

Abstract
Familial hypercholesterolemia (FH) is a hereditary disease primarily due to mutations in the low‐density lipoprotein receptor (LDLR) that lead to elevated cholesterol and premature development of cardiovascular disease. Homozygous FH patients (HoFH) with two dysfunctional LDLR alleles are not as successfully treated with standard hypercholesterol therapies, and more aggressive therapeutic approaches to control cholesterol levels must be considered. Liver transplant can resolve HoFH, and hepatocyte transplantation has shown promising results in animals and humans. However, demand for donated livers and high‐quality hepatocytes overwhelm the supply. Human pluripotent stem cells can differentiate to hepatocyte‐like cells (HLCs) with the potential for experimental and clinical use. To be of future clinical use as autologous cells, LDLR genetic mutations in derived FH‐HLCs need to be corrected. Genome editing technology clustered‐regularly‐interspaced‐short‐palindromic‐repeats/CRISPR‐associated 9 (CRISPR/Cas9) can repair pathologic genetic mutations in human induced pluripotent stem cells. Conclusion: We used CRISPR/Cas9 genome editing to permanently correct a 3‐base pair homozygous deletion in LDLR exon 4 of patient‐derived HoFH induced pluripotent stem cells. The genetic correction restored LDLR‐mediated endocytosis in FH‐HLCs and demonstrates the proof‐of‐principle that CRISPR‐mediated genetic modification can be successfully used to normalize HoFH cholesterol metabolism deficiency at the cellular level. (Hepatology Communications 2017;1:886–898)
Abbreviations
- bp
base pair
- Cas9n
Cas9 nickase
- CRISPR/Cas9
clustered‐regularly‐interspaced‐short‐palindromic‐repeats/CRISPR‐associated 9
- CVD
cardiovascular disease
- DiI‐LDL
low‐density lipoprotein labeled with 1,1′‐dioctadecyl‐3,3,3′,3′‐tetramethyl‐indocarbocyanine perchlorate
- DU
densitometry units
- FACS
fluorescence‐activated cell sorting
- FH
familial hypercholesterolemia
- FIU
fluorescence intensity units
- GFP
green fluorescent protein
- HDR
homology‐directed repair
- HLC
hepatocyte‐like cell
- HoFH
homozygous FH
- ICG
indocyanine green
- iPSC
induced‐pluripotent stem cell
- LDL‐C
low‐density lipoprotein cholesterol
- LDLR
low‐density lipoprotein receptor
- MACS
magnetic‐activated cell sorting
- OCT4
octamer‐binding transcription factor 4
- PAM
protospacer adjacent motif
- PCR
polymerase chain reaction
- RFLP
restriction fragment length polymorphism
- RFP
red‐fluorescent protein
- sgRNA
single‐guide RNA
- SOX2
sex determining region Y‐box 2
- ssODN
single‐stranded oligodeoxynucleotide
- XmnI
Xanthomonas manihotis 7AS1
Introduction
Risks for the development of cardiovascular disease (CVD) include lifestyle choices and environmental factors, but a subgroup of the population develop early CVD primarily due to inheritable genetic mutations in the low‐density lipoprotein receptor (LDLR). Familial hypercholesterolemia (FH) is an autosomal dominant disease causing extremely elevated low‐density lipoprotein‐cholesterol (LDL‐C) and premature CVD.1, 2, 3, 4 Over 1,200 LDLR mutations have been identified that lead to a wide spectrum of disease severity depending on the mutation's effect on LDLR activity.4, 5 Patients with receptor‐defective homozygous FH (HoFH), which can be a true homozygous or compound heterozygous mutation (both referred to as homozygous, 1:250,000) have < 2% LDLR activity and are generally nonresponsive to standard hypercholesterol therapies,6, 7 relying on more aggressive costly approaches, such as weekly/biweekly lipid apheresis.8 Transplantation of a normal liver in patients with HoFH has shown to restore normal LDL‐C levels; however, the most severely affected patients can present as adolescents in their first decade.1, 4 Because of the shortage of donated organs and delayed clinical identification,9, 10 patients with HoFH often have already developed severe CVD and may additionally require heart transplant. Therefore, mitigating the effects of genetically induced hypercholesterolemia at the earliest time possible is critical for these patients.
Because of the multiple clinical challenges related to HoFH cholesterol regulation, alternative approaches for providing therapy are being investigated. Hepatocyte cell transplantation could be an option, but as with solid liver organs, there is a shortage of high‐quality hepatocytes. Because FH is a monogenic disease, it has long been the target of gene therapy, which has shown promise in animal models11, 12, 13, 14 but with mixed results in humans.15 The discovery of cellular reprogramming to generate patient‐specific induced pluripotent stem cells (iPSCs) presents the potential for generating unlimited autologous therapeutic cells.16 iPSCs have been generated from FH patients,17, 18, 19 but the LDLR genetic mutations are retained and require modifications for receptor‐mediated LDL‐C internalization.18, 19 Genome editing system clustered‐regularly‐interspaced‐short‐palindromic‐repeats/Cas‐associated 9 (CRISPR/Cas9) is a relatively simple technology capable of permanent genetic modifications and has been used to permanently repair endogenous disease‐causing genetic mutations in other disease states.20, 21, 22 Although many cell types could be used for correction with CRISPR, an advantage of correcting at the iPSC level is the theoretical unlimited expansion postcorrection of iPSCs without concern for cell senescence and the capability to differentiate to the cell source of choice. This is also critical to the hepatocyte, which is well known to lose its phenotype and poorly proliferate in culture.
As a proof‐of‐concept, we reprogrammed FH fibroblasts carrying a homozygous 3‐base pair (bp) deletion in LDLR exon 4 (GM03040), resulting in a class II mutation with <5% receptor activity (FH‐Piscataway).23 To introduce a permanent correction, we used Cas9 nickase (Cas9n) with paired single‐guide RNAs (sgRNAs) to generate adjacent off‐set nicks in the selected genomic target with reduction of off‐target mutations.24, 25 For the repair template, we used a single‐stranded oligodeoxynucleotide (ssODN) to increase efficiency of homology‐directed repair (HDR).24, 26 After double‐positive transfection and magnetic sorting of the final isolated clones, we estimated an overall efficiency of homozygous correction of 0.2%; however, enriched clones were 83% homozygous corrected with this approach. The hepatocyte‐like cells (HLCs) derived from the corrected FH‐iPSCs confirmed restoration of the mature LDLR protein and normalization of receptor‐mediated LDL internalization compared to noncorrected FH‐HLCs. This demonstrates the feasibility to generate permanently restored endogenous LDLR activity in FH‐HLCs by CRISPR technology.
