

SUMMARY
The de novo evolution of protein-coding genes from noncoding DNA is emerging as a source of molecular innovation in biology. Studies of random sequence libraries, however, suggest that young de novo proteins will not fold into compact, specific structures typical of native globular proteins. Here we show that Bsc4, a functional, natural de novo protein encoded by a gene that evolved recently from noncoding DNA in the yeast S. cerevisiae, folds to a partially specific three-dimensional structure. Bsc4 forms soluble, compact oligomers with high β-sheet content and a hydrophobic core, and undergoes cooperative, reversible denaturation. Bsc4 lacks a specific quaternary state, however, existing instead as a continuous distribution of oligomer sizes, and binds dyes indicative of amyloid oligomers or molten globules. The combination of native-like and non-native-like properties suggests a rudimentary fold that could potentially act as a functional intermediate in the emergence of new folded proteins de novo.
Keywords: De novo protein, structural evolution, conformational specificity, molten globule, amyloid oligomer
eTOC blurb
Recent studies have shown that new protein-coding genes can arise “de novo” from noncoding DNA. The properties of the brand new proteins encoded by these genes remain poorly understood. Here, Cordes et al. show that a very young de novo protein from yeast folds to a partially ordered three-dimensional structure.
INTRODUCTION
Protein folding is difficult and poses a potential roadblock to evolving protein structures from scratch. In classic textbook views, natural proteins such as myoglobin fold cooperatively into specific, stable, soluble, globular structures; these elegant, intricate native states then serve as scaffolds for biological functions such as oxygen binding. Such native structures are, however, rare among amino-acid sequences. Soluble proteins with significant secondary structure content have been recovered from unevolved random amino-acid sequence libraries, but they do not have specific, well-defined tertiary structures (Chiarabelli et al., 2006; Davidson et al., 1995; Doi et al., 1998). Even when such libraries are biased toward compositions or patterns found in natural proteins, the structures recovered tend to have “rudimentary” or “molten globule” characteristics lacking clearly specific tertiary structure (Graziano et al., 2008; Labean et al., 2011; Matsuura et al., 2002). Only in a single well-known case, in which random sequence libraries were subjected to extensive in vitro functional evolution, have clearly native-like structures been recovered (Keefe and Szostak, 2001; Lo Surdo et al., 2004; Mansy et al., 2007). Even among ancient, highly evolved sequences with specific native states, chaperones are often necessary to avoid pitfalls such as aggregation. The difficulty of folding is one justification for the common perception that evolution conservatively “tinkers” with an ancient repertoire, for example by duplicating and modifying existing protein-coding genes, instead of inventing entirely new proteins (Jacob, 1977; Zuckerkandl, 1975).
Not all proteins, however, require specific folding to function, and the relaxation of classic assumptions about structure/function relationships could make more radical mechanisms for molecular innovation far more plausible. Intrinsically disordered proteins (IDPs) that cannot fold independently are a sizeable minority of proteins and serve a variety of biological functions (Dyson and Wright, 2005; Meszaros et al., 2007; Schlessinger et al., 2011; Tompa and Kovacs, 2010). IDPs vary widely in the level of disorder, from random coils to “pre-molten globules” to molten globule states, which are able to fold compactly and have high levels of secondary structure but lack specific tertiary structures (Dunker and Obradovic, 2001; Habchi et al., 2014; Uversky, 2002). Molten globule states may be functional (DeGrado, 1993; Pervushin et al., 2007; Vamvaca et al., 2004), and even the most intrinsically disordered proteins can function through short linear binding motifs (Davey et al., 2012). A functional protein might therefore evolve de novo even if it cannot fold specifically. A protein born as an IDP might later evolve more native-like properties (Zhu et al., 2016) or continue indefinitely as a partially or completely disordered protein.
Until the last decade, there was no clear evidence that whole functional protein-coding genes could evolve de novo, e.g. from previously noncoding DNA; any suggestion that evolution does anything other than “tinkering” with existing scaffolds was speculative. Since 2006 (Levine et al., 2006), however, numerous studies in a variety of organisms have suggested that some genes trace their origins to the appearance and expression of a new open reading frame in noncoding DNA (Andersson et al., 2015; McLysaght and Guerzoni, 2015; Schlotterer, 2015; Tautz and Domazet-Loso, 2011). Such cases provide opportunities to test whether very young proteins emerge as IDPs and if so, whether they nonetheless have some level of order, for example a molten globule state, that might constitute a nascent folded structure.
Despite plenty of genetic evidence for de novo proteins, however, there has been almost no reported experimental characterization of their structures (Schmitz and Bornberg-Bauer, 2017). Recently born de novo proteins have been predicted by sequence analysis to have high levels of intrinsic disorder on average (Wilson et al., 2017), but no systematic experimental study of their ability to fold has been done. The antifreeze protein AFGP from Antarctic Notothenioid fishes contains a tripeptide repeat that evolved de novo from a splice junction in a trypsinogen gene (Chen et al., 1997), and the unusual structure of the repeat region, matching its unusual function as an antifreeze protein, has been probed by numerous methods (Urbanczyk et al., 2017). Some structural data also exists for the special category of de novo proteins encoded by viral “overprinted” genes, which evolved from an alternative reading frame of an existing coding gene, rather than from noncoding DNA. Overprinting proteins are generally predicted to have high intrinsic disorder (Kovacs et al., 2010; Rancurel et al., 2009), but least two are known to fold into specific, compact, novel structures (Meier et al., 2006; Pavesi et al., 2013; Shukla and Hilgenfeld, 2015; Vargason et al., 2003). It is not clear how old these proteins are, and they may retain little signature of their de novo origins. Their structures do, however, point to the existence of pathways for evolving native-like folds de novo. No experimental structure of any de novo protein (young or old), verified to have evolved from a new open reading frame in noncoding DNA, has been reported.
As a step toward structural characterization of young de novo proteins, we present a case study of the yeast protein Bsc4. A serious issue with case studies of individual newborn genes is the difficulty in proving, in the absence of evolutionary conservation, that they are both protein-coding and functional, in addition to proving that they arose from non-coding sequences (McLysaght and Hurst, 2016). The yeast gene BSC4 is an exceptionally well-supported case of an entire functional protein-coding gene that recently evolved de novo from an ancestral noncoding sequence (Cai et al., 2008). The name BSC4 (‘bypass of stop codon’) derives from belonging to a set of Saccharomyces cerevisiae genes with 9–25% stop codon bypass efficiency (Namy et al., 2003). BSC4 is conserved in all strains of S. cerevisiae, but no homologous open reading frame is present in other fungal species, and the hypothetical Bsc4 protein sequence is not similar to any other known protein sequence. A thorough analysis of synteny and phylogeny among numerous fungal species demonstrated that BSC4 is homologous to, and evolved recently from, a region of noncoding DNA in the intergenic region between LYP1 and ALP1 (Cai et al., 2008). BSC4 is nonessential but has two synthetic lethal partners (RPN4 and DUN1) (Pan et al., 2006). Its sequence is >90% conserved across known S. cerevisiae strains (Figure 1) and shows a low dN/dS ratio indicating purifying selection. RT-PCR and mass spectrometry data demonstrate expression of BSC4 at the RNA and protein level, respectively, under normal culture conditions (Cai et al., 2008). Heightened expression of BSC4 is observed in stationary phase (Aragon et al., 2008; Gasch et al., 2000), and both synthetic lethal partners function in DNA damage repair pathways, suggesting that BSC4 plays a role in DNA damage repair during stationary phase (Cai et al., 2008). Bsc4 is a functional, whole de novo protein-coding gene and, given its presence in only a single yeast species, a notably young one that can provide a window into de novo gene origin.
Figure 1. Alignment of unique Bsc4 sequences from S. cerevisiae strains.

Perfectly conserved residues are shown in color. Bsc4 sequences and strain names were obtained from Blastp searches using the S288C sequence, and aligned using ClustalX. The alignment is annotated with JRONN and IUPRED disorder prediction for S288C.
We predict that the Bsc4 protein has at least some folded structure despite the de novo origin and youth of the BSC4 gene. The Bsc4 protein from S. cerevisiae reference strain S288C has 131 amino-acid residues, easily long enough to form a domain. Its sequence is rich in positively charged residues, which disfavors folding, but also rich in hydrophobic residues, which favors folding (Uversky et al., 2000). Based on a weighting of these two factors, the program FoldIndex predicts that Bsc4 will fold (Prilusky et al., 2005). IUPRED (Dosztanyi et al., 2005) and JRONN (Troshin et al., 2011; Yang et al., 2005), also predict relatively low disorder except near the termini (Figure 1).
