

Abstract
An emerging class of long noncoding RNAs (lncRNAs) function as decoy molecules that bind and sequester proteins thereby inhibiting their normal functions. Titration of proteins by lncRNAs has wide-ranging effects affecting nearly all steps in gene expression. While decoy lncRNAs play a role in normal physiology, RNAs expressed from alleles containing nucleotide repeat expansions can be pathogenic due to protein sequestration resulting in disruption of normal functions. This review focuses on commonalities between decoy lncRNAs that regulate gene expression by competitive inhibition of protein function through sequestration and specific examples of nucleotide repeat expansion disorders mediated by toxic RNA that sequesters RNA binding proteins and impedes their normal functions. Understanding how noncoding RNAs compete with various RNA and DNA molecules for binding of regulatory proteins will provide insight into how similar mechanisms contribute to disease pathogenesis.
Keywords: Long noncoding RNA, repeat expansion disorders, decoy lncRNAs, protein sequestration
Introduction
Nucleotide repeat expansion disorders, also known as microsatellite expansion disorders, are caused by a genetic change within a single gene in which a repeated nucleotide sequence, typically 3–10 nucleotides, expands to a number of copies that has detrimental effects. The vast majority of these diseases are dominantly inherited due to a gain of function for the expanded allele. For a subset of these diseases, the pathogenic mechanism includes a toxic gain of function for the RNA transcribed from the expanded allele in which RNA binding proteins are sequestered and the physiological functions of these proteins are disrupted (Schmidt and Pearson 2016, Krzyzosiak et. al. 2012).
Interestingly, one emerging function of long noncoding RNAs (lncRNAs) is to titrate proteins away from their normal biological targets and act as competitive inhibitors of genes or gene products normally targeted by the sequestered proteins. In this review, we focus on parallels between the normal physiological functions of lncRNAs that act as a decoy for proteins to modulate their activity by functional sequestration (Table 1) and the pathological effects of expanded repeat-containing RNAs that bind and disrupt the normal functions of RNA binding proteins (Table 2).
Table 1.
LncRNAs that function by sequestering proteins to regulate target gene activity.
| lncRNA | Function | Protein(s) sequestered | Repeat motif/secondary structure | References |
|---|---|---|---|---|
| Carbon storage regulator B (CsrB)a | Carbon metabolism, motility, biofilm production, epithelial cell invasion, quorum sensing | CsrAa | GGA repeat motif/Forms multiple hairpin structures | Liu et. al. 1997 |
| Repressor of secondary metabolites B (RsmB)a | Plant pathogenesis, exoproducts, motility, quorum sensing | RsmAa | GGA repeat motif/Forms multiple hairpin structures | Liu et. al. 1998 |
| Noncoding repressor of NFAT (NRON) | Regulation of subnuclear localization of NFAT | NFAT, members of the importin family | Willingham et. al. 2005, Sharma et. al. 2011 | |
| Growth-arrest specific 5 (Gas5) | Regulation of steroid hormone activity, regulation of apoptosis and cell cycle | Glucocorticoid receptor, Androgen receptor, Progesterone receptor, Mineralcorticoid receptor | GRE mimic sequences/Forms multiple hairpin structures | Kino et. al. 2010, Pickard and Williams 2016 |
| Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) | Pre-mRNA metabolism, RNA splicing | Serine/Arginine splicing factor 1 (SRSF1) | Tripathi et. al. 2010 | |
| Gomafu | Alters kinetics of splicing reaction for a limited set of target genes | Splicing factor 1 (SF1), Celf3, Quaking protein (QKI) | UACUAAC repeat motif | Tsuijii et. al. 2011, Barry et. al. 2014, Ishizuka et. al. 2014, Ip et. al. 2016 |
| Growth-arrested DNA damage-inducible gene 7 (gadd7) | Regulation of cell cycle genes during DNA damage response | TAR DNA-binding protein (TDP-43) | UG/GU repeat motif | Liu et. al. 2012 |
| p21-associated ncRNA DNA damage activated (PANDA) | Cell cycle regulation during DNA damage response, inhibition of apoptotic gene expression program, establishment and maintenance of senescence phenotype | Nuclear transcription factor Y subunit alpha (NF-YA) | Hung et. al. 2013, Puvvula et. al. 2014 | |
| Lethe | Regulation of NF-κB signaling in inflammatory response | p65/RelA large subunit of NF-κB | Rapicavoli et. al. 2013 | |
| Nuclear paraspeckle assembly transcript 1 (NEAT1) | Formation of paraspeckles, regulation of transcription in stress response | Paraspeckle proteins NONO, SFPQ, and CPSF6 | Hirose et. al. 2014, Imamura et. al. 2014 | |
| lincRNA-p21 | Regulates hypoxia-enhanced glycolysis | von Hippel-Lindau ubiquitin E3 ligase (VHL), hypoxia-inducible factor 1 (HIF-1α) | Yang et. al. 2014 | |
| p50-associated COX-2 extragenic RNA (PACER) | Regulates of COX-2 expression, cell viability | p50 small subunit of NF- κB | Krawczyk and Emerson 2014, Qian et. al. 2016 | |
| NF-κB interacting lncRNA (NKILA)b | Stabilizes inhibitor of NF-κB complex | NF-κB/p65:IκBα complex | Forms multiple hairpin structures | Liu et. al. 2015, Huang et. al. 2016 |
| Noncoding RNA activated by DNA damage (NORAD) | Regulation of genomic stability | Pumilio 2 (PUM2) | UGURUAUA repeats, stem-loop structures | Lee et. al. 2016, Tichon et. al. 2016 |
| 5’ snoRNA capped and 3’ polyadenylated lncRNAs 1 and 2 (SPA1 and SPA2) | Regulation of alternative splicing in Prader-Willi syndrome | TDP-43, RBFOX2, and hnRNP M | Wu et. al. 2016 |
CsrB and RsmB homologs in different bacterial species have been reviewed by Babitzke and Romeo (2007).
A competing hypothesis has been proposed by Dijkstra and Alexander (2015) which brings into question the mechanism proposed by Liu et. al. (2015) by which NKILA regulates NF-κB signaling. In this competing hypothesis, Dijkstra and Alexander suggest a protein coding gene transcribed antisense to NKILA may be responsible for the effects on NF-κB signaling. More studies are necessary to tease apart these potential mechanisms of NF-κB regulation.
Table 2.
