

Abstract
Treating KRAS-mutant lung adenocarcinoma (LUAD) remains a major challenge in cancer treatment given the difficulties associated with directly inhibiting the KRAS oncoprotein1. One approach to addressing this challenge is to define frequently co-occurring mutations with KRAS, which themselves may lead to therapeutic vulnerabilities in tumors. Approximately 20% of KRAS-mutant LUAD tumors carry loss-of-function (LOF) mutations in Kelch-like ECH-associated protein 1 (KEAP1)2-4, a negative regulator of nuclear factor erythroid 2-like 2 (NFE2L2; hereafter NRF2), which is the master transcriptional regulator of the endogenous antioxidant response5-10. The high frequency of mutations in KEAP1 suggests an important role for the oxidative stress response in lung tumorigenesis. Using a CRISPR/Cas9-based approach in a mouse model of Kras-driven LUAD we examined the effects of Keap1 loss in lung cancer progression. We show that loss of Keap1 hyper-activates Nrf2 and promotes Kras-driven LUAD. Combining CRISPR/Cas9-based genetic screening and metabolomic analyses, we show that Keap1/Nrf2-mutant cancers are dependent on increased glutaminolysis, and this property can be therapeutically exploited through the pharmacological inhibition of glutaminase. Finally, we provide a rationale for sub-stratification of human lung cancer patients with KRAS-KEAP1 or -NRF2-mutant tumors as likely to respond to glutaminase inhibition.
Genetically engineered mouse models (GEMMs) of lung cancer have greatly assisted in the functional characterization of genes implicated in human lung cancers. The KrasLSL-G12D/+; p53flox/flox (KP) GEMM of human LUAD faithfully mimics human KRAS-driven LUAD, displaying similarities at the molecular and histopathological level following intratracheal administration of viral vectors expressing Cre-recombinase11. We recently developed a CRISPR/Cas9-based in vivo genome engineering method to rapidly interrogate putative genetic driver events cooperating with oncogenic Kras to promote lung tumorigenesis in the KP model12-14.
Based on the fact that KEAP1 is the third most frequently mutated gene in LUAD and on the high coincidence of KEAP1 inactivating mutations and KRAS-mutation in human lung cancers3, we chose to target this gene in the KP model using CRISPR/Cas9 technology. KP mice were intratracheally infected with pSECC lentiviral vectors expressing sgRNAs against Keap1 or tdTomato as a control (Supplementary Fig 1a). Mice infected with pSECC vectors expressing different sgRNAs targeting Keap1 (hereafter, sgKeap1 mice) had significantly increased tumor burden and faster growth kinetics compared to sgTom mice, as determined by longitudinal micro-computed tomography (micro-CT; p < 0.05, Fig 1a). Consistent with the micro-CT data, histological assessment of tumor burden revealed a significant increase in sgKeap1 mice compared to controls (p < 0.05, Fig 1b). This analysis also showed a dramatic increase in high-grade tumors in sgKeap1 mice compared to controls (Fig 1c and Supplementary Fig 1b, p < 0.0001 for sgKeap1.2 grade 3 and p < 0.001 for sgKeap1.4 grade 4). Furthermore, sgKeap1 tumors displayed increased proliferation as gauged by an increase in mitotic index (phospho-Histone H3; p < 0.05, Fig 1d).
Figure 1. Loss of Keap1 stabilizes Nrf2 and accelerates lung tumorigenesis.
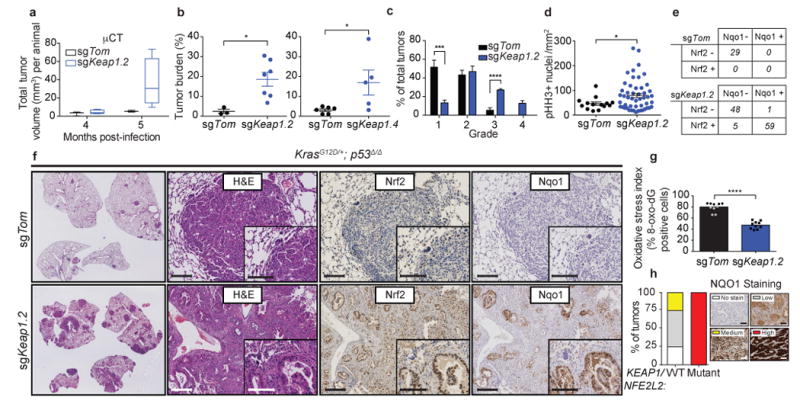
a) Micro-computed tomography (micro-CT) quantification of total tumor volume (mm3) of tumors from sgKeap1.4 (n = 5) or sgTom (n = 3) infected animals at 4 and 5 months post infection. b) Combined quantification of tumor burden (total tumor area/total lung area) in KrasLSL-G12D/+; p53fl/fl (KP) animals after infection with pSECC lentiviruses. Left panel: tumor burden 21 weeks post infection of animals infected with control sgTom (n = 3) or sgKeap1.2 (n = 7). Right panel: tumor burden 21 weeks post infection of animals infected with control sgTom (n = 6) or sgKeap1.4 (n = 5). The asterisks indicate statistical significance obtained from comparing KP-sgKeap1 samples to KP-sgTom samples. c) Distribution of histological tumor grades in KP animals 21 weeks after infection with pSECC lentiviruses expressing: control (sgTom, KP; n = 7 mice), sgKeap1.2 (KP; n = 3 mice). d) Quantification of phospho-Histone H3 (pHH3) positive nuclei per mm2 to assess the mitotic index of tumor cells from lung tumors in KP animals 21 weeks after infection with pSECC lentiviruses expressing: control (sgTom, n = 14 tumors), or sgKeap1.2 (n = 50 tumors). e) Contingency tables demonstrating correlation between nuclear Nrf2 expression and Nqo1 expression. Top panel: quantified tumors obtained from control sgTom infected mice. Bottom panel: quantified tumors obtained from sgKeap1.2 infected mice (two-sided Fisher's exact test, ****p < 0.0001). f) Representative hematoxylin and eosin (H&E) and immunohistochemistry (IHC) staining of serial sections from lung tumors of mice 21 weeks after infection with pSECC-sgTom (top panel) or pSECC-sgKeap1.2 (bottom panel). First panels: representative overall lung tumor burden. Second panel: higher magnification H&E of representative tumors. Third panel: Nuclear Nrf2 IHC. Fourth panel: Nqo1 IHC. Note the accumulation of Nrf2 and Nqo1 occurs only in tumors from pSECC-sgKeap1.2 mice. Inset represents higher magnification. Scale bars are 100um. g) Oxidative stress index as judged by % 8-oxo-dG positive nuclei (n = 10 per genotype). All error bars denote s.e.m. Obtained from two-sided Student's t-test unless otherwise noted. *p < 0.05, ***p < 0.001, ****p < 0.0001. h) KEAP1/NRF2-mutant versus WT human LUAD biopsy IHC for NQO1. All tumor samples were confirmed to be KEAP1/NRF2 mutant via targeted exome sequencing (See Supplementary Table 1). Right legend depicts examples of staining criteria.
To determine the status of the Keap1/Nrf2 pathway in sgKeap1 tumors, we performed immunohistochemical (IHC) analyses to assess whether loss of Keap1 led to both increased nuclear localization of Nrf2 protein and cytoplasmic levels of its target gene Nad(p)h dehydrogenase quinone 1 (Nqo1). The majority (60%) of sgKeap1 tumors had increased nuclear localization of Nrf2 and dramatically higher levels of Nqo1 as compared to controls (p < 0.0001, Fig 1e,f). Importantly, nearly all tumors that stained positively for nuclear Nrf2 also contained higher levels of Nqo1 (p < 0.0001, Fig 1e). Furthermore, the increased levels of Nrf2 in sgKeap1 tumors correlated with significantly lower ROS-dependent oxidation of DNA as compared to control sgTom tumors (Fig 1g). High throughput DNA sequencing of micro-dissected sgKeap1 tumors (sgKeap1.2 and sgKeap1.4) that stained positively for nuclear Nrf2 and Nqo1 revealed that these tumors predominantly contained frameshift LOF insertions or deletions (indels) in Keap1, supporting the IHC analysis indicating Nrf2 pathway activation (Supplementary Fig 1c-e). Additionally, we observed a clonal enrichment of such Keap1 LOF alleles in a lymph node metastasis compared to its paired primary tumor15,16 (Supplementary Fig 1f-h).
We next asked if NQO1 could act as a marker for NRF2 activated human KEAP1/NRF2 mutant LUAD tumors. Targeted exome capture (top 50 mutated LUAD genes based on TCGA3) of 88 LUAD tumors from the NYU Center for Biospecimen Research and Development identified 10 KEAP1 (11%), and 2 NRF2 (2%) mutant tumors, as well as a significant correlation between KEAP1/ NRF2 mutations and increased NQO1 staining (Figure 1h; p = 0.0002; Supplementary Table 1). These data suggest that NQO1 is a suitable biomarker for NRF2 activation in human LUAD.
To determine the role of Nrf2 and Keap1 in regulating proliferation and antioxidant pathways in LUAD, we used CRISPR/Cas9-mediated genome editing to develop isogenic KP-derived lung tumor cell lines with LOF mutations in Nrf2 (KPN), Keap1 (KPK), and sgTom controls (KP) (Supplementary Fig 2a,b; n = 2 cell lines per genotype). As expected, KPK cells had increased nuclear localization of Nrf2 and increased levels of Nrf2 transcriptional targets as assessed by both protein analysis (Gclc; Supplementary Fig 2c) and gene expression analysis (Nqo1, Hmox1 and Gclc; Supplementary Fig 2d). These changes were also observed in KP but not KPN cells upon treatment with Nrf2 activators (Supplementary Fig 2e-h). To validate these results, we performed whole transcriptome analyses (RNA sequencing) and identified transcriptional signatures that clearly distinguished KP from KPK cell lines based on the activation of the Nrf2 transcriptional program (Supplementary Fig 2i; Supplementary Table 2).