RESULTS
Recombinant overexpression, purification and refolding of two Bsc4 variants
As noted above, extant Bsc4 sequences are highly conserved (Figure 1). The most significant variation is the presence or absence of a 10-residue hydrophobic C-terminal tail, IVII(YC)VVRFH, which is predicted by TANGO (Fernandez-Escamilla et al., 2004) and AGGRESCAN (Conchillo-Sole et al., 2007) to be an aggregation hotspot. For our experiments we selected Bsc4 sequences from strains EC1118 and S288C, which are identical except that S288C has the C-terminal tail whereas EC1118 does not. The BSC4 gene is expressed, at least at the transcript level, in both strains (Rossouw et al., 2009).
We overexpressed Bsc4 EC1118 and S288C in E. coli from synthetic, codon-optimized BSC4 genes in T7-based plasmids supplying hexahistidine tags, purified them with denaturing nickel affinity chromatography, and refolded them by dialysis. Both sequences contain four cysteine residues, which in principle may form disulfide bonds. Because Bsc4 lacks clear secretion signals and the yeast cytosol is a reducing environment under normal conditions (Lopez-Mirabal and Winther, 2008), and because incorrect disulfide pairings could complicate refolding, we chose to focus on Bsc4 in reduced form. We thus included 1 mM TCEP as a reducing agent during refolding (we briefly return to this issue in the Discussion, however).
To optimize the affinity tag position and control for its influence, we tested both N-terminal and C-terminal tags. Here, we encountered a dilemma. N-terminally tagged Bsc4 EC1118 showed poor overexpression, leading us to favor a C-terminal tag. But we also found the tag position to have a measurable, though limited, influence on some biophysical properties of Bsc4 S288C. We elected to present primary data on C-terminally tagged variants, while noting any important influence of tag position on the behavior of Bsc4 S288C, and including relevant Supplemental Information.
For both C-terminally tagged Bsc4 variants, we obtained yields of >10 mg soluble protein per liter culture following refolding. Mass spectra confirmed the expected protein mass, accounting for the expected removal of the N-terminal methionine residue (in both bacteria and yeast), since serine is the second residue (Figure S1). Interestingly, the refolded proteins, despite being soluble, were highly resistant to SDS denaturation.
Bsc4 forms oligomers with compact structures and a range of stoichiometries
Both Bsc4 variants refold mainly to soluble oligomers, rather than monomers, under various refolding conditions (pH 5.5–7.5, 100–250 mM NaCl, 1 mM TCEP), as judged by size exclusion chromatography (Figure 2). The apparent oligomer size depends on solution conditions, with higher pH and salt concentration favoring larger oligomers or even aggregates. Under most conditions Bsc4 elutes as a single peak, but the peak is broader than expected based on calibration standards and irregular in shape under some conditions. Under all refolding conditions tested, the apparent molecular weight corresponding to the major peak is at least a dimer, though some traces for Bsc4 S288C also contain a minor peak consistent with monomer (elution volume of 20–21 mL, Figure 2). These findings suggest that Bsc4 refolds to a distribution of multiple oligomers rather than to a single oligomeric state or monomer.
Figure 2. Bsc4 refolds to oligomers of variable size.

Size exclusion chromatograms of affinity-purified A) Bsc4 S288C or B) Bsc4 EC1118, refolded by dialysis from 6 M guanidine into 50 mM MES (pH 5.5), 100 mM KCl (red); 50 mM MES (pH 5.5), 250 mM KCl (purple); 50 mM Tris (pH 7.5), 100 mM KCl (green); or 50 mM Tris (pH 7.5), 250 mM KCl (cyan). All solutions contained 0.2 mM EDTA, plus 1 mM TCEP as a reducing agent. To show estimated oligomer sizes, peaks are annotated with nearest integral number of Bsc4 subunits, based the molecular weight calculated from a five-protein calibration curve (see Materials and Methods). Initial concentration for refolding of Bsc4 EC1118 was 250 μM, while that of Bsc4 S288C was 158 μM. Based on separate experiments to measure concentration dependence (Figure S5), initial protein concentration differences of this magnitude for either protein lead to only small differences in elution volume of refolded protein. See also Figure S4.
Native mass spectrometry (Figure 3 and Figures S2 and S3) confirms a narrow, continuous distribution of compactly folded oligomers. A sample of Bsc4 EC1118 supplied at ~250 μM concentration in 10 mM HEPES (pH 7.5), 100 mM NaCl, 1 mM TCEP buffer was estimated to be hexameric based on size exclusion calibration. Ion mobility - mass spectrometry (IM-MS) plots of Bsc4 in 500 mM ammonium acetate and 1 mM TCEP clearly show oligomers ranging from tetramer to heptamer (Figure 3). Distinct features in the IM-MS plots (Figure 3) demonstrate narrow distributions of drift times, indicating compact conformations of the ions. The mass spectrometry data do not suggest bias toward oligomers with even or odd numbers of subunits (Figure S2). A second sample supplied in 100 mM ammonium acetate, 1 mM DTT was estimated to be tetrameric by size exclusion; mass spectra of this sample show oligomers from dimer to hexamer (Figure S3).
Figure 3. Ion mobility-mass spectrometry (IM-MS) plots of the mass spectrum of Bsc4 oligomers.
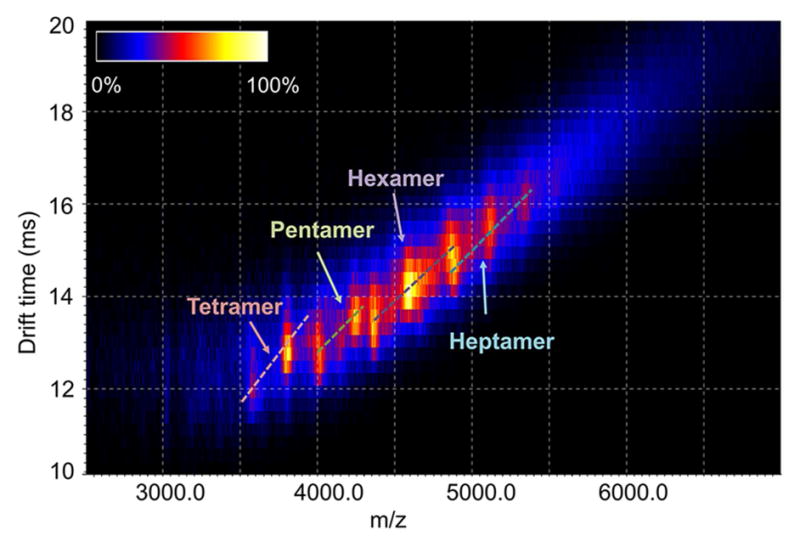
The spectrum of Bsc4 EC1118 (266 μM) in 500 mM ammonium acetate and 1 mM TCEP indicates a continuous distribution of oligomers from tetramer to octamer. The relative abundance of the species is shown in linear scale (color bar inset top left). Distinct spots in the IM-MS plot demonstrate narrow distributions of drift times, indicating relatively compact conformations of the ions. Similar distribution can be observed with Bsc4 concentration diluted to 18 μM. The spectrum of each of the species is extracted and shown in Supplementary Figure 2. The stoichiometry of each of the peaks was confirmed by collision-induced dissociation and surface induced dissociation. See also Figures S1–S3.
Long (S288C) and short (EC1118) versions of Bsc4 give different oligomer distributions (Figure 2), suggesting that the hydrophobic C-terminal tail (IVIIYVVRFH) has some influence on oligomer formation. Oligomer sizes for S288C show a stronger size dependence on refolding conditions. At low salt and low pH, the apparent molecular weight for S288C is slightly smaller (dimer vs. tetramer), while at high pH and high salt large aggregates are seen, even at the void volume (~8 mL) corresponding to the column exclusion limit (~40 MDa). The apparent size of Bsc4 S288C aggregates at high pH also increases over time upon storage of refolded protein at 4 °C. These observations agree with the prediction that the C-terminal tail is aggregation prone (see above). We also note, however, that this tendency is less pronounced in N-terminally tagged S288C (Figure S4).
The oligomerization of Bsc4 is not simply aggregation resulting from high protein concentration (~250 μM) during refolding. Bsc4 refolds predominantly to oligomers even at more modest concentrations (~50 μM) under solution conditions where large oligomers are least favored (low salt/low pH) (Figure S5). The elution volume of the peak maximum shows only a small increase across a four-fold dilution of Bsc4 during refolding. The narrow, continuous distribution of oligomers seen by mass spectrometry would, in fact, be expected to give rise to such dependence, since higher protein concentration should gradually shift the distribution upwards.