Repeat expansion disorders with RNA-mediated toxicity through protein binding and sequestration.
| Repeat expansion disorder | Gene associated with primary mutation | Repeated RNA unit | Length of repeats in affected individuals | Proteins sequestered | References |
|---|---|---|---|---|---|
| Fragile X associated tremor/ataxia syndrome (FXTAS) | Fragile X mental retardation 1 (FMR1) | CGG | 55–200 | hnRNP A2/B1, CELF1, Purα, Sam68, MBNL1, hnRNP G | Verkerk et. al. 1991, Brouwer et. al. 1991, Tassone et. al. 2004, Sofola et. al. 2007, Jin et. al. 2007, Sellier et. al. 2011, Muslimov et. al. 2011 |
| Myotonic dystrophy type 1 (DM1) | Dystrophia myotonica protein kinase (DMPK) | CUG | 50–3,000 | MBNL family, DDX6 | Brook et. al. 1992, Fu et. al. 1992, Mankodi et. al. 2001, Mooers et. al. 2005, Sobczak et. al. 2010, Tian et. al. 2000, Miller et. al. 2000, Pettersson et. al. 2014 |
| Myotonic dystrophy type 2 (DM2) | Cellular nucleic acid binding protein (CNBP) | CCUG | 75–11,000 | MBNL1 | Liquori et. al. 2001, Mankodi et. al. 2001, Jiang et. al. 2004, |
| Huntington disease like- 2 (HDL2) | Junctophilin-3 (JPH) | CUG | 41–58 | MBNL1 | Rudnicki et. al. 2007, Rudnicki et. al. 2008, Holmes et. al. 2001 |
| Huntington’s disease (HD) | Huntingtin (HTT) | CAG | >36 | MBNL1, NCL | Rudnicki et. al. 2008, Ha and Fung 2012, Tsoi et. al. 2013, Banez-Coronel et. al. 2012 |
| Spinocerebellar ataxia 8 (SCA8) | Ataxin-8 opposite strand (ATXN8OS)/Ataxin-8 (ATXN8) | CUG/CAG | 107–1,300 | MBNL1 | Day et. al. 2000, Moseley et. al. 2006, Koob et. al. 1999, Kanadia et. al. 2003, Chen et. al. 2009, Daughters et. al. 2009 |
| Spinocerebellar ataxia 3 (SCA3) | Ataxin-3 (ATXN3) | CAG | 61–84 | MBNL1, Orb2 | Orr and Zoghbi 2007, Orr 2012, Paulson et. al. 1997, Hsu et. al. 2011, Li et. al. 2008, Wang et. al. 2010, Mykowska et. al. 2011, Shieh and Bonini 2011 |
| Spinocerebellar ataxia 10 (SCA10) | Ataxin-10 (ATXN10) | AUUCU | 800–4,500 | hnRNP K | Matsuura et. al. 2000, Tieve et. al. 2011, White et. al. 2010, White et. al. 2012, Bomsztyk et. al. 2004 |
| Spinocerebellar ataxia 31 (SCA31) | Thymidine kinase 2 (TK2), Brain expressed, associated with Nedd4 (BEAN) | UGGAA | >250 | SRSF1, SRSF9 | Sato et. al. 2009, Niimi et. al. 2013, Ring et. al. 1994, Longman et. al. 2000, Xu et. al. 2005 |
| Spinocerebellar ataxia 36 (SCA36) | Nucleolar protein 5a (NOP56) | GGCCTG | 650–2,500 | SRSF2 | Kobayashi et. al. 2011, Ikeda et. al. 2012, Liu et. al. 2014 |
| Amyotrophic lateral sclerosis/Frontotemporal dementia (ALS/FTD) | C9ORF72 | GGGGCC | 700–1,600 | hnRNP A2/B1, RBM45, hnRNP A3, SRSF1, Purα, ADARB2, hnRNP K, PCBP2, SRSF2, hnRNP A1, hnRNP H/F, ALYREF, NCL, hnRNP U, RPL7, RanGAP1 | DeJesus-Hernandez et. al. 2011, Renton et. al. 2011, reviewed in Jazurek et. al. 2016, Zhang et al. 2015 |
LncRNAs
LncRNAs are greater than 200 nucleotides in length and lack protein coding potential (Mercer et. al. 2009). LncRNA expression is typically regulated in a cell-specific manner, for example, examination of human cell lines revealed that only 10% of lncRNAs were expressed in all cell types with 29% expressed in a single cell type (Djebali et. al. 2012). Often lncRNAs are tightly regulated throughout development (reviewed in Mercer et. al. 2009 and Wilusz et. al. 2009). With no evolutionary pressure to conserve open reading frames, lncRNAs may not be subjected to the same constraints as protein coding genes. Instead, lncRNAs may contain short stretches of conserved sequence and functional repeat sequences (Mercer et. al. 2009). While it has been noted that some lncRNA nucleotide sequences are partially conserved, it is more likely that secondary structures are conserved and aid in carrying out specific functions (Yang et. al. 2015).
LncRNAs play roles in multiple biological processes, including neural and muscle development (reviewed in Roberts et. al. 2014 and Nie et. al. 2015) and immunity (reviewed in Yu et. al. 2015, and Sigdel et. al. 2015) and have been implicated in human disease, including cancer and neurodegenerative and muscle diseases (Roberts et. al. 2014, Nie et. al. 2015, Kung et. al. 2013, and Schmitt and Chang 2016). LncRNAs perform roles in all steps of gene expression, from chromatin remodeling and allelic imprinting to post-transcriptional and post-translational processing (reviewed in Mercer et. al. 2009 and Wilusz et. al. 2009). LncRNAs are present in both the nucleus and cytoplasm and individual lncRNAs can by localized to specific subcellular compartments (reviewed in Kung et. al. 2013). Subcategories of lncRNAs are defined based on function. A class of lncRNAs function by recruiting transcription factors to specific sites within the promoter to enhance or silence gene expression. Some lncRNAs function as scaffolds on which protein complexes assemble in nuclear subdomains or affect mRNA stabilization by competing for miRNA binding (reviewed in Nie et. al. 2015, Sigdel et. al. 2015, Yang et. al. 2015, Yu et. al. 2015, and Schmitt and Chang 2016). Our focus in this review is on the lncRNAs that serve as decoy molecules and function to titrate and regulate the proteins that bind to the lncRNA. These lncRNAs are presented in Table 1 with selected examples described below.