We next used this panel of genetically-defined cell lines to further explore the role of the Nrf2/Keap1 pathway in regulating the antioxidant response program. KPN cells had dramatically decreased cell viability in response to multiple agents known to cause oxidative stress compared to KP cells. By contrast, KPK cells showed resistance to all agents tested (Supplementary Fig 3a-e). These effects correlated with the total levels of the major cellular antioxidant glutathione in the different cell lines (Supplementary Fig 3f,g). The loss of viability of KPN cells in response to oxidative stress agents was rescued by antioxidant treatments (Supplementary Fig 3h) or by ectopic expression of a gain-of-function (GOF) allele of Nrf217 (KPN-ix; Supplementary Fig 3i-o). Consistent with these results, both mouse and human Keap1/KEAP1-mutant cells displayed markedly lower ROS levels compared to wild-type (WT) cells (Supplementary Fig 3p,q). Interestingly, KPK cells grew faster than KP cells in vivo but not in vitro (Supplementary Fig 4a-f), suggesting a differential requirement of the Nrf2-antioxidant response during tumorigenesis in vivo. In addition, loss of Keap1/KEAP1 in tumors and cells with WT p5318 accelerated tumorigenesis and growth suggesting that Keap1 is a tumor suppressor in lung cancer progression independent of p53 mutation status (Supplementary Fig 5a-p). These data indicate that Nrf2 levels dictate the differential antioxidant response to oxidative stress, which may provide a selective growth advantage in vivo.
To assess the relevance of these data derived from GEMM studies for human lung cancer, we performed an integrative analysis using a dataset of human LUAD patient samples (n = 548) from The Cancer Genome Atlas (TCGA)3, published Nrf2 datasets7,19,20 and our GEMM-derived Nrf2-driven transcriptional signature. First, we derived a core signature of 108 high confidence NRF2 target genes (Supplementary Table 3) using published datasets. TCGA human LUAD tumors across various disease stages were investigated; the core NRF2 target genes were significantly upregulated in tumors from advanced stage IV disease (p = 0.028, Fig 2a). Additionally, patients whose tumors were most associated with the NRF2 core target signature had significantly worse survival when compared to the rest of the TCGA LUAD cohort (p = 0.008, Fig 2b). In order to evaluate the association between KEAP1 mutations and NRF2 pathway activation, we used gene expression data from all TCGA human LUAD primary tumors to derive a KEAP1-mutant transcriptional signature (Supplementary Fig 6a). This signature was enriched in the core NRF2 target genes, multiple antioxidant pathways, and the NRF2 oncogenic signature18 (NFE2L2.V2; Supplementary Fig 6b,c and Supplementary Table 4). Ranking tumors by the strength of their correlation with this signature allowed for stratification of all LUAD TCGA patients into two sub-populations (n = 91 most-correlated 20%, n = 367 rest of the cohort). These sub-populations exhibited significantly different survival times (p = 0.012, Fig 2c). Similar results were observed within the set of KRAS-mutant patients (n = 24 most-correlated 20%, n = 99 rest of cohort, p = 0.00013; Supplementary Fig 6d). We did not observe significant co-occurrence of KEAP1-mutant and KRAS-mutant patients within the TCGA cohort (p = 0.418). Additionally, within the top 20% of patients that correlate with our KEAP1-mutant signature and exhibit poor survival, we did not observe an enrichment for KRAS-mutant patients (p = 0.816) when compared to the background prevalence of KRAS-mutant patients in the TCGA cohort. Taken together, these data suggest that the poor survival of patients most correlated with the KEAP1-mutant signature cannot be attributed to an over-representation of KRAS-mutant patients.
Figure 2. A NRF2 target gene signature and a human derived KEAP1-mutant and predict human LUAD patient survival.