Materials and Methods
CELL CULTURE
Human fetal fibroblasts (IMR90) were cultured in Dulbecco's modified Eagle's medium‐high glucose (Invitrogen, Carlsbad, CA), 2 mM L‐glutamine (Invitrogen), and 10% fetal bovine serum (Invitrogen). Cells were maintained at 37°C and 5% CO2. Cells were passaged with 0.05% trypsin‐ethylene diamine tetraacetic acid (Invitrogen). Skin fibroblasts from an FH patient (GM03040; Coriell Cell Repositories) were cultured in FH growth medium comprised of minimum essential medium (Invitrogen), 15% fetal bovine serum, 2 mM L‐glutamine, and 0.1 mM nonessential amino acids (Invitrogen). Cells were maintained at 37°C and 5% CO2. Cells were passaged with 0.05% trypsin‐ethylene diamine tetraacetic acid. We cultured 3040‐iPSCs on human embryonic stem cell‐qualified Matrigel‐coated plates (BD Biosciences, San Jose, CA) in mTeSR1 media (STEMCELL Technologies, Vancouver, Canada), with media changed daily.20 Cells were passaged using gentle cell dissociation buffer (STEMCELL Technologies) with 10 mM ROCK inhibitor (Selleck Chemical, Houston, TX) and maintained at 37°C and 5% CO2.20 The human embryonic stem cell line H1 (WiCell, Madison, WI) was cultured as the iPSCs.
CRISPR EDITING DESIGN
sgRNA Design and Cloning
The Massachusetts Institute of Technology (MIT) CRISPR Design Tool (crispr.mit.edu) was used to design paired guides in close proximity to the 3‐bp (TGG) deletion in exon 4 of the LDLR gene (Chr19:11,105,170‐11,105,650) (5′‐…TTCCACTG CCTAAGTGGCGAGTGCATCCACTCCAGCT GGCGCTGTGATGGTGGCCCCGACTGCAA GGACAAATCTGACGAGGAAAACTGCGCT GTGGCCAC‐3′). We selected paired guides 1 and 9 to use with Cas9n. The guides were cloned into pHL‐H1‐ccdB‐mEFα‐RIH (plasmid‐human promoter 1‐toxin ccdB‐mouse elongation factor‐1 alpha‐red fluorescent protein IRES hygromycin) (#60601; Addgene, Cambridge, MA) deposited by the Hotta Laboratory.21 Briefly, primers were designed as described27; for guide‐specific forward primers, 19 bp (bases 2‐20) of MIT sgRNA design were used, and the first base pair was changed to a G to enable H1 polymerase III transcription. Infusion homology arms (in blue; Supporting Fig. S6A) were added to the 5′ end of each primer pair to enable cloning into the vector when cut with Bacillus amyloliquefaciens H1 (BamH1) and Escherichia coli R1 (EcoRI). A polymerase chain reaction (PCR) was performed using Phusion Hot Start Polymerase (Thermo Fisher, Walton, MA) and either guide primer paired with sgUniversal Reverse primer (Supporting Fig. S6A). The PCR product was cloned into pHL‐H1‐ccdB‐mEFa‐RIH with the In‐fusion Cloning kit (Clonetech Laboratories, Inc.). Clones were sequence confirmed with our H1‐forward primer (5′‐GCATGTCGCTATGTGTTCTG‐3′).
Cas9n‐Green Fluorescent Protein Design
We obtained a humanized Streptococcus pyogenes D10A Cas9 nickase plasmid, pHL‐EFlα‐SphcCas9(D10A)‐iP‐A (plasmid‐human elongation factor 1 alpha promoter‐Cas9 (D10A)‐IRES Puromycin) (#60600; Addgene), deposited by Hotta Laboratory.21 The puromycin gene, along with a portion of the internal ribosomal entry site, was removed by digesting with Klebsiella pneumoniae OK8 (KpnI) and BamH1. In‐fusion cloning was used to reinstate the missing internal ribosomal entry site sequence and concurrently insert a green fluorescent protein (GFP) gene. The primers for PCR are listed in Supporting Fig. S6B; sequences in blue are infusion homology arms, while an underlined sequence is that of GFP. The Cas9n‐GFP clone was sequence confirmed.
ssODN Design
A 157‐bp antisense ssODN repair template with 50‐bp left and right homology arms was designed to insert the missing three nucleotides (CCA) while simultaneously adding silent mutations within the protospacer adjacent motif (PAM) site and guide target site (57 bp) to generate a novel Xanthomonas manihotis 7AS1 (XmnI) restriction site (GAATGCATTC) and prohibit further Cas9n editing by blocking the guide recognition sequence. The full nucleotide sequence can be found in Supporting Fig. S4A. The oligonucleotide was obtained from IDT (Coralville, IA) as a 4‐nM ultramer oligonucleotide and was not high‐performance liquid chromatography purified. The ssODN sequence has been modified from chromosome 19, gene NM_00527.4 corresponding to nucleotides 750‐896. Further Materials and Methods can be found in the Supporting Information.
Results
IDENTIFYING THE FH MUTATION IN PATIENT FIBROBLASTS GM03040
We acquired the FH skin fibroblasts (GM03040), which are described as homozygous receptor defective, from the Coriell Cell Repository (Camden, NJ), but no genetic information was provided. We initially confirmed the inability of GM03040 to internalize cholesterol by culturing those fibroblasts and a normal fibroblast control (IMR90) overnight in 5% lipoprotein‐deficient serum media supplemented with either lovastatin or excess sterols and followed the next morning with incubation in LDL labeled with 1,1′‐dioctadecyl‐3,3,3′,3′‐tetramethyl‐indocarbocyanine perchlorate (DiI‐LDL).19 Lovastatin inhibits 3‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase, blocking cholesterol synthesis and leading to an up‐regulation of LDLR. The excess sterols act independent of the LDLR, leading to a decrease in LDLR expression. LDL binding and internalization by the LDLR‐normal IMR90 was robust in response to lovastatin and was abrogated in the presence of excess sterol treatment (Fig. 1A). In contrast, GM03040 showed virtually no DiI‐LDL binding and internalization, even under the same lovastatin treatment conditions that induced robust internalization in IMR90. We next wanted to know if any LDLR was produced by GM03040. Under the same conditions for the LDL binding and internalization assay, we collected cell lysates for analysis by western blot (Fig. 1B). Lovastatin treatment of IMR90 showed strong expression of the mature LDLR protein with low expression of the immature proteins. No LDLR protein was detected when IMR90 cells were exposed to excess sterols. In the GM03040, lovastatin induced low‐level LDLR expression with the greatest increase in immature LDLR protein and very little mature LDLR (Fig. 1B). Although GM03040 expresses little LDLR, even with lovastatin treatment, excess sterols down‐regulated LDLR, indicating these FH fibroblasts are physiologically responsive to cholesterol feedback control, as described by Brown and Goldstein.3 Sequencing of the GM03040 (and IMR90 control) LDLR identified a pathologic homozygous three‐nucleotide deletion in exon 4 (c.654_656delTGG [pG219del]; Fig. 1C), reflecting a class IIB mutation known as FH‐Piscataway that results in less than 5% LDLR activity and misfolding of immature LDLR, which is degraded by the proteasome pathway.23, 28 Together, these data confirm the FH cell line contains a three‐nucleotide deletion in exon 4 of the LDLR, resulting in little to no LDLR‐mediated LDL internalization.
Figure 1.