Bsc4 oligomers have β-sheet secondary structure and a hydrophobic core
Far ultraviolet circular dichroism spectra under conditions that favor smaller oligomers show the presence of β-sheet secondary structure (Figure 4). The combination of mean residue ellipticity values at 200 nm (near +2000 deg•cm2•dmol-1) and 222 nm (near -7000 deg•cm2•dmol-1) is also directly inconsistent with a highly unfolded structure, such as a random coil or “pre-molten globule” (Uversky, 2002). Analysis of secondary structure content using the program K2D3 (Louis-Jeune et al., 2012) gives ~30% β-strand, ~10% α-helix content for both variants. The CD results also agree with our observation (see above) that Bsc4 is SDS-resistant. SDS-resistance correlates with a combination of oligomerization and high β-strand content (Manning and Colon, 2004).
Figure 4. Bsc4 has β-sheet secondary structure.
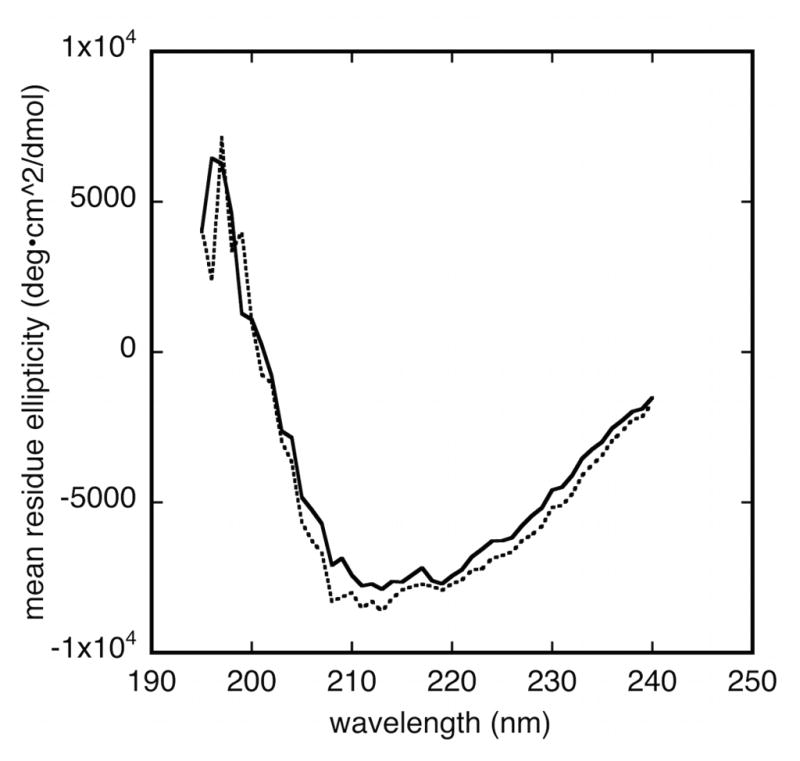
Far ultraviolet circular dichroism spectra of Bsc4 oligomers (S288C, solid line; EC1118, dashed line) from 195–240 nm at 20 °C, at 100 μM protein concentration in a 0.1 mm pathlength cell, in 50 mM MES (pH 5.5), 100 mM KCl, 1 mM TCEP.
Tryptophan fluorescence spectra show strong evidence for burial of the single tryptophan residue (Trp 47). Spectra obtained in native buffers (Figure 5) exhibit maximum fluorescence near 328 nm, consistent with tryptophan burial. Spectra obtained in 6 M guanidine (Figure 5) show maxima near 351 nm, indicating that guanidine denaturation exposes the tryptophan to solvent. These data suggest that tertiary and/or quaternary interactions between side chains form a hydrophobic interior in refolded Bsc4, in agreement with the compact structure inferred from mass spectra.
Figure 5. Tryptophan fluorescence and near ultraviolet circular dichroism of Bsc4.

(A) Tryptophan fluorescence emission spectrum of Bsc4 S288C (S288C, solid line; EC1118, dashed line) at 50 μM in 50 mM MES (pH 5.5), 100 mM KCl, 1 mM TCEP with (red) or without (blue) 6 M guanidine; (B) Near ultraviolet circular dichroism spectra of Bsc4 oligomers (S288C reduced, solid line; EC1118, dashed line) from 310–260 nm at 20 °C, at 100 μM protein concentration in a 1 cm pathlength cell, in 50 mM MES (pH 5.5), 100 mM KCl, 1 mM TCEP. The tryptophan fluorescence spectra show a maximum near 328 nm for folded Bsc4 and 351 nm for guanidine denatured Bsc4.
Near ultraviolet CD spectra of Bsc4 (Figure 5) are somewhat weak in intensity and show less fine structure relative to those of many native proteins (Kelly et al., 2005), suggesting that the hydrophobic core of Bsc4 could have a partially “molten” character (Price et al., 2005; Ptitsyn, 1995). As one point of comparison, the Cε3 domain of IgE, a molten globule of similar subunit size (110 residues vs. 120–130 residues) as the two Bsc4 variants, and with about the same number of Trp/Tyr residues (4 vs. 4–5), has a near ultraviolet CD spectrum that is similar in shape and intensity (in mean residue ellipticity), though with less fine structure (Price et al., 2005). Precise structural interpretation of near ultraviolet CD spectra is not possible, however, so the structure of Bsc4 cannot be conclusively classified as either molten or native-like on this basis.
The folded structure of Bsc4 EC1118 does not appear to include the regions near the N- and C-termini. An HSQC spectrum of a 13C/15N/2H-labelled sample of Bsc4 EC1118 (Figure S6), refolded under conditions that favor small oligomers, shows at least 50 resolvable amide proton resonances. We were able to assign the strongest resonances to regions near the N terminus (3–14) and C terminus (95–98,105–121). TALOS analysis of chemical shifts (Figure S6) shows low S2 values (<0.7) for these residues, indicative of highly dynamic character, which is also consistent with the low spectral dispersion and high intensity of the amide peaks. In addition, peptides released during limited trypsinolysis correspond primarily to these regions of sequence, while other regions appear to be protected (Figure S6). These findings support the predictions by JRONN and IUPRED that the termini are the least ordered regions (Figure 1). The rest of the sequence (residues 15–95, approximately) likely contains a folded domain, and the apparent resistance to proteolysis suggests that it may have a high level of structural order. The lack of clearly assignable resonances, and the lack of wide chemical shift dispersion, may reflect the absence of a unique quaternary and/or tertiary structure for the folded regions; alternatively, or additionally, it may reflect line broadening due to the high molecular weight of the oligomers (a pentamer of Bsc4 EC1118 has Mr ~75 kDa, for example). We return to this subject in the Discussion.
Bsc4 oligomers undergo cooperative, reversible thermal and chemical denaturation
Bsc4 oligomers are highly resistant to thermal denaturation, but both S288C and EC1118 can be melted at least partially under low salt/low pH conditions (Figure 6). Bsc4 S288C is more resistant to thermal denaturation than Bsc4 EC1118 and does not unfold completely even at 98 °C, suggesting that the hydrophobic C-terminal tail contributes stabilizing interactions (this enhanced stability is less pronounced for N-terminally affinity tagged S288C, however; see Figure S7). For both variants, the unfolding transition is reversible, and the protein can be melted and refolded at least twice with a similar apparent denaturation midpoint. Consistent with an oligomeric folded state, the denaturation midpoint is concentration dependent, showing a 5 °C increase for Bsc4 EC1118 over the concentration range 25–100 μM (Figure S8).
Figure 6. Cycled reversible thermal denaturation of small oligomers of Bsc4.

A) Bsc4 S288C, B) Bsc4 EC1118 in 50 MES (pH 5.5), 100 mM KCl, 1 mM TCEP, at 50 μM protein concentration in a 1 mm pathlength cell from 20–98 °C (293–371 K), monitored by circular dichroism at 222 nm. Filled and unfilled red circles represent the forward and reverse phases, respectively, of a first melt, while filled and unfilled blue squares represent forward and reverse melts of a second melt (remelt) of the same sample. Solid and dashed lines represent fits of forward and reverse denaturation curves (see Materials and Methods for details of fitting). For Bsc4 S288C, upper baselines are particularly poorly defined, and the data could not be fit to any unique solution. The fits shown for S288C are therefore for illustrative purposes only. See also Figures S7–S9.