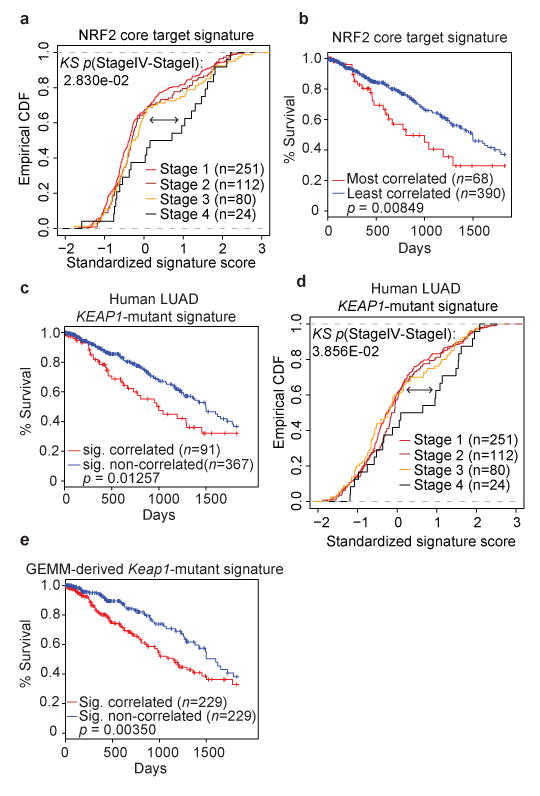
a) Empirical cumulative distribution function (CDF) plot showing correlation of individual tumors with the NRF2 core target signature across various clinical stages within the TCGA LUAD cohort. Each curve in the plot represents a unique clinical stage as depicted in the figure legend. Clinical stage IV tumors (n = 24) are highly correlated with the NRF2 core target signature and are significantly different compared to lower stage I tumors (n = 251; p = 0.028; KS = Kolmogorov-Smirnov test). b) Kaplan-Meier (KM) survival curves comparing LUAD TCGA patients stratified by their correlation with the NRF2 core target signature. Patient tumor samples were binned according to their gene expression correlation with the NRF2 signature. The top 15% (n = 68) correlated tumors exhibit significantly decreased survival compared to the rest (n = 390) of the TCGA LUAD cohort (p = 0.008, log-rank test). c) KM survival curves comparing TCGA LUAD patients stratified by their correlation with the KEAP1-mutant signature derived from TCGA patient expression profiles. The top 20% correlated patients (n = 91) exhibit decreased survival compared to the rest (n = 367) of the TCGA LUAD cohort (p = 0.012, log-rank test). d) Empirical cumulative distribution function (CDF) plot showing expression correlation of individual tumors with the KEAP1-mutant signature across various clinical stages within the TCGA LUAD cohort. Each curve represents a unique clinical stage as depicted in the figure legend. Clinical stage IV tumors (n = 24) are highly correlated with the KEAP1-mutant signature and are significantly different compared to stage I tumors (n = 251; p = 0.038, KS=Kolmogorov-Smirnov test). e) KM survival curves comparing TCGA LUAD patients stratified by their correlation with the murine-derived Keap1-mutant signature. The top 50% correlated tumors (n = 229) exhibit significantly decreased survival compared to the rest (n = 229) of the TCGA LUAD cohort (p < 0.003, log-rank test).
Furthermore, high grade tumors (grades III/IV) and late stage tumors (clinical stage IV disease) were significantly enriched for the human KEAP1-mutant transcriptional signature (Supplementary Fig 6e; grade III/IV: p = 0.02; Fig 2d stage IV: p = 0.038). Importantly, this signature was found to be independently prognostic in the TCGA LUAD cohort while controlling for other clinical covariates in a Cox proportional hazards model (HR = 1.22; univariate p = 0.029, multivariable p = 0.04, Supplementary Table 5) where higher enrichment for the signature was associated with significantly worse survival. We also did not detect an enrichment for TP53 mutated patients in the KEAP1-mutant signature correlated cohort. Likewise, we did not observe a significant co-occurrence of KEAP1-mutant and TP53-mutant patients in the TCGA LUAD cohort (p = 0.115). To assess the translational potential of the GEMM results to human LUAD with KEAP1 mutations, we performed a cross-species comparison of the Keap1-mutant transcriptional signatures. The GEMM Keap1-mutant signature (Supplementary Fig 2i) was significantly enriched in the human KEAP1-mutant signature (Supplementary Fig 6f). Furthermore, the GEMM-based signature could also stratify human patients with significantly different survival times (with correlated patients showing poor survival (p = 0.003, Fig 2e)).
Having established the importance of KEAP1 mutations in mouse and human KRAS-driven LUAD, we sought to uncover potential therapeutic vulnerabilities in this genetic subtype of lung cancer. To this end, we performed a focused CRISPR/Cas9-based genetic screen to identify synthetic genetic interactions with Keap1 mutations. A pool of lentiviruses expressing a focused CRISPR/Cas9 library was engineered to express sgRNAs against a panel of Nrf2 transcriptional targets and genes implicated in the Nrf2 antioxidant response (17 genes and 3 controls, 3-4 sgRNAs/gene, 65 sgRNAs total, Supplementary Table 6 Fig 3a and Supplementary Fig 7a). We infected KP or KPK cell lines (n = 2 per genotype) and assessed the relative depletion of sgRNAs after 14 population doublings to identify genes in which mutations selectively affected the growth of KPK compared to KP cells in culture (average relative depletion score threshold <-0.3). Notably, out of 60 experimental sgRNAs across 17 genes, three out of four sgRNAs against solute carrier family 1 member 5 (Slc1a5), a glutamine transporter21, fell below our threshold values and were depleted in KPK but not KP cells, suggesting that Slc1a5 mutation selectively impairs the growth of Keap1-mutant cells (Fig 3a and Supplementary Fig 7a). We next generated Slc1a5-mutant derivatives of KPK and human lung cancer cells with KRAS and KEAP1 mutations (A549 and H2030). These cells displayed markedly decreased growth, while we observed no effect in Keap1-WT mouse (KP1, KP2) and human (H2009) cell lines upon mutation of Slc1a5 (Fig 3b-d and Supplementary Fig 7b). Furthermore, KPK cell lines were more sensitive to GPNA, a small molecule inhibitor of Slc1a5, compared to KP cell lines (Fig 3e and Supplementary Fig 7c). The selective requirement of Slc1a5 function in KPK cell lines suggested a possible metabolic dependency of KPK cells on glutamine. Indeed, decreasing glutamine concentration in the media led to a robust suppression of growth in KPK cell lines with little effect on KP cell lines (Fig 3f and Supplementary Fig 7d). The dependency of KPK cell lines on Slc1a5 and glutamine could be via the fueling of the tricarboxylic acid (TCA) cycle in the context of an increased glycolytic state22. Consistent with this possibility, we found that both KPK cell lines had higher glucose (Supplementary Fig 7e) and glutamine consumption (Fig 3g) coupled with a marked increase in lactate excretion compared to KP cells (Supplementary Fig 7e). KPK cells also showed increased sensitivity to the glycolysis inhibitor 2-deoxy-D-glucose (2-DG; Supplementary Fig 8a,b). In addition, isotopic carbon labeled glucose (U13-Glucose) tracing revealed decreased contribution of glucose-derived carbons to TCA cycle intermediates in KPK cells compared to KP controls (Supplementary Fig 8c-e), which is not due to differences in the expression of pyruvate carboxylase (Pcx) and glutamine synthetase (Glul) between KPK and KP cells (Supplementary Data Table 2).
