

Abstract
Eicosanoids are biologically active lipid signaling molecules derived from polyunsaturated fatty acids. Many of the actions of eicosanoid metabolites formed by cyclooxygenase and lipoxygenase enzymes have been characterized, however, the epoxy-fatty acids (EpFAs) formed by cytochrome P450 enzymes are newly described by comparison. The EpFA metabolites modulate a diverse set of physiologic functions that include inflammation and nociception among others. Regulation of EpFAs occurs primarily via release, biosynthesis and enzymatic transformation by the soluble epoxide hydrolase (sEH). Targeting sEH with small molecule inhibitors has enabled observation of the biological activity of the EpFAs in vivo in animal models, greatly contributing to the overall understanding of their role in the inflammatory response. Their role in modulating inflammation has been demonstrated in disease models including cardiovascular pathology and inflammatory pain, but extends to neuroinflammation and neuroinflammatory disease. Moreover, while the EpFA demonstrate activity against inflammatory pain, interestingly, this action extends to blocking chronic neuropathic pain as well. This review outlines the role of modulating sEH and the biological action of EpFA in models of pain and inflammatory diseases.
Keywords: soluble epoxide hydrolase (sEH), epoxy-fatty acids (EpFA), inflammatory pain, neuropathic pain, depression, Alzheimer’s Disease
1. Introduction
Eicosanoids are a group of lipid mediators generated from arachidonic acid (ARA) by activity of cyclooxygenases (COX), lipoxygenases (LOX) and cytochrome P450 (CYP450) enzymes. These fatty acid metabolites are implicated in critical biological processes throughout the body in most cells, tissues and organs (Funk, 2001; X. Xu, et al., 2016). Eicosanoids have been intensely investigated for their role in the inflammatory response and more recently the complexity of the pro and anti-inflammatory as well as other non-inflammatory roles for these metabolites have been recognized (Dennis & Norris, 2015). Knowledge of the complex signaling networks that the eicosanoids comprise now extends to include the metabolites of other long chain polyunsaturated acids (LC-PUFA) such as docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) that are recognized to flow through the same enzymatic pathways. Much is known about the bioactivity of prostaglandin metabolites of ARA, and similarly, the leukotriene metabolites have well described, potent biological action. There is less known about the metabolites of the CYP450s, the epoxy-fatty acids (EpFA), though the body of knowledge regarding their bioactivity is growing. Moreover, when addressing the EpFA specifically as a class of lipid mediators, the epoxide metabolites of all LC-PUFAs can be included. It is in examining the biology of this EpFA class that the importance of the soluble epoxide hydrolase (sEH) enzyme was revealed because it is a major regulator of EpFA biology. Uncovering the physiologic role of the EpFA has been greatly aided by the ability to inhibit the sEH enzyme. Because the effects of maintaining EpFA titers in vivo has been largely beneficial, small molecule inhibitors of sEH (sEHI) have become a novel approach to altering disease pathologies including cardiovascular diseases, inflammation, neurodegenerative disorders and chronic pain among others.
a. EpFA Biosynthesis and regulation
LC-PUFA are 14–26 long carbon chains with several double bonds imparting their polyunsaturated nature. The term “eicosa” refers to 20 carbon length fatty acids formed mostly from 20:4(n-6) ARA which, along with the omega-3 metabolites of EPA (20:5, n-3) and longer chain DHA (22:6, n-3) fatty acids, are the major focus of this review. The CYP450 enzymes act on LC-PUFA to form EpFA by epoxidation of the double bonds (Konkel & Schunck, 2011). Multiple regioisomers of EpFA are produced from the parent LC-PUFA depending on the location of the epoxidized double bond. There is also a high degree of enantiofacial selectivity (R/S regioisomer) conferred in this process (Spector, et al., 2004). The epoxidized metabolites, epoxyeicosatrienoic acids (EETs) from omega-6 ARA, epoxyeicosatetraenoic acids (EEQs) from omega-3 EPA, and epoxydocosapentaenoic acids (EDPs) from omega-3 DHA are all classed as EpFA and are principally anti-inflammatory eicosanoids (Morisseau, et al., 2010). The relative contribution of different CYP450s to the total production of the EpFA will vary with substrate availability and concentration. Also, the expression of the CYP450 monooxygenases that produce them vary depending on sex, species, organ and proportion of the regioisomer of epoxide they produce. However, both the CYP450s that produce the EpFA and the sEH that is their principal regulatory enzyme are expressed at some level in most tissues. This demonstrates the biological relevance of these metabolites because all types of EpFA are transformed by the sEH into diols (Figure 1) and in the case of EETs the diols are less active (Spector, 2009).
Figure 1. Long chain polyunsaturated acid metabolism through the CYP450 pathway.
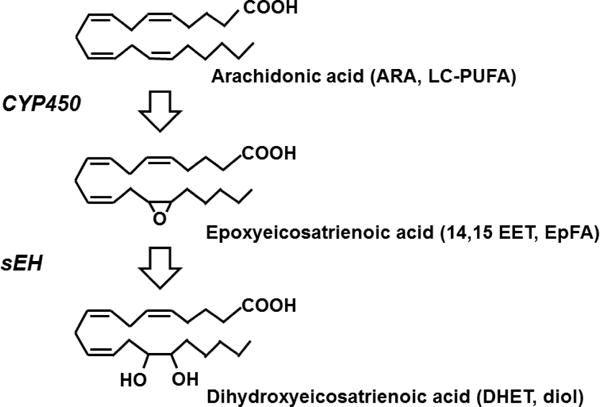
Arachidonic acid (ARA) and other long chain polyunsaturated fatty acids (LC-PUFA) are metabolized by cytochrome P450 enzymes (CYP450) into the epoxy-fatty acids (EpFA). For simplicity, the metabolism of omega-6 ARA is depicted here as an example of LC-PUFA metabolism. A class of EpFA, the epoxyeicosatrienoic acids (EETs), are formed from ARA. Four individual regioisomers can be formed by the epoxidation of any one of the four double bonds with the 14,15 EET depicted. In addition to the epoxides from LC-PUFA, any fatty acids with an olefinic bond may form epoxidized metabolites. The soluble epoxide hydrolase (sEH) adds water to the oxirane ring to yield the diol, in the case of ARA metabolites are termed dihydroxyeicosatrienoic acids (DHETs). This process is the same for omega-3 LC-PUFA including DHA and EPA which form potent biologically active classes of EpFA.
sEH (EC:3.3.2.10) is part of the α/β hydrolase fold super family and is a 120 kD homodimer enzyme with a C-terminal hydrolase and N-terminal phosphatase (Beetham, et al., 1993; Cronin, et al., 2003). The phosphatase domain hydrolyzes phosphorylated lipids such as isoprenoid phosphates and lysophosphatidic acid that stimulate cell growth but far less is known about the biological role of this activity (Oguro & Imaoka, 2012; Oguro, et al., 2009). The C-terminal domain hydrolyzes the epoxides by addition of water to the three membered oxirane ring (Spector, 2009). sEH expression is well conserved among species from simple chordates to preclinical rodents and all mammals tested to date indicating its fundamental role in biology (Harris & Hammock, 2013). sEH is widely distributed throughout the body with the most concentrated expression in the liver, kidney, intestine and vasculature in mammals (Enayetallah, et al., 2004). However, sEH is also found in the brain and in C57Bl/6 mouse is observed more strongly in the cortex, hippocampus, amygdala and striatum (Marowsky, et al., 2009). sEH expression has been found in neurons along with the CYP450 enzymes that produce EpFA (Iliff, et al., 2009) and in astrocytes including astrocytic end feet (Marowsky, et al., 2009). In human naïve brain, sEH is expressed in neurons, oligodendrocytes, astrocytes and ependymal cells (Sura, et al., 2008).
Potent selective inhibitors of sEH were first described in the early 1980’s as a mechanism to identify the biological importance of the enzyme (Mullin & Hammock, 1982). The diols formed from sEH action generally lack the activity of the epoxidized precursors however they are dramatically more polar, move rapidly out of cells, and are easily conjugated and excreted (Greene, et al., 2000). Yet, there is some evidence PUFA diols are chemoattractant for monocytes (Kundu, et al., 2013) and that linoleic diols specifically act as lipokines in the regulation of brown adipose tissue and thermogenesis (Lynes, et al., 2017). There is no overt phenotype with the whole body knockout animals not subjected to physiologic stress (Spector & Kim, 2015), however, knockout mice had lower survival following ischemic events (Hutchens, et al., 2008). In general, the biology of the knockout animals is mimicked by treatment with sEHI.
b. EpFA role in inflammation
Inflammation is the biological response to insult that includes cardiovascular dilation of arterioles and capillaries, increased permeability of the microvasculature, and leukocyte infiltration. The leukocyte infiltration is typically in response to release of chemokines and cytokines (small cell signaling proteins), resulting in redness, heat and pain (Rot & von Andrian, 2004). Several hallmarks of inflammation have been exploited as biomarkers of the condition including cytokines such as interleukins 1β and 6 (IL-1β, IL-6), tumor necrosis factor alpha (TNFα) and chemokines such as vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule 1 (I-CAM 1), endothelial cell selective adhesion molecule (E-selectin), prostaglandins such as prostaglandin E2 (PGE2) and activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB). TNFα is a cytokine which is a central mediator of both acute phase reaction and systemic inflammation. VCAM-1 is activated by TNFα via NFκB and mediates chemotaxis of typically monocytes and lymphocytes and is essential to their adhesion to and infiltration of inflamed tissues. The inflammatory response removes an injury or insult and resolves but when resolution fails it becomes a chronic condition and chronic inflammation is related to numerous major disease states.
EETs and other EpFA are known to reduce inflammation through multiple mechanisms which have been demonstrated in animal models (Figure 2). One such mechanism of EET regioisomers in cardiovascular biology is to inhibit VCAM-1, E-selectin and ICAM-1 expression (Node, et al., 1999; Zhao, et al., 2012). EETs also decrease TNFα secretion from monocytic cells (Bystrom, et al., 2011) and may also inhibit their adherence (Node, et al., 1999). Other mechanisms by which EETs reduce inflammation include blocking the nuclear translocation of NFκB (Bystrom, et al., 2011; Fife, et al., 2008; Node, et al., 1999) which in turn downregulates several enzymes including calcium-insensitive nitric oxide synthase (iNOS), lipoxygenase-5 (LOX-5), and cyclooxygenase-2 (COX-2) that are upregulated in inflammation (Schmelzer, et al., 2006; Schmelzer, et al., 2005). sEHI administration also blocked increases in phospho-IκBα levels which activate NFκB and thus inhibited NFκB signaling in a murine model (D. Xu, et al., 2006). Activation of signal transducer and activator of transcription 3 (STAT3) (Williams, et al., 2004) and other nuclear receptor activation such as peroxisome proliferator activated receptor (PPAR) alpha and gamma are additional mechanisms that have been described for EETs (Fang, 2006; Ng, et al., 2007). In vivo sEH gene deletion and the resulting increase in EETs lowered inflammatory gene expression and neutrophil recruitment, though these effects displayed some organ specificity being more robust in lung (Deng, et al., 2011). EpFA derived from omega-3 LC-PUFA are less well described but recent studies demonstrate they also have generally anti-inflammatory properties (Isobe & Arita, 2014; Morin, et al., 2010). However, it is critical that EpFA and their regio and optical isomers be treated as distinct compounds.
Figure 2. EpFA block inflammation through several mechanisms.
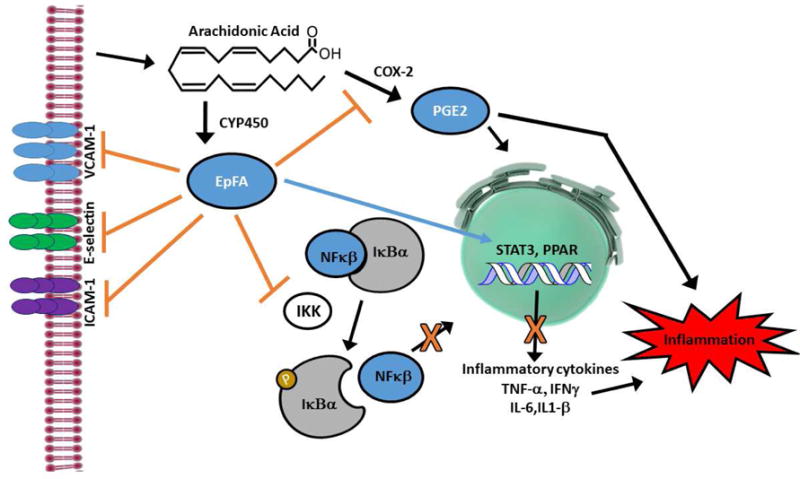
Regioisomers of the EET class of EpFA inhibit VCAM-1, E-selectin and ICAM-1 expression in endothelial cells which blocks the adherence and infiltration of activated monocytes. EETs also reduce inflammation by blocking the nuclear translocation of NFκB, and sEHI administration which elevates all EpFA, blocks the increase of phospho-ΙκΒα levels which activate NFκB and thus inhibit NFκB signaling. This results in the downregulation of several enzymes including calcium-insensitive nitric oxide synthase (iNOS), lipoxygenase-5 (LOX-5), and cyclooxygenase-2 (COX-2, pictured) that are upregulated in inflammation. The downregulation of COX-2 also limits the production of prostaglandin E2 (PGE2) which is a potent inflammatory agent. EETs also decrease TNFα secretion from monocytic cells and inflammatory cytokines in several tissues. Activation of signal transducer and activator of transcription 3 (STAT3) and other nuclear receptors such as peroxisome proliferator activated receptor (PPAR) alpha and gamma are additional anti-inflammatory mechanisms that have been described for EpFA in blocking downstream inflammation.
Determining the mechanisms of EpFA action has been complicated by the lack of a defined receptor. There has been a considerable effort in the last decade to identify a receptor, or more likely receptors, for the EETs with little progress. The COX and LOX systems are perhaps better exploited because prostaglandins and leukotrienes have identified G protein coupled receptors (GPCR) and selective compounds for pharmacological agonism and antagonism. Although lacking a defined receptor, G proteins have been implicated in the action of EETs in coronary smooth muscle (Li & Campbell, 1997) and EETs have been antagonized with a synthetic antagonist (Gauthier, et al., 2002; Gross, et al., 2008). In cerebral artery smooth muscle cells, EETs bind to transient receptor potential cation channel subfamily V member 4 (TRPV4) channels (Earley, et al., 2005; Vriens, et al., 2005; Watanabe, et al., 2003). However, there is evidence that EETs can act on more than one TRP channel (Loot & Fleming, 2011), and that they have effects that are independent of calcium signaling (reviewed in Sudhahar et al. (Sudhahar, et al., 2010)). Thus, there are multiple possible actions of EpFA and their mode of action may differ depending on compound, tissue type and receptor expression.
The sEHI reduce the severity of a variety health problems in animal models. In many cases inflammation could be seen as a common mechanism as introduced above, but in other cases it is hard to understand how a single mechanism could address such diverse illnesses as atrial fibrillation and pancreatitis. It now appears that modulation of endoplasmic reticulum stress and specifically the pathological axis from mitochondrial dysfunction through ROS generation and activation of the ER Stress pathway leading to cell damage is an event common to many of the beneficial effects of EpFA and sEHI.
The bioactivity of EpFA is transient in vivo principally due to the action of sEH. The primary route of EpFA transformation to inactive diols is blocked by inhibiting the sEH enzyme to increase their residence time and observe their bioactivity. Several commonly used sEHI are outlined in Table 1 including their chemical structures (Table 1). Even with sEH inhibited or removed, EpFA are metabolized at a somewhat slower rate by beta oxidation or chain elongation, CYP450 oxidation, reincorporation into glycerides and other pathways (Spector, et al., 2004). Inhibiting sEH has demonstrated anti-inflammatory action in several studies using animal models (J. Y. Liu, et al., 2009a; J. Y. Liu, Yang, et al., 2010; Schmelzer, et al., 2005). Anti-hyperalgesic activity in nociceptive assays has also been correlated with increased concentration of EpFA in vivo (Inceoglu, et al., 2012). In addition the demonstrated bioactivity of EpFA has been supported by advances made with the use of EET analogues in vivo as an alternative experimental strategy (Sudhahar, et al., 2010).
