

Abstract
The mRNA lifecycle is driven through spatiotemporal changes in the protein composition of mRNA particles (mRNPs) that are triggered by RNA-dependent DEAD-box protein (Dbp) ATPases. As mRNPs exit the nuclear pore complex (NPC) in Saccharomyces cerevisiae, this remodeling occurs through activation of Dbp5 by inositol hexakisphosphate (IP6)-bound Gle1. At the NPC, Gle1 also binds Nup42, but Nup42’s molecular function is unclear. Here we employ the power of structure-function analysis in S. cerevisiae and human (h) cells, and find that the high-affinity Nup42-Gle1 interaction is integral to Dbp5 (hDDX19B) activation and efficient mRNA export. The Nup42 carboxy-terminal domain (CTD) binds Gle1/hGle1B at an interface distinct from the Gle1-Dbp5/hDDX19B interaction site. A nup42-CTD/gle1-CTD/Dbp5 trimeric complex forms in the presence of IP6. Deletion of NUP42 abrogates Gle1-Dbp5 interaction, and disruption of the Nup42 or IP6 binding interfaces on Gle1/hGle1B leads to defective mRNA export in S. cerevisiae and human cells. In vitro, Nup42-CTD and IP6 stimulate Gle1/hGle1B activation of Dbp5 and DDX19B recombinant proteins in similar, non-additive manners, demonstrating complete functional conservation between humans and S. cerevisiae. Together, a highly conserved mechanism governs spatial coordination of mRNP remodeling during export. This has implications for understanding human disease mutations that perturb the Nup42-hGle1B interaction.
Phrases: nuclear pore complex, mRNA export, Gle1, Dbp5, Nup42, inositol hexakisphosphate, mRNP remodeling
INTRODUCTION
Eukaryotic cellular function is dependent on the proper and efficient export of mRNAs from the nucleus through nuclear envelope (NE)-embedded NPCs to the cytoplasm. An essential rate-limiting step in this pathway occurs at the cytoplasmic face of the NPC on a structural platform over the NPC central channel1,2. At this site, the ATPase cycle of the S. cerevisiae Dbp5 (DDX19B in human) mediates release of the export factor Mex67-Mtr2 (NXF1 in human) and other RNA-binding proteins (RBPs) from the translocating mRNPs3,4. Several essential factors regulate the Dbp5 ATPase cycle at the NPC cytoplasmic face: the nucleoporin Nup159 (Nup214 in human) which facilitates ADP release5, and the protein Gle1 bound to IP6 which enhances ATP loading for triggering RNA-dependent ATP hydrolysis5–8. Control of the Dbp5 ATPase by Gle1-IP6 and Nup159 at the NPC cytoplasmic face results in spatially coordinated mRNP remodeling of Mex67-Mtr2 and RBPs from exporting mRNPs. As such, directionality is conferred on the export mechanism, allowing release of the mRNP to the cytoplasm for protein synthesis.
Although first identified as an NPC-associated mRNA export factor in S. cerevisiae9, Gle1 is conserved in higher eukaryotes10,11. In addition to regulating Dbp5 at the NPC, Gle1 is required for the function of distinct Dbps at non-NPC subcellular locales. In S. cerevisiae, Gle1-IP6 regulates Dbp5 activity during translation termination, and Gle1 impacts the ATPase activity and RNA binding of Ded1, another DEAD-box protein, for efficient translation initiation independently of IP612–15. In humans, hGle1 shuttles between the nucleus and cytoplasm with at least two isoforms, hGle1A and hGle1B, produced from a single gene11,16. hGle1A and hGleB are identical except for the C-terminal region wherein differential splicing results in an extended 43 amino acids in hGle1B (Figure 1A)16. The hGle1B isoform localizes predominantly at the NPC and is required for mRNA export16,17 whereas hGle1A is not functional at the NPC and has cytoplasmic roles at stress granules and in regulating the human Ded1 homologue DDX3 during translation18. Most recently, hGle1 has also been implicated in centrosome assembly and cilia function, potentially regulating an unidentified DEAD-box protein for localized mRNA metabolism19. Taken together, Gle1 and hGle1 play critical roles in regulating Dbps during multiple stages in gene expression at distinct subcellular locations.
Figure 1. Gle1-Nup42 interaction domains.
(A) Domain structure of Gle1, hGle1B, and hGle1A. For Gle1, K377 K378 indicates IP6-coordinating residues identified in10, V513 A516 I520 indicates residues essential for Dbp5 ATPase regulation7, and Q491 K494 E501 E502 indicates residues essential for interaction with Nup42 identified in this paper. For hGle1B, K526 K527 are homologous to S. cerevisiae Gle1 K377 K378, with K479 K486 as additional positively-charged residues at the IP6 interface (based on homologous residues in the Gle1 structure7, PDB 3RRM), and Q640 K643 E650 D651 indicates residues essential for interaction with hNup42 identified in this paper. Symbols denote changes from disease-associated mutations25,27: asterisks are ALS-associated mutations, filled circle indicates LCCS1 Finmajor, open circles indicate other LCCS1 or LAAHD mutations. From amino to carboxy-terminus: S70X, ALS-associated nonsense mutation; T144_E145insPFQ, LCCS1 Finmajor; R569H, LCCS1; V617M, LAAHD; 654/IVS14-2A>C, ALS (alternate exon use results in novel C-terminal extension from this residue); I694T, LAAHD; R697C, ALS. (B) Domain structure of Nup42 and hNup42. For Nup42, 408–424 indicates minimal Gle1 interaction domain defined in this paper. (A & B) Colored regions indicate truncations used in biochemistry experiments. (C) S. cerevisiae Gle1 contains a conserved patch of residues opposite the Dbp5 binding site. From a Clustal Omega35 sequence alignment between Gle1 and hGle1B, identical (red) and similar (orange) residues were mapped onto the Gle1-Dbp5 structure7, PDB 3RRM). (D) Q491, K494, E501, and E502 are surface accessible residues in the conserved patch on S. cerevisiae Gle1. Residues are indicated on zoomed region boxed in C.
It is speculated that Gle1’s multi-functional capabilities are dictated by unique protein interaction partners at different subcellular sites20. Thus, to understand how spatial regulation of Dbp5 is controlled at the NPC cytoplasmic face, it is important to precisely pinpoint the roles of Gle1 interactions at the NPC. The amino terminal half of Gle1 contains a coiled-coil region that promotes Gle1 self-association and is required for NPC localization (Figure 1A)17. Intriguingly, this region of Gle1 crosslinks to several members of the Nup82 holo-complex1, suggesting a potential mechanism for Gle1 localization at the NPC through direct interaction with this cytoplasmically-oriented complex. The region comprising the first 29 amino acids of the human Gle1 amino-terminus also binds to human Nup155, and is required for NPC localization (Figure 1A)21. Finally, the carboxy-terminal domain (CTD) of Gle1/hGle1B binds the CTD of Nup42/hNup42 (also known as Rip1 in S. cerevisiae and hCG1 or NUPL2 in human) (Figure 1B)9,22,23. The recent structural model of the cytoplasmic NPC face proposes that Gle1, Dbp5, and Nup42 are possibly oriented toward the NPC central channel1, positioning these factors for interaction with exporting mRNPs. However, the molecular details of the interactions that allow this Gle1 positioning are still undetermined. Revealing how Gle1 acts at the NPC is also needed to give insight into human disease mechanisms. Disease mutations linked to hGLE1 that alter hGle1 self-association, the hGle1-hNup42 interaction, and/or the respective pools of hGle1 at the NPC versus in the cytoplasm are associated with devastating pathologies including lethal congenital contracture syndrome 1 (LCCS1)17,24,25 and amyotrophic lateral sclerosis (ALS)26,27.
With respect to Nup42, some discrete functions at the NPC have been defined for its different domains. Our prior studies revealed a role for the FG domain in recruiting the mRNP to be in proximity to Gle1 and Dbp5 for remodeling28, via the Nup42 FG domain interacting with Mex67-Mtr2 (Figure 1B)29. In fact, a fusion between Gle1 and the FG domain of Nup42 (gle1-FGnup42) bypasses the requirement for Nup42 in some genetic contexts28. The Nup42 CTD is also required for specific functions. Nup42/hNup42 mediates the export of heat shock transcripts22,30, and deletion of NUP42 together with IPK1, the kinase that produces IP6, results in a temperature sensitive mRNA export defect31. Nup42 CTD expression is sufficient for the function in heat shock mRNA export32 and rescues nup42△ ipk1△ temperature sensitivity31; whereas, expression of the gle1-FGnup42 fusion does not restore these defects in nup42△mutants28. Since proper Gle1 and Dbp5 function are likewise required for heat shock mRNA export30,33, and IP6 is required for normal Gle1 regulation of Dbp5, we propose that the Nup42 CTD might also impact their interaction.
Nup42 was among the first identified NPC constituents34, but molecular details of its role at the NPC remain unresolved. Current models propose that the Nup42 CTD serves as a docking site for Gle1 at the NPC, and hNup42 for hGle1B. However, hGle1A lacks efficient binding to hNup42 but localizes to the NPC upon hGLE1 knockdown18,22. More importantly, cells with only hGle1A at the NPC are defective for mRNA export18. Thus, Gle1 interaction with Nup42 might be required for proper Gle1 function beyond any role in NPC localization. To further define the mRNA export mechanism mediated by the Gle1-Nup42 interaction at the NPC, we conducted a series of biochemical, genetic and cell biological studies with both the S. cerevisiae and human proteins and respective cells. We find here that Nup42/hNup42 and IP6 have conserved functions in enhancing Gle1/hGle1B stimulation of Dbp5/DDX19B for mRNA export.