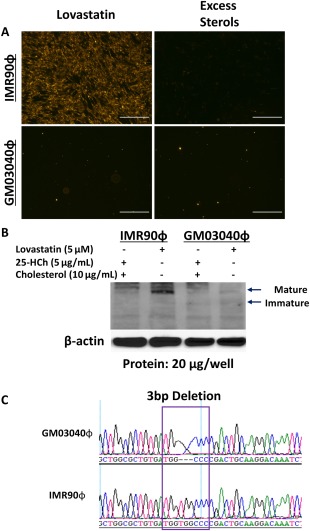
Mutation identification in HoFH fibroblasts. (A) HoFHϕ and control fetal IMR90ϕ were treated overnight in 5% lipoprotein‐deficient serum media supplemented with either lovastatin or excess sterols. A significant amount of fluorescent DiI‐LDL was visualized in IMR90ϕ treated with lovastatin that was abrogated with excess sterols. GM03040ϕ showed impaired DiI‐LDL internalization with lovastatin treatment (scale bars, 200 μm). (B) Western blot for LDLR shows IMR90ϕ up‐regulate LDLR in lovastatin and suppress LDLR when exposed to sterols. In contrast, HoFH GM03040ϕ express comparatively little LDLR under the same conditions. (C) Sanger sequencing revealed a homozygous three‐nucleotide deletion in exon 4 of LDLR in GM03040ϕ. Abbreviation: ϕ, Fibroblasts.
REPROGRAMMING FIBROBLASTS TO iPSCs AND THEIR DIFFERENTIATION INTO HLCs
Before reprogramming the parental GM03040, we confirmed the normal karyotype (Supporting Fig. S1A) and DNA fingerprinting (not shown). We reprogrammed the fibroblasts to 3040‐iPSCs, using a modified synthetic messenger RNA cocktail containing octamer‐binding transcription factor 4 (OCT4), sex determining region Y‐box 2 (SOX2), Kruppel‐like factor 4 (KLF4), c‐mycproto‐oncogene (C‐MYC), and Lin‐28 homolog A (LIN‐28)19and began seeing colonies that were morphologically compact and refractile at approximately day 16. Manual picking of TRA‐1‐81+ colonies was initiated around day 20 for expansion into feeder‐free culture (Supporting Fig. S2A). The 3040‐iPSCs demonstrated immunoreactivity with pluripotence markers TRA‐1‐81 (Podocalyxin carbohydrate binding antibody 1‐81) and Ulex europaeus agglutinin‐1 (UEA1) (Supporting Fig. S2B), TRA‐1‐60 (Podocalyxin carbohydrate binding antibody 1‐60), stage‐specific embryonic antigen 4 (SSEA4), Oct4, and Sox2, while being negative for SSEA1 (Supporting Fig. S2C). We confirmed that the reprogrammed cells were still karyotypically normal (Supporting Fig. S1B), and DNA fingerprinting (not shown) established that the 3040‐iPSCs originated from the parent GM03040 fibroblasts. Histologic examination of harvested teratomas confirmed 3040‐iPSC pluripotence with differentiation into derivatives of all three germ layers in vivo (Supporting Fig. 2D).
Most LDLR are found on liver hepatocytes and are primarily responsible for LDL regulation3; therefore, we wanted to confirm 3040‐iPSC capability to differentiate toward HLCs. Following a described five‐stage protocol,19, 29 3040‐iPSCs showed expression of stage‐specific markers after completion of each differentiation stage, as demonstrated by quantitative PCR (Supporting Fig. S3A; Supporting Table S1). As we have shown,19 3040‐iPSCs undergo differentiation as demonstrated by progression of expression of pluripotence marker OCT4 at stage 0, definitive endoderm marker SOX1729 in stage 1, hepatic lineage specification as indicated by expression of hepatocyte nuclear factor 4a in stages 2‐5, the early hepatocyte marker alpha fetal protein29 beginning in stage 2, and standard albumin29 expression initiated during stage 3. To examine protein expression, we differentiated 3040‐iPSCs to the end of stage 4 and used immunocytochemistry for detection of commonly associated hepatocyte markers, albumin, alpha fetal protein, and cytokeratin 18 (Supporting Fig. S3B).19, 29 Hepatocytes exclusively express a transporter organic anion transport protein in their basolateral membrane that is used by bilirubin. Indocyanine green (ICG) is an anion that is taken up by hepatocytes and excreted into the bile canaliculi.27 We tested 3040‐HLC functionality by an ICG exclusion assay and found that 3040‐HLCs demonstrated the ability to take up and clear ICG dye within 24 hours (Supporting Fig. S3C). These data confirm that the reprogrammed GM03040 FH fibroblasts are pluripotent and can specifically differentiate to functioning HLCs.
CORRECTING THE HOMOZYGOUS 3‐NUCLEOTIDE LDLR DELETION IN 3040‐iPSCs WITH CRISPR
Our laboratory has previously restored physiologic function of the LDLR in FH‐iPSCs, using an episomal transgene; however, plasmid retention is compromised without continuous antibiotic selection.19 We opted to use CRISPR/Cas9 to permanently correct the 3‐bp pathogenic deletion in the 3040‐iPSCs. A schematic outline of our genome editing approach is presented in Fig. 2A. Using the MIT CRISPR Design Tool (crispr.mit.edu), we input the exon 4 LDLR sequence of the FH patient for identification of sgRNAs. Thirteen possible sgRNAs were found that could be used with wild‐type Cas9 and that were arranged in suggested pairs for use with Cas9n. We used the Cas9n with a pair of offset sgRNAs because this grouping had been successful in facilitating specific genome editing in human cells.21 The double‐offset nicking approach has also shown a higher likelihood of forcing HDR to improve specific repairs and reduce off‐target mutations. Our guide pairs (sgRNA1 and sgRNA9) were selected based on Ran et al. criteria.21, 30 The guides generate 5′ overhangs with a + 2 offset and create at least one nick within 6 bp of the target mutation site to increase the probability of HDR.21, 24 HDR requires a repair template for the DNA to use and precisely modify the genome.21, 24, 25, 26 We designed an antisense ssODN template with right and left 50‐bp homology arms surrounding the deletion in exon 4 of LDLR. The repair template contained the deleted three nucleotides for inserting the missing 3 bp (ACC) and silent mutations in the PAM of sgRNA1 within the first 10 bp upstream of the PAM site in sgRNA9 to minimize rebinding after genome repair and to introduce a novel XmnI restriction site (Fig. 2B; Supporting Fig. S4A).21, 24, 25, 26
Figure 2.