Table 1.
Commonly Used sEH Inhibitors
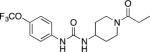 TPPU, UC1770 1-Trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea |
Most commonly used sEHI in recent publications. High activity on the primate sEH, good activity with rodent sEH and often poor activity on sEH of other species. High oral availability and good PK-ADME with PK data available in many species. Centrally active in mice. Lipophilic and high melting requiring a long dissolution time in water and careful formulation (Inceoglu, et al., 2013; Rose, et al., 2010). |
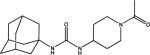 APAU, UC1153, AR9281 1-(1-Acetypiperidin-4-yl)-3-adamantanylurea |
No toxicity at high doses in human Phase I and II trials. No clinically useful efficacy in a human hypertension trial. Short half-life in man and rodents. PK data available in multiple species. Surprisingly high water solubility and rapid dissolution. Potent inhibitor of rodent sEH, less active on primate sEH and poor activity in many other species. Poor target occupancy with human sEH (D. Chen, et al., 2011). |
|
t-AUCB, UC1471 trans-4-[4-(3-Adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid |
Good potency on sEH from a variety of species. PK-ADME known in multiple species. Half-life is longer than AEPU but shorter than TPPU. Good water solubility off-sets lower potency and shorter half-life compared to TPPU (S. H. Hwang, et al., 2007; Shaik, et al., 2013). |
|
t-TUCB, UC1728 trans-4-{4-[3-(4-Trifluoromethoxy-phenyl)-ureido]-cyclohexyloxy}-benzoic acid |
Good potency on sEH from a variety of species. PK-ADME known in multiple species. Longer half-life than t-AUCB above (S. H. Hwang, et al., 2007; J. Y. Liu, et al., 2009a). |
|
AEPU, UC950 1-Adamantanyl-3-{5-[2-(2-ethoxyethoxy)ethoxy]pentyl]}urea |
Moderate potency on sEH from a wide variety of species. Short half-life but bioactive metabolites in mice increase efficacy. Very water soluble and dissolved directly in water. Most water soluble of the potent sEHI (J.-Y. Liu, et al., 2015). |
|
AUDA, UC700 12-(3-adamantan-1-yl-ureido) dodecanoic acid |
Moderate potency on sEH from a wide variety of species. Short half-life, commercially available (A. N. Simpkins, et al., 2009). |
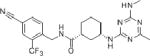 GSK 2256294A (1R,3S)-N-(4-cyano-2-(trifluoromethyl)benzyl)-3-((4-methyl-6-(methylamino)-1,3,5-triazin-2-yl)amino)cyclohexanecarboxamide. |
Tested in human trials. Good PK-ADME in human. High melting with poor water solubility. Careful formulation is needed. Activity on multiple species not reported (Lazaar, et al., 2016; Podolin, et al., 2013). |
 Sorafenib 4-(4-(3-(4-chloro-3- 4-[4-[[4-chloro-3-(trifluoromethyl)phenyl]carbamoylamino]phenoxy]-Nmethylpyridine-2-carboxamide |
Potent inhibitor of the human and rodent sEH. A registered drug to treat cancer possibly as a Raf-1 or pan kinase inhibitor. Poor solubility needing complex formulation. Numerous side effects at high doses (J.-Y. Liu, et al., 2009). |
|
Regorafenib 4-[4-[[4-chloro-3-(trifluoromethyl)phenyl]carbamoylamino]-3-fluorophenoxy]-N-methylpyridine-2-carboxamide |
Roughly an order of magnitude more potent on the human sEH than Sorafenib. Similar side effect spectrum and physical properties (Sung Hee Hwang, et al., 2013). |
|
Triclocarban 3-(4-chlorophenyl)-1-(3,4-dichlorphenyl)urea or 3,4,4′-trichlorocarbanilide |
A reported anti-microbial used in some solid soaps. Approved for topical use only. Selective inhibitor of the human sEH and of similar potency to APAU above on the human sEH. Poorly active in other species. Short half-life in vivo following topical application. Rapidly forms N-glucuronides (Schebb, et al., 2012). |
For full data sheets on the above compounds, contact authors.
2. sEH as a Target for Inflammatory Diseases
Eicosanoids play a fundamental role in inflammation, and classical pharmaceutical approaches have focused on blocking the formation of metabolites or antagonising their receptor mediated action. This is the case with most non-steroidal anti-inflammatory drugs (NSAIDs) and leukotriene receptor antogonists. However, while formally described as inflammatory, the plietropic effects of the eicosanoids are now more deeply appreciated, and it is understood that many of the side effects of these therapies are due to blocking their production. More recently, attention has been focused on alternative eicosanoid or docosanoid metabolites that are anti-inflammatory or pro-resolving including the EpFA (for a review of specialized pro-resolving mediators (SPMs) see Chaing and Serhan, 2017). Even compounds considered as predominantly proinflammatory seem to regulate a series of events leading to resolution of inflammation. Targeting the sEH is a novel strategy in this scheme because inhibiting sEH sustains the endogenous EpFA to attain their biological effects. As mentioned above, other degredation routes for the EpFA exist such as β-oxidation (Spector, et al., 2004). Thus, inhibiting sEH is unlikely to build a large or chronic increase in EpFA, enabling the beneficial effects of their modulation without a large side effect profile.
a. Inflammatory bowel disease (IBD) and chronic peptic ulcer
EpFA are also active against inflammatory disorders of the gastrointestinal tract. Both pharmacological inhibition and gene ablation of sEH were investigated for their ability to reduce chronic active inflammatory bowel disease (W. Zhang, et al., 2012). In this study using a genetically engineered IL-10 null mouse model of IBD, both approaches of limiting sEH activity lowered the number of ulcers and transmural inflammation. Quantitative real time PCR demonstrated an increase in inflammatory cytokines and chemokines including IFN-γ, TNF-α, MCP-1 and VCAM-1 in IL-10 null mice compared to double IL-10/sEH null and sEHI reduced IFN-γ, MCP-1 and VCAM-1 mRNA. Western blot analysis also showed that phosphorylated NFκB was downregulated and oxylipin analysis revealed increased EET to diol ratios, and a decrease in both leukotriene B4 (LTB4) and 5-hydroxyeicosatetraenoic acid (5-HETE) metabolites (W. Zhang, et al., 2012).
The anti-inflammatory efficacy and physiological improvement found in the IBD model are unique to sEH inhibition because the clinical use of NSAIDs including COX-2 selective inhibitors exacerbates IBD (Berg, et al., 2002; Kaufmann & Taubin, 1987; Reuter, et al., 1996). In a later study it was determined that sEH gene deletion decreased adenocarcinoma tumors in the IL-10 null mouse model of IBD (W. Zhang, et al., 2013). In this context, the absence of sEH is anti-inflammatory and therefore limits the transition of IBD to tumor formation in the bowel. However, the role of EpFA in tumor formation is still poorly understood and likely complex. EETs have demonstrated angiogenic activity which is an important contribution to their regulatory role in hyperemia and modulation of neurovascular coupling in the cerebral vasculature. Additionally, their angiogenic activity may be useful in improving wound repair mechanisms (Sander, et al., 2011; Sander, et al., 2013). Angiogenesis is a critical biological process but pathological angiogenesis can lead among other things to enhanced tumor growth. However, in some preclinical cancer models, particularly with solid tumors, sEHI can increase tumor growth. For example, with a solid tumor a 10× therapeutic dose of a sEHI for 10 weeks resulted in slightly increased tumor growth, angiogenesis and metastasis (Panigrahy, et al., 2012). In addition to the effects of inhibiting sEH, when EETs were increased using other techniques including genetic manipulation, multi-organ metastasis and tumor dormancy escape were observed in mice. An interesting enigma is that, in Lewis lung xenographs, high doses of sEHI led to angiogenesis and tumor growth but, if sEHI were given with celecoxib (a NSAID) or an omega-3 LC-PUFA, sEHI demonstrated dramatically reduced angiogenesis and tumor growth in both lung and breast tumor xenographs (G. Zhang, et al., 2014; G. Zhang, et al., 2013). A partial hypothesis to explain this is that an angiogenic metabolite of 8,9-EET can be formed by COX enzymes but the metabolite does not form in the presence of COX inhibitors or competing omega-3 lipids (Rand, et al., 2017). The EDPs which cannot form an analogous angiogenic metabolite are inherently antiangiogenic and the EETs, in the presence of celecoxib, are as well. At this point the proangiogenic effects of sEHI appear minor and the antiangiogenic effects in the presence of celecoxib or an omega-3 dietary supplement appear major. Thus, the homeostatic balance of angiogenesis in relation the EpFA needs to be further investigated for effects in cancer biology. In contrast, the role of sEH inhibition and EpFA in blocking inflammation in the bowel and the subsequent reduction of adenocarcinoma appears more robust. In addition to genetically induced models of IBD, sEH inhibition has also demonstrated positive results in models of NSAID-induced intestinal ulcer. Diclofenac induced ulcers were reduced by pretreatment of sEHI with efficacy comparable to proton pump inhibitors but at a far lower dose (Goswami, et al., 2016). The effect on ulcer correlated with increased EpFA of both omega-6 and omega-3 classes. Furthermore, the efficacy of sEHI in this study suggests that reported beneficial effects of PUFA for ulcer relief (Pineda-Pena, et al., 2012) are potentially mediated by the EpFA metabolites. It also appears that part of the efficacy of proton pump inhibitors such as omeprazol may be due to a reduction in inflammation caused by a dramatic increase in EpFA (Goswami, et al., 2016).
b. Destructive bone diseases
Arthritis
Recently, an sEHI was evaluated in a randomized, blind preclinical study in aged dogs with natural osteoarthritic pain compared to vehicle control and is first reported on here. The pain was evaluated with a questionnaire adapted from the canine brief pain inventory (CBPI). The CBPI was originally developed for owners to assess the pain level of their pets with existing painful conditions. The questionnaire used for this experiment had 12 total questions to assess pain based on activities such as initiation of movement, walking, climbing stairs or jumping for a reward scored with a scale of 0 (not due to pain), 1 (not able to determine), and 2 (obviously due to pain) for the first 11 questions. The 12th question regarding overall pain was scored on a scale from 0 (no pain) to 4 (unwilling to participate). The dogs were assessed for five consecutive days prior to treatment to establish a baseline measurement of their pain. Treatments were with 5 mg/kg of the sEHI t-TUCB (S. H. Hwang, et al., 2007) in an oral capsule or placebo capsule for 5 days. Scores are reported as the percent of maximum possible pain score (score = observed/maximum*100, maximum = 26 for the entire questionnaire). The 5 days of baseline measurements were pooled to compare 5 days of treatment with t-TUCB or placebo. This study revealed that in naturally occurring arthritis sEH inhibition was effective in blocking pain (Figure 3). The sEHI significantly lowered pain scores compared to both the pooled vehicle and placebo control (Kruskal-Wallis One Way Analysis of Variance on Ranks, H = 21.438 with 2 degrees of freedom, p ≤ 0.001, n=7 dogs/group). Importantly the results in dogs reported here represent efficacy against naturally occurring arthritis and not an induced model. While the study is limited due to the number of dogs naturally presenting arthritis and therefore the absence of a positive control, the result of sEHI treatment was statistically significant and biologically meaningful by increasing activity in the dogs. This efficacy in a chronic condition is meaningful, particularly in dogs, because they are highly sensitive to the side effects of NSAIDs which can be lethal (Mathews, 2000). Inhibition of sEH has been suggested as an anti-inflammatory strategy for chronic use in arthritis over NSAIDs and steroidals that have dose limiting side effects (Pillarisetti & Khanna, 2012). Schmelzer et al. demonstrated that sEHI and the resulting increased EETs transcriptionally down regulate a number of enzymes involved in mediation of inflammation including induced COX-2 (Schmelzer, et al., 2005). sEHI have also been found by a number of studies to synergize with NSAIDs. Since sEHI reduce hypertension which is often associated with long term use of NSAIDs, as well as gastrointestinal erosion (Goswami, et al., 2016) and NSAIDs mediated cardiovascular side effects, (J. Y. Liu, Li, et al., 2010; Schmelzer, et al., 2006) sEHI may act as safeners of NSAIDs when the two are combined.
Figure 3. sEH inhibition and EpFA blocks natural arthritic pain.
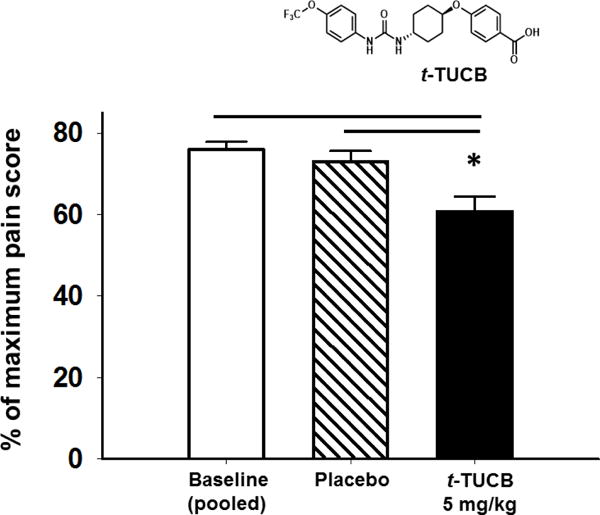
Male and female beagle dogs (ages 8–14 years) with naturally occurring osteoarthritis were administered the sEHI t-TUCB at 5 mg/kg orally in a capsule or placebo control for five days (n=7 dogs/group). The results are presented as an average of 5 days of testing pre-treatment (pooled baseline) compared to an average of 5 days of testing under treatment (placebo or t-TUCB). The sEHI significantly lowered pain scores compared to both pooled baseline and placebo control (p ≤ 0.001).
Osteoporosis
Osteoporosis differs from arthritis in the amount of asymptomatic bone resorption often resulting in fragility fractures (Rachner, et al., 2011). In a recent study, EETs blocked bone loss by effecting osteoclast differentiation in vitro via downregulation of ROS. In addition, EETs administration also reduced bone loss in ovariectomized mice as a model of estrogen deficiency-induced osteoporosis (Guan, et al., 2015). In these experiments EETs decreased ROS release, TNFα levels and NFκB activation. These results support the observation that sEHI inhibition has broad efficacy including lytic bone disease.