RESULTS
Gle1-Nup42 interaction is required for heat shock mRNA export
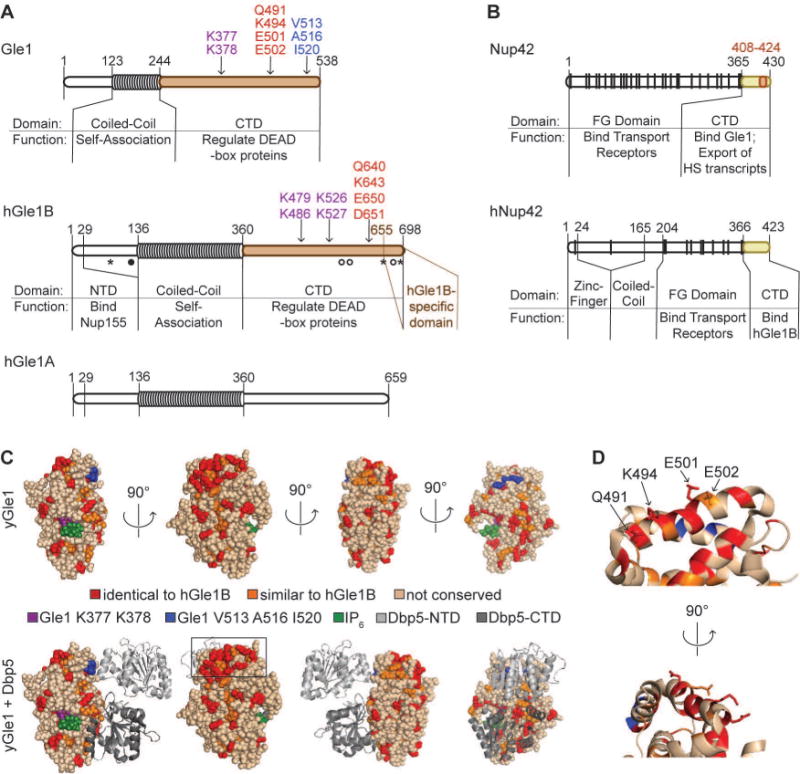
To further investigate the Gle1-Nup42 function, we used a structural homology approach to pinpoint residues critical for the interaction. Gle1/hGle1B binding to Nup42/hNup42 is so highly conserved that the hNup42 CTD interacts with S. cerevisiae Gle1 and rescues the heat shock mRNA export defect of nup42Δ mutants23. Thus, we hypothesized that the amino acid residues involved in the Gle1/hGle1B-Nup42/hNup42 interaction are conserved. From a Clustal Omega35 alignment between S. cerevisiae Gle1 and hGle1B, we mapped the conserved human residues onto the S. cerevisiae protein structure of gle1H337R-IP6-dbp5L327V, PDB 3RRM7). A patch of conserved residues was apparent on the surface of Gle1 that was distinct from the Dbp5 binding interface (Figure 1C). Four polar residues were surface accessible on this patch: Q491, K494, E501, and E502 (Figure 1D). We altered these residues to alanine, first as pairs, then all together, and analyzed the interaction with nup42-CTD via the yeast two-hybrid assay (Y2H). Strains expressing each of the pairwise gle1 mutants (GBD-gle1-CTDQK>AA and GBD-gle1-CTDEE>AA) showed growth with GAD-nup42-CTD, but altering all four residues (GBD-gle1-CTDQKEE>AAAA) resulted in no growth on quadruple dropout media, indicating defective interaction with GAD-nup42-CTD (Figure 2A).
Figure 2. Functional analysis of the proposed Nup42 binding site on Gle1.
(A) QKEE>AAAA disrupts the Gle1-Nup42 Y2H interaction. Indicated plasmids were transformed into the Y2H reporter strain, grown to early log phase at 23°C, and plated on the synthetic media lacking indicated amino acids for growth at 23°C. (B) gle1QKEE>AAAA ipk1Δ phenocopies the growth defect of nup42Δ ipk1Δ. Indicated strains were grown to mid-log phase, serially diluted, and plated on YPD for growth at the indicated temperatures. (C) Gle1 is localized at NPCs when interaction with Nup42 is disrupted. Indicated strains containing carboxy-terminal GFP-tagged GLE1-GFP plasmids were grown to mid-log phase in YPD at 23°C and live cells were imaged by wide-field microscopy. Scale bar = 5μm. (D) gle1QKEE>AAAA disrupts heat-shock mRNA export. Indicated strains were grown at 25°C to early-log phase, kept at 25°C or shifted to 42°C for 15 min, labeled with [35S]methionine for an additional 15 min, and lysed. Lysates were separated by SDS-PAGE, and proteins were visualized by autoradiography. The positions of Hsp proteins, induced upon heat shock, are indicated by asterisks.
Next, we analyzed the function of gle1QKEE>AAAA in vivo. The altered proteins were expressed similar to wild-type Gle1 levels (Figure S1A), rescued a gle1Δ mutant, and displayed no obvious growth defects at all temperatures analyzed, similar to the growth of nup42Δ (Figure 2B, top panel). When expressed as the only copy in cells (Figure S1B), GFP-tagged gle1QKEE>AAAA localized at the NE rim to the same relative extent as wild-type Gle1-GFP. Similarly, Gle1-GFP was localized to the NE rim in nup42Δ cells (Figure 2C). However, gle1QKEE>AAAA mutants demonstrate a temperature-sensitive growth defect when combined with ipk1Δ mutants, phenocopying the growth defect previously observed with nup42Δ ipk1Δ mutants (Figure 2B, bottom panel)31. Finally, we analyzed heat shock mRNA export by testing35S methionine incorporation to protein synthesis after shifting to growth at 42°C. Under these conditions, poly (A)+ RNA is retained in the nucleus, but heat shock transcripts are permitted to export, resulting in preferential translation of heat shock proteins36. Previous analysis had determined that nup42Δ mutants are defective in heat shock mRNA export and protein production30,32, so we anticipated that loss of nup42 binding by the gle1QKEE-AAAA protein would likewise impact the heat shock response. Indeed, the gle1QKEE>AAAA mutant demonstrated a loss of heat shock protein production similar to nup42Δ mutants (Figure 2D, lanes 8, 10). Therefore, we concluded that the Gle1-Nup42 interaction is required for heat shock mRNA export, gle1QKEE>AAAA disrupts this interaction, and association with Nup42 is not required for Gle1 localization at the NPC.
A minimal region of Nup42 that binds Gle1 is not sufficient for function
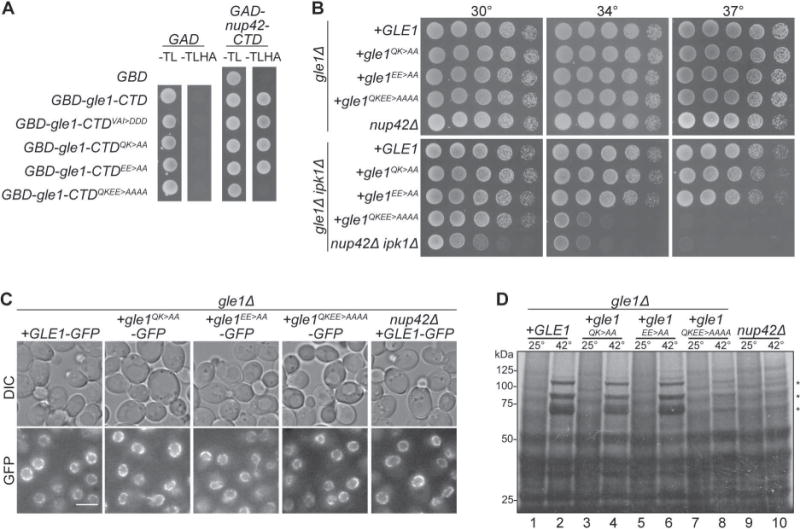
Prior studies defined the Nup42 CTD (residues 365–430) as sufficient for Y2H interaction with Gle123. Using XtalPred-RF37, the Nup42 CTD was predicted to have significant regions of disorder (data not shown). To more narrowly define the required Gle1 interaction domain in the Nup42 CTD, we generated a series of nup42-CTD truncations in the Y2H bait construct (GAD-nup42-CTD) and assayed with GBD-gle1-CTD. Both the GAD-nup42405–424 and GAD-nup42408–425 strains showed growth with GBD-gle1-CTD, revealing a region of seventeen residues (408–424) as necessary for the Gle1-Nup42 interaction (Figure 3A). Moreover, GAD-nup42 truncations lacking residues 408–424 did not grow with GBD-gle1-CTD. From a Clustal Omega alignment of nup42-CTD and hnup42-CTD, residues 408–424 were in the most highly conserved region (Figure 3B). Observing conservation in the minimal Nup42 region required for Nup42-Gle1 binding is consistent with the fact that altering residues found on the conserved patch Gle1 disrupts interaction with Nup42 (Figure 2). BioLayer Interferometry analysis demonstrated a high affinity interaction between recombinant purified MPB-gle1-CTD and a biotinylated peptide corresponding to nup42408–424, with a KD of 0.0915 μM (Figure 3C). Purified recombinant MBP-gle1-CTDQKEE>AAAA had significantly reduced association with the biotin-nup42408–424 peptide with a KD of 0.599 μM. Thus, the nup42408–424 region was necessary and sufficient for physically binding Gle1.
Figure 3. Analysis of minimal Gle1-binding domain in Nup42.
(A) nup42408–424 is required for interaction with Gle1. Indicated plasmids were transformed into the Y2H reporter strain, and struck to –Trp –Leu –His –Ade synthetic media for growth at 23°C. Growth on quadruple drop-out media is indicated as “+”. (B) nup42408–424 lies in a conserved region of the protein. Clustal Omega35 alignment between nup42-CTD and hnup42-CTD. Identical (black) and similar (grey) residues are indicated. The black line above the sequence indicates residues 408–424 of Nup42. (C) Affinity measurements between gle1-CTD and nup42408–424. BioLayer Interferometry was performed with a biotin-nup42408–424 peptide and recombinant gle1-CTD protein. Calculated KD and R2 correlation is indicated. (D) GFP-nup42408–424 localization at the nuclear envelope is disrupted in gle1QKEE>AAAA mutants. Strains containing the indicated plasmids were grown to mid-log phase at 23°C and live cells were imaged by wide-field microscopy. Scale bar = 5μm. (E) nup42408–424 does not rescue the growth defect of nup42Δ ipk1Δ mutants. nup42Δ ipk1Δ was transformed with the indicated GFP-tagged constructs, grown to mid-log phase, and plated on –Leu synthetic media for growth at the indicated temperatures. (F) nup42408–424 is not sufficient for heat shock mRNA export. nup42Δ mutants were transformed with the indicated GFP-tagged constructs, grown at 25°C to early-log phase in synthetic media, kept at 25°C or shifted to 42°C for 15 min, labeled with [35S]methionine for an additional 15 min, and lysed. Lysates were separated by SDS-PAGE, and proteins were visualized by autoradiography. Hsp proteins are indicated by asterisks.
To determine whether this nup42408–424 motif was sufficient for NE rim localization, GFP-tagged Nup42 proteins were expressed in nup42△ and gle1QKEE>AAAA nup42△ cells. Direct fluorescence microscopy showed full-length GFP-Nup42 and GFP-nup42-CTD both localized comparably at the NE rim, indicative of NPC incorporation (Figure 3D). Although a higher background of pan-cellular signal was observed, GFP-nup42408–424 also localized to the NE rim, and, importantly, GFP-nup42408–424 localization was absent from the NE in gle1QKEE>AAAA mutant cells (Figure 3D). This suggested that interaction with Gle1 is sufficient for localizing GFP-nup42408–424 to the NPC. However, GFP-Nup42 and GFP-nup42-CTD were NE rim-localized in gle1QKEE>AAAA mutants, indicating that additional NPC interactions besides Gle1 contribute to Nup42 localization. Interestingly, Nup42 proteins lacking the Gle1 interaction site (GFP-nup42Δ408–424 and GFP-nup42-CTDΔ408–424) were not detected by fluorescence microscopy or immunoblotting, suggesting that loss of interaction with Gle1 reduces Nup42 stability (Figure S2B).
