

SUMMARY
Whereas phosphorylation of serine, threonine, and tyrosine is exceedingly well characterized, the role of histidine phosphorylation in mammalian signaling is largely unexplored. Here we show that phosphoglycerate mutase family 5 (PGAM5) functions as a phosphohistidine phosphatase that specifically associates with and dephosphorylates the catalytic histidine on Nucleoside Diphosphate Kinase B (NDPK-B). By dephosphorylating NDPK-B, PGAM5 negatively regulates CD4+ T cells by inhibiting NDPK-B mediated histidine phosphorylation and activation of the K+ channel KCa3.1, which is required for TCR stimulated Ca2+ influx and cytokine production. Using recently developed monoclonal antibodies that specifically recognize phosphorylation of nitrogens at the N1 (1-pHis) or N3 (3-pHis) positions of the imidazole ring, we detect for the first time phosphoisoform specific regulation of histidine phosphorylated proteins in vivo, and link these modifications to TCR signaling. These results represent an important step forward in studying the role of histidine phosphorylation in mammalian biology and disease.
Keywords: Histidine phosphorylation, protein histidine phosphatases, PGAM5, T cell activation
eTOC Blurb
Protein histidine phosphorylation in mammals has been poorly defined. Panda et al. identify phosphoglycerate mutase 5 (PGAM5) as a mammalian histidine phosphatase that specifically dephosphorylates NDPK-B on H118 thereby inhibiting 3-pHis phosphorylation and activation of the K+ channel KCa3.1. By inhibiting KCa3.1 channel activation, PGAM5 functions to negatively regulate TCR stimulated Ca2+ influx and pro-inflammatory cytokine production in CD4+ T cells.
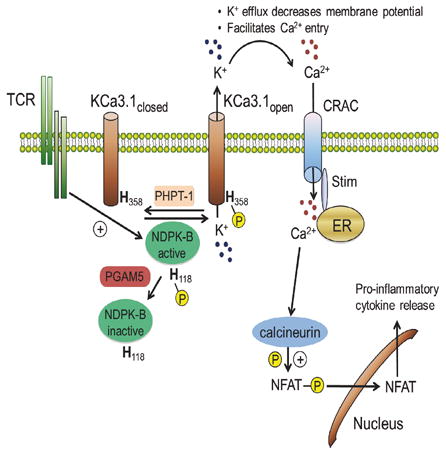
INTRODUCTION
Reversible phosphorylation of proteins modulates their function and thereby regulates virtually all cellular processes. Whereas phosphorylation of serine, threonine and tyrosine are exceedingly well characterized, relatively little is known about phosphorylation of histidine, which may account for as much as ~6% of all incorporation of phosphate into mammalian proteins(Attwood, 2013; Besant and Attwood, 2005; Matthews, 1995; Tan et al., 2002). The identity of the kinases and phosphatases that regulate histidine phosphorylation, their protein targets, and their biological functions have remained obscure.
In contrast to prokaryotes, mammals do not contain two-component histidine kinases and to date only two histidine kinases, nucleoside diphosphate kinase (NDPK)-A (NME1) and NDPK-B (NME2), have been identified(Attwood and Wieland, 2015). NDPKs are encoded by the Nme (non-metastatic cell) gene family and are comprised of 10 family members that are mostly between 16-20 kDa(Boissan et al., 2009). Although most family members are > 80% conserved at the amino acid level, only NDPK-A and -B have been demonstrated to function as histidine kinases(Attwood and Wieland, 2015). Critical to their function as histidine kinases is their ability to auto-phosphorylate on histidine 118, which serves as a high-energy phosphate intermediate that can be transferred either to nucleotide diphosphates or to histidine residues on target proteins, in a manner similar to two-component histidine kinases in prokaryotes. Both nitrogens in the imidazole ring of histidine can be phosphorylated to generate two distinct biologically relevant isomers and include phosphorylation at the N1 position to generate 1-pHis and phosphorylation at the N3 position to generate 3-pHis (Fuhs et al., 2015). The only reported mammalian phosphatase specific for phosphohistidine is the 14 kDa phosphohistidine phosphatase (PHPT-1) (Klumpp and Krieglstein, 2009). NDPKs and PHPT-1 are evolutionarily conserved small proteins that bear no resemblance to serine/threonine or tyrosine kinases or phosphatases.
To gain further insight into the biologic functions and regulation of NDPKs, we sought to identify proteins that interact with NDPK-B. Here, we identify phosphoglycerate mutase family 5 (PGAM5) as an interacting partner of NDPK-B. PGAM5 has previously been shown to localize to the mitochondria where it can function as a serine/threonine phosphatase to dephosphorylate mitochondrial proteins (Chen et al., 2014; Wang et al., 2012). In addition to its mitochondrial localization, PGAM5 also undergoes intramembranous proteolytic cleavage to release a cytosolic pool of PGAM5 (Sekine et al., 2012). In this study, we show that PGAM5 functions as a phosphohistidine phosphatase, which specifically binds and dephosphorylates H118 on NDPK-B leading to inhibition of NDPK-B histidine phosphorylation and activation of KCa3.1, and subsequent T cell receptor (TCR) stimulated Ca2+ influx.
RESULTS
PGAM5 is a histidine phosphatase that specifically binds and dephosphorylates NDPK-B
To identify proteins that regulate NDPK-B, we used a two-step immuno-precipitation (Strep II-IP followed by FLAG-IP) to find proteins that specifically interact with NDPK-B in human embryonic kidney (HEK) 293T cells. Proteins pulled-down exclusively in the NDPK-B IP but not in the control IP (with at least two unique identifying peptides) were identified by mass spectrometry. Among these potential candidates, phosphoglycerate mutase family 5 (PGAM5) was identified (with over 40% sequence coverage) as a top hit that specifically associated with NDPK-B (see Supplemental text for details on mass spectrometry results). PGAM5 is one of 10 members of the phosphoglycerate mutase family that shares a conserved PGAM motif (Jedrzejas, 2000). Many members of this family function as mutases in metabolic pathways and catalyze the transfer of a phosphate group from one position to another on the same metabolite molecule via a phosphohistidine intermediate formed on a conserved histidine residue in the PGAM domain. In contrast, PGAM5 along with STS-1 and 2 are divergent and do not exhibit mutase activity but rather have been shown to function respectively as serine/threonine and tyrosine phosphatases (Carpino et al., 2004; Sadatomi et al., 2013; Wang et al., 2012). PGAM5 and STS-1 and STS-2 utilize a conserved histidine as a phospho-acceptor, with subsequent hydrolyis of the phosphohistidine releasing the free phosphate. This mechanism is very similar to how histidine is used as the phospho-acceptor residue in PHPT-1 (Busam et al., 2006).
We first confirmed the association between PGAM5 and NDPK-B by over-expression in HEK 293T cells. PGAM5 exists in two alternatively spliced isoforms: a longer form (PGAM5-L) and shorter form (PGAM5-S). Both forms contain an amino terminal mitochondrial targeting sequence that localizes PGAM5 to the mitochondria. These isoforms also undergo cleavage in the transmembrane domain between AA 24 and 25 to further generate ∆24 fragment that localizes to the cytosol (Figure S1A) (Sekine et al., 2012). As the myc tag was inserted after AA 56, anti-Myc antibody detected both FL and ∆24 forms of PGAM5, as previously reported (Wang et al., 2012). PGAM5-L (hereafter referred to as PGAM5, unless otherwise stated) specifically interacted with NDPK-B when overexpressed in HEK 293T cells (Figures 1A and S1B). Endogenous PGAM5 and NDPK-B also co-immunopreciptated in HEK 293T cells using specific antibodies but not an isotype control antibody (Figure 1B). The interaction between PGAM5 and NDPK-B was specific. PGAM5 did not interact with the closely related isoforms of NDPK-B, NDPK-A or C, with which NDPK-B shares 88% and 66% amino acid sequence identity, respectively (Figure 1C). In contrast to PGAM5-L, PGAM5-S was localized to a NP-40 insoluble pool as previously reported(Wang et al., 2012), that did not contain NDPK-B protein (Figure S1B). Consistent with this finding, PGAM5-S did not bind or dephosphorylate NDPK-B (Figure S1B).
Figure 1. PGAM5 specifically interacts with NDPK-B and functions as a histidine phosphatase to dephosphorylate H118 on NDPK-B.