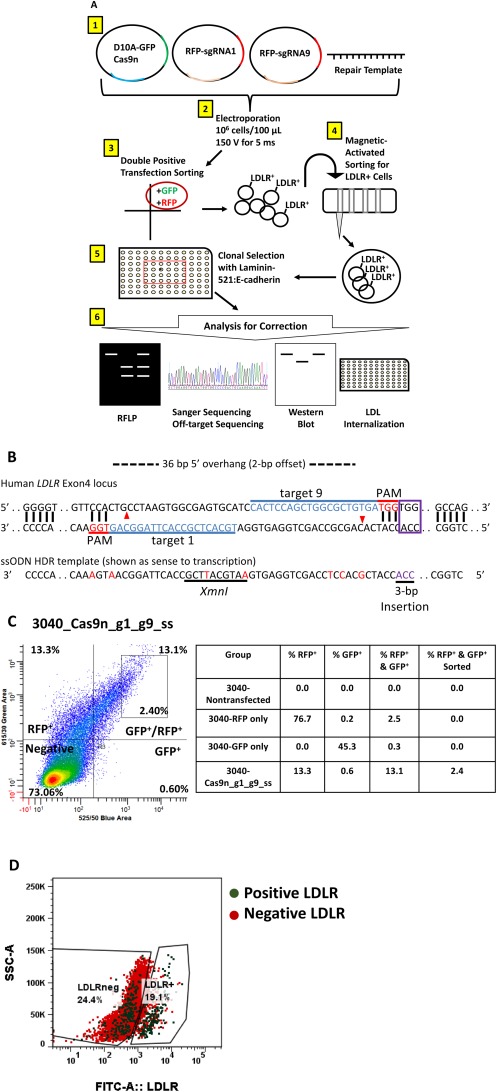
CRISPR correction strategy. (A) Schematic of the methods: 1 3040‐iPSCs were transfected with 5 μg Cas9n, 5 μg sgRNA1, 5 μg sgRNA9, and 5 μg ssODN, using 2 a NEPA21 square‐wave electroporator. 3 Double‐positive (GFP+/RFP+) cells were selected for expansion by fluorescence sorting. 4 LDLR + cells were enriched by magnetic sorting and expanded in culture. 5 Clones were isolated on laminin‐521 + E‐cadherin‐Fc substrate then 6 analyzed for correction of the LDLR by RFLP, sequencing, western blot, and LDL internalization. (B) Diagram of the CRISPR/Cas9 design to target the mutation site of the LDLR. Paired single‐guide RNAs (target 1 and target 9) were selected to target the mutation site, and an ssODN template was created to mediate HDR. The ssODN contains a 10‐nucleotide sequence (underlined) as a novel XmnI restriction enzyme site, additional silent mutations (red nucleotides) at the PAM for sgRNA 1 or within 10 bp upstream of the PAM for sgRNA 9 to prevent rebinding of guides and cleavage with Cas9n following repair, and the 3‐bp insertion (purple). (C) 3040‐iPSCs were sorted for double‐positive RFP and GFP cells by FACS after transfection; 13.1% of cells were dual positive and 2.4% of the population was collected and expanded to increase specificity of positively transfected cells. (D) MACS was performed on the FACS‐sorted 3040‐iPSCs to enrich for LDLR+ expressing cells. Pre‐MACS‐sorted cells had a mix of both positive (green dots) and negative (red dots) LDLR+ cells. After MACS sorting, a clear shift to the right showed 19.1% of LDLR+ cells were collected. Abbreviations: FITC, fluorescein isothiocyanate; SSC‐A, side‐scatter.
We obtained Cas9n and sgRNA plasmids (Addgene #60600 and #60601, respectively) deposited by the Hotta Laboratory20 and cloned in our selected sgRNA (Fig. 2A, step 1). The sgRNA plasmid contains a red fluorescent protein (RFP) that can be used to select for cells positively transfected with the sgRNA. We opted to use double‐fluorescence cell sorting posttransfection rather than antibiotic selection to potentially maximize the selection of transfected cells. The Cas9n plasmid deposited by Hotta was modified to incorporate GFP into the vector. Before transfection, we optimized the electroporation voltage necessary for highest transfection efficiency, referencing the NEPA21 electroporator conditions by the Hotta Laboratory20 (Supporting Fig. S4B). We then transfected the 3040‐iPSCs with 20 μg total nucleic acids (5 μg Cas9n, 5 μg sgRNA1, 5 μg sgRNA9, and 5 μg ssODN) (Fig. 2A, step 2). After 48 hours, we sorted for dual‐positive (i.e., GFP and RFP) cells to subculture only cells transfected with both Cas9n and guide plasmids (Fig. 2A, step 3). We collected 2.4% of the 13.1% population expressing both RFP and GFP (Fig. 2C). Because we used two plasmids for each guide, we could not guarantee that sorted cells contained both guide plasmids for correction. To enrich the population of double‐positive transfected cells that actually contained the 3‐bp insertion, we treated the culture with lovastatin to up‐regulate LDLR expression and targeted the cell surface receptor for magnetic‐activated cell sorting (MACS) (Fig. 2A, step 4). Using a biotinylated‐C7‐LDLR antibody, we enriched for LDLR+ cells within the bulk of the transfected cells. We analyzed the percentage of LDLR+ cells collected (green dots) after MACS by fluorescence‐activated cell sorting (FACS). FACS analysis showed a mix of LDLR+ (green dots) and LDLR– (red dots) before sorting (left box). After MACS, we confirmed a collection of 19.1% LDLR+ cells or corrected cells with a clear shift of green dots to the right (Fig. 2D). After expanding the bulk LDLR+ cells, we adopted the clonal isolation protocol developed by the Tryggvason Laboratory using laminin‐521 and E‐cadherin‐Fc (fragment crystallizable) substrates in mTeSR1‐albumin media (Fig. 2A, step 5).31 Using a clonal dilution approach, we initially plated 120 cells; after 7 days of culture, we identified 16 wells with single colonies growing (empty and multicolony wells were counted as null clones) for a cloning efficiency of 13%. The clonal colonies were expanded for analysis and cryopreservation.
Because we created a donor repair template that contains a unique XmnI restriction site (Fig. 2B; Supporting Fig. S4A), we could use restriction fragment length polymorphism (RFLP) analysis to check for potential correction of the 3040‐iPSC clones. Additionally, we could see if it was a homozygous or heterozygous clone by band size (Fig. 2A, step 6). The expected LDLR PCR product without any enzyme or correction is 576 bp, as indicated by the 3040‐iPSC nontransfected control (Fig. 3A). A homozygous corrected clone would have two bands at 384 bp and 192 bp, while a heterozygous clone would also retain the 576‐bp band. RFLP of the first clone (3040‐C) tested indicated homozygous correction by the presence of only two bands of the correct size (Fig. 3A). RFLP analysis of the other clones showed an additional nine homozygous corrections and six heterozygous corrections (Supporting Fig. S5). To confirm the RFLP results, we performed Sanger sequencing on all clones. Four clones were found to be homozygous corrected by sequencing; however, RFLP demonstrated retention of the 576‐bp band (clones 2, 3, 4, 11). We presumed these were indeed not clonal and removed them from clonal consideration. For the total 12 clones, the RFLP and Sanger sequencing results agreed. Interestingly, sequencing indicated that not only did the heterozygous corrected clones (C12 and C14) only correct a single allele but they also obtained different indels in the noncorrected allele proximal to the target sequence of sgRNA1s. In addition, our selected clone (3040‐C) was determined to be heterozygous in two silent mutation points denoted by the green box (Fig. 3B). From these successfully isolated clonal colonies, our efficiency of homozygous correction from the final isolation of 10 (from 12) homozygous corrected clones is 83%. Because sgRNA can target multiple regions within the genome,25 we investigated for off‐target mutations. We selected three nongenic and three genic regions identified by the CRISPR Design Tool for both sgRNA1 and sgRNA9 with the highest potential for off‐targeting (Supporting Table S2). Sequencing of 3040‐C compared to noncorrected 3040‐iPSCs indicated no changes in the selected regions of the genome (not shown). While we were fortuitous in not detecting off‐targets within the highest selected regions, this may not be the case with other targeted mutations. These results illustrate that the three‐nucleotide deletion was homozygous corrected without additional mutations in the homozygous FH‐iPSCs.
Figure 3.