Because nup42408–424 bound Gle1 and localized to the NE rim, we speculated that the nup42408–424 minimal domain might also be sufficient for functions attributed to the Nup42 CTD. Genetic tests were conducted to determine whether expression of nup42408–424 alone could rescue defects associated with absence of the Nup42 CTD. Centromeric GFP-NUP42, GFP-nup42-CTD, and GFP-nup42408–424 expression vectors were transformed into the temperature sensitive nup42Δ ipk1Δ mutant and growth was assessed at various temperatures. Similar to previously published results31, GFP-NUP42 and GFP-nup42-CTD rescued the temperature-sensitivity of nup42Δ ipk1Δ. However, GFP-nup42408–424 did not rescue the growth defects (Figure 3E; land 6, 8). The Nup42 CTD is also required for export of heat-shock transcripts following a temperature shift to 42°C23. Therefore, we tested whether GFP-nup42408–424 cells produced heat shock proteins as an assay for heat-shock mRNA export. Although GFP-nup42-CTD cells produced heat-shock proteins, GFP-nup42408–424 cells did not (Figure 3F). Untagged nup42408–424 also did not rescue nup42△ growth and heat-shock mRNA export defects (data not shown). Thus, although nup42408–424 was sufficient for interaction with Gle1 and localization to the NE, this region was not sufficient to rescue the function of the Nup42 CTD.
Nup42 interacts with IP6-Gle1-Dbp5 complexes
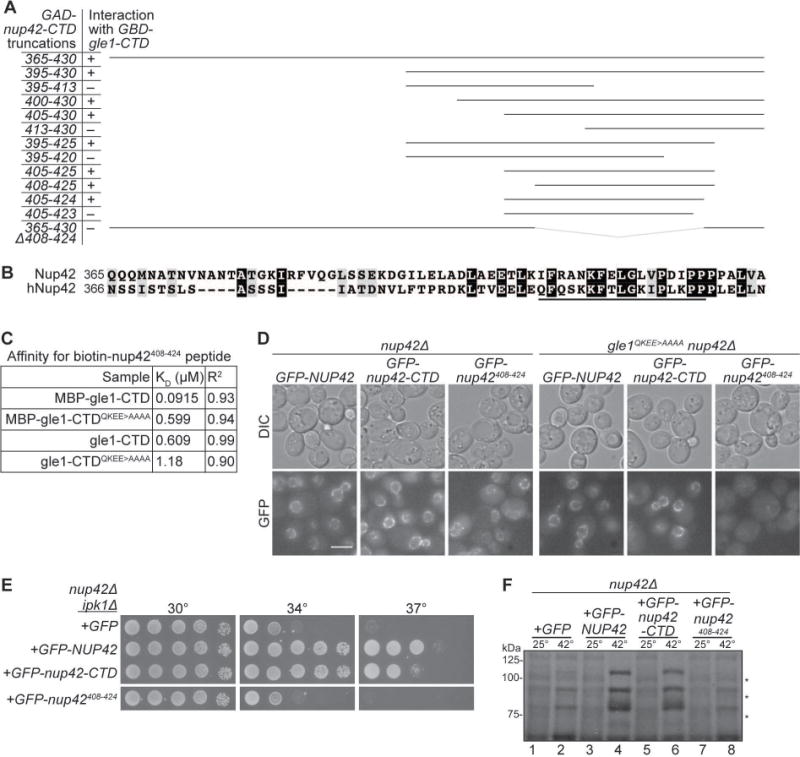
The Gle1 CTD interacts with both Nup42 and Dbp5 and our mutation analysis indicated that Nup42 and Dbp5 interact at distinct Gle1 interfaces. To reveal the role of the Nup42 CTD, we tested whether Gle1 can simultaneously bind both Nup42 and Dbp5. Soluble binding assays were performed with purified recombinant proteins. We used the gain-of-function proteins MBP-gle1-CTDH337R and/or dbp5L327V from prior structural studies7 and GST or GST-nup42-CTD bound to resin. A trimeric GST-nup42-CTD/MPB-gle1-CTDH337R/dbp5L327V complex was observed only in the presence of IP6 (Figure 4A; lane 13, 14). In this assay, binding of GST-nup42-CTD to dbp5L327V was not observed in the absence of MBP-gle1−CTDH337R (Figure 4A; lane 11, 14). Thus, Gle1 bound Nup42, Dbp5, and IP6 at the same time forming an IP6-trimeric protein complex.
Figure 4. Nup42 coordinates Gle1-Dbp5 interaction.
(A) Nup42, Gle1, and Dbp5 interact in a trimeric protein complex with IP6. Soluble binding assays were conducted with purified recombinant proteins immobilized on glutathione coupled Sepharose. Input and bound fractions were analyzed by SDS-PAGE and Coomassie staining. Note: GST-nup42-CTD is proteolytically sensitive and resolves as several bands by SDS-PAGE (lane 2), as observed in23. (B) GBD-gle1-CTDQKEE>AAAA does not interact with GAD-Dbp5 via Y2H. Indicated plasmids were transformed into the Y2H reporter strain, grown to early log phase at 23°C, and plated on the synthetic media lacking indicated amino acids for growth at 23°C. (C) GBD-gle1-CTD does not interact with GAD-Dbp5 in nup42Δ mutants. Indicated plasmids were transformed into wild-type (wt) or nup42Δ Y2H reporter strains, grown to early log phase at 23°, and plated on the synthetic media lacking indicated amino acids for growth at 23°C. (D) Purified recombinant S. cerevisiae proteins used in ATPase assays. 1μg indicated proteins were resolved by SDS-PAGE and Coomassie stained. Asterisk indicates GST cleaved (not removed) from Dbp5 sample. (E) nup42-CTD enhances Gle1 stimulation of Dbp5 ATPase activity in the absence of IP6. PK/LDH-coupled ATPase assays were performed with Dbp5 (500 nM) in the presence of 1 μM A30 RNA and 2 mM ATP gle1-CTD or gle1-CTDQKEE>AAAA (250 nM), IP6 (1 μM), and varying amounts of nup42-CTD or nup42408–424 peptide (250, 500, or 750 nM) were added as indicated. Reactions were incubated at 37°C for 40 min with the A340 monitored every 40 seconds to calculate Kcat. Mean shown for n=3, and standard error is indicated by error bars.
To investigate whether Nup42 impacts the interaction between wild-type Gle1 and Dbp5 in cells, we used the Y2H assay. Notably, although GAD-DBP5 and GBD-gle1-CTD cells grew on selective media, GAD-DBP5 and GBD-gle1-CTDQKEE>AAAA cells did not (Figure 4B). Thus, in the Y2H assay, Nup42 interaction was required for Gle1 to bind Dbp5. As a further test of this, we compared Y2H results in a reporter strain with NUP42 deleted. The nup42△ reporter strain expressing GAD-DBP5 and GBD-gle1-CTD did not grow (Figure 4C). Notably, the GBD-gle1-CTDVAI>DDD mutant (shown previously to disrupt Gle1 stimulation of Dbp57) also did not interact with GAD-DBP5 by Y2H (Figure 4B), even though GBD-gle1-CTDVAI>DDD did grow with GAD-nup42-CTD (Figure 2A). These results provided further support that Gle1 binds Nup42 and Dbp5 at different interfaces and that Nup42 might impact the interaction between Gle1 and Dbp5.
Gle1 stimulation of Dbp5 ATPase activity is enhanced by Nup42 or IP6
To directly determine whether Nup42 CTD has an effect on Gle1 stimulation of the RNA-dependent Dbp5 ATPase activity, in vitro assays were conducted using our established methods and bacterially-expressed recombinant proteins (Figure 4D). Upon addition of IP6, Gle1 stimulates the ATPase activity of Dbp5 ~6-fold in a colorimetric enzyme coupled ATPase rate assay (Figure 4E; lane 2, 7, 8), as previously determined6,8. Under these conditions, when recombinant purified H6-nup42-CTD was added at 750nM into the assay, no obvious change in ATPase activity was observed (Figure 4E, lane 12). Because our hypothesis is that Nup42 enhances the interaction between Gle1 and Dbp5, and IP6 performs a similar function6,8, we then tested whether H6-nup42-CTD affected the Dbp5 ATPase activity in the absence of IP6. Indeed, dose-dependent enhancement of gle1-CTD -stimulated Dbp5 ATPase activity was observed when H6-nup42-CTD was added to this assay without IP6 present (Figure 4E, lanes 9–11). Of note, the full Nup42 CTD (residues 365–430) was required for this stimulation as the nup42408–424 peptide did not enhance ATPase activity when titrated at the same molar concentrations (Figure 4E, lanes 19–21). H6-nup42-CTD did not stimulate Dbp5 ATPase activity in the absence of gle1-CTD (data not shown). Finally, although gle1-CTDQKEE>AAAA showed somewhat reduced stimulation of Dbp5 in the presence of IP6 (Figure 4E; lane 8, 14), H6-nup42-CTD demonstrated no dose-dependent enhancement of Dbp5 ATPase activity in the presence of gle1-CTDQKEE>AAAA. Therefore, through interaction with Gle1, Nup42 and IP6 each augmented Gle1-mediated stimulation of Dbp5 in manners that were not additive.