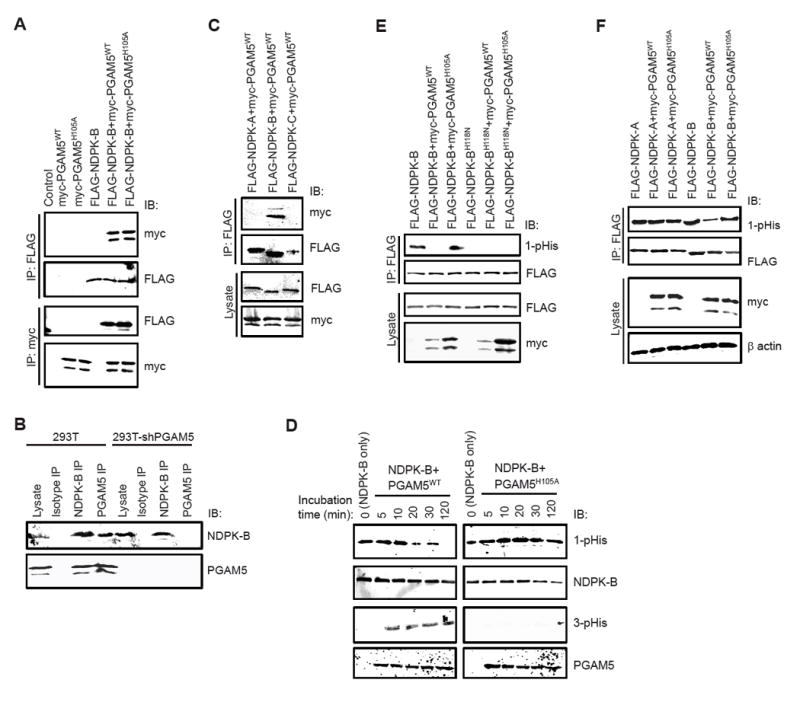
(A) Whole cell lysates from HEK 293T cells over-expressing FLAG-NDPK-B and myc-PGAM5 (WT or H105A mutant) were immuno-precipitated (IP) with anti-FLAG or anti-myc antibodies followed by immunoblotting (IB) with anti-myc or anti-FLAG antibodies as indicated.
(B) Co-IP of endogenous NDPK-B and PGAM5 in HEK 293T cells. Lysates from control or PGAM5 shRNA transfected cells were subjected to IP with isotype control antibody or NDPK-B or PGAM5 antibodies and blots were probed as indicated. The association was specific as demonstrated by the failure of either NDPK-B or PGAM5 to be immunoprecipitated with isotype control antibodies and the failure of anti-PGAM5 antibodies to co-IP NDPK-B in PGAM5 siRNA transfected cells.
(C) FLAG-NDPK-A, NDPK-B, or NDPK-C were transfected into HEK 293T cells together with myc-PGAM5WT and lysates were subjected to anti-FLAG IP followed by IB with anti-myc or anti-FLAG antibodies as shown.
(D) Purified GST-tagged NDPK-BWT was auto-phosphorylated with GTP and then incubated with His-tagged PGAM5WT or PGAM5 mutant (PGAM5H105A) for various times as indicated. Purity of His-tagged PGAM5 proteins was assessed by coomassie blue staining (Figure S2A). Samples were resolved by SDS/PAGE under basic conditions and immunoblotted with anti-1-pHis, 3-pHis, NDPK-B and PGAM5 antibodies as indicated.
(E) FLAG-NDPK-B (WT or H118N) was transfected into 293T cells along with myc-PGAM5WT or PGAM5H105A. Lysates were IP with anti-FLAG antibodies and IB with anti-1-pHis and anti-NDPK-B antibodies. Relative levels of over-expressed PGAM5 and NDPK-B were 3-5 fold higher compared to the endogenous protein levels (Figure S2B).
(F) 293T cells were transfected with FLAG-NDPK-A or NDPK-B along with myc-PGAM5 (WT or H105A mutant). Lysates were probed with anti-myc and anti-β actin antibodies as indicated. Lysates were subjected to anti-FLAG IP and IB with anti-FLAG and anti-1-pHis antibodies (to detect NDPK phosphorylation).
Data are representative of three independent experiments.
See also Figures S1, S2 and S7.
To assess whether PGAM5 can function as a histidine phosphatase and dephosphorylate H118 on NDPK-B, GST-tagged NDPK-B (wild type, WT) was auto-phosphorylated in vitro and then incubated with purified His-tagged PGAM5WT or a phosphatase dead PGAM5 mutant, in which the catalytic histidine was mutated to alanine (PGAM5H105A). Phosphorylation of H118 on NDPK-B was then assessed by immunoblotting with a recently developed anti-1-phosphohistidine (1-pHis) monoclonal antibody (clone SC1-1) (Fuhs et al., 2015); nitrogen (N) at the N1 and N3 position in the imidazole ring of histidine can undergo phosphorylation to generate either 1- or 3-phosphohistidine (1-pHis, 3-pHis)(Fuhs et al., 2015). PGAM5WT, but not PGAM5H105A, dephosphosphorylated NDPK-B (Figure 1D). Consistent with the hypothesis that H105 on PGAM5 functions as the phospho-acceptor that mediated NDPK-B dephosphorylation, we observed time-dependent phosphorylation of H105 on PGAM5WT but not PGAM5H105A, concomitant with the dephosphorylation of NDPK-B using a anti-3-phosphohistidine (3-pHis) monoclonal antibody (clone SC56-2) (Figure 1D). NDPK-B and PGAM5 did not react with a panel of anti-3- and anti-1-pHis antibodies respectively, indicating that each protein was specifically phosphorylated at either the N1 or N3 postion (data not shown). PGAM5WT, but not PGAM5H105A, also dephosphorylated NDPK-B in vivo when co-overexpressed in HEK 293T cells (Figure 1E). The anti-1-pHis antibodies specifically recognized the phosphate group on H118 of NDPK-B as a mutant NDPK-B in which H118 was mutated to asparagine (H118N) was not detected with the anti-1-pHis antibodies (Figure 1E). Dephosphorylation of H118 on NDPK-B by PGAM5 was specific as PGAM5 did not dephosphorylate H118 on NDPK-A under the same conditions (Figure 1F). While PGAM5H105A did not dephosphorylate NDPK-B, it still interacted with NDPK-B (Figure 1A), indicating the interaction between PGAM5 and NDPK-B was independent of PGAM5 enzymatic activity.
By generating a number of PGAM5 truncated mutants (Figure S1C), we identified amino acids 77-88 amino terminal to the PGAM domain to be critical for the inteaction between PGAM5 and NDPK-B (Figure S1D). The inability of PGAM5∆77-88 to bind and dephosphorylate NDPK-B (Figure S1D) or to subsequently inhibit KCa3.1 channel activity (Figure S1E), coupled with our finding that PGAM5 failed to bind and dephosphorylate NDPK-A (Figure 1E), indicates that interaction between PGAM5 and NDPK-B is pre-requisite for PGAM5 to dephosphorylate and inhibit NDPK-B activity.
PGAM5 inhibits NDPK-B mediated 3-pHis phosphorylation and activation of KCa3.1
NDPK-B and PHPT-1 have previously been shown to regulate three protein targets in mammals including the Ca2+-activated K+ channel KCa3.1, TRPV5, and the β subunit of heterotrimeric G proteins (Cai et al., 2014; Hippe et al., 2003; Srivastava et al., 2006b). One of the most important functions for KCa3.1 is to promote an increase in Ca2+ influx via Ca2+ release activated channels (CRAC) after T-cell-receptor (TCR) activation of CD4+ T cells and FcεRI activation of mast cells (Feske et al., 2015). K+ efflux via KCa3.1 maintains a negative membrane potential, which provides the electrical gradient for sustained Ca2+ influx via CRAC and subsequent transcription of cytokines (Feske et al., 2015). Both biochemical and genetic evidence have indicated that NDPK-B is required for KCa3.1 channel activity, by functioning as a histidine kinase that directly phosphorylates H358 located near the carboxyl terminus (CT) of KCa3.1, leading to KCa3.1 activation (Srivastava et al., 2006b). On the other hand, PHPT-1 has been shown to negatively regulate KCa3.1 channel activity and thus activation of CD4+ T cells and mast cells, by dephosphorylating histidine H358 on KCa3.1 (Srivastava et al., 2008). While PHPT-1 inhibits KCa3.1, it does not inhibit NDPK-B (Klumpp and Krieglstein, 2009; Srivastava et al., 2008). This finding raises the possibility that PGAM5, by dephosphorylating NDPK-B, negatively regulates TCR activation of KCa3.1 at a more proximal point in the signaling pathway, by preventing phospho-transfer of the high-energy pH118 intermediate on NDPK-B to H358 in the CT of KCa3.1. Consistent with this hypothesis, shRNA knockdown of PGAM5 in HEK 293T cells overexpressing GFP-KCa3.1 (293-KCa3.1), led to increased 1-pHis phosphorylation of NDPK-B (Figure S3A), which was associated with an increase in KCa3.1 channel activity (Figure S3B), while overexpression of GFP-PGAM5WT but not GFP-PGAM5H105A inhibited KCa3.1 channel activity (Figure S3C).
Similar observations were made in Jurkat T cells overexpressing GFP-KCa3.1 (Jurkat-KCa3.1) (Srivastava et al., 2009), where PGAM5-knockdown increased NDPK-B 1-pHis (but not 3-pHis) phosphorylation (Figure 2A) and increased KCa3.1 channel activity (Figure 2B). The amount of K+ channel current contributed by the K+ channels KCa3.1 and Kv1.3 was quantified by adding the specific KCa3.1 inhibitor TRAM-34 and Kv1.3 inhibitor Shk, as previously reported (Srivastava et al., 2006b). The increase in KCa3.1 channel activity in PGAM5-knockdown Jurkat-KCa3.1 cells was restored to similar levels of control siRNA transfected cells by re-expressing a PGAM5 (WT), but not a PGAM5 phosphatase dead (H105A) siRNA-resistant cDNA, confirming that loss of PGAM5 phosphatase activity accounted for the increase in channel activity (Figure 2C). Increased KCa3.1 channel activity in PGAM5-knockdown cells also resulted in increased TCR-stimulated Ca2+ influx (Figure 2D) and pro-inflammatory cytokine production (Figure 2E). In addition, we now detect for the first time 3-pHis phosphorylation of KCa3.1 in vivo with anti-3-pHis (Figure 2A), but not anti-1pHis antibodies. 3-pHis (but not 1-pHis) KCa3.1 phosphorylation was also markedly increased in PGAM5-knockdown cells, which would be predicted from the increase in KCa3.1 channel activity (Figure 2A).