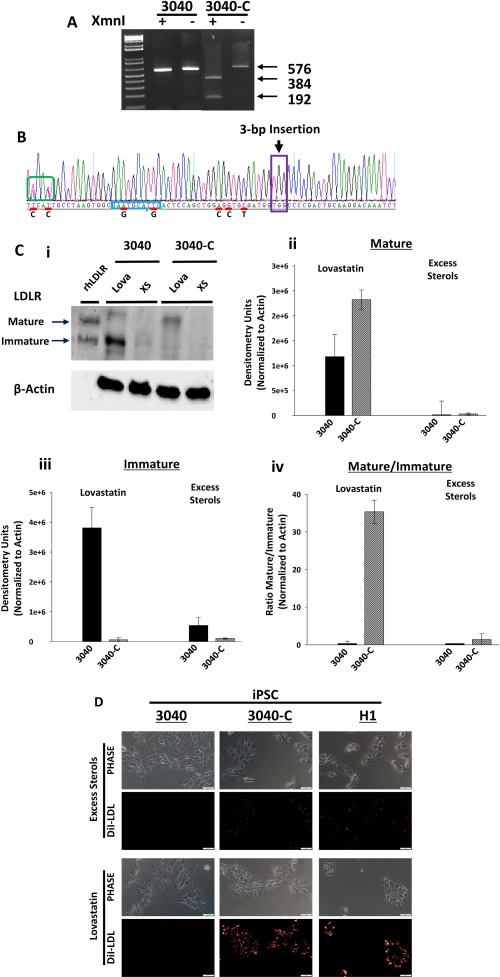
Analysis of 3040‐iPSC LDLR correction by CRISPR/Cas9. (A) RFLP analysis was used to determine 3040‐iPSC correction status by restriction digest with XmnI. Nontransfected 3040‐iPSCs showed no XmnI cleavage (576 bp) in the presence or absence of enzyme. 3040‐iPSC clone 1 (3040‐C) contained two bands at 384 bp and 192 bp in the presence of XmnI, which remained uncut (576 bp) without enzyme. (B) Sanger sequencing of 3040‐C confirmed the permanent insertion of three nucleotides in exon 4 of the LDLR in both alleles (purple box). Silent mutations (red line), including the novel XmnI site (blue box), were also integrated into the gene. Bold letters beneath red lines indicate original native nucleotide. Two silent mutations (green box) were introduced in only one allele. (C) 3040‐C‐iPSCs and 3040‐iPSCs were incubated overnight in lovastatin or excess sterols. Western blot (representative image, C, i) showed 3040‐iPSCs respond with an up‐regulation of an immature LDLR after treatment with lovastatin that is reduced with excess sterols. 3040‐C‐iPSCs had almost no immature LDLR with lovastatin and increased mature LDLR. (C, ii) Quantification (n = 2 independent experiments) showed a 2‐fold increase of mature LDLR in 3040‐C (2.3e6 ± 1.9e5) treated with lovastatin compared to 3040 (1.1e6 ± 4.5e5). (C, iii) Immature LDLR was highly expressed in 3040 (3.8e6 ± 6.7e5) that was almost lost in 3040‐C (6.5e4 ± 6.3e4). (C, iv) Mature/immature LDLR was over 30 times greater in 3040‐C (35 ± 3.1) than 3040 (0.3 ± 0.6) in lovastatin that was abrogated in excess sterols (1.4 ± 1.5 and 0.3 ± 0.1, respectively). Bars shown as mean ± SEM. (D) LDL internalization showed 3040‐C‐iPSCs respond with an increased DiI‐LDL internalization similar to control H1 cells that is almost nonexistent in noncorrected 3040‐iPSCs. LDL uptake is decreased with excess sterols. scale bars, 50 μm. Abbreviations: Lova, lovastatin; rh, recombinant human; XS, excess sterols.
DETERMINING LDLR FUNCTION NORMALIZATION IN 3040‐C‐HLCs AFTER GENETIC CORRECTION
Genomic mutations of the LDLR in FH patients can vary from a single nucleotide change to large deletions that will denote the patient into a certain class.2, 3, 32 The 3‐bp deletion in 3040‐iPSCs are classified as class IIB FH‐Piscataway because the LDLR does not reach maturity and instead is retained in the endoplasmic reticulum as an immature protein that eventually is degraded by the proteosomal pathway.23, 28, 32 After treatment with lovastatin, we observed a robust induction of LDLR expression that was primarily in the form of immature protein in the noncorrected 3040‐iPSCs (3.8e6 ± 6.7e5 densitometry units [DU]) but very little mature protein being detected (1.1e6 ± 4.5e5 DU; Fig. 3C, i‐iii). Excess sterols down‐regulated LDLR protein production in these cells as expected (5e5 ± 2.7e5 DU). In contrast, lovastatin treatment of the corrected 3040‐C‐iPSCs showed a dramatic shift from predominantly immature protein (6.5e4 ± 6.3e4 DU) to predominantly mature protein (2.3e6 ± 1.9e5 DU; Fig. 3C, i‐iii). Again as expected, excess sterol treatment of the corrected cells down‐regulated LDLR protein expression (1e4 ± 2e4 DU). When comparing the ratio of mature to immature LDLR with lovastatin treatment, the corrected cells (35 ± 3.1 DU) were more than 30‐fold higher than the noncorrected cells (0.3 ± 0.6 DU; Fig. 3C, iv), suggesting the CRISPR modification restored the normal LDLR structure; this would allow LDLR to proceed to the Golgi for processing and movement to the plasma membrane. Comparing DiI‐LDL internalization under the same conditions with the human embryonic stem cell line H1, noncorrected 3040‐iPSCs internalize very little DiI‐LDL even with lovastatin treatment (perhaps indicating that the function of “mature” LDLR in these cells is questionable) (Fig. 3D). Confirming the results of the western blot, corrected 3040‐C‐iPSCs bound and internalized DiI‐LDL when exposed to lovastatin, and this receptor‐mediated endocytosis was abrogated by excess sterols to a comparable state to normal H1. These data strongly support the complete genetic correction of the LDLR pathologic mutation and normalization of LDL‐C receptor‐mediated endocytosis.
The hepatocyte is responsible for production and metabolism of cholesterol but is defective in FH; therefore, we differentiated noncorrected and corrected FH‐iPSCs to HLCs to determine if receptor‐mediated endocytosis is corrected and functional in HLCs. After differentiation, HLCs were treated overnight with either lovastatin or excess sterols as before. Fluorescence microscopy of 3040‐C‐HLCs is consistent with 3040‐C‐iPSCs indicating an up‐regulation in DiI‐LDL endocytosis relative to 3040‐HLCs. The H1‐HLC control was similar to 3040‐C‐HLCs (Fig. 4). Quantification of DiI‐LDL fluorescence intensity showed 3040‐C‐HLCs (8.2 ± 1.3 fluorescence intensity units [FIU]) contained a substantially increased fluorescence intensity compared to 3040‐HLCs (1.8 ± 0.8 FIU; Fig. 4) that was comparable to H1‐HLCs (11 ± 2 FIU). For all three cell lines, excess sterols reduced DiI‐LDL binding and internalization to a basal level. Taken together, these data demonstrate our CRISPR design successfully corrected the 3‐bp deletion, restoring LDLR protein and LDLR‐mediated endocytosis in the homozygous FH cells.