Chronic periodontitis
Some of the available data in periodontitis is focused on the role of eicosanoids in general. A recent study examined the oxylipins from human periodontitis samples and found inflammatory markers including increased prostaglandins and leukotrienes (Huang, et al., 2014). EETs and diols were also measured in this study but did not exhibit as much change. In a preclinical model of periodontitis in mouse, both sEHI and sEH null mice showed reduced bone loss when exposed to A. actinomycetemcomitans. In these animals, the chief osteoclastogenic molecules RANK/RANKL/OPG and the chemokine MCP-1 were downregulated and downstream inflammatory JNK and p38 kinase signaling was abated. In addition, this study demonstrated that ER Stress was upregulated with periodontal disease but was blocked by both sEHI administration and in the sEH knockout mice (Trindade da Silva, et al., 2017). Thus, given the role of sEHI and EpFA action against inflammation combined with the effects on other bone loss conditions, sEH inhibition may be a useful strategy to combat periodontitis.
c. Sepsis
Sepsis or endotoxemia in humans is a diverse syndrome of physiological (i.e. systemic arterial hypotension) and immunological responses to microbial infection that include increased circulating levels of cytokines TNFα and IL-6 and high mobility group protein B1 (HMGB1). Historically, therapeutic approaches using antibodies to combat sepsis have had poor results (reviewed in Fink et al. (Fink, 2014)) and blunting of the innate immune response shows more promise. The approach of using sEHI to block the inflammatory response in modeled sepsis prevented lipopolysaccharide (LPS) induced death, reduced pro-inflammatory cytokines, chemokines and prostaglandins (Schmelzer, et al., 2005) and restored systolic blood pressure to near normal levels while increasing EETs to diol ratios (J. Y. Liu, et al., 2009b). One can envision sEHI as muting the cytokine storm and speeding resolution. The level of linoleic acid diols in the plasma may be a valuable marker for the onset of sepsis. Since they cause the may be causative as well (Moghaddam, et al., 1997; Slim, et al., 2001). Thus, although animal models of sepsis are inadequate to completely mirror the human immune response in this condition, they remain a critical path to the development of new therapeutic strategies and sEH inhibition shows great promise. The difficulty of designing a clinical trial for sepsis is due to its often unpredictable and rapid progression as well as defining a profitable clinical path makes sepsis a difficult target. Possibly treating other conditions leading to a cytokine storm such as viral infections or the severe inflammation initiated by modern immunotherapy such as CAR-T for cancer represent more reasonable paths for agents as sEHI that can moderate the cytokine release syndrome (DeFrancesco, 2014).
d. Cardiovascular Disease
The largest literature base on the anti-inflammatory properties of EpFA is in the field of cardiovascular physiology and pathology. By the early 1980s there was a shift in cardiovascular research from a focus solely on the description of anatomic structures (e.g. the endothelium of blood vessels) to their actual function (Furchgott & Zawadzki, 1980). The action of aspirin had been elucidated by John Vane who shared the 1982 Nobel Prize in Physiology or Medicine with laureates Sune Bergstrom and Bengt Samuelson who described the prostaglandin lipid metabolites. As the biological activity of select prostaglandins in vasculature was further elucidated (Moncada & Vane, 1979) a series of papers on the sEH enzyme was published (Gill & Hammock, 1980). It was later found that sEH is a master regulatory enzyme of the endogenous EETs which were revealed to have vasodilatory effects (Singer & Peach, 1983). The effect of EETs on the modulation of vascular tone is more thoroughly reviewed in Sudhahar et al. (Sudhahar, et al., 2010). In the 1990s the relationship between atherosclerosis and inflammation was recognized (Boring, et al., 1998). This underscored the importance of the EpFA action in the cardiovasculature and implied that the anti-inflammatory properties may be a vital part of their antihypertensive action (Libby, et al., 2002). This was investigated in preclinical models where CYP2J2 epoxygenase overexpression and direct EETs administration inhibited VCAM-1 induced with TNFα independent of hyperpolarizing effects (Node, et al., 1999). Anti-inflammatory effects including inhibiting activation of NFκB, cellular adhesion molecules, cytokine expression, and neutrophil infiltration were demonstrated in vivo in mice overexpressing CYP2J2, CYP2C8 and sEH knockouts (Deng, et al., 2011). Additionally, an EET analog decreased inflammation and oxidative stress which attenuated renal injury in Dahl-salt sensitive rats (Hye Khan, et al., 2013) and EpFA action, demonstrated by an enriched omega-3 diet combined with administration of an sEH inhibitor, decreased renal inflammation in an angiotensin- II model of hypertension (Ulu, et al., 2013).
The main mechanisms of this anti-inflammatory action in the cardiovasculature is via endothelial cells and monocytes and several putative receptors including PPAR, GPCRs and TRP channels. Shear stress was demonstrated to attenuate inflammation through PPAR gamma mediated signaling (Y. Liu, et al., 2005). A radiolabeled EET analog was used to determine that EETs have the capacity to bind to a cell surface receptor (Y. Chen, et al., 2011) and this may occur through a G protein (Li & Campbell, 1997). TRPV4 channels are active in regulating vascular tone and this is one of the suggested mechanisms of EETs action in vasodilation (Sonkusare, et al., 2012; Vriens, et al., 2005; Watanabe, et al., 2003). However, it should be cautioned that the action of EETs in the cardiovasculature is complex and a single regioisomer can act selectively in different vascular beds and tissues (Hercule, et al., 2009; Quilley & McGiff, 2000). Additionally, there is some evidence of EET action directly at the TRP channels (Watanabe, et al., 2003), but there is also evidence that EETs act via increased translocation of other TRPC receptors to the cell surface (Fleming, et al., 2007). There is also some evidence of TRPA1 activity in excitable cells with one regioisomer (Brenneis, et al., 2011). Overall the EET-TRP axis (Loot & Fleming, 2011) is incompletely described in vasculature, and it is even more poorly understood in the central nervous system. Despite this incomplete mechanistic knowledge, evidence in multiple models across different types of biology and in several species demonstrates the anti-inflammatory action of EpFA. The bioactivity of the EpFA points out the potential of targeting sEH to modulate inflammation.
3. sEH as a Target for Neurodegenerative Diseases
With the general population ageing, the incidences of neurodegenerative diseases and chronic inflammatory conditions are both on the rise. Thus, there is an urgent need for new approaches to mitigate neuro-inflammation. The role of the EpFA in regulating inflammatory conditions particularly in the brain is a potential target and inhibiting sEH as a strategy to sustain their biological activity is a novel approach with great promise. Here we outline the role of EpFA and sEH in several modeled neurological diseases with an inflammatory component.
a. Stroke
EpFA, the EETs in particular, are known to be protective against cell death in ischemic stroke (Alkayed, et al., 1996; Iliff, et al., 2010; Alexis N. Simpkins, et al., 2009). Importantly, evidence about sexually dimorphic gene expression and stroke outcomes demonstrate the protective effects of lowering sEH enzyme and increasing EpFA producing CYP450s (Alkayed, et al., 1996; Gupta, et al., 2012; Koerner, et al., 2007; W. Zhang, et al., 2008). A sEH genetic polymorphism in humans has been linked to stroke risk as well as cardiovascular disease (Fornage, et al., 2005) and notably a G860A sEH polymorphism that reduces sEH activity and not CYP2J2 gene expression had a protective effect in nonsmoker stroke in China (L. Zhang, et al., 2008). Overexpression of sEH with a K55R polymorphism in Swedish men correlated with hypertension and increased ischemic stroke risk (Fava 2010). In addition to expression of sEH in endothelial cells, sEH is expressed in astrocytes, oligodendrocytes, and neurons in human brain (Sura, et al., 2008). In mice, sEH expression is found in astrocytes and neurons and has been characterized in brain regions providing background for preclinical models of stroke and other neurological disorders (Marowsky, et al., 2009; W. Zhang, et al., 2007). In rat, neurons and astrocytes produce EETs (Alkayed, et al., 1996; Amruthesh, et al., 1992) suggesting there is a fundamental regulation of EpFA bioactivity in the cells of the brain. As an example, it has been demonstrated that EETs are involved in neurovascular coupling (Farr & David, 2011; Higashimori, et al., 2010). Inhibiting sEH has also demonstrated protective effects against ischemic brain damage independent of effects on cerebral blood flow (Iliff, et al., 2009; W. Zhang, et al., 2007). It is important to note that using sEHI as a strategy has often been favored over exogenous EETs administration in these studies because EETs are quickly metabolized (Imig, et al., 2011). Thus, these results reflect on the elevation of potentially several classes of EpFA.
b. Seizure
Early attention for treating epilepsy was focused on anticonvulsants which regulate ion channels and affect mediator proteins to control seizures. While this approach greatly improved the quality of life for epileptics, they did little prevent seizures. Current research is focused on neuroinflammation and neural plasticity as potential targets to prevent recurrent seizures. The observation that blocking leukocyte infiltration blocked epileptogenesis clarified the role of inflammation in initiating epilepsy (Fabene, et al., 2008). However, inflammation is also a consequence of seizure and plays a role in the development of epilepsy after the first seizure event (Vezzani, et al., 2011). Thus, preventing neuroinflammation that promotes seizures or treating neuroinflammation post seizure event are both current therapeutic targets.
The literature on the benefits of EpFA for seizure reveals there is a substantial release of PUFA in the seizure events and neuroinflammation occurs secondary to them (Bazan, et al., 1986). An approach to mitigate the seizures and subsequent neuroinflammation by using sEHI, sEH gene ablation and direct administration of EETs to the brain demonstrated delayed onset of GABA mediated seizure (Inceoglu, et al., 2013). Furthermore, sEHI are efficacious in both chemical and electrical models of temporal lobe epilepsy. In pilocarpine induced status epilepticus (SE), sEHI increased EETs and EET/DHET ratios, reduced neuroinflammation, as well as seizure frequency and duration (Hung, et al., 2015). In the same study, sEHI increased seizure-induction thresholds in epileptogenesis induced by electrical basolateral amygdala (BLA) kindling. In addition to controlling neuroinflammation, sEH inhibition has been shown to influence synaptic transmission and neuroplasticity by upregulating NMDA receptors (Wu, et al., 2015). These data suggest that increasing EpFA levels by inhibiting sEH has potential to halt seizures by reducing neuroinflammation and increasing neural plasticity.
c. Alzheimer’s Disease
There is consensus in the current literature that a large component of the pathogenesis of Alzheimer’s disease (AD) is neuroinflammation (Wyss-Coray & Rogers, 2012). The pathogenesis of AD is also related to oxidative stress and mitochondrial dysfunction (Sultana & Butterfield, 2010). These mechanisms are viewed as either contributing to the formation of or subsequently adding to the damage caused by β-amyloid plaques and tau-protein tangle deposition (Bronzuoli, et al., 2016; Minter, et al., 2016). It has also been demonstrated that PUFA concentrations decline with age in vivo and much study has been made of supplementation in humans (Dacks, et al., 2013; Freund-Levi, et al., 2006). It has been hypothesized that the anti-inflammatory benefits of fish oil, which contains high levels of omega-3 fatty acids, are beneficial in limiting the damage of neurological inflammation leading to amyloid plaque formation (Barberger-Gateau, et al., 2002; Fiala, et al., 2015). Unfortunately, the results have been mixed with little improvement seen with omega-3 fatty acids for established dementia but some improvement for mild cognitive impairment (MCI) as a precursor to AD (Boudrault, et al., 2009). These inconsistent results might be due, in part, to poor characterization of dietary components or high levels of lipid peroxide in the omega-3 LC-PUFA used. It has been recently observed that this may relate to the role of APOE4 and delivery of DHA to the brain (Yassine, et al., 2017). Despite this, omega-3 supplementation is still regarded as a meritorious strategy given that it is well tolerated and has minimal side effects.
There are opposing biological effects of omega-6 versus omega-3 LC-PUFAs (Schmitz & Ecker, 2008), but there are opposing effects within only the omega-6 derived metabolites as well. Several of the omega-6 generated prostaglandins have been implicated in contributing to inflammation including in the brain. Lowering amyloid precursor protein levels and limiting production of pro-inflammatory mediators by using NSAIDs to block COX metabolite production has been newly suggested as a strategy to protect against AD (Herbst-Robinson, et al., 2015). However, this type of pharmacological intervention has been limited by the lack of central nervous system (CNS) penetration in addition to the well-known gastrointestinal and hematological side effects of NSAIDs.
The EETs, on the other hand, demonstrated interaction with mitochondria in the presence of β-amyloid. Select regioisomers of EETs were able to reduce mitochondrial membrane depolarization and fragmentation improving cellular respiration (Sarkar, et al., 2014). The 14,15 EET regioisomer and sEHI both increased cell viability in modeled reactive oxygen stress induced by hydrogen peroxide challenge. A CYP450 inhibitor, miconazole, decreased cell viability in co-cultured astrocytes and dopaminergic neurons (Terashvili, et al., 2012). β-amyloid also reduced EET synthesis in hippocampal astrocytes and neurons (Sarkar, et al., 2011), thus, supporting the levels of EETs by inhibiting sEH holds great promise by improving mitochondrial function in this pathological condition. More recently molecular mechanisms of this action have been revealed in human embryonic kidney 293 (HEK293)/Tau cells activated by hydrogen peroxide (Yao, et al., 2016). Cell viability was increased with sEHI corresponding to a lower phosphorylation of tau protein, upregulation of p-AKT and greater GSK-3β phosphorylation. Related to AD the investigation into the relationship of EpFA and sEH, EETs specifically, in vascular cognitive impairment in humans revealed increased DHETs in VCI patients (Nelson, et al., 2014). EETs and sEHI have also increased axonal outgrowth in primary cultures of cortical, sympathetic and sensory neurons from rat (Abdu, et al., 2010). Thus, both EpFA administration and sEH inhibition are viable strategies in blocking deleterious events contributing to AD.
d. Parkinson ’s Disease
Parkinson’s disease (PD) is a neurodegenerative disorder that has both genetic and environmental etiologies. There is substantial evidence for reactive oxygen species and oxidative stress in the damage that occurs to dopaminergic signaling neurons that give rise to the motor dysfunction characteristic of the disease (Dauer & Przedborski, 2003). PD is the second leading age related neurodegenerative disease after AD and the hallmarks of the disease are oxidative stress, mitochondrial dysfunction, and abnormal protein aggregation such as alpha-synuclein in Lewy bodies (Dauer & Przedborski, 2003; Halbach, et al., 2004). More recently the role of secondary neuroinflammation related to oxidative stress has been investigated as the route to neuronal death in PD (Hirsch & Hunot, 2009; Hirsch, et al., 2012). In addition to the anti-inflammatory effect of EpFA, EETs have demonstrated effects in preventing mitochondrial dysfunction (L. Liu, et al., 2011). An early investigation of allylic human sEH polymorphism and risk of developing PD was inconclusive (Farin, et al., 2001), however, preclinical testing has demonstrated efficacy of both sEHI and sEH gene knockout against MPTP-induced Parkinsonism (Qin, et al., 2015). In this model, sEH deficiency and inhibition prevented dopamine (tyrosine hydroxylase-positive) neuronal loss and improved motor performance. Furthermore 14,15 EET administration protected dopaminergic neurons in mice treated with MPTP. Thus, because of the role neuroinflammation and mitochondrial dysfunction play in PD and the efficacy in preclinical models, it is hypothesized that sEH inhibition may have potential benefit in this condition in humans (Lakkappa, et al., 2016).
e. Depression
There is ample evidence that LC-PUFAs have a role in depression (P. Y. Lin, et al., 2012; Martins, et al., 2012). It is also understood that inflammation can be an integral part of depression. The rate limiting enzyme indoleamine 2,3-dioxygenase (IDO) that metabolizes tryptophan and thereby limits serotonin production is under regulation of inflammatory cytokines (O’Connor, et al., 2009; Oxenkrug, 2010). In addition, increased inflammatory cytokines have been observed in human clinical patients as well as in post mortem brains from individuals with major depressive disorder (Dean, et al., 2010; Haapakoski, et al., 2015; Strawbridge, et al., 2015). This is the background rationale for the use of inflammatory agents to induce depressive states in preclinical models (Frenois, et al., 2007; Fu, et al., 2010; A. H. Miller, et al., 2009).
Recently, to exploit the anti-inflammatory properties of EpFA, the effects of sEH inhibition and genetic ablation have been demonstrated in depression models. Use of the sEHI lowered TNFα in LPS treated but not control mice (Ren, et al., 2016). These experiments also investigated the effects of 14,15 EET and sEHI on neuronal outgrowth in PC12 cells. Other antidepressants have an effect on neuronal plasticity and the results of elevating EpFA in either manner showed increased NGF-induced neurite outgrowth in these cells (Ren, et al., 2016). In behavioral tests, sEHI administered both prophylactically and therapeutically resulted in less inactivity in LPS induced depression in mice. sEH inhibition was also effective against social defeat stress in these studies. sEH inhibition did not alter body weight in control mice but increased sucrose preference in social defeat mice. sEH gene deletion conferred resilience to social defeat stress with sEH knockout mice exhibiting similar social interaction time to non-stressed wild type controls. Importantly, when brain-derived neurotrophic factor - tropomyosin receptor kinase B (BDNF-TrkB) signaling was investigated for its role in the modeled depression, it was revealed that sEH knockout mice have higher BDNF levels in several brain regions compared to wild type controls. Additionally, though all groups had similar tissue levels of TrkB, there was a higher p-TrkB to TrkB protein ratio in sEH null mice as well as synaptogenesis markers (Glutamate receptor subunit GluA1 and post synaptic density protein PSD-95) by Western blot. Thus, these mechanisms may be involved in the resilience to social stress in sEH null mice.