Figure 2. PGAM5 negatively regulates KCa3.1 activity in Jurkat T cells.
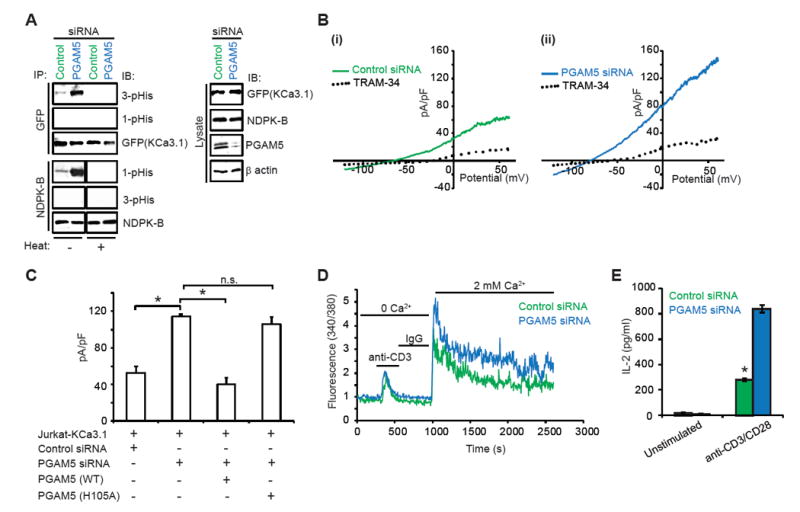
(A) Jurkat T cells over-expressing GFP-KCa3.1 (Jurkat-KCa3.1 cells) were transfected with PGAM5 or control siRNA. Lysates were IP with anti-NDPK-B or anti-GFP (KCa3.1) antibodies and IB with antibodies to 1-pHis, 3-pHis, NDPK-B and GFP antibodies as indicated. Half of the IPs were also heated at 80°C for 2 min prior to loading to ensure specificity of detection of histidine phosphorylation (histidine phosphorylation is heat-sensitive). Lysates were probed with antibodies to GFP (KCa3.1), NDPK-B, PGAM5 (to demonstrate knockdown of PGAM5), and β actin (as a loading control).
(B) KCa3.1 channel activity (TRAM-34 sensitive) was determined by whole-cell patch clamp on Jurkat-KCa3.1 T cells transfected with (i) control or (ii) PGAM5 siRNA. Shown are I/V plots of the cells, as previously described (Srivastava et al., 2006a). TRAM-34 is a specific inhibitor of KCa3.1 channel activity.
(C) Bar graph summary of TRAM-34-sensitive current in cells transfected with control or PGAM5 siRNA and PGAM5-knockdown cells rescued with siRNA-resistant PGAM5 (WT or H105A mutant), plotted at +60 mV (n = 15 to 20 cells each).
(D) Calcium flux was performed in cells re-stimulated with anti-CD3, loaded with Fura-2 AM (5 μM) and attached to a poly-L-lysine-coated coverslip for 20 min. Ca2+ imaging was done with an IX81 epifluorescence microscope (Olympus) and data were analyzed using OpenLab imaging software (Improvision) (n= 80–100 cells for each series), as previously described (Srivastava et al., 2009).
(E) ELISA to quantify IL-2 in the supernatants of control or PGAM5 siRNA transfected cells at 48h post transfection.
Data are representative of three independent experiments. Data are shown as mean±SEM. Statistical significance was calculated using Student’s t test; *(p<0.05); **(p<0.01); n.s. (p>0.05).
See also Figure S3.
To rule out the possibility that PGAM5 may also dephosphorylate and inhibit KCa3.1 and subsequent TCR signaling, we assessed whether PGAM5 could directly dephosphorylate and inhibit KCa3.1 channel activity. We found that PGAM5 did not dephosphorylate KCa3.1 in an in vitro phosphatase assay (Figure 3A) or inhibit KCa3.1 channel activity following activation by NDPK-B in inside-out (I/0) patch experiments (Figure 3B). In contrast, PHPT-1 (a KCa3.1-specific histidine phosphatase) (Srivastava et al., 2008), dephosphorylated KCa3.1 and inhibited KCa3.1 channel activity in the same assays. Taken together, our findings strongly support the hypothesis that endogenous PGAM5 negatively regulates NDPK-B and TCR signaling by specifically dephosphorylating N1 of the imidazole ring of the catalytic H118 on NDPK-B, thereby indirectly inhibiting 3-pHis phosphorylation of KCa3.1and its subsequent activation.
Figure 3. PGAM5 does not directly dephosphorylate or inactivate KCa3.1.
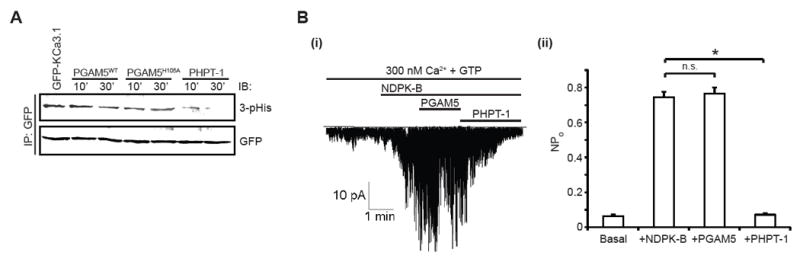
(A) GFP-tagged KCa3.1 was immuno-precipitated by GFP antibody from 293-KCa3.1 GFP cells and incubated with purified His-tagged PGAM5 (WT or H105A mutant) or His-tagged PHPT-1 for various time points as indicated. Samples were resolved by SDS/PAGE under basic conditions and immunoblotted with 3-pHis and GFP antibodies as indicated.
(B) (i) Inside-out (I/O) patches were isolated from 293-KCa3.1 cells. Single channel activity was then recorded in I/O patches that were first incubated with GST-NDPK-B in the presence of 300 nM Ca2+ and GTP. This was followed by addition of His-tagged PGAM5 and His-tagged PHPT-1 to the same patch as indicated in the trace. (ii) Effect of NDPK-B, PGAM5 and PHPT-1 on the open channel probability, NPo. Bar graph represents KCa3.1 NPo as described in (Bi). All recordings in (B) were at +100 mV.
Data are representative of three independent experiments. Data are shown as mean±SEM. Statistical significance was calculated using Student’s t test; *(p<0.05); not significant (n.s.) (p>0.05).
PGAM5 negatively regulates human and mouse CD4+ T cells
Negative regulators of T cells are critical to both set a minimal threshold for T cell activation as well as to provide negative feedback and limit T cell activation (Rangachari and Penninger, 2004). KCa3.1 channels are expressed at low levels on naïve CD4+ T cells (hereafter referred to as naïve cells) but are transcriptionally up-regulated following activation by anti-CD3/CD28 antibodies (Th0 CD4+ T cells; hereafter referred to as Th0 cells) (Ghanshani et al., 2000; Srivastava et al., 2006b). Upon activation, KCa3.1 is then required for TCR-stimulated Ca2+ influx and cytokine production by Th0 cells (Di et al., 2010; Feske et al., 2015). To assess whether PGAM5 limits T cell activation, primary human naïve cells were isolated from buffy coats, transfected with PGAM5 siRNA or scrambled control siRNA, and then activated for 48 hours with anti-CD3/CD28 antibodies to generate Th0 cells. Similar to the results in Jurkat-KCa3.1 T cells, PGAM5-knockdown by siRNA in primary human Th0 cells resulted in increased NDPK-B 1-pHis phosphorylation (Figure 4A), leading to increased KCa3.1 channel activity (Figure 4B), TCR-stimulated Ca2+ influx (Figure 4C) and pro-inflammatory cytokine production (Figures 4D and 4E). Similar results were obtained by transfecting cells with two individual PGAM5 targeting siRNAs (Figure S4). The increase in KCa3.1 channel activity and Ca2+ influx was NDPK-B dependent, as co-transfection of a NDPK-B siRNA together with a PGAM5 siRNA abrogated the increase in KCa3.1 channel activity and TCR mediated Ca2+ influx seen in the PGAM5-knockdown cells (Figures 5A-C).
Figure 4. PGAM5 controls activation of primary human CD4+ T cells.