Curiously, both variants, especially S288C, show a gain in dichroism signal after the first recooling cycle (Figure 6). Difference spectra from before and after thermal denaturation show maximum signal gain near 215 nm, consistent with gain of β-strand secondary structure (Figure S9). Comparisons of size exclusion traces before and after melting show that the oligomer distribution shifts to larger size following the melt (Figure S9). Thus, the gain in dichroism signal is probably attributable to renaturation of the protein to larger oligomers with enriched β-sheet content.
The observation of different elution volumes in the same sample, before and after a thermal melt, suggests that equilibration of oligomers may be slow at room temperature. To investigate this idea further, we injected samples of refolded Bsc4 EC1118 and isolated different size exclusion fractions representing approximately the low and high elution volume halves of the major peak. Reinjection of these fractions within a few hours led to different peak elution volumes; incubation of the samples at 35 °C, however, led to apparent equilibration to a common elution volume over 1 to several days, depending upon conditions. Thus, different oligomers of Bsc4 do equilibrate slowly at ambient temperature.
Bsc4 oligomers undergo cooperative chemical denaturation by guanidine (Figure 7). Bsc4 S288C has a slightly higher denaturation midpoint than Bsc4 EC1118 (3.2 M vs. 2.8 M), consistent with its greater resistance to thermal denaturation (this is also observed with N-terminally affinity tagged Bsc4 S288C; see Figure S7). Free energies of unfolding at zero denaturant, derived from the data fitting, are +5.4 kcal/mol and +3.6 kcal/mol for Bsc4 S288C and EC1118, respectively. Fitted guanidine m values are 1.7 and 1.3 kcal/mol•M, respectively, lower than expected for a typical globular protein of this molecular weight (specifically, a guanidine m value of 1.5 corresponds to 2600 Å2 of surface area burial, which is typical of a ~40-residue protein rather than a 120-residue protein) (Myers et al., 1995). One possible contributor to a low m value is that the folded region does not include the termini.
Figure 7. Cooperative guanidine denaturation of small oligomers of Bsc4.

A) Bsc4 S288C, B) Bsc4 EC1118 in 50 MES (pH 5.5), 100 mM KCl, 1 mM TCEP, at 60 μM protein concentration in a 0.5 mM pathlength cell at 20 °C, monitored by circular dichroism from 250 nm to 215 nm. Guanidine concentrations range from 0 M (purple) to 6 M (maroon) in 0.75 M increments. Note the transition in spectral shape toward a random coil like spectrum with increasing guanidine concentration. Insets show fitting of the ellipticity at 222 nm to a standard two-state chemical denaturation model. See also Figure S7.
Bsc4 oligomers bind dyes indicative of amyloids or molten globules
The formation of compact β-sheet rich oligomers without a specific quaternary state (or specific tertiary interactions, perhaps) led us to wonder whether the folded state of Bsc4 can be compared to oligomeric intermediates in the formation of amyloid fibrils, or perhaps to molten globule intermediates in protein folding. We tested the binding of both Bsc4 variants to Congo Red, Thioflavin T, and ANS, all of which have been reported to bind oligomeric amyloid intermediates (Fandrich, 2012). Changes in ANS fluorescence upon binding have also classically been used as a measure of molten globule character (Ptitsyn, 1995). Bsc4 oligomers of both variants bind all three dyes (Figure 8), showing a shift in the absorbance maximum of Congo Red, with the largest difference near 550 nm; enhancement of fluorescence at 480 nm for thioflavin T; and a large enhancement of fluorescence plus a blue shift of the maximum from ~535 nm to ~480 nm for ANS. The dye binding behavior of Bsc4 suggests that its structure may indeed resemble amyloid oligomers or molten globules.
Figure 8. Dye binding by Bsc4 oligomers.

Binding of Bsc4 S288C (solid blue) and Bsc4 EC1118 (dashed blue) to (A) 20 μM Congo Red (B) 5 μM thioflavin T and (C) 50 μM ANS. Black lines in panels B and C show dye signal alone, with maximum fluorescence normalized to 1, while for Congo Red the data are plotted as absorbance difference spectra. Protein concentrations are 6 μM, 10 μM and 5 μM in panels A, B and C, respectively.
DISCUSSION
We have demonstrated for the first time that a young, naturally functional protein, encoded by a gene that evolved recently de novo from noncoding DNA, folds to a structure with some properties found in native globular proteins. These properties include compactness, stable secondary structure, side chain burial, cooperative denaturation, and some resistance to proteolysis. The structure of Bsc4 is not entirely native-like, however, lacking a specific quaternary state. In addition, we found no conclusive evidence for specific tertiary interactions, and the behavior of Bsc4 in dye-binding experiments is similar to that of amyloid oligomers or molten globules. In sum, Bsc4 is neither an intrinsically disordered protein (at least not a highly unfolded one), nor does it appear likely to be a uniquely folded globular protein. Some observations support a molten globule state, but protease resistance suggests a higher level of order, so its placement in common classifications of structural order, such as the Uversky quartet model, remains in some doubt (Uversky, 2002). Bsc4 might be conservatively described as having a “rudimentary fold” (Labean et al., 2011), and it also bears some comparison to a folding or misfolding intermediate. In any case, a nascent structure with such an unusual combination of properties seems reasonable for the “birth” of folding in a de novo evolved protein. Whether such proteins can later evolve more specific, native-like structures remains speculative.
If some de novo proteins can fold, even partially, then in principle they could be a source of structural innovation, namely new protein domain folds or novel modes of oligomerization. For very young, nascent proteins like Bsc4, it may prove difficult to characterize structure at a resolution sufficient to assess structural novelty. Bsc4 has steadfastly resisted our attempts at crystallization thus far, and does not appear to be a good candidate for structure determination by NMR. The apparent lack of dispersed amide resonances in NMR spectra despite deuteration (Figure S6) may reflect dynamic conformational averaging within a “molten” structure. Alternatively, it could reflect static conformational heterogeneity, either among subunits within individual, low-symmetry oligomeric states, or in different high-symmetry oligomeric states (or some combination thereof). For dispersed NMR peaks in folded regions, static conformational heterogeneity could split peaks and thereby weaken their intensity to the point where they cannot be observed. If a single oligomeric state of Bsc4 could be isolated under some set of conditions, there might be more hope for high-resolution structural studies.
Bsc4 forms smaller oligomers at lower pH and salt concentration (Figure 2), a behavior that we speculate may be due to electrostatic repulsions. Bsc4 S288C and EC1118 are very positively charged proteins near neutral pH, with predicted pI values of 11.2–11.3 owing to 25–26 Lys/Arg residues compared to 4 Glu/Asp residues. Bsc4 EC1118 also contains three His residues, while Bsc4 S288C has four owing to an additional His residue at the C-terminus. Lowering the pH from 7.5 to 5.5 is expected to lead to protonation of the histidines and an increase in the already large net positive charge, potentially increasing electrostatic repulsions between subunits in oligomers. Lowering the salt concentration from 250 mM to 100 mM would be expected to exacerbate such repulsions. Bsc4 may respond by forming smaller oligomers, which would have lower overall positive charge and potentially less internal electrostatic repulsion.
The structural properties of the Bsc4 variants are not likely to be artifacts of simple covalent modifications such as affinity tagging, N-terminal processing or cysteine oxidation state. As we have noted, the N-terminal methionine is processed in E. coli, but this is also expected to occur in native yeast. The affinity tag does have some effect on oligomer distribution, but the qualitative properties of Bsc4 S288C were largely independent of tag location. We further note that removal of the N-terminal tag for Bsc4 S288C by thrombin cleavage does not strongly affect the oligomerization behavior or secondary structure (Figure S10). As to cysteine oxidation, we focused on reduced Bsc4 based on the lack of secretion signals in the sequence and the reducing nature of the yeast cytosol under normal conditions (Lopez-Mirabal and Winther, 2008). However, to test whether disulfide bonding could impart a more specific structure to the protein, we also tried refolding Bsc4 under nonreducing conditions (see STAR Methods). We did observe limited loss of free cysteine, but these samples showed no changes in near UV circular dichroism or in size exclusion chromatography that would indicate a change to a more specific tertiary or quaternary structure.