LncRNAs that function by sequestering proteins
Carbon storage regulator B (CsrB) and Repressor of secondary metabolites (Rsm)
Utilization of decoy RNAs to sequester proteins as a mode of gene regulation was established early on in prokaryotes. Studies in bacteria demonstrated a class of noncoding RNAs, known as small RNAs (sRNAs), that functions as RNA decoys. Liu et. al. (1997) characterized CsrB, the first example of an sRNA in E. coli that functions by sequestering proteins. The CsrB family and the homologous Rsm family of sRNAs function in multiple bacterial species and control diverse biological processes such as carbon metabolism, cell motility, biofilm formation, quorum sensing, and pathogenesis (Table 1) (Liu et. al. 1997, Liu et. al. 1998). CsrB/Rsm families act as global regulators of these processes by binding and sequestering RNA binding proteins CsrA (or RsmA) that function to post-transcriptionally activate or repress target genes. CsrB contains multiple RUACARGGAUGU repeat sequences, which function as 22 potential binding sites for approximately nine CsrA dimers (Fig. 1A). The repeat binding sites of CsrB sRNA form multiple conserved short RNA hairpins; the RNA primary sequence is critical for binding to CsrA proteins while the conserved secondary structure increases the RNA:protein binding affinity (reviewed in Babitzke and Romeo 2007).
Figure 1.
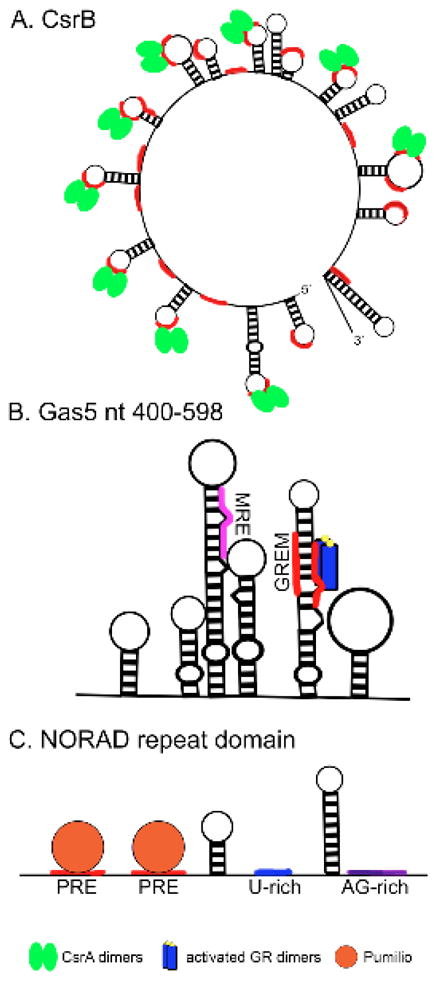
Protein sequestration by lncRNAs. A. E. coli CsrB sRNA contains 22 GGA repeats (highlighted red) within secondary structures that bind and sequester up to nine CsrA dimers, inhibiting activation or repression of CsrA-responsive genes. B. Nucleotides 400–598 of the Gas5 lncRNA forms six hairpin structures, one of which contains two glucocorticoid response elements (GREs) that function as a GRE mimic (GREM, highlighted red) to bind and sequester activated glucocorticoid receptor (blue with yellow ligands), inhibiting transcriptional activity of GR. A mineralcorticoid response element (MRE) that potentially binds mineralcorticoid receptor is present in one of the six hairpin structures. C. Representation of one repeat domain found in the NORAD lncRNA. Each domain contains one or two Pumilio response elements (PRE, highlighted red) that bind and sequester Pumilio proteins to regulate genomic stability.
Noncoding repressor of NFAT (NRON)
An early example of a eukaryotic lncRNA that binds and sequesters proteins is NRON, identified by Schultz and colleagues (2005) as a modulator of nuclear factor of activated T-cells (NFAT). NRON is alternatively spliced with 300–400 base pairs of near perfect conservation between rodents and primates. Specific isoforms of NRON exhibit tissue-specific distribution and are enriched in the placenta, muscle, and lymphoid tissues (Willingham et. al. 2005), consistent with critical NFAT activity in heart, muscle and nervous tissue development and activation of T-cell receptor–mediated immune response (Hogan et al 2003). ShRNA-knockdown of NRON in human embryonic kidney (HEK) 293 cells with a chemically-stimulated increase of intracellular calcium showed dramatic NRON-dependent activation of an NFAT luciferase reporter. This response was specific to NFAT activation and was reproduced in two other mouse cell lines (Willingham et. al. 2005, Imam et. al. 2015). NRON regulates localization of NFAT to the nucleus and increases transcriptional activity of four NFAT isoforms while not affecting other nuclear translocating transcription factors (Willingham et. al. 2005). Four proteins were identified that specifically bind NRON (Table 1) and that significantly activated NFAT upon NRON knockdown and repressed NFAT when NRON was over-expressed. Three of these four proteins; IQGAP1 (a calmodulin-binding protein), KPNB1 (nuclear transport factor importin-β1), and PPP2R1A (a phosphatase structural unit), are members of the importin-β superfamily that directly mediate nuclear-cytoplasmic transport. Direct interaction was demonstrated between NRON and KPNB1 (Willingham et. al. 2005) and a complementary study showed that NRON forms a complex with NFAT and IQGAP1. SiRNA-mediated knockdown of both NRON and IQGAP1 led to more efficient IQGAP1 depletion than did IQGAP1 knockdown alone, suggesting NRON binds and stabilizes IQGAP1 within the cytoplasm (Sharma et. al. 2011). The results strongly suggest that the interactions between the NRON lncRNA and importin family members, as well as NFAT itself, modulate NFAT nuclear translocation and activation of transcription targets.