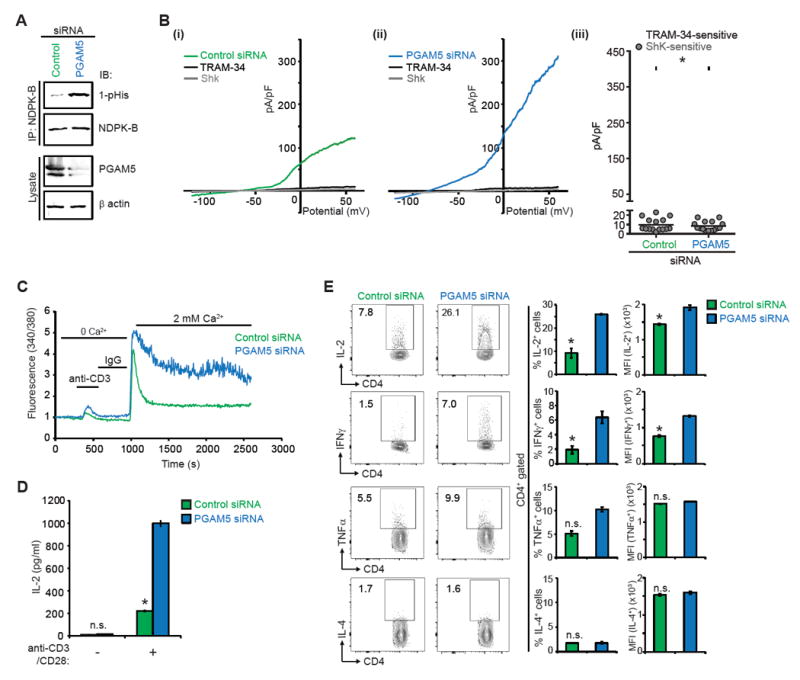
(A-E) Primary human naïve CD4+ T cells were transfected with PGAM5 or control siRNA, rested overnight, and then activated with anti-CD3 and anti-CD28 antibodies for 48h to generate Th0 CD4+ T cells, unless otherwise stated.
(A) Lysates were IP with anti-NDPK-B and NDPK-B phosphorylation was assessed by IB with anti-1-pHis antibodies. Lysates were also probed with anti-PGAM5 and anti-β actin antibodies to check for PGAM5 knockdown.
(B) Whole cell patch clamp was performed on cells to determine KCa3.1 and Kv1.3 activity. Shown are I/V plots of the cells transfected with: (i) control or (ii) PGAM5 siRNA. (iii) Scatter plot of TRAM-34-sensitive current plotted at +60 mV (n = 15 to 20 cells each). TRAM-34 and ShK are specific inhibitors of KCa3.1 and Kv1.3 channel activity, respectively(Srivastava et al., 2006a).
(C) Th0 cells were rested overnight, re-stimulated with anti-CD3, loaded with Fura-2 AM (5 μM) and attached to a poly-L-lysine-coated coverslip for 20 min. Ca2+ imaging was done with an IX81 epifluorescence microscope (Olympus) and data were analyzed using OpenLab imaging software (Improvision) (n= 80–100 cells for each series), as previously described (Srivastava et al., 2009).
(D) Quantification of soluble IL-2 secreted by Th0 cells rested overnight and re-stimulated with anti-CD3/CD28 antibodies for 6h.
(E) Representative intracellular flow cytometry detecting cytokine expression in cells that were rested overnight and re-stimulated with PMA/ionomycin for 4h (left panel). Shown are %positive (middle panel) and mean fluorescence intensity (MFI) (right panel) of cytokine producing cells.
Data are representative of three independent experiments. Data are shown as mean±SEM. Statistical significance was calculated using Student’s t test; *(p<0.05); **(p<0.01); not significant (n.s.) (p>0.05).
See also Figure S4.
Figure 5. PGAM5 counteracts human CD4+ T cell activation through NDPK-B de-phosphorylation.
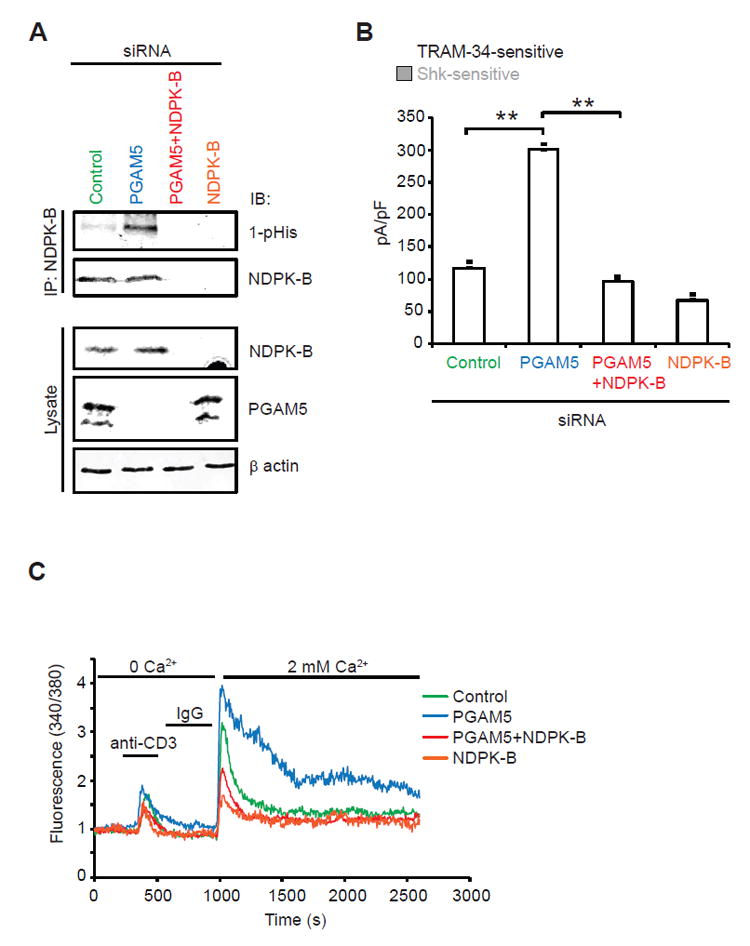
(A-C) Primary human naïve CD4+ T cells were transfected with a control siRNA, siRNA pool to PGAM5, NDPK-B or both, rested overnight, and then activated with anti-CD3 and anti-CD28 antibodies for 48h (to generate Th0 CD4+ T cells).
(A) NDPK-B was IP from cell lysates and IB with antibodies to 1-pHis and NDPK-B to assess NDPK-B 1-pHis phosphorylation. Lysates were IB with anti-NDPK-B and anti-PGAM5 to demonstrate knockdown of each protein and anti-β actin as a loading control.
(B) Whole cell patch clamp of cells as described in Figure 4B. Shown is a bar graph summary of TRAM-34-sensitive and Shk-sensitive currents plotted at +60 mV (n = 15 to 20 cells each).
(C) Ca2+ flux was determined as shown in Figure 4C (n= 80–100 cells for each series). Data are representative of three independent experiments.
Data are shown as mean±SEM. Statistical significance was calculated using Student’s t test; *(p<0.05); **(p<0.01); n.s. (p>0.05).
To further verify the critical negative regulatory role of PGAM5 in TCR signaling in CD4+ T cells, we isolated CD4+ T cells from WT and Pgam5-/- mice. Pgam5-/- mice are born at the expected Mendelian ratio and are phenotypically normal, although they display some features consistent with a parkinsonian movement disorder at 1 year of age due to stabilization of the mitophagy-inducing protein PINK1(Lu et al., 2014). Analysis of lymphocyte subsets from thymus, spleen, and lymph nodes demonstrated that T and B cell development is normal in Pgam5-/- mice (Figure S5). We found that endogenous PGAM5 and NDPK-B also formed a complex in both naïve and Th0 WT CD4+ cells as demonstrated by the co-IP of the two proteins using anti-PGAM5 and NDPK-B antibodies (Figure 6A). Similar to the findings in Jurkat-KCa3.1 and primary human CD4+ T cells, we found increased NDPK-B 1-pHis and KCa3.1 3-pHis phosphorylation (Figure 6B), increased KCa3.1 channel activity (Figure 6C), increased TCR-mediated Ca2+ influx (Figure 6D) and pro-inflammatory cytokine production (Figure 6E) in Pgam5-/- CD4+ T cells when compared with WT CD4+ T cells.
Figure 6. PGAM5 negatively regulates activation of mouse CD4+ T cells.
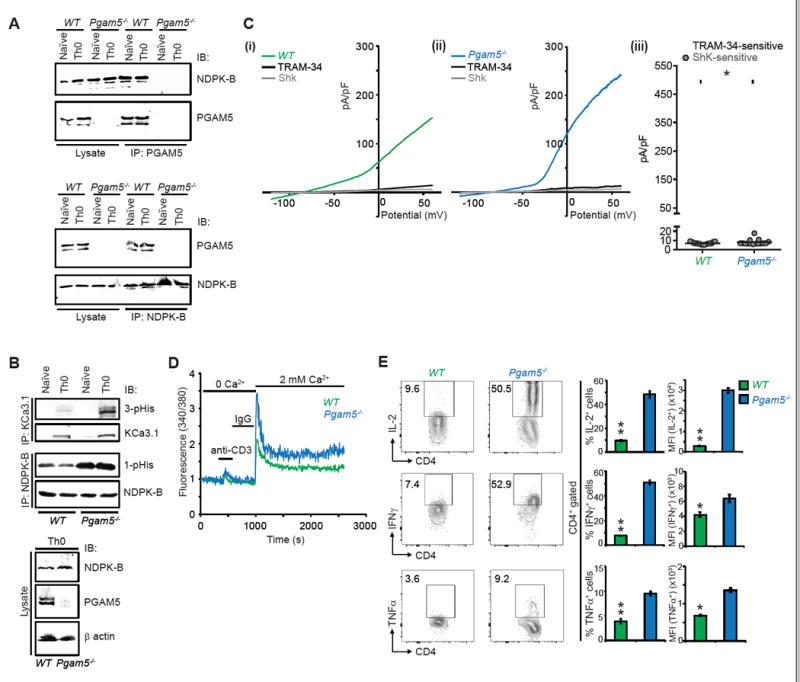
(A-E) Naïve CD4+ T cells were isolated from spleens of wild type (WT) or Pgam5-/- mice and activated with anti-CD3 and anti-CD28 antibodies for 48h to generate Th0 cells.