This study also quantified sEH protein levels which were higher in in both murine induced stress models compared to controls. This result paralleled with increases in post mortem human brain (parietal cortex) samples from depressed, bi-polar or schizophrenic humans examined for sEH protein levels. An important finding in this work that merits highlighting is the rapid onset of anti-depressive action of sEHI in both models of depression. This is noteworthy because pre-clinically as well as clinically, current anti-depressant therapies take several weeks to be at full effect (Gaynes, et al., 2009; Krishnan & Nestler, 2010). This rapid anti-depressive action is without any observable side effects unlike ketamine which is now known to have rapid effects against depression but has major psychotomimetic side effects and abuse potential (Kirby, 2015). Thus, it is now suggested that sEHI may be a promising new strategy to combat depression (Hashimoto, 2016).
Depression is the mental disorder with the highest numbers of effected individuals world-wide but there are other mental conditions that reveal similar changes in sEH and EpFA biology. Increased sEH expression has been observed with anorexia nervosa in human clinical patients which may be associated with depression and anxiety (Scott-Van Zeeland, et al., 2014; Shih, et al., 2016). Additionally, there may be a role neuroinflammation plays in the pathogenesis of anxiety, post-traumatic stress disorder and obsessive compulsive disorder (Furtado & Katzman, 2015) and thus an opportunity for sEH inhibition to improve mental health as well as physical ailment is very broad.
4. sEH as a target for pain
The sEHI and EpFA have demonstrated greater potency than NSAIDs and synergism with inhibitors of both COX and LOX enzymes in reducing inflammation. It is therefore not surprising that this efficacy extends to pain, one of the hallmarks of inflammation. Pain is a complex signaling network that stems from noxious insult or tissue injury and release of inflammatory mediators such as cytokines, ions, bradykinins, prostaglandins and leukotrienes among others. These act on nociceptors directly and drive action potentials signaling the sensation of pain (Woolf & Ma, 2007). Typically, pain is a signal to avoid further damage until the crisis resolves. However, there are situations where the alteration to nociceptors allows pain to persist beyond the inflammation or insult, and this signaling in the absence of stimulation is a pathological pain termed neuropathy.
The mechanisms driving neuropathic pain are still poorly understood and perhaps therefore also poorly treated. New approaches are urgently needed. While there are more available approaches to treat inflammatory pain, most of these therapies have dose or use limiting side effects and new approaches are desirable. Here we outline targeting the sEH as novel strategy with the unique property of being efficacious against both inflammatory and neuropathic pain.
a. Inflammatory Pain
Targeting sEH for pain relief arose from initial investigations of the anti-inflammatory roles of sEHI. In studies that investigated LPS induced sepsis a sophisticated mass spectrometry analysis revealed that inhibiting sEH altered not only the EETs and diol metabolite levels, but also several other metabolites in the COX and LOX metabolism pathways of the ARA cascade (Schmelzer, et al., 2005). In particular, targeting the sEH with small molecule inhibitors lowered levels of PGE2, a potent inflammation mediator and algogen, in addition to NO and other cytokines. This finding was groundbreaking because it demonstrated that stabilizing endogenous bioactive lipids was a novel strategy to limit inflammation compared to other enzyme inhibitors that blocked production of inflammatory metabolites. The observed shift in other prostaglandin metabolites and decrease in COX-2 protein levels with inhibiting sEH was the basis for hypothesizing that sEH inhibition would limit inflammatory pain which was investigated in several subsequent studies (Inceoglu, et al., 2006; Schmelzer, et al., 2006).
sEHI were first tested in a model of inflammatory pain for their ability to synergize with COX-2 selective NSAIDs due to the previously observed effect of sEHI suppressing the COX-2 enzyme (Schmelzer, et al., 2006). This resulted in synergistic decreases in PGE2; reduced COX-2 expression and increased thermal withdrawal latencies in mice. Importantly, these improvements occurred without an observable change in the prostacyclin to thromboxane ratio. Changes in the homeostatic balance of these COX metabolites is suspected to initiate thrombotic events that are adverse side effects of COX-2 selective NSAIDs (Fitzgerald, 2004).
The sEHI were then tested as single administrations to determine if they were anti-hyperalgesic by themselves. Topical administration of two different sEHI was effective in increasing both thermal withdrawal latencies and mechanical thresholds in an LPS inflammatory pain model in rat (Inceoglu, et al., 2006). This study also investigated the direct topical administration of EETs which revealed the EpFA metabolites were anti-hyperalgesic by increasing thermal withdrawal latencies against LPS pain. Interestingly EETs demonstrated a slight pro-nociceptive effect in naïve rats compared to the sEHI which had no effect in the thermal assay. The anti-hyperalgesic action was demonstrated to have a multifactorial mechanism. Interestingly, in this study COX-2 expression levels in spinal cord did not correlate with decreased pain behavior but sEHI elicited anti-hyperalgesia which distinguishes them from glucocorticoids which repress inducible COX-2 to produce anti-hyperalgesia (Brostjan, et al., 1997; Inceoglu, et al., 2008). To further explore this, EETs were screened against a panel of cell receptors and were found to bind the translocator protein (TSPO) which transports cholesterol through mitochondrial membranes. The TPSO cooperates with steroidogenic acute regulatory protein (StARD1) which is upregulated by EETs (Inceoglu, et al., 2008; W. L. Miller, 2007; Wang, et al., 2006). It was evident from these experiments that the activity of sEH inhibition depends on a cellular factor present in inflammation that has a role in activating the TSPO/StARD1 pathway; this was identified as elevated cyclic adenosine monophosphate (cAMP).
Individual regioisomers of the EpFA have also been examined for anti-hyperalgesic activity (Morisseau, et al., 2010). This study demonstrated that all classes of EpFA were efficacious against inflammatory pain, and that the EpFA had higher activity than the parent LC-PUFA. Importantly, the DHA derived EDPs appear to be more potent anti-hyperalgesics than the other classes. This merits using sEHI as a strategy because it elevates the levels of several EpFA as opposed to single administering of one type of epoxidized metabolite or an epoxide mimic.
Importantly, sEHI can also block the pain produced by PGE2 administration (Inceoglu, et al., 2011). This was observed in examining the pain dependence of sEHI action which revealed several pieces of information. First, sEHI are distinct from NSAIDs and steroids which act upstream of PGE2 and prevent its production, and second, that sEHI activity depends on the amount of pain present and does not alter pain thresholds in the absence of a painful condition. Because the action of sEHI was previously hypothesized to require the presence of cAMP, a phosphodiesterase inhibitor (PDEI) used to block the inactivation of cAMP was co-administered with sEHI in this pain model. This strategy resulted in an enhanced response compared to single administration of the sEHI or PDEI. Oxylipin analysis revealed that PDEI increased EpFA which may contribute to their action in the CNS (Inceoglu, et al., 2011). Thus, the EpFA are analgesic in several models of induced inflammatory pain and the anti-hyperalgesia relates to direct administration of the EpFA but also to sEH inhibition; both producing anti-hyperalgesic activity only in active pain states.
b. Neuropathic Pain
The initial investigation of sEHI efficacy in neuropathy was intended to compare a pain model with relatively low COX-2 induction to inflammatory pain. It was a great surprise when sEH inhibition blocked diabetic neuropathy, a chronic pain model that was proposed as the negative control model (Inceoglu, et al., 2008). This was surprising because most NSAIDs that block inflammation have little efficacy against neuropathic pain (Gore, et al., 2007; Wagner, et al., 2013).
Exploring this efficacy against diabetic neuropathy in a preclinical model revealed that there were dose dependent improvements in mechanical pain thresholds and that the sEHI outperformed the standard of care gabapentin (Inceoglu, et al., 2012). This anti-hyperalgesia was independent of changes in glucose tolerance, insulin tolerance and glucose stimulated insulin secretion. Interestingly, the sEH enzyme activity was increased although the synthetic capacity to form EETs was similar between diabetic and control rats. There is debate as to the relevance of testing evoked responses to stimulus in a chronic neuropathy that has the hallmarks of pain in the absence of stimulation and tonic pain. Therefore, the conditioned place preference paradigm was used to further examine the efficacy of the sEHI in diabetic neuropathic pain. This assay combines compound administration with environmental cues during training to assess an acquired place preference associated with pain relief. Studies employing this technique demonstrated the dose dependent efficacy of the sEHI, and importantly, that as potent analgesics, the sEHI lacked rewarding effects in the absence of pain (Wagner, et al., 2014). Importantly, direct administration of EpFA also induced a conditioned response in diabetic mice (Wagner, et al., 2016). Further investigation in a naturally occurring Type I diabetic mouse model (Akita) demonstrated the efficacy of the sEHI (Wagner, et al., 2017). Use of the Akita mice showed the sEHI are effective against diabetic neuropathy and moreover that sEH activity correlates with disease severity in this strain.
Recently the chronic constriction injury (CCI) model (Bennett & Xie, 1988) was used as another paradigm to test the efficacy of sEHI against chronic pain. Surgical models of neuropathic pain allow the testing of the chronic nature of the developed pain state in contrast to acute nociceptive pain and offer an alternative to chemical agents that may alter metabolism. Both male and female adult Sprague Dawley rats underwent the surgical ligature with 4 loose ligatures of the sciatic nerve. Several weeks after surgery when the incision had healed and the neuropathy due to the ligatures had developed, the rats were dosed by oral gavage with vehicle (PEG300) or 3mg/kg of the sEHI TPPU (Rose, et al., 2010) and tested with the von Frey assay for their sensitivity to mechanical touch. An electronic aesthesiometer attached to a rigid tip probe was used to measure the response of both the ipsilateral (Figure 4) and contralateral (not shown) hind paws. The points depicted in the graph represent the average score and standard error of mean for a group of rats that were assayed each 3–5 times per time point with a 1-minute interval between repeated stimulation. The TPPU treatment was highly significant in males (p<0.001) and females (p<0.010) compared to their respective vehicle controls (Two Way ANOVA, Holm-Sidak method post hoc, n=3/group). The time course demonstrated that there is both long lasting efficacy and rapid action relieving pain by 30 min after oral gavage. The males also had a significantly more robust response (p<0.001) than the females possibly due to the higher dimorphic sEH expression observed in male versus female rodents (Gill & Hammock, 1980; Wagner, et al., 2017). Overall, the sEHI was an effective strategy in both male and female CCI neuropathic rats which supports earlier data in streptozocin induced diabetes in rat and both induced and natural diabetes in mouse (Inceoglu, et al., 2012; Wagner, et al., 2017; Wagner, et al., 2014).
Figure 4. sEH inhibition and EpFA blocks chronic neuropathic pain.
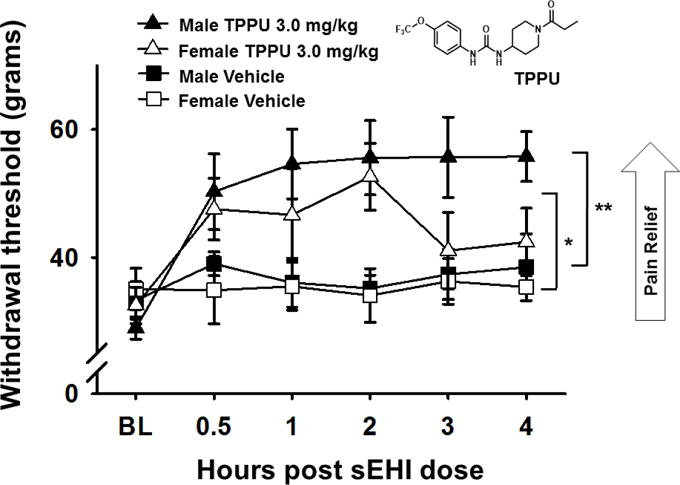
The use of surgical models of neuropathic pain allow the testing of the chronic nature of the developed pain state. In the chronic constriction injury (CCI) model a 3.0 mg/kg oral gavage dose of the sEHI TPPU was effective in male and female rats against neuropathy assessed with a von Frey assay. TPPU treatment was highly significant compared to PEG300 vehicle control in males (**p<0.001) and was also significant in females compared to vehicle control in females (*p<0.010).
Adding to the evidence in preclinical species, sEH inhibition is successful in blocking neuropathic pain in veterinary patients presenting with pathological painful disease. The sEHI t-TUCB was used to treat severe equine laminitis, a condition that often requires humane euthanasia of the horses. The first use of sEHI in this condition was concurrent with failing standard of care therapy and allowed the horse to stand, eat and later recover (A. G. Guedes, et al., 2013). Subsequent studies have demonstrated the continued success of sEHI as a strategy to combat this condition (A. Guedes, et al., 2016). Thus, sEH inhibition is active against severe neuropathic pain under conditions where standard of care therapies have failed and has broken the species barrier.
The efficacy of EpFA mediated analgesia in neuropathic pain revealed another independent mechanism of action. Diabetic rats were assessed for activation of ER Stress markers and the effect of sEH inhibition (Inceoglu, et al., 2015). ER Stress occurs when the homeostatic protein folding and trafficking in the cell is overwhelmed or unbalanced and leads to the unfolded protein response (UPR) and often to apoptosis (Hotamisligil, 2010). Investigation into the beginning pathogenesis of diabetes has long implicated misfolded proteins and ER Stress as precursors to the UPR and pancreatic beta islet apoptosis (Kozutsumi, et al., 1988; Oyadomari, et al., 2002). ER Stress is regulated by three main membrane associated sensors, protein kinase R-like endoplasmic reticulum kinase (PERK), inositol-requiring enzyme 1α (IRE1α) and activating transcription factor 6 (ATF6) which induce the UPR when activated (Figure 5). Administration of sEHI to diabetic rats was able to suppress these markers in skin and spinal cord and elicit anti-hyperalgesic effects in the animals (Inceoglu, et al., 2015).
Figure 5. sEH inhibition and EpFA block Endoplasmic Reticulum Stress (ER Stress).
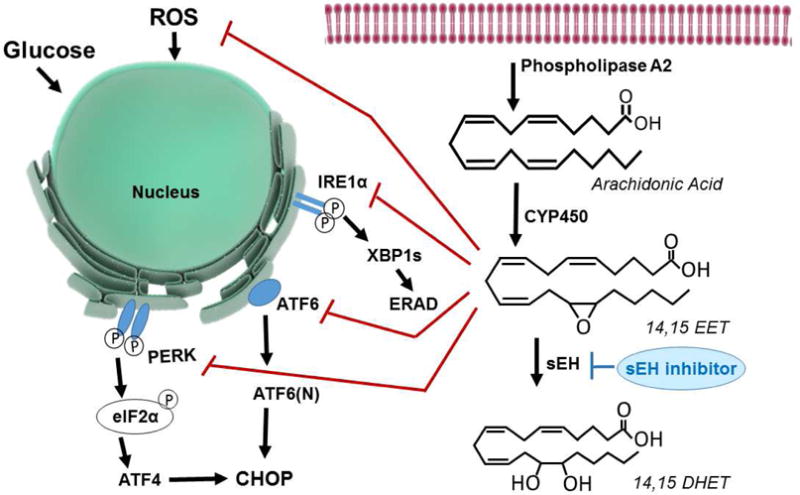
A variety of biological signals can influence the ER Stress pathway such as unfolded and miss folded proteins, high glucose as in diabetes, or reactive oxygen species (ROS) which can be contributed by mitochondrial dysfunction and other sources. EETs reduce the effects of ROS on the ER Stress pathway and stabilize mitochondria (not shown). The three key protein sensors of the ER Stress are inositol-requiring enzyme 1α (IRE1α), activating transcription factor 6 (ATF6) and PKR-like endoplasmic reticulum-resident kinase (PERK). Downstream of IRE1α X-box binding protein-1 when spliced (XBP1s) is activated, similarly ATF6 is cleaved to release the active NH2-terminal domain ATF6(N) and both enter the nucleus as transcription factors. Phosphorylated PERK results in the phosphorylation of eukaryotic initiation factor 2 (eIF2α), ATF4 activation and transcription of C/EBP homologous protein (CHOP) a major participant in genes involved in apoptosis. Apoptotic responses occur when ER Stress is excessive, prolonged, or insufficiently neutralized and is initiated through downstream pathways such as ER-associated protein degradation (ERAD) and CHOP. The enzymatic flow of a single EET regioisomer is depicted on the right. ARA released by phospholipase A2 from the phospholipid bilayer is acted on by CYP450 epoxygenase to form 14,15 EET and would be metabolized by the sEH into a less active 14,15 DHET. Inhibiting sEH maintains the EpFA which block phosphorylation of PERK, elF2α, and IRE1α and significantly decrease XBP1s and ATF6(N) expression. In addition to this action, sEHI also normalize phospho-p38 and phospho-JNK, kinase mediators of neuropathic pain. However, in healthy rats, sEH inhibition does not lead to changes in ER Stress pathways.