Growth-arrest specific 5 (Gas5)
LncRNA Gas5 is induced by starvation and growth arrest. In an experiment to identify factors that regulate glucocorticoid activity, Kino et. al. (2010) showed that Gas5 binds directly to the glucocorticoid receptor (GR) DNA binding domain (DBD) (Table 1) and inhibits its ability to regulate target genes. In the presence of a GR agonist and Gas5, association of GR and Gas5 was markedly increased and GR transcriptional activity was repressed. GR activation resulted in translocation of Gas5 from a primarily cytoplasmic distribution to nuclear accumulation, while GR binding-defective Gas5 mutants and a GR mutant with a defective nuclear localization signal did not show increased nuclear accumulation suggesting that Gas5 translocation is dependent on GR translocation. Gas5 regulates GR activity by directly competing binding of the DBD of activated GR to its target genes such as cellular inhibitor of apoptosis 2 (CIAP2) and serum- and glucocorticoid-regulated kinase 1 (SGK1). Gas5 overexpression inhibited GR binding and transcription of target glucocorticoid response elements (GREs), which was restored upon Gas5 knockdown. Nucleotides 400–598 of Gas5 are necessary and sufficient for the inhibitor activity and contain six hairpin structures, one of which (hairpin 5) contains two GRE mimic sequences (Fig. 1B). Mutations disrupting either the helical structure of the hairpin or conserved nucleotides in either GRE without disrupting the double stranded structure of the hairpin resulted in failure of Gas5 to inhibit GR activity (Kino et. al. 2010). Studies by Hudson et. al. (2014) and Pickard and Williams (2016) demonstrated that GRE mimic (GREM) sequence-containing oligonucleotides were sufficient to induce apoptosis in cancer cell lines, consistent with GR loss of function. GR binds directly to the Gas5 GREMs and binding is competed away with double stranded GRE DNA. Gas5 also suppresses transcriptional activity of androgen receptors (AR), progesterone receptors (PR) and mineralocorticoid receptors (MR), which share the response elements utilized by GR (Kino et. al. 2010). Mutation of the GRE mimic was sufficient to reverse inhibition of AR activity (Hudson et. al. 2014). Regulation of multiple steroid hormone receptors by Gas5 suggests the ability of lncRNA secondary structure to mimic transcription factor binding sites and affect target gene expression could be a more general mechanism of gene regulation.
p21 associated ncRNA DNA damage activated (PANDA)
PANDA was identified in an ultrahigh-resolution tiling microarray across 56 cell cycle regulatory genes in human cells under 54 perturbations, such as cell cycle synchronization, DNA damage, differentiation stimuli, oncogenic stimuli, or carcinogenesis. PANDA is an evolutionarily conserved lncRNA located upstream and divergently transcribed from the CDKN1A transcription start site and is specifically induced by DNA damage. CDKN1A knockdown revealed that PANDA expression is not a CDKN1A-linked transcript nor is its expression dependent on p21CDKN1A, however PANDA expression requires p53. PANDA knockdown revealed induction of 224 genes, which were enriched for genes involved in apoptosis. PANDA RNA specifically brought down the nuclear transcription factor Y subunit alpha (NF-YA) and conversely immunoprecipitation of NF-YA specifically retrieved endogenous PANDA (Table 1). Knockdown of PANDA resulted in increased NF-YA occupancy at target genes and simultaneous knockdown of PANDA and NF-YA led to dramatically reduced induction of apoptotic genes and apoptosis (Hung et. al. 2013). PANDA also plays a critical role in establishing and maintaining senescence by sequestration of NF-YA. In proliferating cells, PANDA is specifically associated with the Scaffold-attachment-factor A (SAFA) protein that interacts with polycomb repressor complex 1 (PRC1) to repress senescence. Loss of SAFA protein results in increased PANDA expression, which switches from association with the SAFA complex to specifically interacting with NF-YA, disrupting the ability of NF-YA to bind its pro-proliferative targets (Puvvula et. al. 2014). The data suggest that PANDA lncRNA sequesters NF-YA following DNA damage, impeding its ability to bind chromatin and activate apoptotic target genes.
Nuclear paraspeckle assembly transcript 1 (NEAT1)
LncRNAs are often localized to specific subcellular compartments (reviewed by Kung et. al. 2013). NEAT is an essential structural component of nuclear paraspeckles (Chen and Carmichael 2009). Human NEAT1 is alternatively spliced producing two transcripts that associate with RNA binding paraspeckle proteins to form the paraspeckles. Hirose et. al. (2014) reported transcription factor sequestration in enlarged paraspeckles by NEAT1 resulting from proteasome inhibition-mediated NEAT1 upregulation (Table 1). Proteasome inhibition led to enlargement of paraspeckles and significantly increased transcription of both NEAT1 isoforms. The authors estimated that enlarged paraspeckles contained four to five times more of the paraspeckle proteins NONO, SFPQ, and CPSF6 corresponding with 50% depletion of nucleoplasmic SFPQ and NONO pools. NEAT1 Knockdown followed by microarray analysis revealed transcriptional upregulation of the RNA editing gene ADARB2. Silencing of each of 32 paraspeckle proteins revealed SFPQ and hnRNP H1 were required for ADARB2 transcription. Proteasome inhibition resulted in 10- to 20-fold reduction in ADARB2 RNA levels that was partially rescued by NEAT1 diminution. SFPQ specifically associated with the ADARB2 promoter region under normal conditions but the interaction was reduced upon proteasome inhibition. Three additional genes identified by NEAT1 knockdown were repressed by proteasome inhibition and dependent on SFPQ for expression (Hirose et. al. 2014). Additionally, Imamura et. al. (2014) demonstrated that viral infections, which increase levels of NEAT1 through activation of TLR3/p38 signaling, result in enlargement of paraspeckles that sequester SFPQ, disrupting SFPQ-mediated regulation of IL-8 transcription. These results, taken together, indicate that NEAT1 regulates gene expression by SFPQ sequestration in enlarged paraspeckles (Hirose et. al. 2014, Imamura et. al. 2014).
lincRNA-p21
Yang et. al. (2014) demonstrated that a long intergenic ncRNA, LincRNA-p21 serves as a decoy by sequestering the von Hippel-Lindau (VHL) ubiquitin E3 ligase protein from binding to hypoxia-inducible factor 1 (HIF-1α) under hypoxic conditions (Table 1). LincRNA-p21 was strongly induced by HIF-1α expression during hypoxia and hypoxia-induced increases in glucose uptake and lactate production were dramatically reversed by lincRNA-p21 depletion. These results suggest lincRNA-p21 is important for regulating hypoxia-enhanced glycolysis. LincRNA-p21 limits HIF-1α expression and modulates HIF-1α transcriptional activity under hypoxic conditions. Additionally, introduction of exogenous HIF-1α into lincRNA-p21 knockdown cells reversed the effects of lincRNA-p21 depletion on glucose uptake, lactate production, and HIF-1α-responsive target gene expression. LincRNA-p21 stabilized the HIF-1α protein without affecting HIF-1α mRNA levels under hypoxic conditions. Binding assays showed that lincRNA-p21 inhibits the interactions between HIF-1α and VHL by directly binding the HIF-1α binding site onVHL, causing its dissociation from HIF-1α and thus preventing subsequent degradation. Additionally, lincRNA-p21 depletion increased the HIF-1α/VHL interactions under hypoxia and VHL knockdown rescued HIF-1α reduction caused by lincRNA-p21 depletion. These results suggest HIF-1α and lincRNA-p21 compete for binding of VHL, limiting degradation of HIF-1α during hypoxia. LincRNA-p21 also binds HIF-1α, leaving open the possibility that lincRNA-p21 could interfere with HIF-1α activity via its titration (Yang et. al. 2014).