Figure 4.
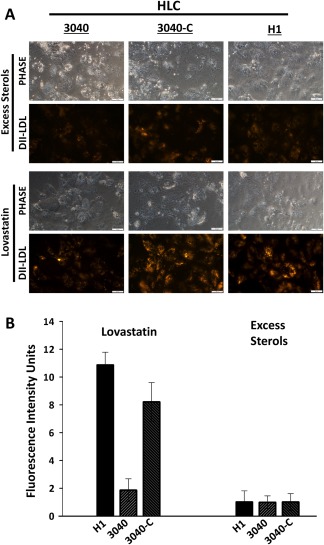
LDLR‐mediated endocytosis is restored in corrected 3040‐iPSC‐derived HLCs. (A) DiI‐LDL uptake was restored in 3040‐C‐HLCs after treatment with lovastatin that was not obvious in 3040‐HLCs. H1‐HLC was used a control for normal LDLR function. (B) Bar graph (n = 2 independent experiments) shows measured fluorescence intensity of DiI‐LDL that correlates with a 4‐fold increase in DiI‐LDL uptake in 3040‐C‐HLCs (8.2 ± 1.3) relative to 3040‐HLCs (1.8 ± 0.8). Excess sterols were normalized to 1 (H1, 1 ± 0.6; 3040, 1 ± 0.5; 3040‐C, 1 ± 0.7) and lovastatin (H1, 11 ± 2; 3040, 1.8 ± 0.8; 3040‐C, 8.2 ± 1.3) normalized to excess sterols. Bars shown as mean ± SEM; scale bars, 50 μm.
Discussion
The novel and important findings of this study are that we have used CRISPR genome editing to permanently correct a homozygous pathologic mutation in patient‐specific iPSCs. We have targeted a class IIB 3‐bp deletion in exon 4 of the LDLR that causes less than 5% receptor activity. The successful insertion of the missing 3 bp has permanently corrected the genetic mutation and restored the physiologic feedback control of receptor expression and cholesterol metabolism. We developed an enrichment protocol to enhance the probability of isolating corrected clones. The placement of the guides with respect to the mutation site likely made our cells more amenable to correction. Double‐positive sorting with GFP and RFP improved selection of dual transfected cells, while MACS sorting increased the yield of LDLR‐expressing cells. Our systematic stepwise enrichment protocol allowed for the final isolation of 10 (from 12) homozygous corrected clones (83%). This procedure is likely adaptable to identifying any corrected surface protein and is a proof‐of‐concept that CRISPR technology can successfully repair LDLR mutations causing FH.
The estimated prevalence of FH is 1:250 and 1:250,000 for heterozygous and homozygous FH, respectively.6, 7, 33 It is not uncommon that receptor‐defective HoFH patients will present with clinically symptomatic CVD even in the first decade of life.34 A critical issue for these patients is early identification and therapeutic intervention.7 Because FH is considered underdiagnosed, screening has been proposed for newborns. For rare HoFH patients, lifestyle changes and statin/ezetimibe therapy may be insufficient to achieve target LDL‐C levels.33 This requires more aggressive treatment, including newly approved pharmacologics (i.e., mipomersen, lomitapide), lipid apheresis, and even liver transplant,8, 9, 10 all of which have their own potential complications. It is also important to recognize that newly approved proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors rely on LDLR expression and have a questionable effect on receptor‐negative HoFH patients.35 This supports the need to develop alternative approaches for treating HoFH.
Gene and cell therapies to restore cholesterol metabolism homeostasis in FH have been investigated for decades.36 Gene therapy to deliver an LDLR transgene to the liver has evolved from retrovirus37 and adenovirus38 to adeno‐associated virus containing liver‐specific promoters.14 The first gene therapy clinical trial using ex vivo LDLR‐transduced autologous hepatocytes showed some benefit to lowering LDL‐C but was halted due to variable metabolic results, in particular in receptor‐defective homozygotes.15, 39 A phase 1/2 clinical trial using an adeno‐associated virus‐based vector to deliver LDLR is currently approved (NCT02651675). Virus‐based gene therapy has demonstrated a capacity to lower cholesterol levels in hypercholesterolemic animal models,40 but issues remain, including transgene persistence, vector delivery efficiency, and lack of LDLR expression regulation because of vector size limitations.19 With regards to cell‐based therapy for FH, multiple reports of liver transplantation demonstrate the effectiveness of providing sufficient LDLR‐normal hepatocytes to resolve FH hypercholesterolemia.9, 10 However, liver availability is limited as are quality hepatocytes. Additionally, transplanted hepatocyte engraftment is very poor unless major insult to the liver is first performed.41 Together, this demonstrates that issues of transgene and cell delivery for treating HoFH have not been completely solved.
CRISPR/Cas9 genome editing has been demonstrated to be a relatively straight forward methodology for creating mutations and for endogenous mutation repair. It has specifically been used in animal models to repair mutations in the dystrophin gene Dmd that causes Duchene muscular dystrophy.20, 42 Pankowicz and colleagues43 used CRISPR/Cas9 in a mouse model of hereditary tyrosinemia type I to delete hydroxyphenylpyruvate dioxygenase (Hpd), which converted the animal to an asymptomatic tyrosinemia type III. Jarrett et al.22 delivered CRISPR/Cas9 in an adeno‐associated virus to disrupt the LDLR and cause hypercholesterolemia. They co‐administered CRISPR/Cas9 to also disrupt the apolipoprotein B (Apob) gene, which rescued the hypercholesterolemic phenotype. In a Chinese clinical trial directed by Lu You, CRISPR was used to inactivate the T‐cell programmed cell death protein 1 (PD1) gene; the inactivated gene was then delivered back to the patient as a treatment for metastatic nonsmall cell lung cancer.40 The National Institutes of Health has now approved a phase I trial for using CRIPSR‐modified T cells in cancer patients.44 Certainly, the immediate benefit of CRISPR genome editing is rapid genetic modification for molecular modeling. Long‐term, although perhaps sooner than later, it may also prove of even greater value for therapeutic applications.
CRISPR technology has also been used to modify iPSCs.30, 45 It can knock in reporters to specific loci, create knockout models, and repair pathologic mutations, as we have demonstrated here. The significance of combining CRISPR genome editing with patient‐specific iPSCs is the ability to generate a completely repaired autologous cell source with the potential to differentiate to any cell lineage. It is widely recognized that lack of therapeutic cell sources is one of the major barriers to cell‐based therapies, and this includes hepatocytes to treat liver diseases.46 Here, we demonstrate the ability to correct a homozygous deletion in the LDLR and normalize receptor‐mediated endocytosis in patient‐derived HoFH‐HLCs.