Is it nonetheless possible that Bsc4 might fold to a different (and perhaps more native-like) structure under some conditions? The formation of oligomers under a range of solution conditions and protein concentrations, and the reversibility of thermal denaturation, strongly suggests that the β-sheet rich oligomers represent the most stable structure for Bsc4, at least at micromolar concentrations. Typical Bsc4 concentrations under normal conditions in yeast are unknown, however, and could be lower. It is possible that while Bsc4 is prone to form oligomers, the native functional form is monomeric, and it is unclear whether such a monomer would fold or be intrinsically disordered. Even if the native form of Bsc4 differs from the structures studied here, however, the results still demonstrate for the first time the rudimentary foldability of a natural de novo evolved sequence.
Some de novo proteins may be born with rudimentary folds that resemble molten globules or amyloid-like states, but such folds need to support function and be nontoxic. Molten globule states can be functional (DeGrado, 1993; Pervushin et al., 2007; Vamvaca et al., 2004), but their potential for cytotoxicity has not been studied systematically. Amyloid cross-β structures are generic, stable folding patterns for polypeptides (Dobson, 2003), and have been proposed as early peptide structures on ancient Earth (Greenwald and Riek, 2012; Maury, 2009). Some amyloid fibrils can support function (Fowler et al., 2007), and some functional natural proteins such as the small heat shock proteins behave like amyloid oligomers (Breydo and Uversky, 2015), exchanging between multiple β-sheet-rich oligomeric states (Delbecq and Klevit, 2013; Haslbeck and Vierling, 2015). Amyloid oligomers can be highly cytotoxic, but toxicity varies considerably with oligomer size and structural features (Breydo and Uversky, 2015), so this danger may be avoidable.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Matthew Cordes (cordes@email.arizona.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bsc4 proteins from Saccharomyces cerevisiae were not obtained from a yeast source, but were overexpressed in Escherichia coli strain BL21(λDE3) using synthetic genes constructed and modified based on sequences in the NCBI nonredundant protein database (Genbank accession CAY82343 in the case of S. cerevisiae strain EC1118, and DAA10291 in the case of strain S288C).
METHOD DETAILS
Cloning
A synthetic, codon-optimized gene encoding the Bsc4 sequence from Saccharomyces cerevisiae strain S288C was supplied by BioBasic (Markham, Ontario, Canada) in a pUC57 cloning vector. The synthetic gene was flanked by NdeI and XhoI restriction sites, which were then used to subclone the gene into a pET-21b expression vector (Novagen), which supplied an in-frame 3′ sequence encoding a C-terminal LEHHHHHH affinity tag. An equivalently tagged expression plasmid encoding Bsc4 from strain EC1118 was subsequently obtained by deletion of 30 bases encoding the last 10 amino-acid residues of the natural S288C sequence (IVIIYVVRFH) using QuikChange mutagenesis (Stratagene). Expression plasmids encoding N-terminally tagged Bsc4 S288C and EC1118 were obtained starting from these constructs in the following way: first, a stop codon was introduced prior to the XhoI site by QuikChange mutagenesis to remove the sequence encoding the C-terminal tag; second, the resulting tagless constructs were digested with NdeI and XhoI; third, the NdeI-XhoI fragment was ligated into a pET-15b backbone produced by digestion with NdeI and XhoI. The pET-15b vector (Novagen) contains a sequence, upstream of the NdeI cloning site, that supplies an N-terminal MGSSHHHHHHSSGLVPRGSH affinity tag.
Overexpression, purification and refolding
Bsc4 variants were overexpressed in Escherichia coli strain BL21(λDE3) and purified by denaturing Ni-NTA affinity chromatography essentially as described (LeFevre and Cordes, 2003). To prevent disulfide bond formation, 15 mM β-mercaptoethanol was used in the lysis and wash buffers and 3–5 mM β-mercaptoethanol in the elution buffer. Proteins were then typically dialyzed into one of several refolding buffers of varying pH and salt concentration: 50 mM MES (pH 5.5), 100 mM KCl; 50 mM MES (pH 5.5), 250 mM KCl; 50 mM Tris (pH 7.5), 100 mM KCl; or 50 mM Tris (pH 7.5), 250 mM KCl. Each refolding buffer also contained 0.2 mM EDTA, and 1 mM TCEP to maintain cysteines in a reduced state. To test whether disulfide bonds could form and affect structure, however, we also conducted refolding experiments without 1 mM TCEP, as well as parallel 20-fold dilution refolding experiments into buffers containing either 1 mM TCEP or mixtures of oxidized (0.4 mM) and reduced (2 mM) glutathione, In both cases, standard Ellman’s tests showed loss of 1–2 (out of 4) free cysteines in samples folded under nonreducing conditions. Such samples showed only minor differences in size exclusion elution volumes or near ultraviolet circular dichroism spectra, suggesting that any disulfide formation does not greatly perturb tertiary or quaternary structure of Bsc4.
Concentrations of refolded purified protein were obtained from A280 values using an estimated extinction coefficient of 9530 M−1 cm−1 for Bsc4 EC1118 and 10810 M−1 cm−1 for Bsc4 S288C, based on the number of tryptophan and tyrosine residues in each sequence.
Size exclusion chromatography
Size exclusion chromatography was carried out on an AKTA FPLC instrument (General Electric) with a Superose 6 10/300 GL column, using an injection volume of 0.5 mL and a flow rate of 0.5 mL/min. This column has a void volume of approximately 8 mL, a total column volume of approximately 24 mL, and an exclusion limit of ~40 MDa. The column was calibrated with 5 standards: ovalbumin (Mr 43000), bovine serum albumin (Mr 67000), aldolase (Mr 158000), ferritin (Mr 440000), and thyroglobulin (Mr 669000). The calibration was found to be insensitive to variations in pH and salt across the ranges used in our experiments. Estimated Mr values and numbers of subunits for Bsc4 oligomers were obtained from observed elution volumes based on a calibration curve obtained at pH 7.5 and 250 mM salt.
Circular dichroism spectroscopy
Circular dichroism spectra, thermal denaturation curves, and guanidine denaturation profiles were obtained on an OLIS DSM-20 CD spectropolarimeter, using 50 MES (pH 5.5), 100 mM KCl, 1 mM TCEP, 0.2 mM EDTA as refolding buffer to generate small Bsc4 oligomers. Far ultraviolet wavelength scans were obtained at 20 °C at a protein concentration of 100 μM in a 0.1 mm pathlength cell, from 240 to 195 nm in 1 nm steps with an integration time of 30 s and with signal averaging from 5 scans. Near ultraviolet wavelength scans were obtained at 20 °C at a protein concentration of 100 μM in a 1 cm pathlength cell, from 310 to 260 nm in 1 nm steps with an integration time of 15 s and with signal averaging from 5 scans. For guanidine denaturation profiles, scans were obtained at 20 °C at a protein concentration of 60 μM in a 0.5 mm pathlength cell, from 250 to 215 nm in 1 nm steps with an integration time of 5 s and with signal averaging from 5 scans. Guanidine concentrations ranging from 0 M to 6 M in 0.75 M increments were obtained by 5x dilution of 300 μM protein stocks into 50 MES (pH 5.5), 100 mM KCl, 1 mM TCEP with guanidine concentrations ranging from 0 M to 7.5 M guanidine. All spectra above were corrected for buffer baseline signals. Thermal denaturation curves were obtained at 50 μM protein concentration in a 1 mm pathlength cell from 20–98 °C (293–371 K), monitored by circular dichroism at 222 nm.
Mass spectrometry of oligomers
One sample of Bsc4 EC1118 (~250 μM) was supplied directly in 100 mM ammonium acetate, 1 mM DTT; a second sample was supplied in 10 mM HEPES (pH 7.5), 100 mM NaCl, 1 mM TCEP, and exchanged with an Amicon Ultra 0.5 mL volume centrifugal filter (with a molecular weight cutoff of 3000 Da) into 500 mM ammonium acetate (pH 7.5) plus 1 mM TCEP prior to analysis. Due to a very slow rate of equilibration between oligomers (see Results), changes in the buffer prior to mass spectrometric analysis are not expected to alter the oligomer distribution significantly, making it possible to compare size distributions from mass spectra with those from size exclusion chromatography on the source samples. The mass spectrometry experiments were performed on a modified Waters Synapt G2S HDMS mass spectrometer (Wilmslow, U.K.) with an surface induced dissociation (SID) device installed between the truncated trap travelling wave ion guide (TWIG) and the ion mobility cell (Zhou et al., 2012). The samples were sprayed with a nanoelectrospray source at a voltage of 1.0 kV. The sampling cone and source offset were set to 20 V. The gas flow rates for Trap, helium cell, and ion mobility cell were set to 10 mL/min, 120 mL/min, and 60 mL/min, respectively. The ion mobility wave velocity and wave height were 350 m/s and 16.0 V. The stoichiometry of the oligomers was confirmed by collision induced dissociation (CID) and SID, which are activation methods providing noncovalent products (Zhou et al., 2012). The CID experiments were conducted in the trap TWIG by accelerating the ions before entering the trap TWIG. The SID experiments were performed in the SID device by steering the ions to collide with the surface by changing the voltage on the front bottom deflector, and the voltages on the other electrodes were tuned to maximize the transmission of the product ions.