The hNup42-hGle1B interaction is conserved in human cells and required for mRNA export
Because the QKEE residues for Nup42 interaction in S. cerevisiae Gle1 are conserved in hGle1B (Figure 1D), we investigated whether hGle1B requires hNup42 interaction in a similar manner. We first examined the localization of mCherry-hGle1B and mCherry-hgle1BQKED>AAAA in HeLa cells. Although mCherry-hgle1BQKED>AAAA was expressed at lower levels than mCherry-hGle1B (Figure S3A), mCherry-hgle1BQKED>AAAA was detected at the NE rim in a subset of transfected cells (Figure 5A). More importantly, the proportion of cells exhibiting NE-localized mCherry-hgle1BQKED>AAAA increased upon siRNA-mediated hGLE1 knockdown (Figure 5B), indicating that endogenous hGle1 outcompetes hgle1BQKED>AAAA for NE rim localization. This is identical to hGle1A behavior wherein it only localizes at the NE rim in the absence of hGle1B18. To analyze whether mCherry-hgle1BQKED>AAAA was functional for mRNA export, we tested for complementation of the mRNA export defect upon hGLE1 knock down (Figure S3B). As previously demonstrated17, knockdown of hGLE1 (A and B) with specific siRNAs resulted in accumulation of poly(A)+ RNA in the nucleus, and expression of siRNA-resistant mCherry-hGLE1BR rescued the mRNA export defect (Figure 5C, 5D). However, expression of siRNA-resistant mCherry-hgle1BR QKED>AAAA did not rescue of mRNA export. No change in the nuclear:cytoplasmic ratio of poly(A)+ RNA was observed in mCherry-hgle1BR-QKED>AAAA transfected cells, compared to vehicle and mCherry (Figure 5D). Indeed, cells with rim-localized mCherry-hgle1BQKED>AAAA maintained a significant mRNA export defect (Figure 5C). These results indicated that the conserved QKED residues are required for hGle1B function, suggesting that interaction with hNup42 is required for mRNA export in human cells.
Figure 5. hGle1B interaction with hNup42 is required for mRNA export.
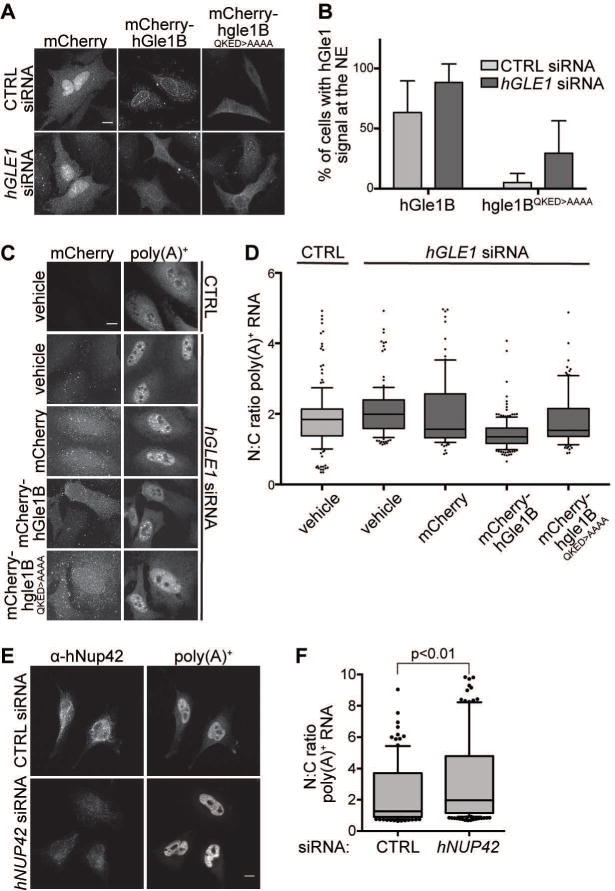
(A–B) mCherry-hgle1BQKED>AAAA is localized to the nuclear rim in a portion of cells. (A) HeLa cells expressing exogenous siRNA-resistant mCherry-hGLE1BR and mCherry-hgle1BR-QKED>AAAA after treatment with CTRL or hGLE1 siRNA were imaged by confocal microscopy. Scale bar=10μm. (B) Quantification of cells with rim-localized mCherry signal from the experiment in A. Approximately sixty cells were quantified from three independent experiments, and the mean is indicated with standard error indicated by error bars. (C-D) mCherry-hgle1BQKED>AAAA does not rescue the mRNA export defect upon hGLE1 knockdown. (C) HeLa cells were transfected with mCherry, mCherry-hGLE1BR or mCherry-hgle1BR-QKED>AAAA plasmids following CTRL or hGLE1 knockdown. After fixation, cells were processed for in situ hybridization using 488-labeled oligo d(T) probe followed by mCherry immunofluoresence. Scale bar=10μm. (D) Quantification of nuclear:cytoplasmic mean fluorescence intensity of oligo d(T) signal. Approximately 100 cells were quantified per condition from three independent experiments with nuclear:cytoplasmic intensity ratio depicted in box and whisker plots (whiskers representing 10%–90%). Statistical analysis includes ANOVA with a post-hoc t-test. (E-F) hNUP42 depletion results in a poly(A)+ mRNA export defect. (E) HeLa cells were transfected with CTRL or hNUP42 siRNA for 72h. After fixation, cells were processed for in situ hybridization using Cy3-labeled oligo d(T) probe followed by hNup42 immunofluorescence. Scale bar=10μm. (F) Quantification of nuclear:cytoplasmic mean fluorescence intensity of oligo d(T) signal. Approximately 200 cells were quantified per condition from three independent experiments with nuclear:cytoplasmic intensity ratio depicted in box and whisker plots (whiskers representing 10%–90%). A t-test was performed to analyze statistical significance.
hNup42 is required for human mRNA export
Prior studies by our laboratory and others tested for roles of the human homologues of S. cerevisiae Gle1, Dbp5, and Ipk1 in mRNA export through NPCs. Knockdown of hGLE1B, DDX19B, and hIPK1 (also known as IPPK) or dominant negative functional perturbation results in nuclear accumulation of poly(A)+ RNA16,17,38,39, reflecting a block to mRNA export. Although we previously showed that hNup42 is required for heat-shock mRNA export22, mRNA export under normal growth conditions was not assessed. To test this directly, siRNA knockdown of hNUP42 in HeLa cells was conducted and poly(A)+ RNA localization analyzed at 37°C. A robust nuclear accumulation of poly(A)+ RNA signal, with loss of cytoplasmic signal, was observed when hNup42 was depleted (Figures 5E, S3C). Quantification demonstrated a statistically significant increase in the nuclear:cytoplasmic signal for poly(A)+ RNA upon hNUP42 knockdown (Figure 5F). Therefore, hNup42 was essential for mRNA export in human cells, corroborating the mRNA export defect observed in mCherry-hgle1BR-QKED>AAAA cells. Further, as depletion of each of the factors all result in mRNA export defects, we concluded that the molecular mechanism for human hGle1B, DDX19B, hNup42, and IP6 function in mRNA export is similar to that of their S. cerevisiae homologues.
hNup42 and IP6 each enhance hGle1B stimulation of DDX19B ATPase activity
To further examine the conservation of hNup42 and IP6 functions in the human system, direct in vitro ATPase assays were conducted with DDX19B. Initial experiments were performed with purified bacterially-expressed DDX19B (denoted DDX19B*) (Figure S4); however, the bacterially-expressed DDX19B* with or without hgle1B-CTD had ~8.3 fold lower enzymatic activity compared to that of S. cerevisiae Dbp5, gle1-CTD and IP6 (Figure S4). We then tested His-tagged DDX19B (H6-DDX19B) purified from a Baculoviral expression system in insect cells based on reported successful DDX19B in vitro protein interaction studies and reports that DDX19B is post-translationally modified in mammalian cells40,41 (Figure 6A), The H6-DDX19B purified from insect cells displayed significant ATPase activity which was stimulated ~4-fold by hgle1B-CTD in the presence of IP6 (Figure 6B; lane 2, 6, 7). Strikingly, titration of hnup42-CTD resulted in dose-dependent enhancement of the hGle1B-stimulated H6-DDX19B ATPase activity in the absence of IP6 (Figure 6B, lanes 8–10). hNup42 and IP6 did not have an additive effect (Figure 6B; lanes 7, 10, 11). Finally, although IP6 further stimulated the hgle1BQKED>AAAA mediated activation of H6-DDX19B (Figure 6B; lane 12, 13), hnup42-CTD did not enhance hgle1BQKED>AAAA stimulation of H6-DDX19B ATPase activity (Figure 6B, lanes 14–16). In sum, in vitro human DDX19B activation was dependent on IP6, hGle1B, and hNup42, with the human and S. cerevisiae proteins showing fully conserved in vitro functions.
Figure 6. hGle1B interactions with hNup42 and IP6 enhances stimulation of DDX19B activity and are required for mRNA export.
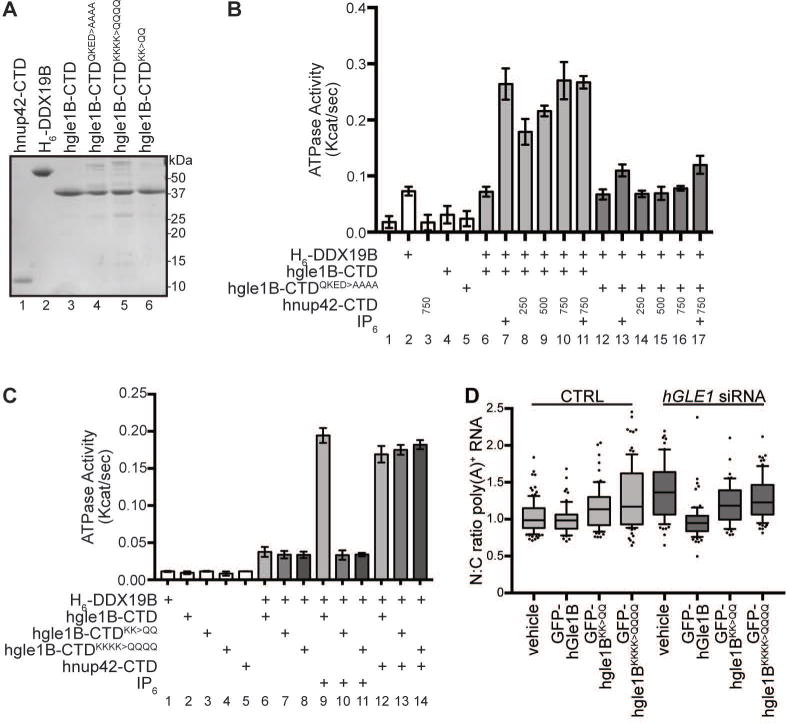
(A) Purified recombinant human proteins used in ATPase assays. 1μg indicated purified proteins were resolved by SDS-PAGE and Coomassie stained. (B) hGle1B and hNup42 stimulate DDX19B ATPase activity in absence of IP6. PK/LDH-coupled ATPase assays were performed with H6-DDX19B (500 nM, expressed in insect cells) in the presence of 1 μM A30 RNA and 2 mM ATP. hgle1B-CTD or hgle1B-CTDQKED>AAAA (250 nM), IP6 (1 μM), and varying amounts of hnup42-CTD (250, 500, or 750 nM) were added as indicated. Reactions preformed as in Figure 3D. Mean shown for n=3, and standard error is indicated by error bars. (C) hGle1B-IP6 interaction is conserved in human proteins. PK/LDH-coupled ATPase assays were performed with H6-DDX19B (500 nM) in the presence of 1 μM A30 RNA and 2 mM ATP, hgle1B-CTD, hgle1B-CTDKK>QQ, or hgle1B-CTDKKKK>QQQQ (250 nM), and hnup42-CTD (750 nM) or IP6 (1 μM) were added as indicated. Purified proteins are shown in 6A. Reactions performed as in Figure 3D. Mean shown for n=3, and standard error is indicated by error bars. (D) hGle1B-IP6 interaction is required for mRNA export. HeLa cells were transfected with vehicle or GFP-hGLE1BR, GFP-hgle1BR-KK>QQ, or GFP-hgle1BR-KKKK>QQQQ plasmids following CTRL or hGLE1 knockdown. Nuclear:cytoplasmic mean fluorescence intensity of oligo d(T) signal was quantified as in Figure 5F.