(A) Naïve or Th0 WT or Pgam5-/- cell lysates were subjected to IP with anti-NDPK-B and anti-PGAM5 antibodies and IB as indicated. The failure of anti-PGAM5 antibodies to co-IP NDPK-B from Pgam5-/- naïve and Th0 cells demonstrates the interaction is specific as PGAM5 needs to be present for anti-PGAM5 antibodies to co-IP NDPK-B.
(B) NDPK-B and KCa3.1 were IP from WT and Pgam5-/- naïve or Th0 cell lysates and probed with antibodies to 1-pHis and NDPK-B for the NDPK-B IP, and 3-pHis and KCa3.1 for the KCa3.1 IP. KCa3.1 is lowly expressed in naïve cells and is induced following activation with anti-CD3/CD28 antibodies (Th0 cells). Th0 cell lysates were blotted with NDPK-B, PGAM5 and β actin as a loading control.
(C) Whole cell patch clamp of Th0 cells to determine KCa3.1 activity as described in Figure 2B. Shown are I/V plots of (i) WT and (ii) Pgam5-/- cells and (iii) Scatter plot of TRAM-34 and Shk sensitive currents (n = 15-20 cells each).
(D) Th0 cells were rested overnight and then re-stimulated with anti-CD3 for measuring calcium flux as described in Figure 2C (n=80–100 cells for each series).
(E) Representative intracellular flow cytometry detecting cytokine expression of WT or Pgam5-/- CD4+ T cells. Th0 cells were rested overnight and re-stimulated with PMA/ionomycin for 4h (left panel). Shown are %positive (middle panel) and mean fluorescence intensity (MFI) (right panel) of cytokine producing cells.
Data are representative of three independent experiments. Data are shown as mean±SEM (n=3 mice per group per experiment). Statistical significance was calculated using Student’s t test; *(p<0.05); **(p<0.01); n.s. (p>0.05).
See also Figure S5.
Pgam5-/- T cells cause an accelerated and more severe GvHD in mice
Graft versus host disease (GvHD) following allogeneic bone marrow transplants provides a rapid and robust preclinical model to assess T cell function in vivo (Vaeth et al., 2015). We found that mice that received allogeneic transfer of CD4+ T cells from Pgam5–/– mice exhibited more severe GvHD pathology (Figures 7A and 7B) that highly correlated with increased serum levels of the cytokines IFN-γ and TNF-α (Figure 7C). As a consequence of augmented cytokine production in Pgam5–/– T cells, Pgam5–/– host mice had significantly lower survival rates (Figure 7D), as compared to the mice that received WT T cells. In addition, Pgam5–/– CD4+ T cells recovered from mice had a marked increase in anti-CD3-mediated Ca2+ flux (Figure 7E) and cytokine production (Figure S6), as compared to WT control CD4+ T cells.
Figure 7. Pgam5-/- T cells cause augmented pathogenicity after allogeneic transplantation.
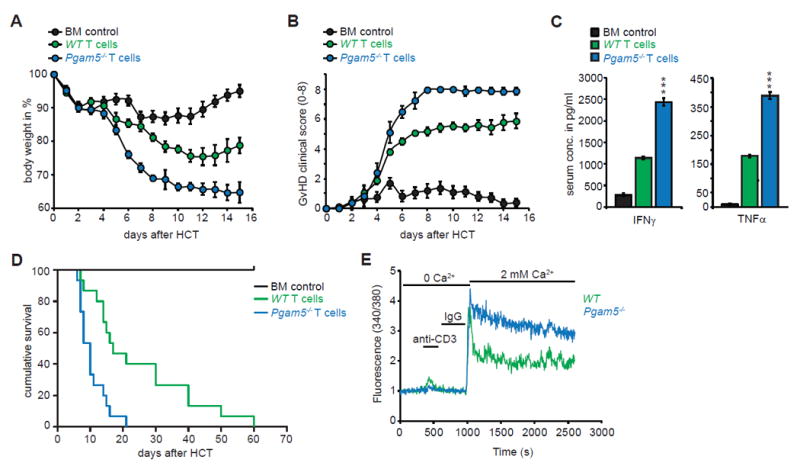
(A-E) Host BALB/c mice (H-2d) were lethally irradiated and transplanted with either 1.5 × 106 allogeneic WT or Pgam5-/- T cells isolated from spleens of donor C57BL/6 (H-2b), along with 5 × 106 bone marrow (BM) cells. BM control group received only BM cells.
(A) Body weight changes of host mice groups post allogeneic-hematopoietic cell transfer (HCT) (n=15 mice per group).
(B) Clinical GvHD scoring (score 0-8) of mice groups post HCT (n=15 mice per group).
(C) Serum IFNγ and TNFα analysis at day 6 post HCT using ELISA. Data are compiled from two independent experiments (n=5 mice per group per experiment).
(D) Survival of mice groups post HCT (n=15 mice per group).
(E) CD4+ T cells were purified from spleens of host mice at day 6 post HCT and calcium flux was performed as described in Figure 2C (n= 80–100 cells for each series).
Data shown is representative of two independent experiments. Statistical significance was calculated using Student’s t test (Figure 4C); ***(p<0.001) and log-rank (Mantel-Cox) test (Figure 4D); ****(p<0.0001).
See also Figure S6.
DISCUSSION
Studying histidine phosphorylation in mammals has been difficult due to challenges in identifying mammalian histidine kinases and phosphatases and the lack of specific reagents to detect and monitor changes in histidine phosphorylation. In contrast to tyrosine and serine/threonine phosphorylation, histidine phosphorylation is heat and acid labile. Consequently, the use of standard approaches to study protein phosphorylation (e.g. the use of phosphatase inhibitors and IMAC or phospho-specific antibodies to preserve and enrich phospho-peptides prior to phospho-site identification by proteomics) is inadequate (Attwood et al., 2007; Besant and Attwood, 2005; Kee and Muir, 2012; Kee et al., 2010). In this report, we provide several insights that further our understanding of histidine phosphorylation and provide a framework that should greatly facilitate future studies to identify new roles for histidine phosphorylation in mammalian biology and disease. We have now identified only the second histidine phosphatase PGAM5, its protein substrate, and provide insight into the mechanisms whereby PGAM5 functions as a histidine phosphatase to specifically dephosphorylate NDPK-B. In addition, using recently generated monoclonal antibodies that specifically recognize 1- and 3-pHis phosphorylated proteins, we have now been able to detect for the first time phosphoisoform specific regulation of pHis phosphorylated proteins in vivo in the context of physiological stimuli.
Two isoforms of PGAM5 exist, with a long form denoted PGAM5-L and a shorter form (PGAM5-S) in which 50 amino acids in the CT are replaced with 16 amino acids from an alternately spliced exon(Lo and Hannink, 2006)(see Figure S1A). Both isoforms contain an amino-terminal mitochondrial targeting motif and have previously been shown to localize to both the outer and inner mitochondria membrane (Chen et al., 2014; Lu et al., 2014; Wang et al., 2012) where PGAM5 has been associated with the regulation of mitochondrial dynamics including mitophagy, mitochondrial fragmentation and fission, and both necrotic and apoptotic cell death. PGAM5 mediates at least some of these effects by functioning as a serine/threonine phosphatase to dephosphorylate mitochondrial localized proteins including the mitophagy receptor FUNDC1(Chen et al., 2014) and dynamin related protein 1 (DRP1), which is required for mitochondrial fission (Wang et al., 2012). In contrast to these known functions, we now demonstrate that PGAM5-L, but not PGAM5-S which is localized to a SDS insoluble fraction in the mitochondria, also acts as a phosphohistidine phosphatase and specifically associates with and inhibits NDPK-B by dephosphorylating the N1 position of pH118. By dephosphorylating NDPK-B, PGAM5 inhibits KCa3.1 channel activity and the subsequent stimulation of TCR-mediated Ca2+ influx and cytokine production by blocking the phospho-transfer of pH118 on NDPK-B to H358 on KCa3.1 which is required for KCa3.1 channel activation. Thus, PGAM5-L is a negative regulator of CD4+ T cell activation.