Figure 3. CRISPR screen reveals that Keap1-mutant cells are glycolytic and sensitive to reduced levels of glutamine.
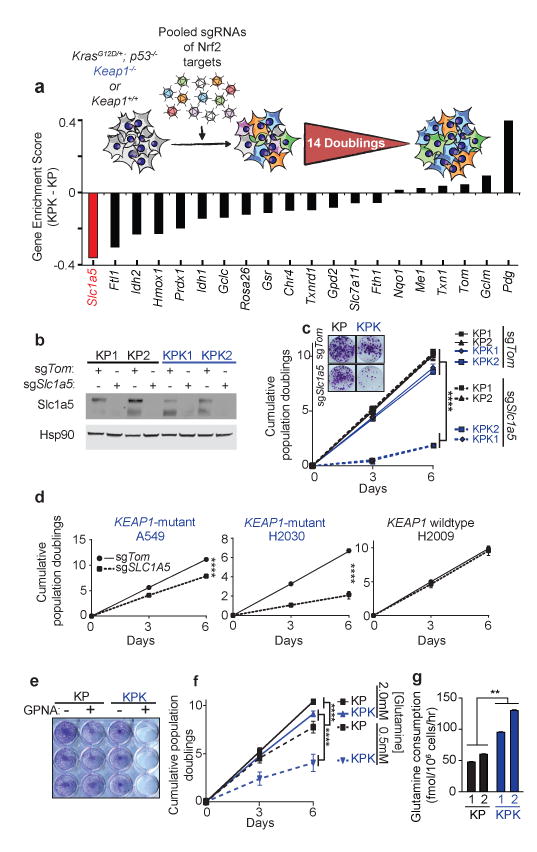
a) Pooled sgRNA library screen. Figure inlet; schematic of experiment. Cells were passaged for 14 doublings before collection. Bars represent the median differential genescore. Full representation in Supplementary Fig 7a. b) Western blot analysis of Slc1a5 in KP and KPK cells post selection infected with sgTom or sgSlc1a5. Hsp90 was used as a loading control. c) Cumulative population doublings of KP and KPK cells after transduction with sgTom or sgSlc1a5 (n = 4). Picture inlet; colony formation assay in KP and KPK cells transduced with sgTom or sgSlc1a5. ****p < 0.0001 obtained from 2-way ANOVA. d) Cumulative population doublings of KRAS-mutant human lung cancer cell line either KEAP1-WT (H2009) or KEAP1-mutant (A549 and H2030) after selection with sgTom or sgSLC1A5 (n = 4). ****p < 0.0001 obtained from 2-way ANOVA. e) Crystal violet stain of KP and KPK cells treated with 1mM GPNA or Vehicle for 72 hrs. f) Cumulative population doublings of KP and KPK cells cultured in 2.0mM or 0.5mM glutamine (n = 4). ****p < 0.0001 obtained from 2-way ANOVA. g) Glutamine consumption in KP and KPK cells measured (n = 3). All samples were normalized to their respective vehicle treated control. **p < 0.01 obtained from 1-way ANOVA with Tukey's post hoc test. All error bars depict s.e.m.