hGle1B requires conserved IP6 binding residues for DDX19B stimulation and mRNA export
To further investigate the IP6 dependence of hGle1B-DDX19B activity in human cells, we tested whether hGle1B coordinates IP6 through the same surface accessible protein interface as the S. cerevisiae Gle1. We previously defined the residues required for IP6 binding to S. cerevisiae Gle1 (K377 and K378) by identification of positive residues that were conserved across fungal and metazoan species10. In prior structural modeling of fungal, plant and human Gle1, it was observed that the IP6 binding pocket of hGle1 and Arabidopsis thaliana Gle1 is less positively-charged than that of S. cerevisiae42. However, the homologous IP6-coordinating lysine (K) residues of hGle1B (K526, K527) are conserved. Altering these lysine (K) residues of hGle1B (K526, K527) to glutamine (Q) (hgle1B-CTDKK>QQ) abolished the enhanced stimulation of H6-DDX19B in the presence of IP6 (Figure 6C, comparing lanes 7, 10). We also tested a hGle1B variant with changes in two additional positively-charged residues (K479, K486) whose homologous residues are found near the IP6 binding pocket of S. cerevisiae Gle1. The hgle1B-CTDKKKK>QQQQ also did not show IP6 dependent stimulation of H6-DDX19B. Importantly, the hgle1B-CTDKK>QQ protein still stimulated H6-DDX19B at a basal level (Figure 6C, comparing lane 1 with 10 and 11), and with the addition of hnup42-CTD, hgle1B-CTDKK>QQ enhanced H6-DDX19B ATPase activity levels comparable to that with wild-type hgle1B-CTD and hnup42-CTD (Figure 6C, lanes 12–14). Thus, the IP6 interface changes on hgle1B-CTDKK>QQ did not perturb the hNup42 interface or overall folding.
Further supporting a conserved role for an IP6 interaction with hGle1B during mRNA export, we found that expression of the siRNA-resistant GFP-hgle1BKK>QQ did not rescue the mRNA export defect seen with siRNA hGLE1-knockdown in human cells, whereas wild-type GFP-hGle1B did rescue (Figure 6D). As such, mRNA export was inhibited in human cells when the IP6-dependent hGle1B function was perturbed by altering the conserved K526 and K527 residues. Thus, although the plant and human IP6 interactions surfaces are less positively charged compared to S. cerevisiae and not fully homologous in terms of protein sequence, these results with the prior studies42 documented a conserved requirement for IP6 during hGle1B function in mRNA export in all tested eukaryotic cells.
DISCUSSION
Here we reveal important insights into the functional organization of the mRNA export machinery at the NPC cytoplasmic face. Our biochemical and physiological assays demonstrate a unique role for Nup42 in mediating Gle1 regulation of the Dbp5 ATPase activity. In both S. cerevisiae and human systems, Nup42 is not critical for localizing Gle1 at the NPC. Rather, Nup42/hNup42 enhances the Gle1/hGle1B stimulation of the RNA-dependent Dbp5/DDX19B ATPase (Figure 4E and 6B). We find that Gle1 utilizes distinct surface accessible binding sites for interaction with IP6, Nup42 and Dbp5 (Figure 1C), and a nup42-CTD/gle1-CTD/Dbp5 trimeric complex forms in the presence of IP6. As some of the hGLE1 disease-associated mutations alter the carboxy-terminal region of hGle1B required for hNup42 binding (Figure 1A)25,27, we predict that perturbed DDX19B activation and mRNP remodeling dynamics at the NPC underlie some of these human pathologies.
An active role for Nup42 and hNup42 during mRNA export is distinct from the historically attributed function of these proteins in localizing Gle1/hGle1 to the NPC. Here, in nup42Δ mutants, we find Gle1-GFP localizes to the NPC as robustly as in wild-type cells, and GFP-gle1QKEE>AAAA is also localized to the NE rim (Figure 2C). In fact, Nup42 is at least partly reliant on Gle1 for its localization since GFP-nup42408–424 is lost from the rim in gle1QKEE>AAAA mutants (Figure 3D). Moreover, the hGle1A isoform which lacks hNup42 binding localizes to the NPC in the absence of hGle1B18, and hGle1 remains localized to the NE rim upon hNUP42 knockdown22. A recent structural analysis of the Nup82 complex (Nup159, Nup82, Nsp1) revealed the interactions that anchor this complex to the S. cerevisiae NPC cytoplasmic side1, with the FG domains for Nup159 and Nsp1 situated across the NPC channel for their function in mRNA export28,43. The Fernandez-Martinez study also reports substantial interactions between the Gle1 amino terminal region (NTD) and the Nup82 complex, which corroborates our prior finding that the hGle1 NTD is required for NPC localization (both its coiled-coil region and the Nup155 interaction region (Figure 1A,17,21;. This also explains why overexpression of GLE1 rescues growth defects of nup82 mutants44. In contrast, Nup42’s only reported crosslinking partner is Gle11. Thus, the Gle1 NTD is the primary determinant of NPC localization whereas the Gle1 CTD interaction with Nup42 and IP6 is required for mRNP remodeling.
Importantly, although a minimal conserved peptide of seventeen amino acids in Nup42 (nup42408–424) is sufficient for interaction with Gle1, additional sequence in the Nup42 CTD is required for function (Figures 3, 4). A conserved patch on Gle1 is essential for interaction with the minimal nup42408–424 peptide (Figure 1D, 2, 3), placing this interface at the opposite face of where Dbp5 binds Gle1 (Figure 1C,7). However, Nup42 is required for the Y2H interaction between Gle1 and Dbp5 in cells (Figure 4C). We predict that the sequence in the Nup42 CTD flanking the minimal Gle1 binding peptide (residues 408–424) is responsible for mediating the Gle1-Dbp5 interaction and the nup42-CTD/gle1-CTD activation of Dbp5. To date, we have not been able to determine whether this occurs directly (with Nup42 interacting with both Gle1 and Dbp5) or indirectly (with Nup42 binding Gle1 and altering its conformation). We did not observe a Nup42-Dbp5 interaction in our soluble binding assay (Figure 4A lane 11), but it is possible that low affinity interactions are not detected by this method. We also analyzed the IP6-trimeric GST-nup42-CTD/MPB- gle1H337R/dbp5L327V protein complex formation in the presence of different nucleotides; however, no obvious differences in gle1H337R-dbp5L327V interaction were observed, potentially due to the use of gain-of-function proteins required for this assay (Figure 4A).
The hypothesis that Nup42 mediates an interaction between Gle1 and Dbp5 suggests that the Nup42 CTD role is similar to that of IP6, explaining why no additive effect is seen with IP6 and nup42-CTD during in vitro ATPase assays (Figure 4E and 6B). The redundant functions also explain why nup42Δ ipk1Δ double mutants are temperature-sensitive with no growth defects observed for single mutants in S. cerevisiae31. IP6 is required for GST-nup42-CTD/MBP-gle1-CTDH337R/dbp5L327V trimeric protein complex formation in soluble binding assays (Figure 4A). We speculate that IP6 is required in vitro for trimeric complex formation but does not enhance Gle1 stimulation of Dbp5 in the presence of Nup42 because the former experiment requires capture of a stable interaction, whereas observation of ATPase activity only relies on transient stimulation. Having two parallel ways to support Gle1-Dbp5 interaction through either Nup42 or IP6 potentially allows combinatorial control over the NPC-localized activity of Dbp5 in cells. Such spatial control is likely especially critical for modulating Gle1 through its multiple subcellular locations and functions.
We find that hGle1B, like S. cerevisiae Gle1, demonstrates enhanced stimulation of DDX19B in the presence of IP6 or hNup42-CTD. However, in contrast to S. cerevisiae, hGle1B interactions with hNup42 and IP6 are each individually required for mRNA export in human cells, indicating stricter control over this process in vivo (Figure 5 and 6D). The molecular requirement for hNup42 during nuclear mRNA export might also be due to the fact that the IP6 binding pocket of hGle1B is less positively-charged42 and potentially binds to IP6 more weakly than S. cerevisiae Gle1 does to IP6. However, as others have reported, the IP6 binding pocket of Arabidopsis thaliana Gle1 has even lower positive charge than hGle1B but demonstrates enhanced stimulation of LOS4 (A. thaliana Dbp5 orthologue) in the presence of IP642. Therefore, a requirement for IP6 in Gle1 stimulation of DEAD-box ATPases during mRNA export is conserved between S. cerevisiae, plants and human cells.
Based on our proposed mechanism for Nup42 CTD function, we posit that Nup42/hNup42 acts at an early step in the ATPase cycle of Dbp5/DDX19B at the NPC, where Gle1 enhances ATP loading onto Dbp5. This model is diagrammed in Figure 7. With the Nup42 FG domain recruiting mRNPs for remodeling by Dbp528, the Nup42 CTD ensures activation of Dbp5 (via the Gle1-Nup42 CTD interaction) when Mex67-bound mRNPs are present (via the Nup42 FG-Mex67 interaction). We propose that Nup42 serves as an important sensor that couples these steps in the mechanism. We anticipate that these interactions might direct specific interaction between Dbp5/DDX19B and mRNA to enable remodeling of specific proteins. As other DEAD-box proteins are activated by interaction partners in a similar manner to Gle1 activation of Dbp5 (eIF4G for eIF4A and THO for Sub245,46), similar mechanisms might exist to spatially regulate these activities. This work also illustrates how Nup42/hNup42 as a specific binding partner at the NPC cytoplasmic face allows specificity to Gle1 and hGle1B function differentially from cytoplasmic Gle1/hGle1A function. With S. cerevisiae Nup42 being specifically required for heat shock mRNA export, we propose that Nup42 binding might also preferentially stabilize Gle1 and confer thermostability on the mRNA export mechanism. As such, this might explain how the Nup42/Gle1-mediated mRNA export mechanism is crucial under heat shock conditions.