Noncoding RNA activated by DNA damage (NORAD)
Two independent groups recently reported on a poorly characterized lncRNA, NORAD, involved in regulation of the DNA damage response. NORAD is a highly conserved, 5.3 kb lncRNA that is abundantly and ubiquitously expressed across tissues and cell lines (Lee et. al 2016, Tichon et. al. 2016). NORAD depletion results in a chromosomal instability phenotype, including stable tetraploidization in some NORAD−/− clones, high mitotic error rate, and presence of chromosomal structural rearrangements. NORAD reactivation in diploid NORAD knockout cells rescued chromosomal instability, suggesting NORAD regulates both ploidy and chromosomal stability (Lee et. al. 2016). The NORAD transcript is localized to the cytoplasm and contains twelve repeated NORAD domains (Lee et. al. 2016, Tichon et. al. 2016). Each NORAD domain contains one or two Pumilio response elements that bind strongly and specifically to Pumilio proteins (PUM1 and PUM2), in addition to one short and one long stem-loop structure separated by a short U-rich stretch (Fig. 1C) (Tichon et. al. 2016). Reanalysis of a previously published PAR-CLIP dataset for PUM2 (Hafner et. al. 2010) indicated that NORAD was the most highly represented PUM2 target. Each NORAD transcript binds multiple PUM proteins at 15–17 conserved Pumilio response elements distributed throughout the NORAD domains of the transcript and mutation of PUM binding sites reduces the PUM/NORAD interaction (Lee et. al. 2016, Tichon et. al. 2016). The high abundance of NORAD RNA results in binding of hundreds to thousands of PUM protein molecules per cell, suggesting NORAD may sequester the majority of PUM proteins, inhibiting their ability to bind and repress target mRNAs. PUM2 targets were downregulated in NORAD−/− cells and enriched for genes involved in cell cycle regulation, mitosis, DNA repair, and DNA replication. PUM1 or PUM2 overexpression reversed the effects of NORAD on target gene expression (Lee et. al. 2016, Tichon et. al. 2016). Single or double knockout of PUM1 and PUM2, followed by NORAD inactivation partially suppressed the chromosome instability and mitotic errors observed in NORAD−/− cells, suggesting Pumilio proteins act downstream of NORAD in regulating genomic stability (Lee et. al. 2016). The data from these two studies strongly suggest that functional sequestration of PUM by NORAD prevents overabundance of PUM proteins and repression of PUM target mRNA and promotes genomic stability (Lee et. al. 2016).
5’ snoRNA capped and 3’ polyadenylated lncRNAs 1 and 2 (SPA1 and SPA2)
Wu et. al. (2016) provided evidence for a previously unidentified type of lncRNA that is 5’ snoRNA capped and 3’ polyadenylated (SPA-lncRNAs). RNA immunoprecipitation, using an antibody against fibrillarin, followed by RNA-seq in a human ovarian carcinoma cell line, led to identification of two SPA-lncRNAs, SPA1 and SPA2, expressed from the imprinted Prader-Willi syndrome (PWS) region. SPA1 is 34 kb in length and is capped by the snoRNA SNORD107. SPA2 is located 5.6 kb downstream of SPA1, is 16 kb in length and is capped by the snoRNA SNORD109A. SPA1 and SPA2 are retained in the nucleus and form nuclear accumulations with sno-lncRNAs also expressed from the PWS region. Three RNA binding proteins, TDP-43, RBFOX2, and hnRNP M, interacted with both SPA1 and SPA2 lncRNAs and SPA1/2 sequestered greater than 1% of each RNA binding protein even though the nuclear accumulations of these lncRNAs occupy only 0.02% to 0.1% of the nuclear volume. All three RNA binding proteins directly and strongly interacted with SPA1, SPA2, and other PWS-region sno-lncRNAs. Nuclear accumulations of SPAs were present in induced pluripotent stem cells (iPSCs) of normal individuals while nuclear accumulations of the SPAs were absent in PWS patient iPSCs. Knockout of the entire 141 kb genomic region encoding SPA1 and SPA2 was generated in human cell lines to model the absence of sno-lncRNAs in PWS. RNA-seq in the SPA-lncRNA knockout cells showed 348 splicing events were altered, with 90 showing corresponding change of RNA binding proteins binding to the pre-mRNAs by individual-nucleotide resolution CLIP. These results suggest a link between mislocalization of RNA binding proteins due to sequestration by SPA-lncRNAs, alternative splicing, and PWS pathogenesis (Wu et. al. 2016).
Role of long noncoding RNAs in disease
In addition to the critical roles for lncRNAs in maintaining multiple biological functions, many lncRNAs, including those discussed above, have been implicated in disease pathogenesis. Thorough reviews of lncRNAs implicated in disease have been published by Huarte (2015), Schmitt and Chang (2016), Nie et. al. (2015), Simionescu-Bankston and Kumar (2016), Roberts et. al. (2014), Sigdel et. al. (2015), and Wan et. al. (2016). Here, we focus on diseases in which nucleotide repeat expansions generate toxic RNA that aggregate and function similarly to decoy lncRNAs, sequestering proteins from their normal biological functions.
To date, 43 genetically-inherited nucleotide repeat disorders associated with a single gene have been identified (Schmidt and Pearson 2016, Krzyzosiak et. al. 2012). Nucleotide repeat disorders typically cause disease by one or more of three mechanisms: 1) gain-of-function of toxic proteins translated from nucleotide expansions located within coding regions of affected genes, observed in the polyglutamine (polyQ) diseases (CAG repeats), 2) loss-of-function, of either the protein containing expanded amino acid repeats or by affecting expression when the repeat expansion is located in the intronic or promoter regions, such as CGG or GAA expansions in Fragile X and Friedreich’s ataxia, and 3) toxic gain-of-RNA function from repeat expansions most often when located in noncoding regions of the affected gene, such as CTG, CAG, and CGG repeat expansions (Krzyzosiak et. al. 2012, Sicot and Gomes-Pereira 2013). Complicating these mechanisms is the observation that pathogenic repeat expansions produce antisense as well as sense transcripts raising questions of the pathogenic contributions of the antisense transcript (Batra et. al. 2010). Additionally, mono- and di-peptide repeat proteins, some with demonstrated toxicity, are produced from both the sense and antisense strands of hairpin-forming transcripts by repeat-associated non-ATG (RAN) translation (Zu et. al. 2011, Kearse and Todd 2014).