In addition to functioning in the mitochondria, a fraction of PGAM5 is cleaved between amino acids 24 and 25 to release a cytosolic form (Sekine et al., 2012). We also detect PGAM5-L (∆24) in all cells we have tested including 293 cells and human and mouse CD4+ T cells used in these studies. We think that cytosolic form of PGAM5-L, PGAM5 (∆24), likely mediates dephosphorylation and inhibition of NDPK-B. This is based on the finding that NDPK-B is predominantly cytosolic and functions at the plasma membrane to phosphorylate and activate KCa3.1. In addition, we find that in contrast to wild type PGAM5, overexpression of protease cleavage resistant mutant PGAM5 (PGAM5S24F) does not dephosphorylate NDPK-B (Figure S7A) or inhibit KCa3.1 channel activity (Figure S7B). The cytosolic PGAM5 (∆24) is likely generated by an intramembranous protease localized to the mitchondria and in this regard the rhomboid protease PARL is one candidate that can function as a PGAM5 protease under some conditions(Sekine et al., 2012). Important questions to be addressed in future studies are the identity of the intramembranous PGAM5 protease(s) in CD4+ T cells that cleaves PGAM5-L and mechanisms for its regulation.
Our studies also provide insight into how histidine phosphatase specificity is governed. While PGAM5 dephosphorylates and inhibits NDPK-B, it is unable to dephosphorylate and inhibit KCa3.1. In contrast, PHPT-1 dephosphorylates and inhibits KCa3.1, but not NDPK-B. Direct binding of each phosphatase to their respective target is critical for determining specificity. With regard to PGAM5, we identified 12 amino acids N-terminal to the PGAM domain that mediates binding to NDPK-B and which is required for PGAM5 to dephosphorylate NDPK-B. Further evidence for the importance of binding for PGAM5 to dephosphorylate pHis phosphorylated proteins is shown by the failure of PGAM5 to both bind and dephosphorylate NDPK-A, despite NDPK-B and NDPK-A sharing 88% sequence identity. We have previously shown that PHPT-1 associates with KCa3.1 by binding 14 amino acids in the carboxy-terminus of KCa3.1 and this interaction is likely critical for PHPT-1 to dephosphorylate KCa3.1(Srivastava et al., 2006b). These findings have several important implications. First, it suggests that approaches to identify proteins that bind each of these phosphatases will be a powerful tool to uncover new pHis containing substrates for both PGAM5 and PHPT-1 and potentially uncover new biological roles for histidine phosphorylation. Second, these findings highlight the critical and non-redundant role for histidine phosphatases to fine tune TCR activation of CD4+ T cells by functioning to regulate distinct targets in the signaling pathway from the TCR to KCa3.1 activation (Figure S7C), which is further supported by the in vivo experiments showing that Pgam5-/- T cells elicit much more severe GVHD in vivo.
The recent generation of monoclonal antibodies that specifically recognize 1- and 3-pHis phosphorylated proteins is a major advance in studying histidine phosphorylation and has enabled us to answer several questions that until now could not be addressed. Our ability to selectively detect N3 phosphorylation of KCa3.1 with anti-3-pHis antibodies is the first case where both a histidine protein kinase substrate as well as the specific phosphoisoform modified has been detected in vivo in the context of a physiological stimulus. In contrast, previous studies of pHis phosphorylation have predominantly identified pHis enzyme intermediates where pHis plays a catalytic role in enzyme activity as has been described for ATP citrate lyase, PGAM1 and succinyl-CoA synthetase (Fuhs et al., 2015; Klumpp et al., 2003; Vander Heiden et al., 2010; Wagner and Vu, 1995). The anti-pHis antibodies have also enabled us to directly measure changes in NDPK-B pHis phosphorylation by PGAM5 in vivo, the specific phosphoisoform dephosphorylated by PGAM5 as well as PHPT-1, and the mechanism whereby PGAM5 functions as a histidine phosphatase. The ability of PGAM5 to dephosphorylate 1-pHis on NDPK-B and PHPT-1 to dephosphorylate 3-pHis on KCa3.1 raises the possibility that each phosphatase will specifically dephosphorylate one or the other phosphoisoform, although assessing the phosphoisoform of other PGAM5 and PHPT-1 substrates when known, as well as co-crystal structures of PGAM5 and PHPT-1 with specific phosphohistidine peptide analogs will ultimately be required to test this idea. In addition, we found that concomitant with NPDK-B dephosphorylation, PGAM5 becomes 3-pHis phosphorylated on the analogous histidine that functions as the catalytic histidine in other PGAM family members that contain mutase activity. It remains to be determined if the phosphate from histidine (pHis) is then spontaneously released from PGAM5 or even whether PGAM5 can function as a phosphate donor to phosphorylate other proteins.
While our studies have focused on the role of PGAM5 and pHis phosphorylation in the regulation of TCR activation of T cells, our discoveries likely represent only the “tip of the iceberg” for new roles of pHis signaling in mammalian biology. It is anticipated that with the identification of additional histidine kinases and phosphatase, such as PGAM5, the mechanisms whereby pHis phosphorylation is regulated, coupled with the potential held by these anti-1- and 3-pHis antibodies to detect pHis phosphorylated proteins, additional roles for pHis phosphorylation in mammalian biology and disease will quickly be realized.
EXPERIMENTAL PROCEDURES
Mice
Pgam5-/- mice have previously been described(Lu et al., 2014). Male BALB/c (H-2d) mice were purchased from Jackson laboratories. WT and Pgam5-/- mice were on C57BL/6 background (H-2b). 6-8 week old male littermates were used for the experiments. All mice were bred and housed in the specific pathogen-free facility at the Skirball Institute of Biomolecular Medicine and used in accordance with institutional guidelines.
Tandem immuno-precipitation and mass spectrometry
Human embryonic kidney (HEK293T) cells were transiently transfected with FLAG-Strep II-tagged NDPK-B or a control plasmid using calcium chloride. Lysis of cell pellets was carried out at 48h post-transfection using the lysis buffer (50 mM Tris, pH 8, 100 mM NaCl, 10% glycerol, 1 mM EDTA, 5 mM MgCl2, 0.5% NP-40 along with freshly added 1 mM DTT, protease and phosphatase inhibitors. The first immuno-precipitation was carried out with Strep-Tactin Sepharose beads (2h, 4°C, rocking). Strep II-tag immuno-precipitates were then incubated with 1X Desthiobiotin elution buffer for 30 min at room temperature to elute the immuno-precipitated proteins. Next, the eluted proteins were incubated with anti-FLAG M2 beads (2h, 4°C, rocking) and FLAG immuno-precipitates were incubated with 3X FLAG peptide (100 μg/ml) for 30 min at room temperature to elute the immuno-precipitated proteins. Two independent tandem immuno-precipitation experiments were performed.
The eluted proteins were reduced with DTT, alkylated with iodoacetamide and loaded onto an SDS/PAGE gel for in gel digestion. The resulting tryptic peptides were analyzed by nanoflow LC-MS using a data dependent acquisition method on an Orbitrap Elite mass spectrometer, as previously described(Drummond et al., 2015). The MS/MS spectra were searched against the Uniprot homo sapiens database using Sequest within Proteome Discoverer (ThermoFisher). The results were filtered using a 1% False Discovery Rate (FDR) searched against a decoy database and proteins with less than two unique peptides were filtered. Proteins identified in the control IP were subtracted from the proteins identified in the NDBK-B IP and a shortened list was interrogated for potential NDBK-B interacting proteins. See also supplemental text for additional details on methodology.
Immunoprecipitation and western blot assays
FLAG-NDPK constructs (-A, -B or -C) and myc-PGAM5 (WT or H105A mutant), GFP-PGAM5 (full-length, ∆24, ∆77, ∆98, ∆77-88) constructs or PGAM5-GFP (WT or S24F) constructs were transfected into HEK293T cells using calcium chloride transfection method. Cells were lysed in the lysis buffer (50 mM Tris, pH 8, 100 mM NaCl, 10% glycerol, 1 mM EDTA, 5 mM MgCl2, 0.5% NP-40 along with freshly added 1 mM DTT, protease and phosphatase inhibitors) for 30 min on ice and spun down at 13,200 rpm for 10 min at 4°C. The supernatant collected was labelled as lysate. The proteins in the precipitate were extracted by SDS containing buffer (labelled as SDS-soluble fraction), as previously described(Wang et al., 2012). Lysates were immuno-precipitated with anti-FLAG or anti-GFP antibodies as indicated. After washing three times with the lysis buffer, bound proteins were eluted using SDS loading buffer (pH 8.8), separated by SDS/PAGE (pH 8.8) and immunoblotted with antibodies as indicated.
In vitro kinase/phosphatase assay
GST-NDPK-B (WT or H118N mutant) and His-PGAM5∆77 (WT or H105A mutant) were expressed in E. coli and purified, as previously described (Lee et al., 1993; Wang et al., 2012). GST-NDPK-B was phosphorylated by incubating with GTP in kinase buffer (50 mM triethanolamine hydrochloride (pH 7.4), 150 mM NaCl, 2 mM MgCl2, 1 mM EDTA, 1 mM DTT) for 30 min at room temperature. Free GTP was removed from the solution by size-exclusion columns (10 kDa molecular weight cutoff). Phosphorylated NDPK-B was then incubated with His-PGAM5 (WT or H105A mutant) in kinase buffer for various periods of time as indicated. The reaction was stopped by adding sample buffer (pH 8.8) and NDPK-B phosphorylation was assessed by immunoblotting with anti-1-pHis and NDPK-B antibodies. Results are representative of three independent experiments.