We next investigated whether increased glutamine utilization in KPK cell lines could be exploited as a metabolic liability. As glutaminase is the rate-limiting enzyme for glutamine utilization in the cell14,23 (Fig 4a), we tested two small molecule inhibitors of glutaminase: BPTES and CB-83921, the latter which is currently in phase I clinical trials for KRAS-mutant lung cancer (Fig 4a,b and Supplementary Fig 9a). KPK cells were markedly more sensitive to both drugs compared to KP cells (Fig 4c). In addition, a panel of human lung cancer cells containing KEAP1 or GOF-NRF2 mutations were sensitive to glutaminase inhibition while KEAP1-WT cells were largely resistant (Fig 4d and Supplementary Fig 9b). Interestingly, pretreatment of KPK cells with glutamate, pyruvate or cell permeable alpha-ketoglutarate, but not the antioxidants Trolox or N-acetyl cysteine (NAC), rescued CB-839 sensitivity (Supplementary Fig 9c-f). These results suggest that glutaminase inhibition suppresses cell growth by blocking anaplerosis and not through loss of antioxidant production. To determine whether the sensitivity of KPK cells to glutaminase inhibition was dependent on hyperactive Nrf2 signaling, we transduced KP cells with lentiviruses expressing a GOF-Nrf2 allele (Supplementary Fig 3i-o; Supplementary Fig 10a,b; KP-ix). Expression of GOF-Nrf2 in KP cells led to increased sensitivity to CB-839 (Supplementary Fig 10c). In addition, genetic complementation of Keap1 in KPK cells reduced Nrf2 protein levels, expression of Nrf2 target genes, reversed the in vivo growth advantage of KPK cells, and rescued the viability of CB-839 treated KPK cells (Supplementary Fig 10d-g).
Figure 4. Keap1-mutant cells display a robust sensitivity to glutaminase inhibition.
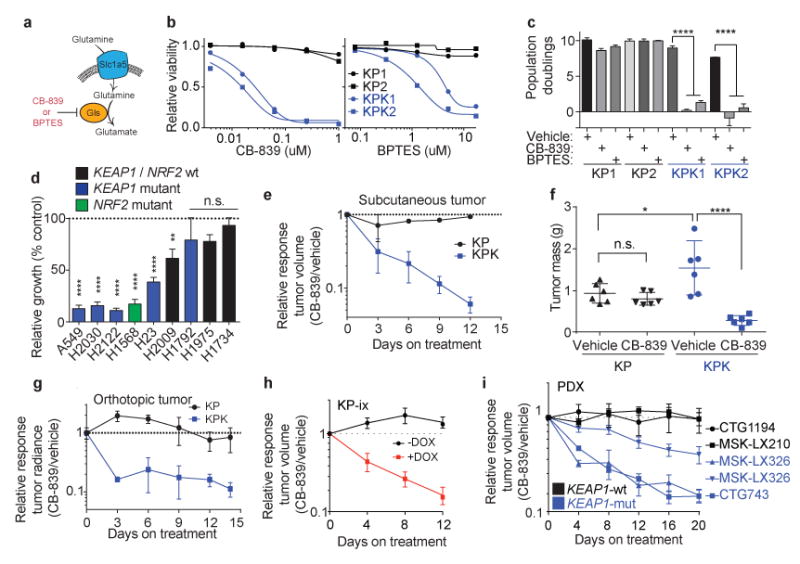
a) Schematic of glutamine uptake by Slc1a5 and hydrolysis of glutamine to glutamate by Gls. Inhibitors of Gls are shown in red. b) Relative viability assayed by cell-titer glo (relative luminescent units) on KP and KPK cells after treatment with CB-839 (left) or BPTES (right) for 72 hrs. All data points are relative to vehicle treated controls (n = 4 technical replicates/data point). c) Cumulative population doublings of KP and KPK cells in the presence of vehicle, CB-839 or BPTES (n = 4 technical replicates/data point) after 6 days in culture. d) Trypan blue exclusion viability counts of indicated human lung cancer cell lines. Each cell line was cultured in the presence of vehicle or 500nM CB-839 (n = 4 technical replicates/cell line). Displayed results are normalized against vehicle treated cell lines after 72 hrs of treatment. A549 and H1975 are TP53-wild type, all others are TP53-mutant. e) Subcutaneous tumor volumes of KP and KPK treated with vehicle or CB-839 starting from day 13 measured over time for 25 days (n = 6 tumors/genotype/treatment). Related to Fig 4f. f) Final tumor masses related to Supplementary Data Fig 11b. *p < 0.05, ****p < 0.0001 obtained from 1-way ANOVA with Tukey's post hoc test. g) Orthotopic growth measurements of KP and KPK cells treated with vehicle or CB-839 starting from day 13 (n = 4 mice/genotype/treatment). Quantitation of luminescence (photon flux) in mice orthotopically transplanted with KP or KPK cells transduced with a vector expressing Luciferase. Relative photon flux calculated by normalizing all time points per animal to initial measurements at 10 days post transplantation. Individual groups depicted in Supplementary Data Fig 11c. ***p < 0.001 obtained from 2-way ANOVA. h) Subcutaneous tumor volumes of KP-ix (inducible GOF-Nrf2) treated with vehicle or CB-839 in the presence or absence of doxycycline (DOX) (n = 6 mice/DOX treatment). Individual groups and full experiment depicted in Supplementary Data Fig 11d. i) Five patient-derived xenograft (PDX) models treated with vehicle or CB-839 for the indicated amount of days. Individual groups and full experiments depicted in Supplementary Data Fig 11g and h. All error bars depict s.e.m.