Fragile X syndrome (FXS) and Fragile X-associated tremor/ataxia syndrome (FXTAS)
The Fragile X syndrome is the most common form of inherited intellectual disability. The disease-causing FXS mutation, a CGG repeat sequence in the fragile X mental retardation 1 (FMR1) gene, was the first identified cause of a nucleotide repeat expansion disorder (Verkerk et. al. 1991). FXS occurs when the CGG repeat in FMR1 is greater than 230 repeats resulting in aberrant epigenetic silencing of FMR1 and loss of protein expression (Coffee et. al. 1999, Sutcliffe et. al. 1992). The premutation containing 55–200 CGG repeats results in Fragile X-associated tremor/ataxia syndrome (FXTAS) (Table 2), characterized by gait ataxia, progressive action tremor, autonomic dysfunction, and neurodegeneration (Brouwer et. al. 1991). In FXTAS patients, FMR1 RNA levels are increased up to eight-fold but protein levels are normal or slightly reduced (Kenneson et. al. 2001). The CGG repeats in FMR1 mRNA expressed in FXTAS form a highly stable hairpin loop (Fig. 2A), accumulate in the nucleus, and colocalize with more than 20 proteins, including the RNA-binding proteins hnRNP A2/B1, Purα, Sam68, MBNL1, and hnRNP G (Sobczak et. al. 2003, Sobczak et. al. 2010, Tassone et. al. 2004, Sofola et. al. 2007, Jin et. al. 2007, Sellier et. al. 2010). Muslimov et. al. (2011) reported that CGG repeat expansions resulted in mislocalization of hnRNP A2/B1 target mRNAs from dendrites to the neuron cell body, which was restored by expression of hnRNP A2/B1 to neurons. hnRNP A2/B1 binds directly to the CGG repeats, tethering CELF1 protein to the repeats (Sofola et. al. 2007). Purα was reported in nuclear inclusions of FXTAS patient brains (Jin et. al. 2007). Furthermore, exogenous expression of hnRNP A2/B1, CELF1 or Purα suppresses neurodegeneration in a Drosophila model of FXTAS (Muslimov et. al. 2011, Sofola et. al. 2007, Jin et. al. 2007). The results are consistent with sequestration and functional loss of hnRNP A2/B1. Similarly, sequestration of the splicing regulator Sam68 by indirect association with CGG expansion RNA foci recruits other proteins such as MBNL1 and hnRNP G, disrupting Sam68-dependent splicing (Sellier et. al. 2010). Early emphasis in FXTAS studies involved the RNA toxicity-mediated pathogenesis; however, a purely RNA-mediated pathology might not fully explain several critical aspects of FXTAS. Recently, multiple studies demonstrated that RAN translation products translated from CGG repeats in at least two open reading frames are detected in Drosophila, mouse, and human cell models of FXTAS (Todd et. al. 2013, Sellier et. al. 2017). Additionally, an FMR1 antisense transcript generated RAN translation products from the CCG repeat in all three reading frames (Krans et. al. 2016). RAN-translated CGG repeats in the FMR1 5’-UTR exhibited increased toxicity and severely impaired locomotor function while isolated CGG repeats forming only expanded RNA were indistinguishable from control animals at three months of age (Sellier et. al. 2017). It remains unclear to what extent RNA-mediated toxicity with protein sequestration and RAN translation play a role in FXTAS pathogenesis.
Figure 2.

Sequestration of proteins by toxic RNA in nucleotide repeat expansion disorders. A. In FXTAS, expansion of 55–200 CGG repeats in the FMR1 5’UTR result in formation of nuclear foci that sequester hnRNP A2/B1, Sam68, CELF1, Purα, MBNL1, and hnRNP G. B. DM1 is caused by expansion of CUG repeats in DMPK mRNAs. Transcripts containing more than 55 repeats results in sequestration of MBNL proteins and DDX6, in addition to stabilization of CELF1 proteins, leading to clinical manifestations of DM1.
Myotonic dystrophy types 1 and 2 (DM1 and DM2)
Myotonic dystrophy type 1 (DM1) is the most common adult onset muscle disease resulting from CTG expansions in the 3’UTR of the dystrophia myotonica protein kinase (DMPK) gene (Table 2) (Brook et. al. 1992, Fu et. al. 1992). DM1 is a multisystemic disease characterized by skeletal muscle weakness, wasting and myotonia, cardiac arrhythmias and conduction defects, cataract formation, and defects in neurological function, such as hypersomnia, executive dysfunction, and cerebral atrophy (Goodwin and Swanson 2013, Thornton 2014). Individuals affected by DM1 have from 50 to 3000 CTG repeats in the DMPK gene (Brook et. al. 1992). The number of repeats is both intergenerationally and somatically unstable with increased instability in non-dividing cells (Yum et. al. 2017). Myotonic dystrophy type 2 (DM2) is caused by a CCTG repeat expansion in the first intron of cellular nucleic acid binding protein (CNBP) (Liquori et. al. 2001). Affected individuals contain between 75 to 11,000 CCTG repeats (Table 2). DM2 shares clinical features with DM1. The CCTG repeats in DM2 are also somatically unstable, but are less likely to have intergenerational instability (Mohan et. al. 2014, Thornton 2014). A key pathogenic feature of both DM1 and DM2 is the presence of nuclear foci containing expanded repeat RNA that form stable hairpins with U-U bulges that allows for interactions with RNA-binding proteins (Fig. 2B) (Liquori et. al. 2001, Mankodi et. al. 2001, Mooers et. al. 2005, Sobczak et. al. 2010, Tian et. al. 2000). At least 20 CUG repeats are necessary for CUG-expanded RNA to form the hairpin structure (Napierala et. al. 1997, Michalowski et. al. 1999, Tian et. al. 2000). Nuclear C(C)UG RNA expansion foci formed in DM1 and DM2 cells sequester RNA-binding proteins to the foci, such as members of the Muscleblind family (Miller et. al. 2000, Jiang et. al. 2004). Another toxic function carried out by CUG expansions, albeit not thought to be through protein sequestration, is hyperphosphorylation of CELF1 proteins (Kuyumcu-Martinez et. al. 2007). Both MBNL and CELF1 proteins regulate alternative splicing during development that is mis-regulated in DM1 and DM2, leading to disease-associated features (Lin et. al. 2006, Kalsotra et. al. 2008). In addition to widespread splicing defects, sequestration also leads to aberrant mRNA localization and transport, mRNA stability, microRNA biogenesis, and polyadenylation (Wang et. al. 2012, Masuda et. al. 2012, Rau et. al. 2011, Kalsotra et. al. 2014, Batra et. al. 2014). In DM1 human primary fibroblasts, the DEAD-box helicase DDX6 interacts with CUG repeat RNA. Over-expression of this protein rescued DM1-associated mis-splicing by partially dispersing sequestered MBNL1 and releasing nuclear foci. DDX6 knockdown in DM1 primary fibroblasts significantly increased the number of nuclear foci and sequestered MBNL1 (Pettersson et. al. 2014). Together these results demonstrate that sequestration of RNA binding factors by expanded CUG RNA produces pathogenic features of DM1.