CD4+ T cell isolation and silencing of PGAM5 and NDPK-B
Human naïve CD4+ T cells were isolated from peripheral blood buffy coats (NY Blood Center) from healthy adult donors using the CD4+ T cell isolation kit from Miltenyi Biotec according to manufacturer’s protocol. Mouse naïve CD4+ T cells were isolated from spleens of 6-8 week old WT and Pgam5-/- mice using the mouse CD4+ T cell isolation kit from Miltenyi Biotec according to manufacturer’s protocol. We routinely obtained >95% CD4+ T cells as assessed by flow cytometry. To silence PGAM5 or NDPK-B in human CD4+ T cells, naïve cells were electroporated with siRNAs as indicated using Amaxa nucleofector kit and reagents (Amaxa Biosystems) according to manufacturer’s protocol. For PGAM5-knockdown in Figure 4, data from co-transfecting two PGAM5 targeting siRNAs are shown (product names: Hs_PGAM5_2 and Hs_PGAM5_10, Qiagen). Data shown in Figure 5 is from transfecting a pool of four siRNAs (product names: Hs_MG5352_3, Hs_PGAM5_2, Hs_PGAM5_9 and Hs_PGAM5_10) and in Figure S4 is from individual siRNAs (Hs_PGAM5_2 or Hs_PGAM5_10, Qiagen). For NDPK-B-knockdown, a SMART pool combination of four siRNAs (Dharmacon) was used as previously described (Srivastava et al., 2006b). Cells were rested overnight to allow recovery, followed by activation with anti-CD3 (5μg/ml) and CD28 (2μg/ml) antibodies for 48h, to generate Th0 cells. PGAM5 or NDPK-B knockdown was assessed in Th0 cells by immunoblotting with anti-PGAM5 and anti-NDPK-B antibodies. All experiments were performed at least three times with independently isolated cells.
Whole cell patch clamp
CD4+ T cells: Whole cell patch clamp was performed on primary human or mouse (isolated from WT or Pgam5-/- mice spleens) naïve cells after stimulation with anti-CD3 (5μg/ml) and CD28 (2μg/ml) antibodies for 48h, to generate Th0 cells, as previously described (Srivastava et al., 2008).
Jurkat-KCa3.1 T cells and 293-KCa3.1 cells: Whole cell patch clamp on cells was performed as previously described(Srivastava et al., 2006a). Data are representative of at least three independent experiments.
Retroviral transduction for siRNA rescue in Jurkat-KCa3.1 cells
Retroviral production and infection were performed as previously described (Volk et al., 2014). PGAM5 (WT or H105A) siRNA-resistant cDNA was cloned into MIGR1 retroviral plasmid using XhoI-EcoRI restriction sites. High viral titer of siRNA-resistant PGAM5 (WT or H105A) expressing retrovirus were produced by co-transfecting 293 cells with the retroviral vector and packaging and envelope vectors using the calcium chloride transfection method. Retroviral supernatants were harvested 24 and 48h post transfection. Jurkat-KCa3.1 cells were transduced with the virus by adding the supernatant and spinning at 2000 rpm, 32°C, for 4 h. Cells were incubated under normal culture conditions (37°C, 5% CO2) and KCa3.1 channel activity was measured by whole cell patch clamp on transduced cells (GFP+) 24h post transduction.
I/O Patch Clamping to assess KCa3.1 channel activity
Single-channel activity was recorded on 293 cells stably transfected with KCa3.1 using the inside-out (I/O) patch-clamp method with an Axopatch-200B amplifier (Axon Instruments, Foster City, California), as previously described(Srivastava et al., 2006b). Data were analyzed using pClamp 9.0 (Axon Instruments) and Origin 7.0 (Microcal) software.
Intracellular calcium activity (Calcium flux)
Cells were loaded with 5 μM fura-2/AM (Molecular Probes) in RPMI 1640 medium and 10% FBS for 30 min at RT. Cells were then attached to a poly-L-lysine-coated coverslip for 20 min, and Ca2+ imaging was done with an IX81 epifluorescence microscope (Olympus) and OpenLab imaging software (Improvision), as described earlier (Srivastava et al., 2009). For single-cell analysis, 340/380 nm fura-2 emission ratios of >100 cells per experiment were analyzed. Background fluorescence obtained from regions containing no cells was digitally subtracted from each image. To compare the different groups, the 340/380 nm ratio was normalized to 1 by dividing the fluorescence values at different time points to the cellular fluorescence at time 0. Experiments were repeated at least 3 independent times.
Bone marrow (BM) and T cell isolation for the allogeneic transfer experiments
For isolating the BM cells, femur and tibia bones were excised from the donor (WT or Pgam5-/- littermates) mice. Bones were cut at the ends and flushed with PBS containing 0.1% BSA through a 70 μm cell strainer to isolate the cells. For isolating T cells, spleen and lymph nodes were isolated, mashed, and passed through a 70 μm cell strainer with erythrocyte lysis buffer (Lonza, Cat # 10-548E). The cells were washed twice with PBS containing 0.1% BSA and T cells were purified using the EasySep™ mouse T cell isolation kit (Stemcell, Cat # 19851) according to manufacturer’s instructions. Purity of T cell population was typically >95%.
Allogeneic hematopoietic cell transplantation (allo-HCT)
BALB/c host mice (H-2d) were conditioned by myeloablative total body irradiation (TBI) using a split dose of total 8.0 Gy. After 2h, mice were injected retro-orbitally with sex-matched 5 × 106 C57BL/6 (H-2b) WT BM cells with or without 1.5 × 106 C57BL/6 T cells (from WT or Pgam5-/- littermates). Mice were treated with antibiotic-containing drinking water for 7 days before and after allo-HCT to prevent non-specific infections. Transplanted mice were monitored daily for weight loss and clinical scoring for GvHD symptoms, as previously described (Vaeth et al., 2015).
Statistical analysis
All data are shown as mean ± SEM and represent combined data from at least two or three independent experiments as mentioned. All data were analyzed using the Graph Pad Prism 5 software. Figures were prepared using Adobe Illustrator. Differences between the individual groups were analyzed for statistical significance by two-tailed Student’s t test (*=p<0.05, **=p<0.01, n.s.=p>0.05; not significant) or log-rank (Mantel Cox) test (****p<0.0001) as indicated in the figure legends.
Supplementary Material
Highlights.
PGAM5 is a mammalian histidine phosphatase that specifically dephosphorylates NDPK-B
pHis antibodies detect NDPK-B and KCa3.1 phosphorylation in vitro and in vivo
PGAM5 decreases KCa3.1 activity, Ca2+ influx, cytokine production in CD4+ T cells
Pgam5-/- T cells cause accelerated and more severe GvHD in mice
Acknowledgments
We thank Dr. Michael Lenardo and Dr. Wei Lu (NIH) for kindly providing the Pgam5–/– mice. We thank Dr. Zhigao Wang (UT Southwestern) for providing the myc-PGAM5 and PGAM5 shRNA constructs. We thank Drs. Stefan Feske (Department of Pathology, NYU) and Susan Schwab (Skirball Institute of Biomolecular Medicine) for providing antibodies and advice. We thank Dr. Beatrix Ueberheide (Proteomics Resource Center, NYU) for mass-spectrometric analysis. This work is supported by a grant from the NIH to E.Y.S. (GM099873), and to T.H. (CA14195, CA80100, CA82683 and CA194584). T.H. is a Frank and Else Schilling American Cancer Society Professor, and holds the Renato Dulbecco Chain in Cancer Research.