The role of ER Stress in the pathogenesis of diabetes is well established but there are also many links between ER Stress and inflammation including the activation of NFκB pathway, ROS production and c-Jun N-terminal kinase (INK) signaling (Hotamisligil, 2010). In addition, there is evidence that the cellular dysfunction of ER Stress has consequences for neurodegeneration and progression of diseases such as AD (J. H. Lin, et al., 2008; Riederer, et al., 2011; Sin & Nollen, 2015). Thus, protecting against the trio of ER Stress, ROS and mitochondrial dysfunction is the unifying action that allows the sEHI to be beneficial in a wider variety of pathological conditions.
c. Potential Side Effects of Targeting sEH
Importantly, the analgesic efficacy of sEH inhibition occurs without the common side effects of NSAIDs or narcotic analgesics. The well described gastrointestinal (GI) ulceration produced by NSAID use is not trivial given the significant rate of hospitalization and death associated with GI bleeding, particularly in elderly patients (Lazzaroni & Bianchi Porro, 2004; Shah & Mehta, 2012). There are also cardiovascular effects of NSAIDs including the selective COX-2 inhibitors that were thought to spare the GI side effects (Lazzaroni & Bianchi Porro, 2004). Interestingly, it has been demonstrated that the anti-inflammatory action of sEH inhibition does not have GI side effects, in fact as mentioned previously, sEH inhibition can mitigate GI ulceration caused by NSAIDs (Goswami, et al., 2016). Inhibiting the sEH does not alter the thromboxane to prostacyclin ratio in vivo (Schmelzer, et al., 2005) or clotting time (J. Y. Liu, Yang, et al., 2010; Schmelzer, et al., 2006) and thus are not anticipated to have the cardiovascular side effects associated with NSAIDs. The other intense side effects of NSAIDs including induced hypersensitivity, NSAID-exacerbated cutaneous disease, respiratory disease and/or induced urticaria/angioedema have not been observed with sEH inhibition in preclinical models, though specific tests for these reactions have not been reported on.
It has also been demonstrated that the analgesia mediated by sEH inhibition does not exhibit the hallmarks of narcotic pain therapies. Typical opioid narcotics cause tolerance, withdrawal and addiction as on-target side effects and also have severe off-target side effects of respiratory depression and constipation. Recent studies of sEH inhibition in chronic pain states have used an operant conditioned preference assay to not only asses pain relief but also the addictive potential of sEHI mediated analgesia (Wagner, et al., 2014). This work demonstrated inhibiting sEH did not produce reward seeking behavior (a conditioned place preference) in naïve and sEH null mice. Additionally, direct administration the EpFA demonstrated both analgesic efficacy against neuropathic pain as well as a lack of rewarding side effects (Wagner, et al., 2016).
5. Conclusion
Inhibiting sEH stabilizes endogenous EpFA that have demonstrated beneficial effects in regulating inflammation including in neurological diseases in addition to combatting chronic and inflammatory pain in preclinical models. sEH inhibitors have been optimized as experimental tools in the past several decades. These molecules designed as transition state mimics of sEH have improved in potency to the single nanomolar range. There are commercially available sEH inhibitors (AUDA, t-AUCB and TPPU) as well as published methods for their synthesis and a large body of literature describing the biological effects of their use in experimental models. In particular, the analgesia mediated by sEHI in preclinical models is novel because it is non-narcotic, out performs NSAIDs without their side effects and has advantages for conditions with inflammatory and hypertensive comorbidities. Additionally, the ability of EpFA to reduce neuroinflammation has great potential to intervene in the progression of neurodegenerative diseases. Overall, the robust analgesia in both inflammatory pain and chronic painful conditions typically refractory to most therapies offers promise for a new approach to alleviate pain. Building on the efficacy of sEHI against pain and inflammation in preclinical models and clinical veterinary patients, sEHI are being developed for use in humans. Previous clinical trials with sEH inhibitors such as GSK2256294 have demonstrated that sEH inhibitors have the potential for broad application and targeting sEH is well tolerated. Optimized inhibitors of sEH are currently being advanced to the clinic for the treatment of diabetic neuropathy.
Acknowledgments
This work was supported by the National Institute of Environmental Health Sciences (NIEHS) Grant R01 ES002710, NIEHS Superfund Research Program P42 ES004699, National Institute of Neurological Disorders and Stroke (NINDS) U54 NS079202-01 and Grants NIEHS T32ES007059, NIH 5T32DC008072-05 and 4T32HL086350-09 (to K.W.). Partial support for clinical developmental of sEH inhibitors for human medicine comes from the NIEHS SBIR Program R44ES025598 and the NIH NINDS Blueprint Neurotherapeutics Network UH2NS094258. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations
- AD
Alzheimer’s Disease
- ARA
arachidonic acid
- COX
cyclooxygenase
- CYP450
cytochrome P450
- DHA
docosahexaenoic acid
- EPA
eicosapentaenoic acid
- EETs
epoxyeicosatrienoic acids
- EpFA
epoxy-fatty acids
- ER Stress
endoplasmic reticulum stress
- IBD
inflammatory bowel disease
- LOX
lipoxygenase
- NSAIDs
non-steroidal anti-Inflammatory drugs
- NFκB
nuclear factor kappa-light-chain-enhancer of activated B cells
- PD
Parkinson’s Disease
- PGE2
prostaglandin E2
- she
soluble epoxide hydrolase
- sEHI
soluble epoxide hydrolase inhibitors
- ROS
reactive oxygen stress
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement: The University of California holds patents on the sEH inhibitors used in this study as well as their use to treat inflammation, inflammatory pain, and neuropathic pain. BD Hammock and CB McReynolds are co-founders and KM Wagner and WK Schmidt are employees of EicOsis L.L.C., a startup company advancing sEH inhibitors into the clinic.
References
- Abdu E, Bruun DA, Yang D, Yang J, Inceoglu B, Hammock BD, Alkayed NJ, Lein PJ. Epoxyeicosatrienoic acids enhance axonal growth in primary sensory and cortical neuronal cell cultures. J Neurochem. 2010;117:632–642. doi: 10.1111/j.1471-4159.2010.07139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkayed NJ, Narayanan J, Gebremedhin D, Medhora M, Roman RJ, Harder DR. Molecular Characterization of an Arachidonic Acid Epoxygenase in Rat Brain Astrocytes. Stroke. 1996;27:971–979. doi: 10.1161/01.str.27.5.971. [DOI] [PubMed] [Google Scholar]
- Amruthesh SC, Falck JR, Ellis EF. Brain synthesis and cerebrovascular action of epoxygenase metabolites of arachidonic acid. J Neurochem. 1992;58:503–510. doi: 10.1111/j.1471-4159.1992.tb09749.x. [DOI] [PubMed] [Google Scholar]
- Barberger-Gateau P, Letenneur L, Deschamps V, Peres K, Dartigues JF, Renaud S. Fish, meat, and risk of dementia: cohort study. BMJ. 2002;325:932–933. doi: 10.1136/bmj.325.7370.932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazan NG, Birkle DL, Tang W, Reddy TS. The accumulation of free arachidonic acid, diacylglycerols, prostaglandins, and lipoxygenase reaction products in the brain during experimental epilepsy. Adv Neurol. 1986;44:879–902. [PubMed] [Google Scholar]
- Beetham JK, Tian TG, Hammock BD. cDNA Cloning and Expression of a Soluble Epoxide Hydrolase from Human Liver. Archives of Biochemistry and Biophysics. 1993;305:197–201. doi: 10.1006/abbi.1993.1411. [DOI] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Berg DJ, Zhang J, Weinstock JV, Ismail HF, Earle KA, Alila H, Pamukcu R, Moore S, Lynch RG. Rapid development of colitis in NSAID-treated IL-10-deficient mice. Gastroenterology. 2002;123:1527–1542. doi: 10.1053/gast.2002.1231527. [DOI] [PubMed] [Google Scholar]
- Birrell MA, Maher SA, Dekkak B, Jones V, Wong S, Brook P, Belvisi MG. Anti-inflammatory effects of PGE2 in the lung: role of the EP4 receptor subtype. Thorax. 2015;70:740–747. doi: 10.1136/thoraxjnl-2014-206592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- Boudrault C, Bazinet RP, Ma DW. Experimental models and mechanisms underlying the protective effects of n-3 polyunsaturated fatty acids in Alzheimer’s disease. J Nutr Biochem. 2009;20:1–10. doi: 10.1016/j.jnutbio.2008.05.016. [DOI] [PubMed] [Google Scholar]
- Brenneis C, Sisignano M, Coste O, Altenrath K, Fischer MJ, Angioni C, Fleming I, Brandes RP, Reeh PW, Woolf CJ, Geisslinger G, Scholich K. Soluble epoxide hydrolase limits mechanical hyperalgesia during inflammation. Mol Pain. 2011;7:78. doi: 10.1186/1744-8069-7-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronzuoli MR, Iacomino A, Steardo L, Scuderi C. Targeting neuroinflammation in Alzheimer’s disease. J Inflamm Res. 2016;9:199–208. doi: 10.2147/JIR.S86958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brostjan C, Anrather J, Csizmadia V, Natarajan G, Winkler H. Glucocorticoids inhibit E-selectin expression by targeting NF-kappaB and not ATF/c-Jun. The Journal of Immunology. 1997;158:3836–3844. [PubMed] [Google Scholar]
- Bystrom J, Wray JA, Sugden MC, Holness MJ, Swales KE, Warner TD, Edin ML, Zeldin DC, Gilroy DW, Bishop-Bailey D. Endogenous Epoxygenases Are Modulators of Monocyte/Macrophage Activity. PLoS ONE. 2011;6:e26591. doi: 10.1371/journal.pone.0026591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Whitcomb R, MacIntyre E, Tran V, Do ZN, Sabry J, Patel DV, Anandan SK, Gless R, Webb HK. Pharmacokinetics and Pharmacodynamics of AR9281, an Inhibitor of Soluble Epoxide Hydrolase, in Single- and Multiple-Dose Studies in Healthy Human Subjects. The Journal of Clinical Pharmacology. 2011 doi: 10.1177/0091270010397049. [DOI] [PubMed] [Google Scholar]
- Chen Y, Falck JR, Manthati VL, Jat JL, Campbell WB. 20-Iodo-14,15-epoxyeicosa-8(Z)-enoyl-3-azidophenylsulfonamide: photoaffinity labeling of a 14,15-epoxyeicosatrienoic acid receptor. Biochemistry. 2011;50:3840–3848. doi: 10.1021/bi102070w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang N, Serhan CN. Structural elucidation and physiologic functions of specialized pro-resolving mediators and their receptors. Molecular Aspects of Medicine. doi: 10.1016/j.mam.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang N, Serhan CN. Structural elucidation and physiologic functions of specialized pro-resolving mediators and their receptors. Mol Aspects Med. 2017 doi: 10.1016/j.mam.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin A, Mowbray S, Durk H, Homburg S, Fleming I, Fisslthaler B, Oesch F, Arand M. The N-terminal domain of mammalian soluble epoxide hydrolase is a phosphatase. Proc Natl Acad Sci U S A. 2003;100:1552–1557. doi: 10.1073/pnas.0437829100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dacks PA, Shineman DW, Fillit HM. Current evidence for the clinical use of long-chain polyunsaturated N-3 fatty acids to prevent age-related cognitive decline and Alzheimer’s disease. Journal of Nutrition, Health and Aging. 2013;17:240–251. doi: 10.1007/s12603-012-0431-3. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson’s Disease: Mechanisms and Models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Dean B, Tawadros N, Scarr E, Gibbons AS. Regionally-specific changes in levels of tumour necrosis factor in the dorsolateral prefrontal cortex obtained postmortem from subjects with major depressive disorder. J Affect Disord. 2010;120:245–248. doi: 10.1016/j.jad.2009.04.027. [DOI] [PubMed] [Google Scholar]
- DeFrancesco L. CAR-T cell therapy seeks strategies to harness cytokine storm. Nat Biotechnol. 2014;32:604. doi: 10.1038/nbt0714-604. [DOI] [PubMed] [Google Scholar]
- Deng Y, Edin ML, Theken KN, Schuck RN, Flake GP, Kannon MA, DeGraff LM, Lih FB, Foley J, Bradbury JA, Graves JP, Tomer KB, Falck JR, Zeldin DC, Lee CR. Endothelial CYP epoxygenase overexpression and soluble epoxide hydrolase disruption attenuate acute vascular inflammatory responses in mice. FASEB J. 2011;25:703–713. doi: 10.1096/fj.10-171488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol. 2015;15:511–523. doi: 10.1038/nri3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley S, Heppner TJ, Nelson MT, Brayden JE. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ Res. 2005;97:1270–1279. doi: 10.1161/01.RES.0000194321.60300.d6. [DOI] [PubMed] [Google Scholar]
- Enayetallah AE, French RA, Thibodeau MS, Grant DF. Distribution of soluble epoxide hydrolase and of cytochrome P450 2C8, 2C9, and 2J2 in human tissues. J Histochem Cytochem. 2004;52:447–454. doi: 10.1177/002215540405200403. [DOI] [PubMed] [Google Scholar]
- Fabene PF, Navarro Mora G, Martinello M, Rossi B, Merigo F, Ottoboni L, Bach S, Angiari S, Benati D, Chakir A, Zanetti L, Schio F, Osculati A, Marzola P, Nicolato E, Homeister JW, Xia L, Lowe JB, McEver RP, Osculati F, Sbarbati A, Butcher EC, Constantin G. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat Med. 2008;14:1377–1383. doi: 10.1038/nm.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X. Soluble epoxide hydrolase: a novel target for the treatment of hypertension. Recent Pat Cardiovasc Drug Discov. 2006;1:67–72. doi: 10.2174/157489006775244227. [DOI] [PubMed] [Google Scholar]
- Farr H, David T. Models of neurovascular coupling via potassium and EET signalling. Journal of Theoretical Biology. 2011;286:13–23. doi: 10.1016/j.jtbi.2011.07.006. [DOI] [PubMed] [Google Scholar]
- Fiala M, Halder RC, Sagong B, Ross O, Sayre J, Porter V, Bredesen DE. ω-3 Supplementation increases amyloid-β phagocytosis and resolvin D1 in patients with minor cognitive impairment. The FASEB Journal. 2015;29:2681–2689. doi: 10.1096/fj.14-264218. [DOI] [PubMed] [Google Scholar]
- Fife KL, Liu Y, Schmelzer KR, Tsai HJ, Kim IH, Morisseau C, Hammock BD, Kroetz DL. Inhibition of soluble epoxide hydrolase does not protect against endotoxin-mediated hepatic inflammation. The Journal of pharmacology and experimental therapeutics. 2008;327:707–715. doi: 10.1124/jpet.108.142398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink MP. Animal models of sepsis. Virulence. 2014;5:143–153. doi: 10.4161/viru.26083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald GA. Coxibs and cardiovascular disease. N Engl J Med. 2004;351:1709–1711. doi: 10.1056/NEJMp048288. [DOI] [PubMed] [Google Scholar]