Antisense transcription of the human DM1 locus was reported by multiple groups (Cho et. al. 2005, Huguet et. al. 2012, Gudde et. al. 2017). Accumulation of CAG-containing RNA foci due to DMPK antisense transcription has been observed in DM1 tissues, revealing increased complexity of RNA toxicity in DM1 (Michel et. al. 2015, Huguet et. al. 2012). It is known that CAG repeats are capable of sequestering MBNL, however, it is unclear if antisense DMPK RNA significantly contributes to disease (Ho et. al. 2005). A recent detailed analysis of antisense transcription in the DM1 locus by Wansink and colleagues (2017) showed that multiple low abundance antisense transcripts are generated from the DMPK locus (termed DM1-AS) from a transcription unit that is much larger than originally defined. Within this population of newly described antisense RNAs, some transcripts contain CAG repeats that can be translated into proteins containing polyserine repeat tracts, while other splice isoforms leave the CAG tract in an intronic region. Despite their low abundance, expression of the antisense transcripts significantly correlated with disease severity but their contribution to DM1 pathogenesis remains undefined (Gudde et. al. 2017).
Huntington disease like-2 (HDL2) and Huntington’s disease (HD)
Huntington disease like-2 (HDL2) is a dominantly inherited disease causing motor coordination defects, dementia, and neurodegeneration. HDL2 is caused by a bidirectionally transcribed expansion of 41–58 CTG repeats in the alternatively spliced exon 2a of the junctophilin-3 (JPH3) gene (Table 2), primarily expressed in the brain (Rudnicki et. al. 2008, Holmes et. al. 2001). The repeat is located in either the coding or 3’-UTR of the JPH3 transcript, depending on whether exon 2a is included or excluded. Expression of the repeat in the coding region produces proteins containing polyleucine or polyalanine tracts while expansion in the 3’-UTR suggests a toxic RNA-mediated pathogenesis (Holmes et. al. 2001, Rudnicki et. al. 2007). CUG repeats of the sense transcript form stable hairpin structures that accumulate into RNA foci and sequester MBNL1 and exhibit mis-splicing in MBNL1-dependent events (Rudnicki et. al. 2007). Additionally, nuclear retention of expanded transcripts may reduce JPH3 proteins levels (Seixas et. al. 2012) contributing to disease pathogenesis.
Expression of the antisense transcript results in nuclear polyQ inclusions, which may account for disease features closely resembling those of patients with Huntington’s disease (HD) (Wilburn et. al. 2011). HD is caused by expansion of CAG repeats in the coding region of the Huntingtin (HTT) gene, resulting in expression of HTT protein containing polyQ expansions (Table 2). Altered protein function is likely to be the primary cause of pathogenesis (Rudnicki et. al. 2008, Ha and Fung, 2012), however, expanded CAG RNA forms RNA foci and partially sequesters MBNL1 and nucleolin (NCL) (Krzyzosiak et. al. 2012, Tsoi et. al. 2013). Sequestration of these proteins leads to dysregulation of alternative splicing by MBNL1 sequestration as well as down-regulation of rRNA transcription and nucleolar stress due to NCL sequestration (Banez-Coronel et. al. 2012). Further exploration into the RNA-mediated toxicity in HD will be important to establish the extent to which toxic RNA affects disease progression.
Spinocerebellar ataxias (SCAs)
Spinocerebellar ataxias (SCAs) are inherited neurological diseases causing motor coordination defects (Hersheson et. al. 2012). Mutations in 37 genes cause the full array of SCAs (SCA1-SCA37), however our focus here will be those SCAs classified as repeat expansion disorders which involve toxic RNA-mediated pathogenesis (Table 2), including SCA8 (CUG repeats), SCA3 (CAG repeats), SCA10 (AUUCU repeats), SCA31 (UGGAA repeats), and SCA36 (GGCCUG repeats) (Matilla-Duenas et. al. 2012).
SCA3
The most common SCA worldwide, SCA3, is caused by a CAG repeat expansion mutation in the coding region (exon 10) of the ataxin-3 (ATXN3) gene (Orr and Zoghbi 2007, Orr 2012). Individuals affected by SCA3 exhibit late onset ataxia and neurodegeneration and carry 61–84 CAG repeats (Paulson et. al. 1997). SCA3 was initially considered a typical polyQ expansion disorder, however, results from SCA3 models in mice, Drosophila, and C. elegans suggest that CAG repeat RNA causes some disease features (Hsu et. al. 2011, Li et. al. 2008, Wang et. al. 2010). Importantly, expressing glutamine repeats using CAA rather than CAG codons mitigated the SCA3 phenotype in Drosophila suggesting a role for the CAG RNA in pathogenesis (Li et. al. 2008). In all three animal models and in human cells, CAG repeat RNA forms MBNL1-containing nuclear foci and in human SCA3 cells, MBNL1-dependent splicing changes were observed (Hsu et. al. 2011, Li et. al. 2008, Wang et. al. 2010, Mykowska et. al. 2011). Over-expression of CeMbnl in C. elegans partially rescued the SCA3 phenotype consistent with MBNL1 sequestration; however, a similar experiment in Drosophila resulted in a more severe neurodegenerative phenotype due to increased polyQ protein levels (Wang et. al. 2010, Li et. al. 2008). The inconsistent results leave open the possible contribution of MBNL1 sequestration by CAG repeats in SCA3 pathogenesis.