Footnotes
AUTHOR CONTRIBUTIONS:
S.P., T.H. and E.Y.S. conceived the study and designed the experiments. S.P. performed most of the experiments. S.S. performed whole cell patch clamp and calcium flux experiments. Z.L. performed sub-cloning to create plasmid constructs used for in vitro experiments. M.V. helped in performing GvHD experiments and scoring the mice phenotype. S.R.F. and T.H. generated anti-phospho His antibodies. S.P. and E.Y.S. wrote the manuscript with input from all the authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Attwood PV. Histidine kinases from bacteria to humans. Biochem Soc Trans. 2013;41:1023–1028. doi: 10.1042/BST20130019. [DOI] [PubMed] [Google Scholar]
- Attwood PV, Piggott MJ, Zu XL, Besant PG. Focus on phosphohistidine. Amino Acids. 2007;32:145–156. doi: 10.1007/s00726-006-0443-6. [DOI] [PubMed] [Google Scholar]
- Attwood PV, Wieland T. Nucleoside diphosphate kinase as protein histidine kinase. Naunyn Schmiedebergs Arch Pharmacol. 2015;388:153–160. doi: 10.1007/s00210-014-1003-3. [DOI] [PubMed] [Google Scholar]
- Besant PG, Attwood PV. Mammalian histidine kinases. Biochim Biophys Acta. 2005;1754:281–290. doi: 10.1016/j.bbapap.2005.07.026. [DOI] [PubMed] [Google Scholar]
- Boissan M, Dabernat S, Peuchant E, Schlattner U, Lascu I, Lacombe ML. The mammalian Nm23/NDPK family: from metastasis control to cilia movement. Mol Cell Biochem. 2009;329:51–62. doi: 10.1007/s11010-009-0120-7. [DOI] [PubMed] [Google Scholar]
- Busam RD, Thorsell AG, Flores A, Hammarstrom M, Persson C, Hallberg BM. First structure of a eukaryotic phosphohistidine phosphatase. The Journal of biological chemistry. 2006;281:33830–33834. doi: 10.1074/jbc.C600231200. [DOI] [PubMed] [Google Scholar]
- Cai X, Srivastava S, Surindran S, Li Z, Skolnik EY. Regulation of the epithelial Ca2+ channel TRPV5 by reversible histidine phosphorylation mediated by NDPK-B and PHPT1. Mol Biol Cell. 2014;25:1244–1250. doi: 10.1091/mbc.E13-04-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpino N, Turner S, Mekala D, Takahashi Y, Zang H, Geiger TL, Doherty P, Ihle JN. Regulation of ZAP-70 activation and TCR signaling by two related proteins, Sts-1 and Sts-2. Immunity. 2004;20:37–46. doi: 10.1016/s1074-7613(03)00351-0. [DOI] [PubMed] [Google Scholar]
- Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, Huang L, Zhou C, Cai X, Fu C, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Molecular cell. 2014;54:362–377. doi: 10.1016/j.molcel.2014.02.034. [DOI] [PubMed] [Google Scholar]
- Di L, Srivastava S, Zhdanova O, Ding Y, Li Z, Wulff H, Lafaille M, Skolnik EY. Inhibition of the K+ channel KCa3.1 ameliorates T cell-mediated colitis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:1541–1546. doi: 10.1073/pnas.0910133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond ES, Nayak S, Ueberheide B, Wisniewski T. Proteomic analysis of neurons microdissected from formalin-fixed, paraffin-embedded Alzheimer’s disease brain tissue. Scientific reports. 2015;5:15456. doi: 10.1038/srep15456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S, Wulff H, Skolnik EY. Ion channels in innate and adaptive immunity. Annu Rev Immunol. 2015;33:291–353. doi: 10.1146/annurev-immunol-032414-112212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhs SR, Meisenhelder J, Aslanian A, Ma L, Zagorska A, Stankova M, Binnie A, Al-Obeidi F, Mauger J, Lemke G, et al. Monoclonal 1- and 3-Phosphohistidine Antibodies: New Tools to Study Histidine Phosphorylation. Cell. 2015;162:198–210. doi: 10.1016/j.cell.2015.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanshani S, Wulff H, Miller MJ, Rohm H, Neben A, Gutman GA, Cahalan MD, Chandy KG. Up-regulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences. The Journal of biological chemistry. 2000;275:37137–37149. doi: 10.1074/jbc.M003941200. [DOI] [PubMed] [Google Scholar]
- Hippe HJ, Lutz S, Cuello F, Knorr K, Vogt A, Jakobs KH, Wieland T, Niroomand F. Activation of heterotrimeric G proteins by a high energy phosphate transfer via nucleoside diphosphate kinase (NDPK) B and Gbeta subunits. Specific activation of Gsalpha by an NDPK B.Gbetagamma complex in H10 cells. The Journal of biological chemistry. 2003;278:7227–7233. doi: 10.1074/jbc.M210305200. [DOI] [PubMed] [Google Scholar]
- Jedrzejas MJ. Structure, function, and evolution of phosphoglycerate mutases: comparison with fructose-2,6-bisphosphatase, acid phosphatase, and alkaline phosphatase. Progress in biophysics and molecular biology. 2000;73:263–287. doi: 10.1016/s0079-6107(00)00007-9. [DOI] [PubMed] [Google Scholar]
- Kee JM, Muir TW. Chasing phosphohistidine, an elusive sibling in the phosphoamino acid family. ACS Chem Biol. 2012;7:44–51. doi: 10.1021/cb200445w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee JM, Villani B, Carpenter LR, Muir TW. Development of stable phosphohistidine analogues. J Am Chem Soc. 2010;132:14327–14329. doi: 10.1021/ja104393t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumpp S, Bechmann G, Maurer A, Selke D, Krieglstein J. ATP-citrate lyase as a substrate of protein histidine phosphatase in vertebrates. Biochem Biophys Res Commun. 2003;306:110–115. doi: 10.1016/s0006-291x(03)00920-3. [DOI] [PubMed] [Google Scholar]
- Klumpp S, Krieglstein J. Reversible phosphorylation of histidine residues in proteins from vertebrates. Sci Signal. 2009;2:pe13. doi: 10.1126/scisignal.261pe13. [DOI] [PubMed] [Google Scholar]
- Lee CH, Li W, Nishimura R, Zhou M, Batzer AG, Myers MG, Jr, White MF, Schlessinger J, Skolnik EY. Nck associates with the SH2 domain-docking protein IRS-1 in insulin-stimulated cells. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:11713–11717. doi: 10.1073/pnas.90.24.11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo SC, Hannink M. PGAM5, a Bcl-XL-interacting protein, is a novel substrate for the redoxregulated Keap1-dependent ubiquitin ligase complex. The Journal of biological chemistry. 2006;281:37893–37903. doi: 10.1074/jbc.M606539200. [DOI] [PubMed] [Google Scholar]
- Lu W, Karuppagounder SS, Springer DA, Allen MD, Zheng L, Chao B, Zhang Y, Dawson VL, Dawson TM, Lenardo M. Genetic deficiency of the mitochondrial protein PGAM5 causes a Parkinson’s-like movement disorder. Nat Commun. 2014;5:4930. doi: 10.1038/ncomms5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews HR. Protein kinases and phosphatases that act on histidine, lysine, or arginine residues in eukaryotic proteins: a possible regulator of the mitogen-activated protein kinase cascade. Pharmacol Ther. 1995;67:323–350. doi: 10.1016/0163-7258(95)00020-8. [DOI] [PubMed] [Google Scholar]
- Rangachari M, Penninger JM. Negative regulation of T cell receptor signals. Curr Opin Pharmacol. 2004;4:415–422. doi: 10.1016/j.coph.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Sadatomi D, Tanimura S, Ozaki K, Takeda K. Atypical protein phosphatases: emerging players in cellular signaling. International journal of molecular sciences. 2013;14:4596–4612. doi: 10.3390/ijms14034596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine S, Kanamaru Y, Koike M, Nishihara A, Okada M, Kinoshita H, Kamiyama M, Maruyama J, Uchiyama Y, Ishihara N, et al. Rhomboid protease PARL mediates the mitochondrial membrane potential loss-induced cleavage of PGAM5. The Journal of biological chemistry. 2012;287:34635–34645. doi: 10.1074/jbc.M112.357509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Di L, Zhdanova O, Li Z, Vardhana S, Wan Q, Yan Y, Varma R, Backer J, Wulff H, et al. The class II phosphatidylinositol 3 kinase C2beta is required for the activation of the K+ channel KCa3.1 and CD4 T-cells. Mol Biol Cell. 2009;20:3783–3791. doi: 10.1091/mbc.E09-05-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Ko K, Choudhury P, Li Z, Johnson AK, Nadkarni V, Unutmaz D, Coetzee WA, Skolnik EY. Phosphatidylinositol-3 phosphatase myotubularin-related protein 6 negatively regulates CD4 T cells. Mol Cell Biol. 2006a;26:5595–5602. doi: 10.1128/MCB.00352-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Li Z, Ko K, Choudhury P, Albaqumi M, Johnson AK, Yan Y, Backer JM, Unutmaz D, Coetzee WA, et al. Histidine phosphorylation of the potassium channel KCa3.1 by nucleoside diphosphate kinase B is required for activation of KCa3.1 and CD4 T cells. Molecular cell. 2006b;24:665–675. doi: 10.1016/j.molcel.2006.11.012. [DOI] [PubMed] [Google Scholar]
- Srivastava S, Zhdanova O, Di L, Li Z, Albaqumi M, Wulff H, Skolnik EY. Protein histidine phosphatase 1 negatively regulates CD4 T cells by inhibiting the K+ channel KCa3.1. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:14442–14446. doi: 10.1073/pnas.0803678105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan E, Besant PG, Attwood PV. Mammalian histidine kinases: do they REALLY exist? Biochemistry. 2002;41:3843–3851. doi: 10.1021/bi012021r. [DOI] [PubMed] [Google Scholar]
- Vaeth M, Bauerlein CA, Pusch T, Findeis J, Chopra M, Mottok A, Rosenwald A, Beilhack A, Berberich-Siebelt F. Selective NFAT targeting in T cells ameliorates GvHD while maintaining antitumor activity. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:1125–1130. doi: 10.1073/pnas.1409290112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, Christofk HR, Wagner G, Rabinowitz JD, Asara JM, et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2010;329:1492–1499. doi: 10.1126/science.1188015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk A, Li J, Xin J, You D, Zhang J, Liu X, Xiao Y, Breslin P, Li Z, Wei W, et al. Co-inhibition of NF-kappaB and JNK is synergistic in TNF-expressing human AML. The Journal of experimental medicine. 2014;211:1093–1108. doi: 10.1084/jem.20130990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner PD, Vu ND. Phosphorylation of ATP-citrate lyase by nucleoside diphosphate kinase. The Journal of biological chemistry. 1995;270:21758–21764. doi: 10.1074/jbc.270.37.21758. [DOI] [PubMed] [Google Scholar]
- Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228–243. doi: 10.1016/j.cell.2011.11.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.