Figure 7. Schematic for Nup42 function at the NPC cytoplasmic face.
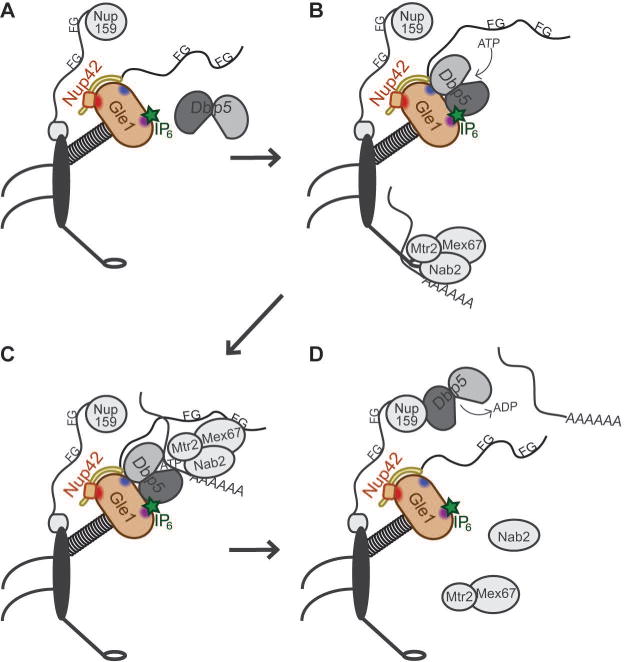
(A) Dbp5 dynamically associates with the NPC. (B) The Nup42 CTD stimulates Gle1-IP6 interaction with Dbp5, with Gle1-IP6 promoting ATP loading onto Dbp5. Mature mRNPs interact with the Mex67-Mtr2 heterodimer via adaptors such as the poly(A)+ binding protein Nab2, permitting export through the NPC. (C) The FG domain of Nup42 recruits the mRNP in close proximity to Dbp5, and Dbp5-ATP binds RNA. Dbp5 might also be a constituent of the exporting mRNP. (D) ATP hydrolysis induces a conformational change in Dbp5 and the bound RNA for release of Nab2, and Mex67-Mtr2, which are recycled into the nucleus for additional rounds of mRNP export. The remodeled mRNP is released into the cytoplasm. Dbp5 interacts with the Nup159 amino terminal domain (NTD) to facilitate ADP release.
One important distinction between the Dbp5 and DDX19B activities might be linked to a role for potential post-translational modifications or small effectors in the human system. A recent study reported roles for DDX19B phosphorylation in altering its nuclear activity during the DNA damage response40. We show here that DDX19B* purified from bacteria is not activated by hgle1-CTD in the presence of IP6 or hnup42-CTD, and demonstrates relatively low enzymatic activity (Figure S4). In contrast, DDX19B purified from insect cells shows full, conserved functionality, including gle1-CTD-mediated stimulation that is enhanced in the presence of hnup42-CTD or IP6 (Figure 6). Our future studies will be focused on pinpointing potential DDX19B modifications needed for activity and regulation during mRNA export.
To further delineate the mRNA export mechanism, additional structural details regarding hNup42 and hGle1B molecular interactions in the context of the NPC are needed, and further analysis is required to fully understand how the mRNP remodeling platform is arranged to interact with exporting mRNPs. Overall, we demonstrate that the NPC protein Nup42 interacts with Gle1/hGle1B to locally regulate Dbp5/DDX19B activity. Importantly, this work demonstrates a remarkable conservation of function for the mRNP remodeling factors Gle1/hGle1B, Nup42/hNup42, Dbp5/DDX19B, and IP6 during mRNA export in human and S. cerevisiae cells. These results also reveal novel unexpected steps in the molecular mechanism for mRNA export at the NPC cytoplasmic face that impact normal and disease cell function.
METHODS
Yeast Strains, Growth, and Y2H Analysis
Table S1 lists the yeast strains used in this study. Yeast genetic methods including mating, sporulation, dissection, and transformations were conducted according to standard procedures47. Yeast strains were grown at indicated temperatures in either YPD (2% peptone, 2% dextrose, 1% yeast extract) or selective minimal media lacking appropriate amino acids and supplemented with 2% dextrose and 5-fluoroorotic acid (5-FOA; United States Biological) as needed at 1.0 mg/mL. For growth analysis on plates, indicated strains were grown in YPD liquid media to mid-log phase (OD600 ~0.5), serially diluted, and equal numbers of cells were spotted onto YPD media for growth at the indicated temperatures. For Y2H analysis, vectors expressing GBD and GAD fusions were transformed into the reporter strain (PJ69-4A) or nup42Δ reporter strain (SWY6432) and selected on –Trp –Leu synthetic media. The resulting strains were then grown to mid-log phase, serially diluted, and equal numbers of cells were spotted onto –Trp –Leu or –Trp –Leu –His –Ade media. The plates were then incubated at 23°C for the indicated number of days.
Vector Construction
Table S2 lists the vectors used in this study. Vectors were cloned using standard molecular biology strategies or by Gibson Assembly (New England Biolabs, Ipswich, MA), and site-directed mutagenesis was performed using QuikChange (Agilent Genomics, Santa Clara, CA), and sequencing confirmed all vectors generated.
Live Cell Microscopy
Yeast strains were grown to mid-log phase in YPD or synthetic media lacking appropriate amino acids at the indicated temperatures. Cultures were collected, re-suspended in synthetic complete media at room temperature, and imaged. Wide-field images were acquired using a microscope (BX50; Olympus) equipped with a motorized stage (Model 999000, Ludl), Olympus 100× NA1.3 UPlanF1 objective, and digital charge coupled device camera (Orca-R2; Hamamatsu). Images were processed with ImageJ (NIH) or Adobe Photoshop CS6.
Heat Shock Protein Production Assay
The [35S]methionine incorporation assay of heat shock proteins was performed as described28,48. Nascently produced radiolabeled proteins from whole-cell lysate were separated by SDS-PAGE, and the resulting gel was dried and exposed to autoradiography film.
Immunoblotting
For immunoblotting of yeast S. cerevisiae whole-cell lysates, cells were cultured and lysates were prepared as previously described in SDS loading buffer28. Proteins were resolved by SDS-PAGE and blotted using affinity-purified guinea pig anti-Gle1 (ASW 4310), rabbit anti-GFP (Thermo Fisher Scientific, Waltham, MA), or anti-yPgk1 (Thermo Fisher Scientific). IRDye 800CW-conjugated goat anti-mouse (LI-COR, Lincoln, NE) or Alexa Fluor 700 goat anti-rabbit (Thermo Fisher Scientific) antibodies (1:5000) were visualized with the Li-Cor Odyssey scanner (Lincoln, NE).
For HeLa cell lysate immunoblotting, cells were grown in 60 mm dishes (Fisher Scientific, Pittsburg, PA) and lysed in RIPA buffer (Sigma, St. Louis, MO) supplemented with EDTA-free cOmplete protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN). Proteins were resolved by SDS-PAGE and blotted with rat anti-mCherry (Sigma), anti-hNup42/NUPL2 (Sigma), mouse anti-beta-actin (Sigma), or anti-hGle117 antibodies. Infrared 700- or 800-conjugated secondary antibodies (Thermo Fisher Scientific) were visualized with the Li-Cor Odyssey scanner.
HeLa cell culture and transfection
HeLa cells were cultured in complete DMEM media (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals, Norcross, GA) at 37°C in 5% CO2. Knockdown add-back of hGLE1 was performed using the previously validated protocol17. Negative control siRNA and hGLE1 siRNAs were purchased from Qiagen (Valencia, CA). hNUP42 SMARTpool siRNA was purchased from Dharmacon (Lafayette, CO). Cells were reverse-transfected with indicated 25 nM siRNAs using HiPerFect (Qiagen) and then transfected 24 h later with relevant constructs (siRNA-resistant mCherry-hGLE1BR or mCherry-hgle1BR-QKED>AAAA for Figure 4; siRNA-resistant GFP-hGLE1BR or GFP-hgle1BR-KK>QQ for Figure 5) using Fugene 6 (Promega, Madison, WI) per manufacturer’s instructions.
HeLa immunofluorescence
HeLa cells expressing mCherry-hGLE1BR and mCherry-hgle1BR-QKED>AAAA were grown on 1.5 mm round coverslips in a 24-well plate (Fisher Scientific) and immunofluorescence was performed as previously described18 using mAb414 (Biolegend, San Diego, CA) and anti-mCherry (Sigma). Cells were imaged using a 63x/1.4 NA oil-immersion objective on a confocal microscope (Leica TCS Sp5). Images were processed with ImageJ (NIH) or Adobe Photoshop CS6.
HeLa mRNA export assay
The previously validated knockdown add back approach (for hGLE117 or described hNUP42 knockdown was employed followed by in situ hybridization of 488-oligo d(T) to poly(A)+ RNA. Cells were imaged using a 63x/1.4 NA oil-immersion objective on a confocal microscope (Leica TCS Sp5). The nuclear:cytoplasmic ratio was determined by measuring mean intensity of these compartments in Image J (NIH) and dividing nuclear by cytoplasmic signal. Images were processed with ImageJ (NIH) or Adobe Photoshop CS6.
Protein Expression and Purification
Proteins were expressed (except DDX19B* and H6-DDX19B) in E. coli Rosetta cells (Novagen) cultured in Terrific Broth under chloramphenicol and corresponding plasmid antibiotic resistance (kanamycin or ampicillin) selection. Protein expression was induced with IPTG (0.2 mM) at an OD600 of 0.5–0.8 for 18 hours at 18°C. Bacteria were lysed by sonication in Buffer B (20 mM HEPES (pH 7.5), 150 mM NaCl, 0.5 mM DTT, and 20% glycerol) supplemented with EDTA-free cOmplete protease inhibitor cocktail (Sigma) and 2 mM PMSF. All purified proteins were snap-frozen in an ethanol-dry ice bath and stored at −80°C.