To investigate the therapeutic potential of targeting glutaminase in Keap1-mutant tumors in vivo, we transplanted KP and KPK cells subcutaneously and orthotopically (lung) in immunodeficient animals. Once tumors were established, we initiated treatment with either vehicle or CB-839 (Supplementary Fig 11a). Consistent with an earlier study14, we found that KP-derived tumors exhibited no response to CB-839 treatment (Fig 4e-g and Supplementary Fig 11b,c). By contrast, KPK-derived subcutaneous and orthotopic tumors had dramatically decreased growth and established smaller final tumor weights in response to CB-839 treatment (Fig 4e-g and Supplementary Fig 11b,c). Furthermore, transplanted KP-ix cells exhibited increased growth upon doxycycline-dependent induction of GOF-Nrf2, which was suppressed by glutaminase inhibition (Fig 4h and Supplementary Fig 11d). Finally, we demonstrated that glutaminase inhibition suppressed the in vivo growth of KRAS-driven human LUAD cancer cell lines and patient-derived xenografts with KEAP1 mutations, but had no effect on the growth of KEAP1-WT tumors (Figure 4i; Supplementary Fig 11e-I; Supplementary Table 7). Taken together, these data suggest that glutaminase or other targets within this metabolic pathway are attractive therapeutic targets in Keap1/Nrf2-mutant LUAD. Furthermore, rational stratification of patients harboring mutations in KEAP1 or NRF2 may predict treatment response to glutaminase inhibitors.
In conclusion, we demonstrate that Keap1 mutations activate the Nrf2 antioxidant program and cooperate with mutant Kras to drive LUAD progression, supporting the requirement for cancer cells to overcome oxidative stress barriers during tumorigenesis24-30. We hypothesize that the metabolic requirement for glutaminolysis in KEAP1/NRF2-mutant LUAD tumors may also present a therapeutic vulnerability in other cancers with genetic31-36, epigenetic37-39 or post-transcriptional17 alterations in the KEAP1/NRF2 pathway. A recent study demonstrated that KEAP1 loss potentiates resistance to multiple targeted therapies in EGFR- and RAS-driven cancers, highlighting the importance of our therapeutic strategy against KRAS-KEAP1-mutant lung cancer40. Furthermore, our findings provide unique insight into the therapeutic potential of targeting metabolic dependencies based on somatic variants by combining genetic and metabolic approaches to identify novel targets in translational oncology. Collectively, our study presents a novel CRISPR/Cas9-based precision medicine platform that can be used to characterize putative cooperating mutations and identify genotype-specific vulnerabilities in cancer.
Supplementary Material
Acknowledgments
We thank D. McFadden, R. Possemato, S. Sayin and T. Gonzalez-Robles for critical reading of the manuscript. We thank T. Tammela, L. Sullivan, G. DeNicola and I. Harris for scientific discussions and feedback. S. Levine and T. Mason for massively parallel sequencing expertise; M. Griffin, M. Jennings and G. Paradis for FACS support; K. Cormier and the Hope Babette Tang (1983) Histology Facility for histology support; I. Baptista, A. Deconinck, J. Teixeira and K. Yee for administrative support; and the Swanson Biotechnology Center for excellent core facilities. This work was supported in part by the Laura and Isaac Perlmutter Cancer Support Grant and NIH S10 awards and the Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute. T.P. was supported by the American Cancer Society and Hope Funds for Cancer Research. T.P. lab is supported by the NIH (K22CA201088-01) and NYU Department of Pathology Bridge Grant. R.R. was supported by the National Science Foundation Graduate Research Fellowship under Grant No. 1122374. V.I.S received support from the Swedish Medical Research Council, the AG Fond, the Wenner-Gren Foundations and is the recipient of EMBO Long Term Fellowship ALTF 1451-2015 co-funded by the European Commission (LTCOFUND2013, GA-2013-609409) with support from Marie Curie Actions. S.E.L. is supported by an NIH training grant (5T32HL007151-38). H.I.P. human tumor collection was supported by NCI EDRN grant (2U01CA 111295-04). T.J. lab research was supported by the Cancer Center Support Grant P30-CA14051 and the Howard Hughes Medical Institute. T.J. is a Howard Hughes Medical Institute Investigator, the David H. Koch Professor of Biology, and a Daniel K. Ludwig Scholar.
Footnotes
Author Contributions: R.R., V.I.S., F.J.S.R., T.J., and T.P. designed the study; R.R., V.I.S., M.R.B., S.M.D., S.X.S., S.E.L., T.R.K, D.C.E, L.S., and B.M. performed experiments; A.B. and I.D. conducted bioinformatic analyses; S.M.D. and M.V.H. provided feedback and interpretation of metabolism data; E.E.S. and J.R.P. provided custom Nrf2 antibody; C.J.T. provided advice and feedback on CB-839 administration; R.T.P performed histopathological analysis of GEMMs; A.D., V.A., J.T.P., and C.M.R. generated and characterized PDX models; I.D., A.H., A.L.M, C.G, and H.I.P. were involved in human tumor collection, sequencing and characterization; R.R., V.I.S., T.J., and T.P., wrote the manuscript with comments from all authors.