- Fleming I, Rueben A, Popp R, Fisslthaler B, Schrodt S, Sander A, Haendeler J, Falck JR, Morisseau C, Hammock BD, Busse R. Epoxyeicosatrienoic Acids Regulate Trp Channel-Dependent Ca<sup>2+</sup> Signaling and Hyperpolarization in Endothelial Cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27:2612–2618. doi: 10.1161/ATVBAHA.107.152074. [DOI] [PubMed] [Google Scholar]
- Fornage M, Lee CR, Doris PA, Bray MS, Heiss G, Zeldin DC, Boerwinkle E. The soluble epoxide hydrolase gene harbors sequence variation associated with susceptibility to and protection from incident ischemic stroke. Hum Mol Genet. 2005;14:2829–2837. doi: 10.1093/hmg/ddi315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenois F, Moreau M, O’Connor J, Lawson M, Micon C, Lestage J, Kelley KW, Dantzer R, Castanon N. Lipopolysaccharide induces delayed FosB/DeltaFosB immunostaining within the mouse extended amygdala, hippocampus and hypothalamus, that parallel the expression of depressive-like behavior. Psychoneuroendocrinology. 2007;32:516–531. doi: 10.1016/j.psyneuen.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund-Levi Y, Eriksdotter-Jönhagen M, Cederholm T, Basun H, Faxén-Irving G, Garlind A, Vedin I, Vessby B, Wahlund LO, Palmblad J. ω-3 fatty acid treatment in 174 patients with mild to moderate Alzheimer disease: OmegAD study - A randomized double-blind trial. Archives of Neurology. 2006;63:1402–1408. doi: 10.1001/archneur.63.10.1402. [DOI] [PubMed] [Google Scholar]
- Fu X, Zunich SM, O’Connor JC, Kavelaars A, Dantzer R, Kelley KW. Central administration of lipopolysaccharide induces depressive-like behavior in vivo and activates brain indoleamine 2,3 dioxygenase in murine organotypic hippocampal slice cultures. J Neuroinflammation. 2010;7:43. doi: 10.1186/1742-2094-7-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- Furtado M, Katzman MA. Neuroinflammatory pathways in anxiety, posttraumatic stress, and obsessive compulsive disorders. Psychiatry Research. 2015;229:37–48. doi: 10.1016/j.psychres.2015.05.036. [DOI] [PubMed] [Google Scholar]
- Gauthier KM, Deeter C, Krishna UM, Reddy YK, Bondlela M, Falck JR, Campbell WB. 14,15-Epoxyeicosa-5(Z)-enoic acid: a selective epoxyeicosatrienoic acid antagonist that inhibits endothelium-dependent hyperpolarization and relaxation in coronary arteries. Circ Res. 2002;90:1028–1036. doi: 10.1161/01.res.0000018162.87285.f8. [DOI] [PubMed] [Google Scholar]
- Gaynes BN, Warden D, Trivedi MH, Wisniewski SR, Fava M, Rush AJ. What did STAR*D teach us? Results from a large-scale, practical, clinical trial for patients with depression. Psychiatr Serv. 2009;60:1439–1445. doi: 10.1176/ps.2009.60.11.1439. [DOI] [PubMed] [Google Scholar]
- Gill SS, Hammock BD. Distribution and properties of a mammalian soluble epoxide hydrase. Biochem Pharmacol. 1980;29:389–395. doi: 10.1016/0006-2952(80)90518-3. [DOI] [PubMed] [Google Scholar]
- Gore M, Dukes E, Rowbotham DJ, Tai KS, Leslie D. Clinical characteristics and pain management among patients with painful peripheral neuropathic disorders in general practice settings. Eur J Pain. 2007;11:652–664. doi: 10.1016/j.ejpain.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Goswami SK, Wan D, Yang J, Trindade da Silva CA, Morisseau C, Kodani SD, Yang GY, Inceoglu B, Hammock BD. Anti-Ulcer Efficacy of Soluble Epoxide Hydrolase Inhibitor TPPU on Diclofenac-Induced Intestinal Ulcers. J Pharmacol Exp Ther. 2016;357:529–536. doi: 10.1124/jpet.116.232108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene JF, Newman JW, Williamson KC, Hammock BD. Toxicity of epoxy fatty acids and related compounds to cells expressing human soluble epoxide hydrolase. Chem Res Toxicol. 2000;13:217–226. doi: 10.1021/tx990162c. [DOI] [PubMed] [Google Scholar]
- Gross GJ, Gauthier KM, Moore J, Falck JR, Hammock BD, Campbell WB, Nithipatikom K. Effects of the selective EET antagonist, 14,15-EEZE, on cardioprotection produced by exogenous or endogenous EETs in the canine heart. Am J Physiol Heart Circ Physiol. 2008;294:H2838–2844. doi: 10.1152/ajpheart.00186.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan H, Zhao L, Cao H, Chen A, Xiao J. Epoxyeicosanoids suppress osteoclastogenesis and prevent ovariectomy-induced bone loss. FASEB J. 2015;29:1092–1101. doi: 10.1096/fj.14-262055. [DOI] [PubMed] [Google Scholar]
- Guedes A, Galuppo L, Hood D, Hwang SH, Morisseau C, Hammock BD. Soluble epoxide hydrolase activity and pharmacologic inhibition in horses with chronic severe laminitis. Equine Veterinary Journal. 2016:n/a–n/a. doi: 10.1111/evj.12603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedes AG, Morisseau C, Sole A, Soares JH, Ulu A, Dong H, Hammock BD. Use of a soluble epoxide hydrolase inhibitor as an adjunctive analgesic in a horse with laminitis. Veterinary anaesthesia and analgesia. 2013;40:440–448. doi: 10.1111/vaa.12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta NC, Davis CM, Nelson JW, Young JM, Alkayed NJ. Soluble epoxide hydrolase: sex differences and role in endothelial cell survival. Arterioscler Thromb Vasc Biol. 2012;32:1936–1942. doi: 10.1161/ATVBAHA.112.251520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haapakoski R, Mathieu J, Ebmeier KP, Alenius H, Kivimaki M. Cumulative meta-analysis of interleukins 6 and 1beta, tumour necrosis factor alpha and C-reactive protein in patients with major depressive disorder. Brain Behav Immun. 2015;49:206–215. doi: 10.1016/j.bbi.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbach OvBu, Schober A, Krieglstein K. Genes, proteins, and neurotoxins involved in Parkinson’s disease. Progress in Neurobiology. 2004;73:151–177. doi: 10.1016/j.pneurobio.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Harris TR, Hammock BD. Soluble epoxide hydrolase: gene structure, expression and deletion. Gene. 2013;526:61–74. doi: 10.1016/j.gene.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K. Soluble epoxide hydrolase: a new therapeutic target for depression. Expert Opinion on Therapeutic Targets. 2016:1–3. doi: 10.1080/14728222.2016.1226284. [DOI] [PubMed] [Google Scholar]
- Herbst-Robinson KJ, Liu L, James M, Yao Y, Xie SX, Brunden KR. Inflammatory Eicosanoids Increase Amyloid Precursor Protein Expression via Activation of Multiple Neuronal Receptors. Scientific reports. 2015;5:18286. doi: 10.1038/srep18286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hercule HC, Schunck WH, Gross V, Seringer J, Leung FP, Weldon SM, da Costa Goncalves A, Huang Y, Luft FC, Gollasch M. Interaction between P450 eicosanoids and nitric oxide in the control of arterial tone in mice. Arterioscler Thromb Vasc Biol. 2009;29:54–60. doi: 10.1161/ATVBAHA.108.171298. [DOI] [PubMed] [Google Scholar]
- Higashimori H, Blanco VM, Tuniki VR, Falck JR, Filosa JA. Role of epoxyeicosatrienoic acids as autocrine metabolites in glutamate-mediated K+ signaling in perivascular astrocytes. American journal of physiology. Cell physiology. 2010;299:C1068–1078. doi: 10.1152/ajpcell.00225.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch EC, Hunot S. Neuroinflammation in Parkinson’s disease: a target for neuroprotection? The Lancet Neurology. 2009;8:382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Vyas S, Hunot S. Neuroinflammation in Parkinson’s disease. Parkinsonism & Related Disorders. 2012;18(Supplement 1):S210–S212. doi: 10.1016/S1353-8020(11)70065-7. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Zhu M, Li Z, Sa R, Chu Q, Zhang Q, Zhang H, Tang W, Zhang M, Yin H. Mass spectrometry-based metabolomic profiling identifies alterations in salivary redox status and fatty acid metabolism in response to inflammation and oxidative stress in periodontal disease. Free Radic Biol Med. 2014;70:223–232. doi: 10.1016/j.freeradbiomed.2014.02.024. [DOI] [PubMed] [Google Scholar]
- Hung YW, Hung SW, Wu YC, Wong LK, Lai MT, Shih YH, Lee TS, Lin YY. Soluble epoxide hydrolase activity regulates inflammatory responses and seizure generation in two mouse models of temporal lobe epilepsy. Brain Behav Immun. 2015;43:118–129. doi: 10.1016/j.bbi.2014.07.016. [DOI] [PubMed] [Google Scholar]
- Hutchens MP, Nakano T, Dunlap J, Traystman RJ, Hurn PD, Alkayed NJ. Soluble epoxide hydrolase gene deletion reduces survival after cardiac arrest and cardiopulmonary resuscitation. Resuscitation. 2008;76:89–94. doi: 10.1016/j.resuscitation.2007.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SH, Tsai HJ, Liu JY, Morisseau C, Hammock BD. Orally bioavailable potent soluble epoxide hydrolase inhibitors. J Med Chem. 2007;50:3825–3840. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SH, Wecksler AT, Zhang G, Morisseau C, Nguyen LV, Fu SH, Hammock BD. Synthesis and Biological Evaluation of Sorafenib- and Regorafenib-like sEH Inhibitors. Bioorganic & Medicinal Chemistry Letters. 2013;23:3732–3737. doi: 10.1016/j.bmcl.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hye Khan MA, Neckar J, Manthati V, Errabelli R, Pavlov TS, Staruschenko A, Falck JR, Imig JD. Orally active epoxyeicosatrienoic acid analog attenuates kidney injury in hypertensive Dahl salt-sensitive rat. Hypertension. 2013;62:905–913. doi: 10.1161/HYPERTENSIONAHA.113.01949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Jia J, Nelson J, Goyagi T, Klaus J, Alkayed NJ. Epoxyeicosanoid signaling in CNS function and disease. Prostaglandins Other Lipid Mediat. 2010;91:68–84. doi: 10.1016/j.prostaglandins.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang R, Zeldin DC, Alkayed NJ. Epoxyeicosanoids as mediators of neurogenic vasodilation in cerebral vessels. Am J Physiol Heart Circ Physiol. 2009;296:H1352–1363. doi: 10.1152/ajpheart.00950.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD, Simpkins AN, Renic M, Harder DR. Cytochrome P450 eicosanoids and cerebral vascular function. Expert Rev Mol Med. 2011;13:e7. doi: 10.1017/S1462399411001773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inceoglu B, Bettaieb A, Trindade da Silva CA, Lee KS, Haj FG, Hammock BD. Endoplasmic reticulum stress in the peripheral nervous system is a significant driver of neuropathic pain. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:9082–9087. doi: 10.1073/pnas.1510137112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inceoglu B, Jinks SL, Schmelzer KR, Waite T, Kim IH, Hammock BD. Inhibition of soluble epoxide hydrolase reduces LPS-induced thermal hyperalgesia and mechanical allodynia in a rat model of inflammatory pain. Life Sci. 2006;79:2311–2319. doi: 10.1016/j.lfs.2006.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inceoglu B, Jinks SL, Ulu A, Hegedus CM, Georgi K, Schmelzer KR, Wagner K, Jones PD, Morisseau C, Hammock BD. Soluble epoxide hydrolase and epoxyeicosatrienoic acids modulate two distinct analgesic pathways. Proc Natl Acad Sci U S A. 2008;105:18901–18906. doi: 10.1073/pnas.0809765105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inceoglu B, Wagner K, Schebb NH, Morisseau C, Jinks SL, Ulu A, Hegedus C, Rose T, Brosnan R, Hammock BD. Analgesia mediated by soluble epoxide hydrolase inhibitors is dependent on cAMP. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:5093–5097. doi: 10.1073/pnas.1101073108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inceoglu B, Wagner KM, Yang J, Bettaieb A, Schebb NH, Hwang SH, Morisseau C, Haj FG, Hammock BD. Acute augmentation of epoxygenated fatty acid levels rapidly reduces pain-related behavior in a rat model of type I diabetes. Proc Natl Acad Sci U S A. 2012;109:11390–11395. doi: 10.1073/pnas.1208708109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inceoglu B, Zolkowska D, Yoo HJ, Wagner KM, Yang J, Hackett E, Hwang SH, Lee KS, Rogawski MA, Morisseau C, Hammock BD. Epoxy fatty acids and inhibition of the soluble epoxide hydrolase selectively modulate GABA mediated neurotransmission to delay onset of seizures. PLoS ONE. 2013;8:e80922. doi: 10.1371/journal.pone.0080922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isobe Y, Arita M. Identification of novel omega-3 fatty acid-derived bioactive metabolites based on a targeted lipidomics approach. J Clin Biochem Nutr. 2014;55:79–84. doi: 10.3164/jcbn.14-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann HJ, Taubin HL. Nonsteroidal anti-inflammatory drugs activate quiescent inflammatory bowel disease. Ann Intern Med. 1987;107:513–516. doi: 10.7326/0003-4819-107-4-513. [DOI] [PubMed] [Google Scholar]
- Kirby T. Ketamine for depression: the highs and lows. The Lancet Psychiatry. 2015;2:783–784. doi: 10.1016/S2215-0366(15)00392-2. [DOI] [PubMed] [Google Scholar]
- Koerner IP, Jacks R, DeBarber AE, Koop D, Mao P, Grant DF, Alkayed NJ. Polymorphisms in the human soluble epoxide hydrolase gene EPHX2 linked to neuronal survival after ischemic injury. J Neurosci. 2007;27:4642–4649. doi: 10.1523/JNEUROSCI.0056-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkel A, Schunck WH. Role of cytochrome P450 enzymes in the bioactivation of polyunsaturated fatty acids. Biochim Biophys Acta. 2011;1814:210–222. doi: 10.1016/j.bbapap.2010.09.009. [DOI] [PubMed] [Google Scholar]
- Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332:462–464. doi: 10.1038/332462a0. [DOI] [PubMed] [Google Scholar]
- Krishnan V, Nestler EJ. Linking molecules to mood: new insight into the biology of depression. Am J Psychiatry. 2010;167:1305–1320. doi: 10.1176/appi.ajp.2009.10030434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu S, Roome T, Bhattacharjee A, Carnevale KA, Yakubenko VP, Zhang R, Hwang SH, Hammock BD, Cathcart MK. Metabolic products of soluble epoxide hydrolase are essential for monocyte chemotaxis to MCP-1 in vitro and in vivo. Journal of Lipid Research. 2013;54:436–447. doi: 10.1194/jlr.M031914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakkappa N, Krishnamurthy PT, Hammock BD, Velmurugan D, Bharath MMS. Possible role of Epoxyeicosatrienoic acid in prevention of oxidative stress mediated neuroinflammation in Parkinson disorders. Medical Hypotheses. 2016;93:161–165. doi: 10.1016/j.mehy.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaar AL, Yang L, Boardley RL, Goyal NS, Robertson J, Baldwin SJ, Newby DE, Wilkinson IB, Tal-Singer R, Mayer RJ, Cheriyan J. Pharmacokinetics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br J Clin Pharmacol. 2016;81:971–979. doi: 10.1111/bcp.12855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzaroni M, Bianchi Porro G. Gastrointestinal side-effects of traditional non-steroidal anti-inflammatory drugs and new formulations. Aliment Pharmacol Ther, 20 Suppl. 2004;2:48–58. doi: 10.1111/j.1365-2036.2004.02037.x. [DOI] [PubMed] [Google Scholar]