Tryptophan fluorescence and dye binding
Proteins for these experiments were refolded in 50 mM MES (pH 5.5), 100 mM KCl, 1 mM TCEP, 0.2 mM EDTA to generate small oligomers. Tryptophan fluorescence spectra were obtained on an ISS PC1 photon counting spectrofluorimeter in L-format with excitation monochromator set at 280 nm and emission wavelengths scanned from 300 to 450 nm. Proteins were scanned at 50 μM in 50 MES (pH 5.5), 100 mM KCl, 1 mM TCEP, 0.2 mM EDTA. For Congo Red binding, a stock solution of Congo Red was made by dissolving the dye to a concentration of 7 mg/mL (10 mM) in stock buffer (10 mM sodium phosphate [pH 7], 100 mM sodium chloride). 2 μL of the stock solution were then diluted into 1 mL of stock buffer in a plastic cuvette with a 1 mL pathlength, for a working concentration of 20 μM Congo Red. Bsc4 was then titrated into the Congo Red sample. Binding was monitored by changes in the absorbance spectrum from 400 to 700 nm, measured using a Cary 50 UV-visible spectrophotometer. The spectra were corrected for wavelength-dependent scattering prior to subtraction to obtain difference spectra. Changes in Congo Red absorbance appeared to saturate at ~5 μM protein concentration, suggesting that binding is quite strong and that there are multiple Congo Red binding sites per Bsc4 subunit. For thioflavin T binding, a 250 μM stock solution of thioflavin T was prepared by dissolving 8 mg of the dye in 10 mL phosphate buffer (10 mM sodium phosphate [pH 7], 150 mM sodium chloride). The stock was then diluted to 5 μM in phosphate buffer in a microcuvette with a 1 cm excitation pathlength. Bsc4 was then titrated into the thioflavin T sample. Binding was monitored using an ISS PC1 photon counting spectrofluorometer in L-format with excitation monochromator set at 440 nm and emission wavelengths scanned from 460 to 560 nm. Thioflavin T showed an approximately linear increase in fluorescence as a function of protein concentration from 10–40 μM. For ANS binding, ANS was diluted from a 1.1 mM stock solution in 50 MES (pH 5.5), 100 mM KCl, 1 mM TCEP, 0.2 mM EDTA to a concentration of 50 μM in the same buffer with or without 5–50 μM Bsc4 EC1118 or S288C. Changes in fluorescence were monitored using an ISS PC1 photon counting spectrofluorometer in L-format with excitation monochromator set at 380 nm and emission wavelengths scanned from 400 to 600 nm. Fluorescence of ANS at 480 nm increased (but not linearly) with protein concentration from 5–50 μM.
NMR spectroscopy
13C/15N/2H-labelled Bsc4 EC1118 was produced by overexpression in M9 minimal media containing 0.8 mg/mL 15NH4Cl as sole nitrogen source, 2.5 mg/mL 13C6-glucose as sole carbon source, and 100% 2H2O as a source of deuterium. To condition cells to 2H-labelled media, a 40 mL starter culture was initially grown in M9 media in 100% 1H2O up to an OD600 of 0.1, then switched to 50% 2H2O until OD600~0.2, then switched to 100% 2H2O until OD600~0.4. At this point the starter culture was transferred to a larger flask, and the cells kept at an OD600 of 0.1–0.4 by gradual addition of deuterated media up to a volume of 950 mL. When this culture reached OD600~0.5, it was induced by addition of 100 μg/mL IPTG, and growth continued for 5 h. Following affinity purification, the protein was refolded by dialysis into 50 MES (pH 5.5), 50 mM KCl, 1 mM TCEP. The refolded protein sample was split and concentrated to 200 μM, 430 μM, 770 μM, and 1 mM. All four samples had highly similar HSQC spectra lacking detectable dispersed resonances (temperature range 15–45 °C). Triple-resonance spectra for assignment of disordered regions were acquired at 15 °C on the 1 mM sample on a Varian Inova-600 spectrometer equipped with a triple-resonance cryogenic probe. The relaxation delay was set to 1.5 s. 1H-15N-HSQC, HNCO and HNCACB spectra were acquired using 8 scans, utilizing the TROSY pulse sequence to enhance sensitivity. NMR data were processed using NMRPipe (Delaglio et al., 1995)and resonances were assigned manually using SPARKY (T. Goddard and D.G. Kneller, SPARKY 3, University of California, San Francisco). Backbone chemical shifts (CA, CB, CO, N, HN) were analyzed by TALOS-N (Shen and Bax, 2015) to predict backbone order parameters (S2).
Limited proteolysis
Mag-trypsin magnetic beads (Clontech Laboratories; 0.3 mL of a 5% suspension) were equilibrated in 10 mM HEPES (pH 7.5), 100 mM NaCl, 1 mM TCEP. Nickel-affinity and size-exclusion purified Bsc4 EC1118 (at a concentration of 4 mg/mL in the same buffer) was mixed with an equal volume of equilibrated Mag-Trypsin beads and incubated for 10 min. To end the reaction, the reaction mixture was placed on a magnetic separator and the protein solution pipetted away from the beads. Prior to analysis, solutions were flash frozen in liquid nitrogen and stored at −80 °C. Tandem mass spectrometric analysis (MS/MS) was conducted using a Thermo Fisher LTQ Orbitrap Velos mass spectrometer at the Arizona Proteomics Core Facility, and tandem mass spectra were extracted. Charge state deconvolution and deisotoping were not performed. All MS/MS samples were analyzed using Sequest (XCorr Only) (Thermo Fisher Scientific, San Jose, CA, USA; version 1.3.0.339). Sequest (XCorr Only) was set up to search a database including E. coli proteins, common contaminants, and the tagged Bsc4 EC1118 sequence (5191 entries), assuming the digestion enzyme trypsin. Sequest (XCorr Only) was searched with a fragment ion mass tolerance of 0.80 Da and a parent ion tolerance of 10.0 PPM. Oxidation of methionine and carbamidomethyl of cysteine were specified in Sequest (XCorr Only) as variable modifications. Scaffold (version Scaffold_4.7.5, Proteome Software Inc., Portland, OR) was used to validate MS/MS based peptide and protein identifications.
QUANTIFICATION AND STATISTICAL ANALYSIS
For chemical denaturation monitored by circular dichroism, unfolding free energies (ΔGu, 20 °C) and denaturant m values were obtained by nonlinear least squares fitting of the ellipticity at 222 nm as a function of guanidine concentration, to a model in which ΔGu was assumed to vary linearly with guanidine concentration and the slopes and intercepts of folded and unfolded baselines were allowed to vary.
For thermal denaturation monitored by circular dichroism, temperature-dependent dichroism data were fitted to the following relationship (Becktel and Schellman, 1987):
Baseline slopes and intercepts for folded and unfolded states were allowed to vary in some fits, though the upper (unfolded) baseline was very poorly defined in some cases and had to be restrained based on values observed in cases where the baseline was well defined. The heat capacity of unfolding (ΔCp) was fixed at 1400 cal mol−1 K−1 based on an estimate of 100 residues of folded sequence (subtracting disordered N and C termini based on NMR data) and 14 cal mol−1K−1 per residue (Myers et al., 1995). Because of the poor definition of the upper baselines for S288C, the thermal denaturation fits were used for illustrative purposes only (see Figure 6), to highlight the cooperativity of the denaturation rather than to extract reliable Tm values.
In MS/MS analysis of limited proteolysis solutions using Sequest and Scaffold (see above), tagged Bsc4 EC1118 was identified with 100% probability based on identification of 22 exclusive unique peptides. Protein probabilities were assigned by the Protein Prophet algorithm (Nesvizhskii et al., 2003). Peptide identifications were accepted if they could be established at greater than 10.0% probability to achieve an FDR less than 0.1% by the Scaffold Local FDR algorithm (with the exception of 15 identifications of the N-Type equation here.terminal peptide SIVLR, all identifications included in Fig. S6B, met a higher standard of 26.0% probability of 0.1% FDR).
DATA AND SOFTWARE AVAILABILITY
Not applicable.
ADDITIONAL RESOURCES
Not applicable.