Competing Financial Interests: The authors report no competing financial interests
References
- 1.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov. 2014;13:828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berger AH, et al. High-throughput Phenotyping of Lung Cancer Somatic Mutations. Cancer Cell. 2016;30:214–228. doi: 10.1016/j.ccell.2016.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh A, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS medicine. 2006;3:e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Itoh K, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 6.Itoh K, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitsuishi Y, et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22:66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 8.Harris IS, et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell. 2015;27:211–222. doi: 10.1016/j.ccell.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 9.DeNicola GM, et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet. 2015;47:1475–1481. doi: 10.1038/ng.3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sullivan LB, Gui DY, Vander Heiden MG. Altered metabolite levels in cancer: implications for tumour biology and cancer therapy. Nat Rev Cancer. 2016;16:680–693. doi: 10.1038/nrc.2016.85. [DOI] [PubMed] [Google Scholar]
- 11.DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc. 2009;4:1064–1072. doi: 10.1038/nprot.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanchez-Rivera FJ, et al. Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature. 2014;516:428–431. doi: 10.1038/nature13906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mazur PK, et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat Med. 2015;21:1163–1171. doi: 10.1038/nm.3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davidson SM, et al. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016;23:517–528. doi: 10.1016/j.cmet.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maresch R, et al. Multiplexed pancreatic genome engineering and cancer induction by transfection-based CRISPR/Cas9 delivery in mice. Nat Commun. 2016;7:10770. doi: 10.1038/ncomms10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKenna A, et al. Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science. 2016;353:aaf7907. doi: 10.1126/science.aaf7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldstein LD, et al. Recurrent Loss of NFE2L2 Exon 2 Is a Mechanism for Nrf2 Pathway Activation in Human Cancers. Cell Rep. 2016;16:2605–2617. doi: 10.1016/j.celrep.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 18.Meylan E, et al. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature. 2009;462:104–107. doi: 10.1038/nature08462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh A, et al. RNAi-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy. Cancer Res. 2008;68:7975–7984. doi: 10.1158/0008-5472.CAN-08-1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malhotra D, et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010;38:5718–5734. doi: 10.1093/nar/gkq212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hassanein M, et al. SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin Cancer Res. 2013;19:560–570. doi: 10.1158/1078-0432.CCR-12-2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Metallo CM, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16:619–634. doi: 10.1038/nrc.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bauer AK, et al. Targeted deletion of Nrf2 reduces urethane-induced lung tumor development in mice. PLoS One. 2011;6:e26590. doi: 10.1371/journal.pone.0026590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeNicola GM, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Satoh H, Moriguchi T, Takai J, Ebina M, Yamamoto M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer Res. 2013;73:4158–4168. doi: 10.1158/0008-5472.CAN-12-4499. [DOI] [PubMed] [Google Scholar]
- 27.Sayin VI, et al. Antioxidants accelerate lung cancer progression in mice. Sci Transl Med. 2014;6:221ra215. doi: 10.1126/scitranslmed.3007653. [DOI] [PubMed] [Google Scholar]
- 28.Chio II, et al. NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell. 2016;166:963–976. doi: 10.1016/j.cell.2016.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Satoh H, et al. NRF2 Intensifies Host Defense Systems to Prevent Lung Carcinogenesis, but After Tumor Initiation Accelerates Malignant Cell Growth. Cancer Res. 2016;76:3088–3096. doi: 10.1158/0008-5472.CAN-15-1584. [DOI] [PubMed] [Google Scholar]
- 30.Kerr EM, Gaude E, Turrell FK, Frezza C, Martins CP. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature. 2016;531:110–113. doi: 10.1038/nature16967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cancer Genome Atlas Research, N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27:2179–2191. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Konstantinopoulos PA, et al. Keap1 mutations and Nrf2 pathway activation in epithelial ovarian cancer. Cancer Res. 2011;71:5081–5089. doi: 10.1158/0008-5472.CAN-10-4668. [DOI] [PubMed] [Google Scholar]
- 34.Shibata T, et al. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology. 2008;135:1358–1368. 1368 e1351–1354. doi: 10.1053/j.gastro.2008.06.082. [DOI] [PubMed] [Google Scholar]
- 35.Kim YR, et al. Oncogenic NRF2 mutations in squamous cell carcinomas of oesophagus and skin. The Journal of pathology. 2010;220:446–451. doi: 10.1002/path.2653. [DOI] [PubMed] [Google Scholar]
- 36.Sato Y, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet. 2013;45:860–867. doi: 10.1038/ng.2699. [DOI] [PubMed] [Google Scholar]
- 37.Fabrizio FP, et al. Keap1/Nrf2 pathway in kidney cancer: frequent methylation of KEAP1 gene promoter in clear renal cell carcinoma. Oncotarget. 2017 doi: 10.18632/oncotarget.14492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muscarella LA, et al. Regulation of KEAP1 expression by promoter methylation in malignant gliomas and association with patient's outcome. Epigenetics. 2011;6:317–325. doi: 10.4161/epi.6.3.14408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hanada N, et al. Methylation of the KEAP1 gene promoter region in human colorectal cancer. BMC cancer. 2012;12:66. doi: 10.1186/1471-2407-12-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krall EB, et al. KEAP1 loss modulates sensitivity to kinase targeted therapy in lung cancer. eLife. 2017;6 doi: 10.7554/eLife.18970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.