- Lee KS, Liu JY, Wagner KM, Pakhomova S, Dong H, Morisseau C, Fu SH, Yang J, Wang P, Ulu A, Mate CA, Nguyen LV, Hwang SH, Edin ML, Mara AA, Wulff H, Newcomer ME, Zeldin DC, Hammock BD. Optimized inhibitors of soluble epoxide hydrolase improve in vitro target residence time and in vivo efficacy. Journal of Medicinal Chemistry. 2014;57:7016–7030. doi: 10.1021/jm500694p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li PL, Campbell WB. Epoxyeicosatrienoic acids activate K+ channels in coronary smooth muscle through a guanine nucleotide binding protein. Circ Res. 1997;80:877–884. doi: 10.1161/01.res.80.6.877. [DOI] [PubMed] [Google Scholar]
- Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- Lin JH, Walter P, Yen TS. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. 2008;3:399–425. doi: 10.1146/annurev.pathmechdis.3.121806.151434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PY, Mischoulon D, Freeman MP, Matsuoka Y, Hibbeln J, Belmaker RH, Su KP. Are omega-3 fatty acids antidepressants or just mood-improving agents? The effect depends upon diagnosis, supplement preparation, and severity of depression. Mol Psychiatry. 2012;17:1161–1163. doi: 10.1038/mp.2012.111. author reply 1163–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JY, Park SH, Morisseau C, Hwang SH, Hammock BD, Weiss RH. Sorafenib Has sEH Inhibitory Activity Which Contributes to Its Effect Profile in vivo. Molecular cancer therapeutics. 2009;8:2193–2203. doi: 10.1158/1535-7163.MCT-09-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JY, Tsai HJ, Morisseau C, Lango J, Hwang SH, Watanabe T, Kim IH, Hammock BD. In vitro and in vivo metabolism of N-adamantyl substituted urea-based soluble epoxide hydrolase inhibitors. Biochemical Pharmacology. 2015;98:718–731. doi: 10.1016/j.bcp.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JY, Li N, Yang J, Li N, Qiu H, Ai D, Chiamvimonvat N, Zhu Y, Hammock BD. Metabolic profiling of murine plasma reveals an unexpected biomarker in rofecoxib-mediated cardiovascular events. Proc Natl Acad Sci U S A. 2010;107:17017–17022. doi: 10.1073/pnas.1011278107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JY, Tsai HJ, Hwang SH, Jones PD, Morisseau C, Hammock BD. Pharmacokinetic optimization of four soluble epoxide hydrolase inhibitors for use in a murine model of inflammation. Br J Pharmacol. 2009a;156:284–296. doi: 10.1111/j.1476-5381.2008.00009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JY, Tsai HJ, Hwang SH, Jones PD, Morisseau C, Hammock BD. Pharmacokinetic optimization of four soluble epoxide hydrolase inhibitors for use in a murine model of inflammation. British Journal of Pharmacology. 2009b;156:284–296. doi: 10.1111/j.1476-5381.2008.00009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JY, Yang J, Inceoglu B, Qiu H, Ulu A, Hwang SH, Chiamvimonvat N, Hammock BD. Inhibition of soluble epoxide hydrolase enhances the anti-inflammatory effects of aspirin and 5-lipoxygenase activation protein inhibitor in a murine model. Biochem Pharmacol. 2010;79:880–887. doi: 10.1016/j.bcp.2009.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Chen C, Gong W, Li Y, Edin ML, Zeldin DC, Wang DW. Epoxyeicosatrienoic Acids Attenuate Reactive Oxygen Species Level, Mitochondrial Dysfunction, Caspase Activation, and Apoptosis in Carcinoma Cells Treated with Arsenic Trioxide. Journal of Pharmacology and Experimental Therapeutics. 2011;339:451–463. doi: 10.1124/jpet.111.180505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhang Y, Schmelzer K, Lee TS, Fang X, Zhu Y, Spector AA, Gill S, Morisseau C, Hammock BD, Shyy JY. The antiinflammatory effect of laminar flow: the role of PPARgamma, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc Natl Acad Sci U S A. 2005;102:16747–16752. doi: 10.1073/pnas.0508081102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loot AE, Fleming I. Cytochrome P450-derived epoxyeicosatrienoic acids and pulmonary hypertension: central role of transient receptor potential C6 channels. J Cardiovasc Pharmacol. 2011;57:140–147. doi: 10.1097/FJC.0b013e3181ed088d. [DOI] [PubMed] [Google Scholar]
- Lynes MD, Leiria LO, Lundh M, Bartelt A, Shamsi F, Huang TL, Takahashi H, Hirshman MF, Schlein C, Lee A, Baer LA, May FJ, Gao F, Narain NR, Chen EY, Kiebish MA, Cypess AM, Bluher M, Goodyear LJ, Hotamisligil GS, Stanford KI, Tseng YH. The cold-induced lipokine 12,13-diHOME promotes fatty acid transport into brown adipose tissue. Nat Med. 2017 doi: 10.1038/nm.4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marowsky A, Burgener J, Falck JR, Fritschy JM, Arand M. Distribution of soluble and microsomal epoxide hydrolase in the mouse brain and its contribution to cerebral epoxyeicosatrienoic acid metabolism. Neuroscience. 2009;163:646–661. doi: 10.1016/j.neuroscience.2009.06.033. [DOI] [PubMed] [Google Scholar]
- Martins JG, Bentsen H, Puri BK. Eicosapentaenoic acid appears to be the key omega-3 fatty acid component associated with efficacy in major depressive disorder: a critique of Bloch and Hannestad and updated meta-analysis. Mol Psychiatry. 2012;17:1144–1149. doi: 10.1038/mp.2012.25. discussion 1163–1147. [DOI] [PubMed] [Google Scholar]
- Mathews KA. Nonsteroidal anti-inflammatory analgesics. Indications and contraindications for pain management in dogs and cats. Vet Clin North Am Small Anim Pract. 2000;30:783–804. vi–vii. doi: 10.1016/s0195-5616(08)70007-x. [DOI] [PubMed] [Google Scholar]
- Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65:732–741. doi: 10.1016/j.biopsych.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller WL. Steroidogenic acute regulatory protein (StAR), a novel mitochondrial cholesterol transporter. Biochim Biophys Acta. 2007;1771:663–676. doi: 10.1016/j.bbalip.2007.02.012. [DOI] [PubMed] [Google Scholar]
- Minter MR, Taylor JM, Crack PJ. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J Neurochem. 2016;136:457–474. doi: 10.1111/jnc.13411. [DOI] [PubMed] [Google Scholar]
- Moghaddam MF, Grant DF, Cheek JM, Greene JF, Williamson KC, Hammock BD. Bioactivation of leukotoxins to their toxic diols by epoxide hydrolase. Nat Med. 1997;3:562–566. doi: 10.1038/nm0597-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada S, Vane JR. The role of prostacyclin in vascular tissue. Federation proceedings. 1979;38:66–71. [PubMed] [Google Scholar]
- Morin C, Sirois M, Echave V, Albadine R, Rousseau E. 17,18-epoxyeicosatetraenoic acid targets PPARgamma and p38 mitogen-activated protein kinase to mediate its anti-inflammatory effects in the lung: role of soluble epoxide hydrolase. Am J Respir Cell Mol Biol. 2010;43:564–575. doi: 10.1165/rcmb.2009-0155OC. [DOI] [PubMed] [Google Scholar]
- Morin C, Sirois M, Echave V, Gomes MM, Rousseau E. EET displays anti-inflammatory effects in TNF-alpha stimulated human bronchi: putative role of CPI-17. Am J Respir Cell Mol Biol. 2008;38:192–201. doi: 10.1165/rcmb.2007-0232OC. [DOI] [PubMed] [Google Scholar]
- Morisseau C, Inceoglu B, Schmelzer K, Tsai HJ, Jinks SL, Hegedus CM, Hammock BD. Naturally occurring monoepoxides of eicosapentaenoic acid and docosahexaenoic acid are bioactive antihyperalgesic lipids. J Lipid Res. 2010;51:3481–3490. doi: 10.1194/jlr.M006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullin CA, Hammock BD. Chalcone oxides—potent selective inhibitors of cytosolic epoxide hydrolase. Arch Biochem Biophys. 1982;216:423–439. doi: 10.1016/0003-9861(82)90231-4. [DOI] [PubMed] [Google Scholar]
- Nelson JW, Young JM, Borkar RN, Woltjer RL, Quinn JF, Silbert LC, Grafe MR, Alkayed NJ. Role of soluble epoxide hydrolase in age-related vascular cognitive decline. Prostaglandins Other Lipid Mediat. 2014;113–115:30–37. doi: 10.1016/j.prostaglandins.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng VY, Huang Y, Reddy LM, Falck JR, Lin ET, Kroetz DL. Cytochrome P450 eicosanoids are activators of peroxisome proliferator-activated receptor alpha. Drug Metab Dispos. 2007;35:1126–1134. doi: 10.1124/dmd.106.013839. [DOI] [PubMed] [Google Scholar]
- Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor JC, Lawson MA, Andre C, Moreau M, Lestage J, Castanon N, Kelley KW, Dantzer R. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry. 2009;14:511–522. doi: 10.1038/sj.mp.4002148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguro A, Imaoka S. Lysophosphatidic acids are new substrates for the phosphatase domain of soluble epoxide hydrolase. J Lipid Res. 2012;53:505–512. doi: 10.1194/jlr.M022319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguro A, Sakamoto K, Suzuki S, Imaoka S. Contribution of hydrolase and phosphatase domains in soluble epoxide hydrolase to vascular endothelial growth factor expression and cell growth. Biol Pharm Bull. 2009;32:1962–1967. doi: 10.1248/bpb.32.1962. [DOI] [PubMed] [Google Scholar]
- Oxenkrug GF. Tryptophan kynurenine metabolism as a common mediator of genetic and environmental impacts in major depressive disorder: the serotonin hypothesis revisited 40 years later. Isr J Psychiatry Relat Sci. 2010;47:56–63. [PMC free article] [PubMed] [Google Scholar]
- Oyadomari S, Araki E, Mori M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis. 2002;7:335–345. doi: 10.1023/a:1016175429877. [DOI] [PubMed] [Google Scholar]
- Panigrahy D, Edin ML, Lee CR, Huang S, Bielenberg DR, Butterfield CE, Barnes CM, Mammoto A, Mammoto T, Luria A, Benny O, Chaponis DM, Dudley AC, Greene ER, Vergilio JA, Pietramaggiori G, Scherer-Pietramaggiori SS, Short SM, Seth M, Lih FB, Tomer KB, Yang J, Schwendener RA, Hammock BD, Falck JR, Manthati VL, Ingber DE, Kaipainen A, D’Amore PA, Kieran MW, Zeldin DC. Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J Clin Invest. 2012;122:178–191. doi: 10.1172/JCI58128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavord ID, Wong CS, Williams J, Tattersfield AE. Effect of inhaled prostaglandin E2 on allergen-induced asthma. Am Rev Respir Dis. 1993;148:87–90. doi: 10.1164/ajrccm/148.1.87. [DOI] [PubMed] [Google Scholar]
- Pineda-Pena EA, Jimenez-Andrade JM, Castaneda-Hernandez G, Chavez-Pina AE. Docosahexaenoic acid, an omega-3 polyunsaturated acid protects against indomethacin-induced gastric injury. Eur J Pharmacol. 2012;697:139–143. doi: 10.1016/j.ejphar.2012.09.049. [DOI] [PubMed] [Google Scholar]
- Podolin PL, Bolognese BJ, Foley JF, Long E, Iii, Peck B, Umbrecht S, Zhang X, Zhu P, Schwartz B, Xie W, Quinn C, Qi H, Sweitzer S, Chen S, Galop M, Ding Y, Belyanskaya SL, Israel DI, Morgan BA, Behm DJ, Marino JP, Jr, Kurali E, Barnette MS, Mayer RJ, Booth-Genthe CL, Callahan JF. In vitro and in vivo characterization of a novel soluble epoxide hydrolase inhibitor. Prostaglandins & Other Lipid Mediators. 2013;104–105:25–31. doi: 10.1016/j.prostaglandins.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Qin X, Wu Q, Lin L, Sun A, Liu S, Li X, Cao X, Gao T, Luo P, Zhu X, Wang X. Soluble Epoxide Hydrolase Deficiency or Inhibition Attenuates MPTP-Induced Parkinsonism. Mol Neurobiol. 2015;52:187–195. doi: 10.1007/s12035-014-8833-3. [DOI] [PubMed] [Google Scholar]
- Quilley J, McGiff JC. Is EDHF an epoxyeicosatrienoic acid? Trends Pharmacol Sci. 2000;21:121–124. doi: 10.1016/s0165-6147(00)01445-0. [DOI] [PubMed] [Google Scholar]
- Rachner TD, Khosla S, Hofbauer LC. Osteoporosis: now and the future. The Lancet. 377:1276–1287. doi: 10.1016/S0140-6736(10)62349-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachner TD, Khosla S, Hofbauer LC. Osteoporosis: now and the future. The Lancet. 2011;377:1276–1287. doi: 10.1016/S0140-6736(10)62349-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand AA, Barnych B, Morisseau C, Cajka T, Lee KSS, Panigrahy D, Hammock BD. Cyclooxygenase-derived proangiogenic metabolites of epoxyeicosatrienoic acids. Proc Natl Acad Sci U S A. 2017;114:4370–4375. doi: 10.1073/pnas.1616893114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Q, Ma M, Ishima T, Morisseau C, Yang J, Wagner KM, Zhang JC, Yang C, Yao W, Dong C, Han M, Hammock BD, Hashimoto K. Gene deficiency and pharmacological inhibition of soluble epoxide hydrolase confers resilience to repeated social defeat stress. Proc Natl Acad Sci U S A. 2016;113:E1944–1952. doi: 10.1073/pnas.1601532113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter BK, Asfaha S, Buret A, Sharkey KA, Wallace JL. Exacerbation of inflammation-associated colonic injury in rat through inhibition of cyclooxygenase-2. J Clin Invest. 1996;98:2076–2085. doi: 10.1172/JCI119013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riederer BM, Leuba G, Vernay A, Riederer IM. The role of the ubiquitin proteasome system in Alzheimer’s disease. Exp Biol Med (Maywood) 2011;236:268–276. doi: 10.1258/ebm.2010.010327. [DOI] [PubMed] [Google Scholar]
- Rose TE, Morisseau C, Liu JY, Inceoglu B, Jones PD, Sanborn JR, Hammock BD. 1-Aryl-3-(1-acylpiperidin-4-yl)urea inhibitors of human and murine soluble epoxide hydrolase: structure-activity relationships, pharmacokinetics, and reduction of inflammatory pain. J Med Chem. 2010;53:7067–7075. doi: 10.1021/jm100691c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rot A, von Andrian UH. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol. 2004;22:891–928. doi: 10.1146/annurev.immunol.22.012703.104543. [DOI] [PubMed] [Google Scholar]