The H6-DDX19B plasmid was transformed into DH10Bac™ (Thermo Fisher Scientific) E. coli cells and plated on Luria agar plates containing tetracycline (10 μg/mL), gentamycin (7 μg/mL), kanamycin (50 μg/mL), IPTG (40 μg/mL), and X-Gal (100 μg/mL). White colonies were re-struck three times before the recombinant bacmid was purified from 1.5 mL of an overnight culture in LB containing tetracycline, gentamycin, and kanamycin. Transposition was verified by PCR with M13/pUC forward and reverse sequencing primers. Sf9 cells were cultured in Sf-900(TM) III SFM (Thermo Fisher Scientific) at 27°C. P0 virus stocks were generated by transfecting Sf9 cells (2 mL at 0.5×106 cells/mL) in a 6-well plate using Cellfectin(R) II Reagent (Thermo Fisher Scientific) and harvested three days post transfection. P1 virus was generated by adding 0.25 mL of P0 stock to 100-mm Petri dishes containing 10 mL of cells at 1×106 cells/mL and harvested three days post infection. P2 virus stocks were generated by adding 0.2 mL of P1 virus stocks to 20 mL of 1×106 cells/mL Sf9 cells in 125 mL shake flasks. P2 virus stocks were harvested after three days post infection with shaking at 140 rpm. Protein expression was achieved using High Five™ cells (Thermo Fisher Scientific), which were cultured in Express Five(R) SFM (Thermo Fisher Scientific) at 27°C with shaking at 140 rpm. P2 virus stocks (10 mL) were used to infect 450 mL of High Five™ cells (1×106 cells/mL) in 1 L shake flasks. High Five™ cells were harvested 48 hrs post infection and lysed using a Dounce homogenizer in the presence of Buffer B and 2 mM PMSF.
For MBP, MBP-gle1-CTDH337R, H6-MBP-TEV-gle1-CTD, H6-MBP-TEV-gle1-CTDQKEE>AAAA, H6-MBP-TEV-hgle1B-CTD, H6-MBP-PPS-hgle1B-CTDQKED>AAAA, H6-MBP-PPS-hgle1B-CTDKK>QQ, and H6-MBP-PPS-hgle1B-CTDKKKK>QQQQ proteins, the soluble fractions were purified with amylose resin (New England Biolabs) according to manufacturer recommendations. The amino-terminal tags on H6-MBP-TEV-gle1-CTD, H6-MBP-TEV-gle1-CTDQKEE>AAAA, H6-MBP-TEV-hgle1B-CTD, H6-MBP-PPS-hgle1B-CTDQKED>AAAA, H6-MBP-PPS-hgle1B-CTDKK>QQ, and H6-MBP-PPS-hgle1B-CTDKKKK>QQQQ were cleaved by addition of H6-TEV or H6-PPS to the amylose elution fractions and incubation overnight at 4°C. His-MBP and protease were removed by collecting flow-through after Ni Immobilized Metal Affinity Chromatography (Ni IMAC) (New England Biolabs). Flow-through was dialyzed overnight in Buffer B.
H6-DDX19B and H6-nup42-CTD were purified by Ni IMAC. Elution fractions were pooled and dialyzed overnight in Buffer B (H6-nup42-CTD) or Buffer B with 400 mM NaCl (H6-DDX19B).
GST, GST-Dbp5, GST-dbp5L327V, GST-nup42-CTD, and GST-hnup42-CTD were purified by glutathione-coupled Sepharose chromatography. For GST-Dbp5, GST-dbp5L327V, and GST-hnup42-CTD, the GST tag was cleaved by Factor Xa (New England Biolabs) digestion and inactivated by the addition of PMSF. The GST tag was removed from dbp5L327V by ion-exchange chromatography as in4. All proteins were dialyzed overnight in Buffer B.
GST-DDX19B* was expressed in E. coli Rosetta cells cultured in Terrific Broth under ampicillin and chloramphenicol antibiotic selection. Protein expression was induced with IPTG (0.1 mM) at an OD600 of 0.8 at 18°C for 18 hours. The harvested cell pellet was suspended in 30 mM HEPES pH 7.5, 400 mM NaCl, 10% Glycerol, 1 mM DTT, 2 mM MgCl2 supplemented with ETDA-free cOmplete protease inhibitor cocktail and 2 mM PMSF and cells were lysed by sonication. Cleared lysate was incubated with buffer equilibrated glutathione-coupled sepharose 4B resin for 1.5 hours at 4°C with rotation. The resin was washed with 12 column volumes of buffer before elution with 10 mM reduced glutathione. The GST tag was removed by Factor Xa incubation for 36 hours at 4°C after the NaCl concentration was reduced to 200 mM by dilution with buffer containing no NaCl. GST was removed by passing the protein over buffer-equilibrated glutathione-coupled sepharose 4B resin and collecting the flow through. Protein homogeneity was verified by SDS-PAGE.
ATPase Assay
Colorimetric enzyme-coupled ATPase rate assays were performed as described5. Briefly, reaction mixtures (100μL), containing 10 mM HEPES (pH 7.5), 45 mM NaCl, 2 mM MgCl2, 1 mM DTT, 20U SUPERase·In™ RNase Inhibitior (Thermo Fisher Scientific), 6 mM PEP (Sigma), 1.2 mM NADH (Sigma), 2.5 mM ATP (Sigma), 2 μL PK/LDH (Sigma), 1 μM polyA RNA, 500 nM Dbp5 or H6-DDX19B, 1 μM IP6 (Sigma, as indicated), 250 nM gle1-CTD or hgle1B-CTD (or mutants), and titration of H6-nup42-CTD or hnup42-CTD as indicated. A340 was monitored every 40 sec for 40 min at 37°C in a BioTek Synergy HT microplate reader. Kcat/sec was calculated as (((OD340/min × 2.5)/6.22×10−3)/μM protein).
BioLayer Interferometry
BioLayer interferometry assays were performed utilizing a ForteBIO Octet-Red96 at the Vanderbilt Antibody and Protein Resource Core. Biotinylated nup42408–424 peptide (GenScript, Piscataway, NJ) was immobilized on a streptavidin conjugated biosensor at a well concentration of 0.5 μM in buffer containing 20 mM HEPES (pH 7.4), 200 mM NaCl, 1 mM DTT, 10% glycerol, 0.25 mM IP6, and 0.01% Nonidet P-40. Immobilization was carried out over 5 minutes, followed by a 5-minute incubation in 100 μg/mL biocytin to cap any free streptavidin sites on the biosensor surface. Following a 5-minute wash and 1-minute baseline, gle1-CTD or gle1-CTDQKEE>AAAA was allowed to associate for 5 minutes. Dissociation was followed for an addition 5 minutes. A single biosensor was used to measure binding at each concentration in parallel and responses in buffer alone were used to control for signal drift. The data analysis software provided by ForteBio was used to normalize all responses to an appropriate baseline, and subtract the buffer only control. The KD was calculated from the determined kon and koff rates for the interaction.
In vitro binding assay
400 pmol GST or GST-nup42-CTD, 250 pmol MBP or MBP-gle1H337R-CTD, and 500 pmol dbp5L327V were incubated with equilibrated glutathione-coupled Sepharose in Buffer B. IP6, ADP, or AMP-PNP, were added at 50 μM. Samples were incubated at 4°C for 2 hours and washed three times with Buffer B (IP6, ADP, or AMP-PNP added in wash buffer where appropriate). Bound proteins were eluted in SDS loading buffer, resolved on a 12% (bottom) 7.5% (top) Tris-Glycine gel, and Coomassie stained.
Supplementary Material
SYNOPSIS.
Activation of the DEAD-box ATPase Dbp5 by Gle1-IP6 is required for mRNA export through nuclear pore complexes (NPCs). In this paper, Adams, et al. demonstrate that a constituent of the NPC, Nup42, impacts the ATPase activity of Dbp5 through interaction with Gle1. This function, as well as IP6-hGle1B interaction, is required for mRNA export in human cells and altered in human diseases. This work reveals how Gle1-Dbp5 activity is locally coordinated for function at the proper place and time.
Acknowledgments
The authors thank the Wente laboratory, particularly Renee Dawson, PhD, for discussions and critical reading of the manuscript; Andre Hoelz, PhD, (California Institute of Technology) for communicating results prior to publication; Rob Carnahan, PhD and Matt Goff (Vanderbilt Antibody and Protein Resource) for BioLayer Interferometry assistance; the Vanderbilt Central for Structural Biology for pSV282; Françoise Stutz, PhD, (Geneva) for pFS507; Brandt Eichman, PhD, (Vanderbilt) for use of equipment in purification from insect cells. This work was supported by grants from the National Institutes of Health (R37GM051219 [S.R.W.] and training position on T32HD007502 [R.L.A.]).
Footnotes
The authors declare no competing financial interests.