The FH‐iPSC used in our study is considered a class IIB FH‐Piscataway mutation. This 3‐bp in‐frame deletion results in a missing Gly197 in the highly conserved binding domain in exon 4 of the LDLR and is highly prevalent in the Ashkenazi Jewish community originating from Lithuania.23 A class II mutation leads to an improperly folded LDLR because the spacing between cysteine residues becomes abnormal. The immature LDLR is retained in the endoplasmic reticulum and eventually degraded by the proteasome pathway.28 Once corrected, it is expected that the immature LDLR expression will decrease as the LDLR will properly fold and transition from the endoplasmic reticulum to the Golgi for glycosylation to a fully mature LDLR before it is transferred to the plasma membrane. Our western blot with fibroblast cells (Fig. 1B) shows a very low level of mature LDLR protein in IMR90 and no immature LDLR in GM03040. This could be because these are fibroblast cell lines and do not express LDLR as hepatocytes do. Western blot of noncorrected 3040‐iPSCs treated with lovastatin shows very little mature and robust immature protein expression resulting in a very small mature/immature ratio of LDLR that is reversed in the corrected FH‐iPSCs. Curiously, the total LDLR protein (immature + mature) was less in the corrected cells (Fig. 3C, i). The precise reason for this is unclear but is likely the result of restoration of feedback regulation, increased receptor recycling, and more efficient LDL‐C processing. Interestingly, the immature LDLR expression appears to be uncontrolled protein production in the noncorrected FH cells. Further investigation would be needed to determine if this is unique to a class II mutation.
What we have shown is a relatively efficient way to fully correct a genetic mutation using Cas9n to reduce chances of off‐target mutations and sgRNA to mediate specific on‐target cleavage. Intriguingly, only the heterozygous corrected clones introduced indels into their gene. In addition to candidate off‐target sites, it is known that other off‐target modifications may occur in places not predicted in the candidate list due to a variety of reasons, including genomic and epigenomic properties that may affect cleavage frequency; therefore, unbiased detection of off‐target cleavage, such as whole genome sequencing, is recommended and would be required before any corrected cell would be delivered to a human patient.47 Although CRISPR‐corrected HoFH‐HLCs by themselves do not solve all cell/gene‐based therapy technical hurdles, they do provide a platform for autologous cell replacement with a permanent genetic correction to the underlying pathology. CRISPR‐corrected HoFH‐HLCs could provide an unlimited source of autologous hepatocytes that could be delivered directly to the liver or engineered into a functional liver‐like tissue with sufficient cell mass to have a significant effect or even restore LDL‐C homeostasis.
These data validate the feasibility to use Cas9n, dual sgRNA, and a repair template to conclusively amend a genetic mutation in a homozygous FH cell line that has the capacity to be derived into functional HLCs and physiologically mediate LDLR endocytosis. In the future, the use of CRISPR‐edited iPSCs could be tested in patients as is currently being performed in the treatment of cancer.44
Authors names in bold designate shared co‐authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1110/full.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgment
We thank Dr. Lukasz Slomnicki and Dr. Michal Hetman (University of Louisville) for their assistance with electroporation, Dr. Jennifer Strange and Dr. Dan Cohn (University of Kentucky) for help with cell sorting, Dr. Paula Chiton and Katlin Stivers for unlimited assistance in FACS analysis and data configuration, Dr. Ron Gregg for laboratory space and materials for RFLP and off‐target analysis, Dr. Sergey Rodin for clonal isolation communications, Jason Beare for image analysis support, and the University of Louisville Cardiothoracic Department for use of the ChemiDoc MP Imaging System (Bio‐Rad).
Potential conflict of interest: Nothing to report.
Supported by National Institutes of Health grant NIH 1R21EB0022185 and the Jewish Heritage Fund for Excellence (to N.L.B.).
REFERENCES
- 1. Gidding SS. The complexities of homozygous familial hypercholesterolemia management. Pediatr Transplant 2016;20:1020‐1021. [DOI] [PubMed] [Google Scholar]
- 2. Goldstein JL, Brown MS. The LDL receptor defect in familial hypercholesterolemia. Implications for pathogenesis and therapy. Med Clin North Am 1982;66:335‐362. [DOI] [PubMed] [Google Scholar]
- 3. Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol 2009;29:431‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Varret M, Rabes J‐P. Missense mutation in the LDLR gene: a wide spectrum in the severity of familial hypercholesterolemia, mutations in human genetic disease. In: Cooper PD, ed. Mutations in Human Genetic Disease, 2012. [Google Scholar]
- 5. Soutar AK, Naoumova RP. Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat Clin Pract Cardiovasc Med 2007;4:214‐225. [DOI] [PubMed] [Google Scholar]
- 6. Sjouke B, Kusters DM, Kindt I, Besseling J, Defesche JC, Sijbrands EJ, et al. Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: prevalence, genotype‐phenotype relationship, and clinical outcome. Eur Heart J 2015;36:560‐565. [DOI] [PubMed] [Google Scholar]
- 7. Wiegman A, Gidding SS, Watts GF, Chapman MJ, Ginsberg HN, Cuchel M, et al. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur Heart J 2015;36:2425‐2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thompson GR. Managing homozygous familial hypercholesterolaemia from cradle to grave. Atheroscler Suppl 2015;18:16‐20. [DOI] [PubMed] [Google Scholar]
- 9. Kakaei F, Nikeghbalian S, Kazemi K, Salahi H, Bahador A, Dehghani SM, et al. Liver transplantation for homozygous familial hypercholesterolemia: two case reports. Transplant Proc 2009;41:2939‐2941. [DOI] [PubMed] [Google Scholar]
- 10. Alim A, Tokat Y, Erdogan Y, Gokkaya Z, Dayangac M, Yuzer Y, et al. Liver transplantation for homozygote familial hypercholesterolemia: the only curative treatment. Pediatr Transplant 2016;20:1060‐1064. [DOI] [PubMed] [Google Scholar]
- 11. Chowdhury JR, Grossman M, Gupta S, Chowdhury NR, Baker JR Jr, Wilson JM. Long‐term improvement of hypercholesterolemia after ex vivo gene therapy in LDLR‐deficient rabbits. Science 1991;254:1802‐1805. [DOI] [PubMed] [Google Scholar]
- 12. Gunsalus JR, Brady DA, Coulter SM, Gray BM, Edge AS. Reduction of serum cholesterol in Watanabe rabbits by xenogeneic hepatocellular transplantation. Nat Med 1997;3:48‐53. [DOI] [PubMed] [Google Scholar]
- 13. Havel RJ, Yamada N, Shames DM. Watanabe heritable hyperlipidemic rabbit. Animal model for familial hypercholesterolemia. Arteriosclerosis 1989;9(Suppl.):I33‐I38. [PubMed] [Google Scholar]
- 14. Kassim SH, Li H, Bell P, Somanathan S, Lagor W, Jacobs F, et al. Adeno‐associated virus serotype 8 gene therapy leads to significant lowering of plasma cholesterol levels in humanized mouse models of homozygous and heterozygous familial hypercholesterolemia. Hum Gene Ther 2013;24:19‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grossman M, Rader DJ, Muller DW, Kolansky DM, Kozarsky K, Clark BJ 3rd, et al. A pilot study of ex vivo gene therapy for homozygous familial hypercholesterolaemia. Nat Med 1995;1:1148‐1154. [DOI] [PubMed] [Google Scholar]