Supplementary Material
Highlights.
The young, functional de novo protein Bsc4 has a rudimentary ability to fold.
Bsc4 forms compact oligomers with high β-sheet content and a hydrophobic core.
Bsc4 lacks a specific quaternary state and binds dyes suggestive of amyloid oligomers.
Young de novo proteins can have some structural order and native-like properties.
Acknowledgments
This work was supported by National Institutes of Health grant GM104040 (R01 to M.H.J.C. and J.M), GM113658 (to V.H.W.), and John Templeton Foundation grant 39667 (to J.M.). The authors declare no competing interests. Mass spectrometry data for limited proteolysis experiments were acquired by the Arizona Proteomics Consortium supported by NIEHS grant ES06694 to SWEHSC, NIH/NCI grant CA023074 to the UA Cancer Center and by the Bio5 Institute of the University of Arizona. The Thermo Fisher LTQ Orbitrap Velos mass spectrometer was provided by grant 1S10 RR028868-01 from NIH/NCRR.
Footnotes
AUTHOR CONTRIBUTIONS. D.B., J.S.C., J.Y., J.C., V.K.K., and M.H.J.C. conducted the experiments; S.G.F. and M.H.J.C. conducted database studies; M.H.J.C., V.H.W. and J.M. designed the experiments; M.H.J.C. and J.M. wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersson DI, Jerlstrom-Hultqvist J, Nasvall J. Evolution of new functions de novo and from preexisting genes. Cold Spring Harb Perspect Biol. 2015:7. doi: 10.1101/cshperspect.a017996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragon AD, Rodriguez AL, Meirelles O, Roy S, Davidson GS, Tapia PH, Allen C, Joe R, Benn D, Werner-Washburne M. Characterization of differentiated quiescent and nonquiescent cells in yeast stationary-phase cultures. Mol Biol Cell. 2008;19:1271–1280. doi: 10.1091/mbc.E07-07-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becktel WJ, Schellman JA. Protein stability curves. Biopolymers. 1987;26:1859–1877. doi: 10.1002/bip.360261104. [DOI] [PubMed] [Google Scholar]
- Breydo L, Uversky VN. Structural, morphological, and functional diversity of amyloid oligomers. FEBS Lett. 2015;589:2640–2648. doi: 10.1016/j.febslet.2015.07.013. [DOI] [PubMed] [Google Scholar]
- Cai J, Zhao R, Jiang H, Wang W. De novo origination of a new protein-coding gene in Saccharomyces cerevisiae. Genetics. 2008;179:487–496. doi: 10.1534/genetics.107.084491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, DeVries AL, Cheng CH. Evolution of antifreeze glycoprotein gene from a trypsinogen gene in Antarctic notothenioid fish. Proc Natl Acad Sci U S A. 1997;94:3811–3816. doi: 10.1073/pnas.94.8.3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarabelli C, Vrijbloed JW, De Lucrezia D, Thomas RM, Stano P, Polticelli F, Ottone T, Papa E, Luisi PL. Investigation of de novo totally random biosequences, Part II: On the folding frequency in a totally random library of de novo proteins obtained by phage display. Chem Biodivers. 2006;3:840–859. doi: 10.1002/cbdv.200690088. [DOI] [PubMed] [Google Scholar]
- Conchillo-Sole O, de Groot NS, Aviles FX, Vendrell J, Daura X, Ventura S. AGGRESCAN: a server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinformatics. 2007;8:65. doi: 10.1186/1471-2105-8-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey NE, Van Roey K, Weatheritt RJ, Toedt G, Uyar B, Altenberg B, Budd A, Diella F, Dinkel H, Gibson TJ. Attributes of short linear motifs. Mol Biosyst. 2012;8:268–281. doi: 10.1039/c1mb05231d. [DOI] [PubMed] [Google Scholar]
- Davidson AR, Lumb KJ, Sauer RT. Cooperatively folded proteins in random sequence libraries. Nat Struct Biol. 1995;2:856–864. doi: 10.1038/nsb1095-856. [DOI] [PubMed] [Google Scholar]
- DeGrado WF. Peptide engineering. Catalytic molten globules. Nature. 1993;365:488–489. doi: 10.1038/365488a0. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Delbecq SP, Klevit RE. One size does not fit all: the oligomeric states of alphaB crystallin. FEBS Lett. 2013;587:1073–1080. doi: 10.1016/j.febslet.2013.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- Doi N, Yomo T, Itaya M, Yanagawa H. Characterization of random-sequence proteins displayed on the surface of Escherichia coli RNase HI. FEBS Lett. 1998;427:51–54. doi: 10.1016/s0014-5793(98)00392-5. [DOI] [PubMed] [Google Scholar]
- Dosztanyi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21:3433–3434. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- Dunker AK, Obradovic Z. The protein trinity--linking function and disorder. Nat Biotechnol. 2001;19:805–806. doi: 10.1038/nbt0901-805. [DOI] [PubMed] [Google Scholar]
- Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- Fandrich M. Oligomeric intermediates in amyloid formation: structure determination and mechanisms of toxicity. J Mol Biol. 2012;421:427–440. doi: 10.1016/j.jmb.2012.01.006. [DOI] [PubMed] [Google Scholar]
- Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol. 2004;22:1302–1306. doi: 10.1038/nbt1012. [DOI] [PubMed] [Google Scholar]
- Fowler DM, Koulov AV, Balch WE, Kelly JW. Functional amyloid--from bacteria to humans. Trends Biochem Sci. 2007;32:217–224. doi: 10.1016/j.tibs.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziano JJ, Liu W, Perera R, Geierstanger BH, Lesley SA, Schultz PG. Selecting folded proteins from a library of secondary structural elements. J Am Chem Soc. 2008;130:176–185. doi: 10.1021/ja074405w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald J, Riek R. On the possible amyloid origin of protein folds. J Mol Biol. 2012;421:417–426. doi: 10.1016/j.jmb.2012.04.015. [DOI] [PubMed] [Google Scholar]
- Habchi J, Tompa P, Longhi S, Uversky VN. Introducing protein intrinsic disorder. Chem Rev. 2014;114:6561–6588. doi: 10.1021/cr400514h. [DOI] [PubMed] [Google Scholar]
- Haslbeck M, Vierling E. A first line of stress defense: small heat shock proteins and their function in protein homeostasis. J Mol Biol. 2015;427:1537–1548. doi: 10.1016/j.jmb.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob F. Evolution and tinkering. Science. 1977;196:1161–1166. doi: 10.1126/science.860134. [DOI] [PubMed] [Google Scholar]
- Keefe AD, Szostak JW. Functional proteins from a random-sequence library. Nature. 2001;410:715–718. doi: 10.1038/35070613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly SM, Jess TJ, Price NC. How to study proteins by circular dichroism. Biochim Biophys Acta. 2005;1751:119–139. doi: 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Kovacs E, Tompa P, Liliom K, Kalmar L. Dual coding in alternative reading frames correlates with intrinsic protein disorder. Proc Natl Acad Sci U S A. 2010;107:5429–5434. doi: 10.1073/pnas.0907841107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labean TH, Butt TR, Kauffman SA, Schultes EA. Protein folding absent selection. Genes (Basel) 2011;2:608–626. doi: 10.3390/genes2030608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeFevre KR, Cordes MH. Retroevolution of lambda Cro toward a stable monomer. Proc Natl Acad Sci U S A. 2003;100:2345–2350. doi: 10.1073/pnas.0537925100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MT, Jones CD, Kern AD, Lindfors HA, Begun DJ. Novel genes derived from noncoding DNA in Drosophila melanogaster are frequently X-linked and exhibit testis-biased expression. Proc Natl Acad Sci U S A. 2006;103:9935–9939. doi: 10.1073/pnas.0509809103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Surdo P, Walsh MA, Sollazzo M. A novel ADP- and zinc-binding fold from function-directed in vitro evolution. Nat Struct Mol Biol. 2004;11:382–383. doi: 10.1038/nsmb745. [DOI] [PubMed] [Google Scholar]
- Lopez-Mirabal HR, Winther JR. Redox characteristics of the eukaryotic cytosol. Biochim Biophys Acta. 2008;1783:629–640. doi: 10.1016/j.bbamcr.2007.10.013. [DOI] [PubMed] [Google Scholar]
- Louis-Jeune C, Andrade-Navarro MA, Perez-Iratxeta C. Prediction of protein secondary structure from circular dichroism using theoretically derived spectra. Proteins. 2012;80:374–381. doi: 10.1002/prot.23188. [DOI] [PubMed] [Google Scholar]