- Sarkar P, Narayanan J, Harder DR. Differential effect of amyloid beta on the cytochrome P450 epoxygenase activity in rat brain. Neuroscience. 2011;194:241–249. doi: 10.1016/j.neuroscience.2011.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar P, Zaja I, Bienengraeber M, Rarick KR, Terashvili M, Canfield S, Falck JR, Harder DR. Epoxyeicosatrienoic acids pretreatment improves amyloid beta-induced mitochondrial dysfunction in cultured rat hippocampal astrocytes. Am J Physiol Heart Circ Physiol. 2014;306:H475–484. doi: 10.1152/ajpheart.00001.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schebb NH, Franze B, Maul R, Ranganathan A, Hammock BD. In vitro glucuronidation of the antibacterial triclocarban and its oxidative metabolites. Drug metabolism and disposition: the biological fate of chemicals. 2012;40:25–31. doi: 10.1124/dmd.111.042283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelzer KR, Inceoglu B, Kubala L, Kim IH, Jinks SL, Eiserich JP, Hammock BD. Enhancement of antinociception by coadministration of nonsteroidal anti-inflammatory drugs and soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci U S A. 2006;103:13646–13651. doi: 10.1073/pnas.0605908103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci U S A. 2005;102:9772–9777. doi: 10.1073/pnas.0503279102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz G, Ecker J. The opposing effects of n-3 and n-6 fatty acids. Progress in Lipid Research. 2008;47:147–155. doi: 10.1016/j.plipres.2007.12.004. [DOI] [PubMed] [Google Scholar]
- Scott-Van Zeeland AA, Bloss CS, Tewhey R, Bansal V, Torkamani A, Libiger O, Duvvuri V, Wineinger N, Galvez L, Darst BF, Smith EN, Carson A, Pham P, Phillips T, Villarasa N, Tisch R, Zhang G, Levy S, Murray S, Chen W, Srinivasan S, Berenson G, Brandt H, Crawford S, Crow S, Fichter MM, Halmi KA, Johnson C, Kaplan AS, La Via M, Mitchell JE, Strober M, Rotondo A, Treasure J, Woodside DB, Bulik CM, Keel P, Klump KL, Lilenfeld L, Plotnicov K, Topol EJ, Shih PB, Magistretti P, Bergen AW, Berrettini W, Kaye W, Schork NJ. Evidence for the role of EPHX2 gene variants in anorexia nervosa. Mol Psychiatry. 2014;19:724–732. doi: 10.1038/mp.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Mehta V. Controversies and advances in non-steroidal anti-inflammatory drug (NSAID) analgesia in chronic pain management. Postgrad Med J. 2012;88:73–78. doi: 10.1136/postgradmedj-2011-130291. [DOI] [PubMed] [Google Scholar]
- Shaik JSB, Ahmad M, Li W, Rose ME, Foley LM, Hitchens TK, Graham SH, Hwang SH, Hammock BD, Poloyac SM. Soluble epoxide hydrolase inhibitor <em>trans</em>-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid is neuroprotective in rat model of ischemic stroke. American Journal of Physiology - Heart and Circulatory Physiology. 2013;305:H1605–H1613. doi: 10.1152/ajpheart.00471.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih PB, Yang J, Morisseau C, German JB, Zeeland AA, Armando AM, Quehenberger O, Bergen AW, Magistretti P, Berrettini W, Halmi KA, Schork N, Hammock BD, Kaye W. Dysregulation of soluble epoxide hydrolase and lipidomic profiles in anorexia nervosa. Mol Psychiatry. 2016;21:537–546. doi: 10.1038/mp.2015.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpkins AN, Rudic RD, Schreihofer DA, Roy S, Manhiani M, Tsai HJ, Hammock BD, Imig JD. Soluble Epoxide Inhibition Is Protective Against Cerebral Ischemia via Vascular and Neural Protection. Am J Pathol. 2009;174:2086–2095. doi: 10.2353/ajpath.2009.080544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpkins AN, Rudic RD, Schreihofer DA, Roy S, Manhiani M, Tsai HJ, Hammock BD, Imig JD. Soluble epoxide inhibition is protective against cerebral ischemia via vascular and neural protection. Am J Pathol. 2009;174:2086–2095. doi: 10.2353/ajpath.2009.080544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sin O, Nollen EA. Regulation of protein homeostasis in neurodegenerative diseases: the role of coding and non-coding genes. Cell Mol Life Sci. 2015;72:4027–4047. doi: 10.1007/s00018-015-1985-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer HA, Peach MJ. Endothelium-dependent relaxation of rabbit aorta. II. Inhibition of relaxation stimulated by methacholine and A23187 with antagonists of arachidonic acid metabolism. J Pharmacol Exp Ther. 1983;226:796–801. [PubMed] [Google Scholar]
- Singh RK, Tandon R, Dastidar SG, Ray A. A review on leukotrienes and their receptors with reference to asthma. J Asthma. 2013;50:922–931. doi: 10.3109/02770903.2013.823447. [DOI] [PubMed] [Google Scholar]
- Slim R, Hammock BD, Toborek M, Robertson LW, Newman JW, Morisseau CH, Watkins BA, Saraswathi V, Hennig B. The role of methyl-linoleic acid epoxide and diol metabolites in the amplified toxicity of linoleic acid and polychlorinated biphenyls to vascular endothelial cells. Toxicol Appl Pharmacol. 2001;171:184–193. doi: 10.1006/taap.2001.9131. [DOI] [PubMed] [Google Scholar]
- Smith KR, Pinkerton KE, Watanabe T, Pedersen TL, Ma SJ, Hammock BD. Attenuation of tobacco smoke-induced lung inflammation by treatment with a soluble epoxide hydrolase inhibitor. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:2186–2191. doi: 10.1073/pnas.0409591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, Hill-Eubanks DC, Nelson MT. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science. 2012;336:597–601. doi: 10.1126/science.1216283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector AA. Arachidonic acid cytochrome 450 epoxygenase pathway. J Lipid Res. 2009;50(Suppl):S52–56. doi: 10.1194/jlr.R800038-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res. 2004;43:55–90. doi: 10.1016/s0163-7827(03)00049-3. [DOI] [PubMed] [Google Scholar]
- Spector AA, Kim HY. Discovery of essential fatty acids. J Lipid Res. 2015;56:11–21. doi: 10.1194/jlr.R055095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strawbridge R, Arnone D, Danese A, Papadopoulos A, Herane Vives A, Cleare AJ. Inflammation and clinical response to treatment in depression: A meta-analysis. Eur Neuropsychopharmacol. 2015;25:1532–1543. doi: 10.1016/j.euroneuro.2015.06.007. [DOI] [PubMed] [Google Scholar]
- Sudhahar V, Shaw S, Imig JD. Epoxyeicosatrienoic Acid Analogs and Vascular Function. Current Medicinal Chemistry. 2010;17:1181–1190. doi: 10.2174/092986710790827843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana R, Butterfield DA. Role of oxidative stress in the progression of Alzheimer’s disease. J Alzheimers Dis. 2010;19:341–353. [Google Scholar]
- Sura P, Sura R, Enayetallah AE, Grant DF. Distribution and expression of soluble epoxide hydrolase in human brain. J Histochem Cytochem. 2008;56:551–559. doi: 10.1369/jhc.2008.950659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashvili M, Sarkar P, Van Nostrand M, Falck JR, Harder DR. The protective effect of astrocyte-derived 14,15-EET on H(2)O(2)-induced cell injury in Astrocyte-dopaminergic neuronal cell line co-culture. Neuroscience. 2012;223:68–76. doi: 10.1016/j.neuroscience.2012.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trindade da Silva CA, Bettaieb A, Napimoga MH, Lee KSS, Inceoglu B, Ueira-Vieira C, Bruun D, Goswami SK, Haj FG, Hammock BD. Soluble epoxide hydrolase pharmacological inhibition decreases alveolar bone loss by modulating host inflammatory response, RANK-related signaling, ER stress and apoptosis. Journal of Pharmacology and Experimental Therapeutics. 2017 doi: 10.1124/jpet.116.238113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulu A, Harris TR, Morisseau C, Miyabe C, Inoue H, Schuster G, Dong H, Iosif AM, Liu JY, Weiss RH, Chiamvimonvat N, Imig JD, Hammock BD. Anti-inflammatory effects of omega-3 polyunsaturated fatty acids and soluble epoxide hydrolase inhibitors in angiotensin-II-dependent hypertension. Journal of Cardiovascular Pharmacology. 2013;62:285–297. doi: 10.1097/FJC.0b013e318298e460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, French J, Bartfai T, Baram TZ. The role of inflammation in epilepsy. Nat Rev Neurol. 2011;7:31–40. doi: 10.1038/nrneurol.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriens J, Owsianik G, Fisslthaler B, Suzuki M, Janssens A, Voets T, Morisseau C, Hammock BD, Fleming I, Busse R, Nilius B. Modulation of the Ca2 permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circ Res. 2005;97:908–915. doi: 10.1161/01.RES.0000187474.47805.30. [DOI] [PubMed] [Google Scholar]
- Wagner K, Gilda J, Yang J, Wan D, Morisseau C, Gomes AV, Hammock BD. Soluble epoxide hydrolase inhibition alleviates neuropathy in Akita (Ins2 Akita) mice. Behav Brain Res. 2017;326:69–76. doi: 10.1016/j.bbr.2017.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner K, Inceoglu B, Dong H, Yang J, Hwang SH, Jones P, Morisseau C, Hammock BD. Comparative efficacy of 3 soluble epoxide hydrolase inhibitors in rat neuropathic and inflammatory pain models. Eur J Pharmacol. 2013;700:93–101. doi: 10.1016/j.ejphar.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner K, Lee KS, Yang J, Hammock BD. Epoxy fatty acids mediate analgesia in murine diabetic neuropathy. Eur J Pain. 2016 doi: 10.1002/ejp.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner K, Yang J, Inceoglu B, Hammock BD. Soluble epoxide hydrolase inhibition is antinociceptive in a mouse model of diabetic neuropathy. The journal of pain : official journal of the American Pain Society. 2014;15:907–914. doi: 10.1016/j.jpain.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Shen CL, Dyson MT, Yin X, Schiffer RB, Grammas P, Stocco DM. The involvement of epoxygenase metabolites of arachidonic acid in cAMP-stimulated steroidogenesis and steroidogenic acute regulatory protein gene expression. J Endocrinol. 2006;190:871–878. doi: 10.1677/joe.1.06933. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003;424:434–438. doi: 10.1038/nature01807. [DOI] [PubMed] [Google Scholar]
- Williams L, Bradley L, Smith A, Foxwell B. Signal Transducer and Activator of Transcription 3 Is the Dominant Mediator of the Anti-Inflammatory Effects of IL-10 in Human Macrophages. The Journal of Immunology. 2004;172:567–576. doi: 10.4049/jimmunol.172.1.567. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Ma Q. Nociceptors–Noxious Stimulus Detectors. Neuron. 2007;55:353–364. doi: 10.1016/j.neuron.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Wu HF, Yen HJ, Huang CC, Lee YC, Wu SZ, Lee TS, Lin HC. Soluble epoxide hydrolase inhibitor enhances synaptic neurotransmission and plasticity in mouse prefrontal cortex. Journal of biomedical science. 2015;22:94. doi: 10.1186/s12929-015-0202-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med. 2012;2:a006346. doi: 10.1101/cshperspect.a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Li N, He Y, Timofeyev V, Lu L, Tsai HJ, Kim IH, Tuteja D, Mateo RK, Singapuri A, Davis BB, Low R, Hammock BD, Chiamvimonvat N. Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci U S A. 2006;103:18733–18738. doi: 10.1073/pnas.0609158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Li R, Chen G, Hoopes SL, Zeldin DC, Wang DW. The Role of Cytochrome P450 Epoxygenases, Soluble Epoxide Hydrolase, and Epoxyeicosatrienoic Acids in Metabolic Diseases. Advances in nutrition. 2016;7:1122–1128. doi: 10.3945/an.116.012245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao ES, Tang Y, Liu XH, Wang MH. TPPU protects tau from H2O2-induced hyperphosphorylation in HEK293/tau cells by regulating PI3K/AKT/GSK-3beta pathway. J Huazhong Univ Sci Technolog Med Sci. 2016;36:785–790. doi: 10.1007/s11596-016-1662-z. [DOI] [PubMed] [Google Scholar]
- Yassine HN, Braskie MN, Mack WJ, Castor KJ, Fonteh AN, Schneider LS, Harrington MG, Chui HC. Association of Docosahexaenoic Acid Supplementation With Alzheimer Disease Stage in Apolipoprotein E epsilon4 Carriers: A Review. JAMA Neurol. 2017 doi: 10.1001/jamaneurol.2016.4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeldin DC, Plitman JD, Kobayashi J, Miller RF, Snapper JR, Falck JR, Szarek JL, Philpot RM, Capdevila JH. The rabbit pulmonary cytochrome P450 arachidonic acid metabolic pathway: characterization and significance. J Clin Invest. 1995;95:2150–2160. doi: 10.1172/JCI117904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeldin DC, Wohlford-Lenane C, Chulada P, Bradbury JA, Scarborough PE, Roggli V, Langenbach R, Schwartz DA. Airway inflammation and responsiveness in prostaglandin H synthase-deficient mice exposed to bacterial lipopolysaccharide. Am J Respir Cell Mol Biol. 2001;25:457–465. doi: 10.1165/ajrcmb.25.4.4505. [DOI] [PubMed] [Google Scholar]
- Zhang G, Panigrahy D, Hwang SH, Yang J, Mahakian LM, Wettersten HI, Liu JY, Wang Y, Ingham ES, Tam S, Kieran MW, Weiss RH, Ferrara KW, Hammock BD. Dual inhibition of cyclooxygenase-2 and soluble epoxide hydrolase synergistically suppresses primary tumor growth and metastasis. Proc Natl Acad Sci U S A. 2014;111:11127–11132. doi: 10.1073/pnas.1410432111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Panigrahy D, Mahakian LM, Yang J, Liu JY, Stephen Lee KS, Wettersten HI, Ulu A, Hu X, Tam S, Hwang SH, Ingham ES, Kieran MW, Weiss RH, Ferrara KW, Hammock BD. Epoxy metabolites of docosahexaenoic acid (DHA) inhibit angiogenesis, tumor growth, and metastasis. Proc Natl Acad Sci U S A. 2013;110:6530–6535. doi: 10.1073/pnas.1304321110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Ding H, Yan J, Hui R, Wang W, Kissling GE, Zeldin DC, Wang DW. Genetic variation in cytochrome P450 2J2 and soluble epoxide hydrolase and risk of ischemic stroke in a Chinese population. Pharmacogenet Genomics. 2008;18:45–51. doi: 10.1097/FPC.0b013e3282f313e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Koerner IP, Noppens R, Grafe M, Tsai HJ, Morisseau C, Luria A, Hammock BD, Falck JR, Alkayed NJ. Soluble epoxide hydrolase: a novel therapeutic target in stroke. J Cereb Blood Flow Metab. 2007;27:1931–1940. doi: 10.1038/sj.jcbfm.9600494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Liao J, Li H, Dong H, Bai H, Yang A, Hammock BD, Yang GY. Reduction of inflammatory bowel disease-induced tumor development in IL-10 knockout mice with soluble epoxide hydrolase gene deficiency. Mol Carcinog. 2013;52:726–738. doi: 10.1002/mc.21918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Otsuka T, Sugo N, Ardeshiri A, Alhadid YK, Iliff JJ, DeBarber AE, Koop DR, Alkayed NJ. Soluble epoxide hydrolase gene deletion is protective against experimental cerebral ischemia. Stroke. 2008;39:2073–2078. doi: 10.1161/STROKEAHA.107.508325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Yang AL, Liao J, Li H, Dong H, Chung YT, Bai H, Matkowskyj KA, Hammock BD, Yang GY. Soluble epoxide hydrolase gene deficiency or inhibition attenuates chronic active inflammatory bowel disease in IL-10(−/−) mice. Dig Dis Sci. 2012;57:2580–2591. doi: 10.1007/s10620-012-2217-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Tu L, Li X, Yang S, Chen C, Xu X, Wang P, Wang DW. Delivery of AAV2-CYP2J2 protects remnant kidney in the 5/6-nephrectomized rat via inhibition of apoptosis and fibrosis. Hum Gene Ther. 2012;23:688–699. doi: 10.1089/hum.2011.135. [DOI] [PubMed] [Google Scholar]