. , & Carbone, I. (2008). Characterization and population analysis of the mating‐type genes in Aspergillus flavus and Aspergillus parasiticus . Fungal Genetics and Biology, 45, 1292–1299. [DOI] [PubMed] [Google Scholar]
- Rank, C. , Nielsen, K. F. , Larsen, T. O. , Varga, J. , Samson, R. A. , & Frisvad, J. C. (2011). Distribution of sterigmatocystin in filamentous fungi. Fungal Biology, 115, 406–420. [DOI] [PubMed] [Google Scholar]
- Raper, K. B. , & Fennell, D. I. (1965). The genus Aspergillus. Baltimore, MD: Williams & Wilkins. [Google Scholar]
- Samson, R. A. , & Varga, J. (2009). What is a species in Aspergillus? Medical Mycology, 47(Suppl 1), S13–S20. [DOI] [PubMed] [Google Scholar]
- Schierup, M. H. , Mikkelsen, A. M. , & Hein, J. (2001). Recombination, balancing selection and phylogenies in MHC and self‐incompatibility genes. Genetics, 159, 1833–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, C. A. , Woloshuk, C. P. , Robertson, D. , & Payne, G. A. (2007). Silencing of the aflatoxin gene cluster in a diploid strain of Aspergillus flavus is suppressed by ectopic aflR expression. Genetics, 176, 2077–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares, C. , Rodrigues, P. , Peterson, S. W. , & Venâncio, N. L. A. (2012). Three new species of Aspergillus section Flavi isolated from almonds and maize in Portugal. Mycologia, 104, 682–697. [DOI] [PubMed] [Google Scholar]
- Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30, 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Y. , Corcoran, P. , Menkis, A. , Whittle, C. A. , Andersson, S. G. E. , & Johannesson, H. (2012). Large‐scale introgression shapes the evolution of the mating‐type chromosomes of the filamentous ascomycete Neurospora tetrasperma . PLOS Genetics, 8, e1002820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford, D. L. (1998). PAUP* phylogenetic analysis using parsimony (*and other methods). Sunderland, MA: Sinauer Associates. [Google Scholar]
- Tibshirani, R. , Walther, G. , & Hastie, T. (2001). Estimating the number of clusters in a data set via the gap statistic. Royal Statistical Society, 63, 411–423. [Google Scholar]
- Tracy, C. , & Widom, H. (1994). Level‐spacing distribution and the Airy kernel. Communications in Mathematical Physics, 159, 151–174. [Google Scholar]
- Wassila, D. , Houda, B. , & Mohamed, B. (2015). Edaphic factors affecting distribution of soil fungi in three chotts located in Algerian desert. Courrier du Savoir, 19, 147–152. [Google Scholar]
- Yelton, M. M. , Hamer, J. E. , & Timberlake, W. E. (1984). Transformation of Aspergillus‐nidulans by using a trpC plasmid. Proceedings of the National Academy of Science USA‐Biological Sciences, 81(5), 1470–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DNA Sequences: GanBank accessions DQ390825‐DQ391160; FJ871513‐FJ878506; KX853136‐KX853999; HM353299‐HM355372; HM745598‐HM745901; HQ000095‐HM002855.