- 16. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861‐872. [DOI] [PubMed] [Google Scholar]
- 17. Cayo MA, Cai J, DeLaForest A, Noto FK, Nagaoka M, Clark BS, et al. JD induced pluripotent stem cell‐derived hepatocytes faithfully recapitulate the pathophysiology of familial hypercholesterolemia. Hepatology 2012;56:2163‐2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fattahi F, Asgari S, Pournasr B, Seifinejad A, Totonchi M, Taei A, et al. Disease‐corrected hepatocyte‐like cells from familial hypercholesterolemia‐induced pluripotent stem cells. Mol Biotechnol 2013;54:863‐873. [DOI] [PubMed] [Google Scholar]
- 19. Ramakrishnan VM, Yang JY, Tien KT, McKinley TR, Bocard BR, Maijub JG, et al. Restoration of physiologically responsive low‐density lipoprotein receptor‐mediated endocytosis in genetically deficient induced pluripotent stem cells. Sci Rep 2015;5:13231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li HL, Fujimoto N, Sasakawa N, Shirai S, Ohkame T, Sakuma T, et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR‐Cas9. Stem Cell Reports 2015;4:143‐154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, et al. Double nicking by RNA‐guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013;154:1380‐1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jarrett KE, Lee CM, Yeh YH, Hsu RH, Gupta R, Zhang M, et al. Somatic genome editing with CRISPR/Cas9 generates and corrects a metabolic disease. Sci Rep 2017;7:44624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hobbs HH, Russell DW, Brown MS, Goldstein JL. The LDL receptor locus in familial hypercholesterolemia: mutational analysis of a membrane protein. Annu Rev Genet 1990;24:133‐170. [DOI] [PubMed] [Google Scholar]
- 24. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013;339:819‐823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sander JD, Joung JK. CRISPR‐Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 2014;32:347‐355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chu VT, Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, et al. Increasing the efficiency of homology‐directed repair for CRISPR‐Cas9‐induced precise gene editing in mammalian cells. Nat Biotechnol 2015;33:543‐548. [DOI] [PubMed] [Google Scholar]
- 27. Ho CM, Dhawan A, Hughes RD, Lehec SC, Puppi J, Philippeos C, et al. Use of indocyanine green for functional assessment of human hepatocytes for transplantation. Asian J Surg 2012;35:9‐15. [DOI] [PubMed] [Google Scholar]
- 28. Li Y, Lu W, Schwartz AL, Bu G. Degradation of the LDL receptor class 2 mutants is mediated by a proteasome‐dependent pathway. J Lipid Res 2004;45:1084‐1091. [DOI] [PubMed] [Google Scholar]
- 29. Song Z, Cai J, Liu Y, Zhao D, Yong J, Duo S, et al. Efficient generation of hepatocyte‐like cells from human induced pluripotent stem cells. Cell Res 2009;19:1233‐1242. [DOI] [PubMed] [Google Scholar]
- 30. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 2013;8:2281‐2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rodin S, Antonsson L, Hovatta O, Tryggvason K. Monolayer culturing and cloning of human pluripotent stem cells on laminin‐521‐based matrices under xeno‐free and chemically defined conditions. Nat Protoc 2014;9:2354‐2368. [DOI] [PubMed] [Google Scholar]
- 32. Russell DW, Lehrman MA, Sudhof TC, Yamamoto T, Davis CG, Hobbs HH, et al. The LDL receptor in familial hypercholesterolemia: use of human mutations to dissect a membrane‐protein. Cold Spring Harb Symp Quant Biol 1986;51:811‐819. [DOI] [PubMed] [Google Scholar]
- 33. de Ferranti SD, Rodday AM, Mendelson MM, Wong JB, Leslie LK, Sheldrick RC. Prevalence of familial hypercholesterolemia in the 1999 to 2012 United States National Health and Nutrition Examination Surveys (NHANES). Circulation 2016;133:1067‐1072. [DOI] [PubMed] [Google Scholar]
- 34. Widhalm K, Binder CB, Kreissl A, Aldover‐Macasaet E, Fritsch M, Kroisboeck S, et al. Sudden death in a 4‐year‐old boy: a near‐complete occlusion of the coronary artery caused by an aggressive low‐density lipoprotein receptor mutation (W556R) in homozygous familial hypercholesterolemia. J Pediatr 2011;158:167. [DOI] [PubMed] [Google Scholar]
- 35. Raal FJ, Honarpour N, Blom DJ, Hovingh GK, Xu F, Scott R, et al. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double‐blind, placebo‐controlled trial. Lancet 2015;385:341‐350. [DOI] [PubMed] [Google Scholar]
- 36. Wilson JM, Chowdhury NR, Grossman M, Wajsman R, Epstein A, Mulligan RC, et al. Temporary amelioration of hyperlipidemia in low density lipoprotein receptor‐deficient rabbits transplanted with genetically modified hepatocytes. Proc Natl Acad Sci U S A 1990;87:8437‐8441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pakkanen TM, Laitinen M, Hippelainen M, Kallionpaa H, Lehtolainen P, Leppanen P, et al. Enhanced plasma cholesterol lowering effect of retrovirus‐mediated LDL receptor gene transfer to WHHL rabbit liver after improved surgical technique and stimulation of hepatocyte proliferation by combined partial liver resection and thymidine kinase‐‐ganciclovir treatment. Gene Ther 1999;6:34‐41. [DOI] [PubMed] [Google Scholar]
- 38. Herz J, Gerard RD. Adenovirus‐mediated transfer of low density lipoprotein receptor gene acutely accelerates cholesterol clearance in normal mice. Proc Natl Acad Sci U S A 1993;90:2812‐2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Grossman M, Raper SE, Kozarsky K, Stein EA, Engelhardt JF, Muller D, et al. Successful ex vivo gene therapy directed to liver in a patient with familial hypercholesterolaemia. Nat Genet 1994;6:335‐341. [DOI] [PubMed] [Google Scholar]
- 40. Cyranoski D. CRISPR gene‐editing tested in a person for the first time. Nature 2016;539:479. [DOI] [PubMed] [Google Scholar]
- 41. Wesolowska A, Gewartowska M, Olszewski WL, Durlik M. Successful transplantation of hepatocytes requires temporary elimination of scavenger and NK cells, partial hepatectomy and ligation of bile duct. Ann Transplant 2004;9:40‐42. [PubMed] [Google Scholar]
- 42. Tabebordbar M, Zhu K, Cheng JK, Chew WL, Widrick JJ, Yan WX, et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016;351:407‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pankowicz FP, Barzi M, Legras X, Hubert L, Mi T, Tomolonis JA, et al. Reprogramming metabolic pathways in vivo with CRISPR/Cas9 genome editing to treat hereditary tyrosinaemia. Nat Commun 2016;7:12642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cyranoski D. Chinese scientists to pioneer first human CRISPR trial. Nature 2016;535:476‐477. [DOI] [PubMed] [Google Scholar]
- 45. Li HL, Gee P, Ishida K, Hotta A. Efficient genomic correction methods in human iPS cells using CRISPR‐Cas9 system. Methods 2016;101:27‐35. [DOI] [PubMed] [Google Scholar]
- 46. Puppi J, Strom SC, Hughes RD, Bansal S, Castell JV, Dagher I, et al. Improving the techniques for human hepatocyte transplantation: report from a consensus meeting in London. Cell Transplant 2012;21:1‐10. [DOI] [PubMed] [Google Scholar]
- 47. Martin F, Sanchez‐Hernandez S, Gutierrez‐Guerrero A, Pinedo‐Gomez J, Benabdellah K. Biased and unbiased methods for the detection of off‐target cleavage by CRISPR/Cas9: an overview. Int J Mol Sci 2016;17pii:E1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1110/full.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information