References
- 1.Fernandez-Martinez J, Kim SJ, Shi Y, et al. Structure and Function of the Nuclear Pore Complex Cytoplasmic mRNA Export Platform. Cell. 2016;167(5):1215–1228.e25. doi: 10.1016/j.cell.2016.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oeffinger M, Zenklusen D. To the pore and through the pore: a story of mRNA export kinetics. Biochim Biophys Acta. 2012;1819(6):494–506. doi: 10.1016/j.bbagrm.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lund MK, Guthrie C. The DEAD-box protein Dbp5p is required to dissociate Mex67p from exported mRNPs at the nuclear rim. Mol Cell. 2005;20(4):645–651. doi: 10.1016/j.molcel.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 4.Tran EJ, Zhou Y, Corbett AH, Wente SR. The DEAD-box protein Dbp5 controls mRNA export by triggering specific RNA:protein remodeling events. Mol Cell. 2007;28(5):850–859. doi: 10.1016/j.molcel.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 5.Noble KN, Tran EJ, Alcázar-Román AR, Hodge CA, Cole CN, Wente SR. The Dbp5 cycle at the nuclear pore complex during mRNA export II: nucleotide cycling and mRNP remodeling by Dbp5 are controlled by Nup159 and Gle1. Genes Dev. 2011;25(10):1065–1077. doi: 10.1101/gad.2040611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alcázar-Román AR, Tran EJ, Guo S, Wente SR. Inositol hexakisphosphate and Gle1 activate the DEAD-box protein Dbp5 for nuclear mRNA export. Nat Cell Biol. 2006;8(7):711–716. doi: 10.1038/ncb1427. [DOI] [PubMed] [Google Scholar]
- 7.Montpetit B, Thomsen ND, Helmke KJ, Seeliger MA, Berger JM, Weis K. A conserved mechanism of DEAD-box ATPase activation by nucleoporins and InsP6 in mRNA export. Nature. 2011;472(7342):238–242. doi: 10.1038/nature09862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weirich CS, Erzberger JP, Flick JS, Berger JM, Thorner J, Weis K. Activation of the DExD/H-box protein Dbp5 by the nuclear-pore protein Gle1 and its coactivator InsP6 is required for mRNA export. Nat Cell Biol. 2006;8(7):668–676. doi: 10.1038/ncb1424. [DOI] [PubMed] [Google Scholar]
- 9.Murphy R, Wente SR. An RNA-export mediator with an essential nuclear export signal. Nature. 1996;383(6598):357–360. doi: 10.1038/383357a0. [DOI] [PubMed] [Google Scholar]
- 10.Alcázar-Román AR, Bolger TA, Wente SR. Control of mRNA export and translation termination by inositol hexakisphosphate requires specific interaction with Gle1. J Biol Chem. 2010;285(22):16683–16692. doi: 10.1074/jbc.M109.082370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watkins JL, Murphy R, Emtage JL, Wente SR. The human homologue of Saccharomyces cerevisiae Gle1p is required for poly(A)+ RNA export. Proc Natl Acad Sci U S A. 1998;95(12):6779–6784. doi: 10.1073/pnas.95.12.6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bolger TA, Wente SR. Gle1 is a multifunctional DEAD-box protein regulator that modulates Ded1 in translation initiation. J Biol Chem. 2011;286(46):39750–39759. doi: 10.1074/jbc.M111.299321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bolger TA, Folkmann AW, Tran EJ, Wente SR. The mRNA export factor Gle1 and inositol hexakisphosphate regulate distinct stages of translation. Cell. 2008;134(4):624–633. doi: 10.1016/j.cell.2008.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gross T, Siepmann A, Sturm D, et al. The DEAD-box RNA helicase Dbp5 functions in translation termination. Science. 2007;315(5812):646–649. doi: 10.1126/science.1134641. [DOI] [PubMed] [Google Scholar]
- 15.Aryanpur PP, Regan CA, Collins JM, et al. Gle1 regulates RNA binding of the DEAD-box helicase Ded1 in its complex role in translation initiation. Mol Cell Biol. 2017 Aug; doi: 10.1128/MCB.00139-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kendirgi F, Barry DM, Griffis ER, Powers MA, Wente SR. An essential role for hGle1 nucleocytoplasmic shuttling in mRNA export. J Cell Biol. 2003;160(7):1029–1040. doi: 10.1083/jcb.200211081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Folkmann AW, Collier SE, Zhan X, Aditi, Ohi MD, Wente SR. Gle1 functions during mRNA export in an oligomeric complex that is altered in human disease. Cell. 2013;155(3):582–593. doi: 10.1016/j.cell.2013.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aditi, Folkmann AW, Wente SR. Cytoplasmic hGle1A regulates stress granules by modulation of translation. Mol Biol Cell. 2015;26(8):1476–1490. doi: 10.1091/mbc.E14-11-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jao L-E, Akef A, Wente SR. A role for Gle1, a regulator of DEAD-box RNA helicases, at centrosomes and basal bodies. Mol Biol Cell. 2017;28(1):120–127. doi: 10.1091/mbc.E16-09-0675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Folkmann AW, Noble KN, Cole CN, Wente SR. Dbp5, Gle1-IP6 and Nup159: a working model for mRNP export. Nucl Austin Tex. 2011;2(6):540–548. doi: 10.4161/nucl.2.6.17881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rayala HJ, Kendirgi F, Barry DM, Majerus PW, Wente SR. The mRNA export factor human Gle1 interacts with the nuclear pore complex protein Nup155. Mol Cell Proteomics MCP. 2004;3(2):145–155. doi: 10.1074/mcp.M300106-MCP200. [DOI] [PubMed] [Google Scholar]
- 22.Kendirgi F, Rexer DJ, Alcázar-Román AR, Onishko HM, Wente SR. Interaction between the shuttling mRNA export factor Gle1 and the nucleoporin hCG1: a conserved mechanism in the export of Hsp70 mRNA. Mol Biol Cell. 2005;16(9):4304–4315. doi: 10.1091/mbc.E04-11-0998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strahm Y, Fahrenkrog B, Zenklusen D, et al. The RNA export factor Gle1p is located on the cytoplasmic fibrils of the NPC and physically interacts with the FG-nucleoporin Rip1p, the DEAD-box protein Rat8p/Dbp5p and a new protein Ymr 255p. EMBO J. 1999;18(20):5761–5777. doi: 10.1093/emboj/18.20.5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jao L-E, Appel B, Wente SR. A zebrafish model of lethal congenital contracture syndrome 1 reveals Gle1 function in spinal neural precursor survival and motor axon arborization. Dev Camb Engl. 2012;139(7):1316–1326. doi: 10.1242/dev.074344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nousiainen HO, Kestilä M, Pakkasjärvi N, et al. Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat Genet. 2008;40(2):155–157. doi: 10.1038/ng.2007.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aditi, Glass L, Dawson TR, Wente SR. An amyotrophic lateral sclerosis-linked mutation in GLE1 alters the cellular pool of human Gle1 functional isoforms. Adv Biol Regul. 2016;62:25–36. doi: 10.1016/j.jbior.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaneb HM, Folkmann AW, Belzil VV, et al. Deleterious mutations in the essential mRNA metabolism factor, hGle1, in amyotrophic lateral sclerosis. Hum Mol Genet. 2015;24(5):1363–1373. doi: 10.1093/hmg/ddu545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adams RL, Terry LJ, Wente SR. Nucleoporin FG domains facilitate mRNP remodeling at the cytoplasmic face of the nuclear pore complex. Genetics. 2014;197(4):1213–1224. doi: 10.1534/genetics.114.164012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Strässer K, Bassler J, Hurt E. Binding of the Mex67p/Mtr2p heterodimer to FXFG, GLFG, and FG repeat nucleoporins is essential for nuclear mRNA export. J Cell Biol. 2000;150(4):695–706. doi: 10.1083/jcb.150.4.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saavedra CA, Hammell CM, Heath CV, Cole CN. Yeast heat shock mRNAs are exported through a distinct pathway defined by Rip1p. Genes Dev. 1997;11(21):2845–2856. doi: 10.1101/gad.11.21.2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller AL, Suntharalingam M, Johnson SL, Audhya A, Emr SD, Wente SR. Cytoplasmic inositol hexakisphosphate production is sufficient for mediating the Gle1-mRNA export pathway. J Biol Chem. 2004;279(49):51022–51032. doi: 10.1074/jbc.M409394200. [DOI] [PubMed] [Google Scholar]
- 32.Stutz F, Kantor J, Zhang D, McCarthy T, Neville M, Rosbash M. The yeast nucleoporin rip1p contributes to multiple export pathways with no essential role for its FG-repeat region. Genes Dev. 1997;11(21):2857–2868. doi: 10.1101/gad.11.21.2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rollenhagen C, Hodge CA, Cole CN. The Nuclear Pore Complex and the DEAD Box Protein Rat8p/Dbp5p Have Nonessential Features Which Appear To Facilitate mRNA Export following Heat Shock. Mol Cell Biol. 2004;24(11):4869–4879. doi: 10.1128/MCB.24.11.4869-4879.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stutz F, Neville M, Rosbash M. Identification of a novel nuclear pore-associated protein as a functional target of the HIV-1 Rev protein in yeast. Cell. 1995;82(3):495–506. doi: 10.1016/0092-8674(95)90438-7. [DOI] [PubMed] [Google Scholar]
- 35.Sievers F, Higgins DG. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol Biol Clifton NJ. 2014;1079:105–116. doi: 10.1007/978-1-62703-646-7_6. [DOI] [PubMed] [Google Scholar]
- 36.Saavedra C, Tung KS, Amberg DC, Hopper AK, Cole CN. Regulation of mRNA export in response to stress in Saccharomyces cerevisiae. Genes Dev. 1996;10(13):1608–1620. doi: 10.1101/gad.10.13.1608. [DOI] [PubMed] [Google Scholar]
- 37.Slabinski L, Jaroszewski L, Rychlewski L, Wilson IA, Lesley SA, Godzik A. XtalPred: a web server for prediction of protein crystallizability. Bioinforma Oxf Engl. 2007;23(24):3403–3405. doi: 10.1093/bioinformatics/btm477. [DOI] [PubMed] [Google Scholar]
- 38.Hodge CA, Tran EJ, Noble KN, et al. The Dbp5 cycle at the nuclear pore complex during mRNA export I: dbp5 mutants with defects in RNA binding and ATP hydrolysis define key steps for Nup159 and Gle1. Genes Dev. 2011;25(10):1052–1064. doi: 10.1101/gad.2041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wickramasinghe VO, Savill JM, Chavali S, et al. Human inositol polyphosphate multikinase regulates transcript-selective nuclear mRNA export to preserve genome integrity. Mol Cell. 2013;51(6):737–750. doi: 10.1016/j.molcel.2013.08.031. [DOI] [PubMed] [Google Scholar]
- 40.Hodroj D, Recolin B, Serhal K, et al. An ATR-dependent function for the Ddx19 RNA helicase in nuclear R-loop metabolism. EMBO J. 2017 Mar; doi: 10.15252/embj.201695131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rajakylä EK, Viita T, Kyheröinen S, Huet G, Treisman R, Vartiainen MK. RNA export factor Ddx19 is required for nuclear import of the SRF coactivator MKL1. Nat Commun. 2015;6:5978. doi: 10.1038/ncomms6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Terry LJ, Wente SR. Nuclear mRNA export requires specific FG nucleoporins for translocation through the nuclear pore complex. J Cell Biol. 2007;178(7):1121–1132. doi: 10.1083/jcb.200704174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hurwitz ME, Strambio-de-Castillia C, Blobel G. Two yeast nuclear pore complex proteins involved in mRNA export form a cytoplasmically oriented subcomplex. Proc Natl Acad Sci U S A. 1998;95(19):11241–11245. doi: 10.1073/pnas.95.19.11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee H-S, Lee D-H, Cho HK, Kim SH, Auh JH, Pai H-S. InsP6-sensitive variants of the Gle1 mRNA export factor rescue growth and fertility defects of the ipk1 low-phytic-acid mutation in Arabidopsis. Plant Cell. 2015;27(2):417–431. doi: 10.1105/tpc.114.132134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ren Y, Schmiege P, Blobel G. Structural and biochemical analyses of the DEAD-box ATPase Sub2 in association with THO or Yra1. eLife. 2017;6 doi: 10.7554/eLife.20070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schütz P, Bumann M, Oberholzer AE, et al. Crystal structure of the yeast eIF4A-eIF4G complex: an RNA-helicase controlled by protein-protein interactions. Proc Natl Acad Sci U S A. 2008;105(28):9564–9569. doi: 10.1073/pnas.0800418105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sherman F, Fink GR, Hicks JB. Laboratory Course Manual for Methods in Yeast Genetics. Cold Spring Harbor Laboratory; 1986. [Google Scholar]
- 48.Carmody SR, Tran EJ, Apponi LH, Corbett AH, Wente SR. The mitogen-activated protein kinase Slt2 regulates nuclear retention of non-heat shock mRNAs during heat shock-induced stress. Mol Cell Biol. 2010;30(21):5168–5179. doi: 10.1128/MCB.00735-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.