- Manning M, Colon W. Structural basis of protein kinetic stability: resistance to sodium dodecyl sulfate suggests a central role for rigidity and a bias toward beta-sheet structure. Biochemistry. 2004;43:11248–11254. doi: 10.1021/bi0491898. [DOI] [PubMed] [Google Scholar]
- Mansy SS, Zhang J, Kummerle R, Nilsson M, Chou JJ, Szostak JW, Chaput JC. Structure and evolutionary analysis of a non-biological ATP-binding protein. J Mol Biol. 2007;371:501–513. doi: 10.1016/j.jmb.2007.05.062. [DOI] [PubMed] [Google Scholar]
- Marty MT, Baldwin AJ, Marklund EG, Hochberg GK, Benesch JL, Robinson CV. Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Anal Chem. 2015;87:4370–4376. doi: 10.1021/acs.analchem.5b00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura T, Ernst A, Pluckthun A. Construction and characterization of protein libraries composed of secondary structure modules. Protein Sci. 2002;11:2631–2643. doi: 10.1110/ps.0215102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maury CP. Self-propagating beta-sheet polypeptide structures as prebiotic informational molecular entities: the amyloid world. Orig Life Evol Biosph. 2009;39:141–150. doi: 10.1007/s11084-009-9165-6. [DOI] [PubMed] [Google Scholar]
- McLysaght A, Guerzoni D. New genes from non-coding sequence: the role of de novo protein-coding genes in eukaryotic evolutionary innovation. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140332. doi: 10.1098/rstb.2014.0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLysaght A, Hurst LD. Open questions in the study of de novo genes: what, how and why. Nat Rev Genet. 2016;17:567–578. doi: 10.1038/nrg.2016.78. [DOI] [PubMed] [Google Scholar]
- Meier C, Aricescu AR, Assenberg R, Aplin RT, Gilbert RJ, Grimes JM, Stuart DI. The crystal structure of ORF-9b, a lipid binding protein from the SARS coronavirus. Structure. 2006;14:1157–1165. doi: 10.1016/j.str.2006.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meszaros B, Tompa P, Simon I, Dosztanyi Z. Molecular principles of the interactions of disordered proteins. J Mol Biol. 2007;372:549–561. doi: 10.1016/j.jmb.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Myers JK, Pace CN, Scholtz JM. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namy O, Duchateau-Nguyen G, Hatin I, Hermann-Le Denmat S, Termier M, Rousset JP. Identification of stop codon readthrough genes in Saccharomyces cerevisiae. Nucleic Acids Res. 2003;31:2289–2296. doi: 10.1093/nar/gkg330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- Pan X, Ye P, Yuan DS, Wang X, Bader JS, Boeke JD. A DNA integrity network in the yeast Saccharomyces cerevisiae. Cell. 2006;124:1069–1081. doi: 10.1016/j.cell.2005.12.036. [DOI] [PubMed] [Google Scholar]
- Pavesi A, Magiorkinis G, Karlin DG. Viral proteins originated de novo by overprinting can be identified by codon usage: application to the “gene nursery” of Deltaretroviruses. PLoS Comput Biol. 2013;9:e1003162. doi: 10.1371/journal.pcbi.1003162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervushin K, Vamvaca K, Vogeli B, Hilvert D. Structure and dynamics of a molten globular enzyme. Nat Struct Mol Biol. 2007;14:1202–1206. doi: 10.1038/nsmb1325. [DOI] [PubMed] [Google Scholar]
- Price NE, Price NC, Kelly SM, McDonnell JM. The key role of protein flexibility in modulating IgE interactions. J Biol Chem. 2005;280:2324–2330. doi: 10.1074/jbc.M409458200. [DOI] [PubMed] [Google Scholar]
- Prilusky J, Felder CE, Zeev-Ben-Mordehai T, Rydberg EH, Man O, Beckmann JS, Silman I, Sussman JL. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics. 2005;21:3435–3438. doi: 10.1093/bioinformatics/bti537. [DOI] [PubMed] [Google Scholar]
- Ptitsyn OB. Molten globule and protein folding. Adv Protein Chem. 1995;47:83–229. doi: 10.1016/s0065-3233(08)60546-x. [DOI] [PubMed] [Google Scholar]
- Rancurel C, Khosravi M, Dunker AK, Romero PR, Karlin D. Overlapping genes produce proteins with unusual sequence properties and offer insight into de novo protein creation. J Virol. 2009;83:10719–10736. doi: 10.1128/JVI.00595-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossouw D, Olivares-Hernandes R, Nielsen J, Bauer FF. Comparative transcriptomic approach to investigate differences in wine yeast physiology and metabolism during fermentation. Appl Environ Microbiol. 2009;75:6600–6612. doi: 10.1128/AEM.01251-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlessinger A, Schaefer C, Vicedo E, Schmidberger M, Punta M, Rost B. Protein disorder--a breakthrough invention of evolution? Curr Opin Struct Biol. 2011;21:412–418. doi: 10.1016/j.sbi.2011.03.014. [DOI] [PubMed] [Google Scholar]
- Schlotterer C. Genes from scratch--the evolutionary fate of de novo genes. Trends Genet. 2015;31:215–219. doi: 10.1016/j.tig.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz JF, Bornberg-Bauer E. Fact or fiction: updates on how protein-coding genes might emerge de novo from previously non-coding DNA. F1000Res. 2017;6:57. doi: 10.12688/f1000research.10079.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Bax A. Protein structural information derived from NMR chemical shift with the neural network program TALOS-N. Methods Mol Biol. 2015;1260:17–32. doi: 10.1007/978-1-4939-2239-0_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla A, Hilgenfeld R. Acquisition of new protein domains by coronaviruses: analysis of overlapping genes coding for proteins N and 9b in SARS coronavirus. Virus Genes. 2015;50:29–38. doi: 10.1007/s11262-014-1139-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz D, Domazet-Loso T. The evolutionary origin of orphan genes. Nat Rev Genet. 2011;12:692–702. doi: 10.1038/nrg3053. [DOI] [PubMed] [Google Scholar]
- Tompa P, Kovacs D. Intrinsically disordered chaperones in plants and animals. Biochem Cell Biol. 2010;88:167–174. doi: 10.1139/o09-163. [DOI] [PubMed] [Google Scholar]
- Troshin PV, Procter JB, Barton GJ. Java bioinformatics analysis web services for multiple sequence alignment--JABAWS:MSA. Bioinformatics. 2011;27:2001–2002. doi: 10.1093/bioinformatics/btr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanczyk M, Gora J, Latajka R, Sewald N. Antifreeze glycopeptides: from structure and activity studies to current approaches in chemical synthesis. Amino Acids. 2017;49:209–222. doi: 10.1007/s00726-016-2368-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11:739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Vamvaca K, Vogeli B, Kast P, Pervushin K, Hilvert D. An enzymatic molten globule: efficient coupling of folding and catalysis. Proc Natl Acad Sci U S A. 2004;101:12860–12864. doi: 10.1073/pnas.0404109101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargason JM, Szittya G, Burgyan J, Hall TM. Size selective recognition of siRNA by an RNA silencing suppressor. Cell. 2003;115:799–811. doi: 10.1016/s0092-8674(03)00984-x. [DOI] [PubMed] [Google Scholar]
- Wilson BA, Foy SG, Neme R, Masel J. Young Genes are Highly Disordered as Predicted by the Preadaptation Hypothesis of De Novo Gene Birth. Nat Ecol Evol. 2017;1:0146–0146. doi: 10.1038/s41559-017-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZR, Thomson R, McNeil P, Esnouf RM. RONN: the bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics. 2005;21:3369–3376. doi: 10.1093/bioinformatics/bti534. [DOI] [PubMed] [Google Scholar]
- Zhou M, Dagan S, Wysocki VH. Protein subunits released by surface collisions of noncovalent complexes: nativelike compact structures revealed by ion mobility mass spectrometry. Angew Chem Int Ed Engl. 2012;51:4336–4339. doi: 10.1002/anie.201108700. [DOI] [PubMed] [Google Scholar]
- Zhu H, Sepulveda E, Hartmann MD, Kogenaru M, Ursinus A, Sulz E, Albrecht R, Coles M, Martin J, Lupas AN. Origin of a folded repeat protein from an intrinsically disordered ancestor. Elife. 2016:5. doi: 10.7554/eLife.16761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckerkandl E. The appearance of new structures and functions in proteins during evolution. J Mol Evol. 1975;7:1–57. doi: 10.1007/BF01732178. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.