SCA8
SCA8 is characterized by motor coordination defects common to the SCAs, and also involves cerebellar atrophy, slurred speech, and abnormal eye movements. SCA8 is caused by expansion of 107–1,300 repeats in the ataxin 8 (ATXN8) gene (Day et. al. 2000). The repeat tract is bidirectionally transcribed leading to generation of an expanded CAG transcript that produces polyglutamine-containing protein and an expanded CUG repeat transcript from the ataxin-8 opposite strand (AXN8OS) gene (Moseley et. al. 2006, Koob et. al. 1999). Toxic CUG RNA folds into a stable hairpin structure forming ribonuclear foci and colocalizes with MBNL1 in neurons. The SCA8 phenotype in transgenic mice was enhanced when combined with a MBNL1 knockout (Kanadia et. al. 2003). Furthermore, splicing defects in SCA8 are rescued by MBNL1 overexpression (Chen et. al. 2009, Daughters et. al. 2009). These results suggest that sequestration of MBNL protein on expanded CUG repeats is at least partially responsible for SCA8 pathology. Adding to the complexity of SCA8 pathogenesis is the discovery that, even in the absence of an ATG codon, RAN translation of the CAG expansion in all three open reading frames results in co-expression of polyglutamine, polyserine, and polyalanine proteins (Cleary and Ranum 2013, and Zu et. al. 2011).
SCA10
SCA10 is caused by an ATTCT repeat in the ninth intron of the ataxin-10 (ATXN10) gene. Affected individuals develop ataxia, seizures, mild peripheral nerve and cognitive impairment (Matsuura et. al. 2000, Teive et. al. 2011). Repeat sizes between 800–4,500 result in the clinical manifestations of SCA10 (Matsuura et. al. 2000). NMR and crystallographic evidence suggest AUUCU repeat RNA forms an unusual hairpin structure composed of a structured A-form helix with A-U and U-U base pairing, UCU•UCU internal loops with two U-U noncanonical pairs and one C-C non-canonical pair (Handa et. al. 2005, Park et. al. 2015). Expanded AUUCU RNA is resistant to degradation and forms both nuclear and cytoplasmic foci that co-localize with hnRNP K in mouse brain and human SCA10 fibroblasts (White et. al. 2010, White et. al. 2012, Walsh et. al. 2015). HnRNP K sequestration leads to abnormal splicing and decreased protein activity of hnRNP K-regulated transcripts. HnRNP K knockout or expression of the AUUCU RNA repeats resulted in translocation of protein kinase Cδ to the mitochondria and activation of caspase-3-mediated apoptosis. The apoptotic phenotype was rescued by over-expression of hnRNP K in repeat-expressing cells (Bomsztyk et. al. 2004, White et. al. 2010). The results support a role for hnRNP K loss of function in SCA10 due to sequestration by AUUCU repeat RNA.
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD)
Amyotrophic lateral sclerosis (ALS) causes selective degeneration of motor neurons, muscle wasting, and paralysis (Van Damme and Robberecht 2013). Some familial forms of ALS also exhibit a clinical presentation of frontotemporal dementia (FTD) (Giordana et. al. 2011, Rademakers et. al. 2012). While the majority of ALS and FTD are sporadic, approximately 10% of ALS patients and 25–50% of FTD patients exhibit familial forms of the diseases (Rademakers et. al. 2012, Robberecht and Philips 2013, Graff-Radford and Woodruff 2007, Gros-Louis et. al. 2006, Rohrer et. al. 2009). The most frequent genetic cause of ALS and FTD is a GGGGCC hexanucleotide repeat expansion in the C9ORF72 first intron (DeJesus-Hernandez et. al. 2011, Renton et. al. 2011). The affected individuals have GGGGCC expansions of 700–1,600 repeats (DeJesus-Hernandez et. al. 2011, Dobson-Stone et. al. 2012, Robberecht and Philips 2013). One proposed pathogenic mechanism is toxicity of the sense and/or antisense repeat-containing RNA. Total levels of C9ORF72 sense and antisense RNA increase 7 to 8-fold in patients (Mori et. al. 2013) and the expanded repeat sense and antisense transcripts accumulate into RNA foci in human brain and spinal cord cells (DeJesus-Hernandez et. al. 2011, Gendron et. al. 2013). Dipeptide repeat RAN translation products are readily detected in C9-ALS tissues and proteotoxicity has been demonstrated in C9-ALS/FTD patients (reviewed in Goodwin and Swanson 2013). The GGGGCC repeats are predicted to form both hairpin and G-quadruplex secondary structures (Ash et. al. 2013, Fratta et. al. 2012, Reddy et. al. 2013) and can bind a large number of RNA binding proteins with proposed roles in dysregulation of the normal biological function of these sequestered proteins (Table 2) (reviewed in Jazurek et. al. 2016). Recent studies have presented strong evidence that expression of dipeptide repeats contribute to C9-ALS by disruption of nucleocytoplasmic transport (reviewed in Taylor 2017), suggesting that the contribution of RNA toxicity due to protein sequestration is not a singular mechanism.
Conclusion and Perspectives
A shared functional mechanism exists between noncoding toxic RNAs in repeat expansion disorders and decoy lncRNAs. Decoy lncRNAs tend to contain repetitive elements, form secondary structures, and facilitate protein binding. This class of long noncoding RNAs regulate gene expression by titrating RNA-binding proteins and transcription factors, sequestering them into either nuclear or cytoplasmic foci, and preventing them from carrying out downstream functions. Protein sequestration is also a pathogenic mechanism of nucleotide repeat disorders, by expression of RNAs containing the expanded repeats commonly resulting in mis-regulation of RNA-binding proteins and RNA metabolism. These disorders predominantly affect the nervous system and musculoskeletal system and these disorders have a more complex mechanism of pathogenicity than previously thought, combining proteotoxicity with toxic RNA-mediated pathology.
This review focused on toxic RNA-repeat expansion disorders involving interference of RNA or DNA binding protein function. Given what is currently known about repeat expansion disorders, several conclusions can be made. First, although repeat elements are common throughout the genome, repeats reaching critical threshold length are capable of becoming unstable and expanding. Second, instability occurs in regions near annotated genes that are transcribed at a level high enough to cause observable pathological phenotypes (Lee and McMurray 2014). Third, pathogenesis resulting from RNA containing repeat expansions require secondary structures formed by the expansion (Budworth and McMurray 2013). Hairpin forming repeats tend to be over-represented in repeat expansion disorders, suggesting the hairpin structure is important for gene expression regulation (Krzyzosiak et. al. 2012). Hairpin structures are dynamic with specific nucleotides determining hairpin stability (Sobczak et. al. 2003, Sobczak et. al. 2010). The hairpin structures become more thermodynamically stable with increasing length, increasing the likelihood that disease severity also increases with increasing repeat length (Lee and McMurray 2014). Hexanucleotide repeats, such as the GGGGCC repeats found in ALS patients, and potentially the GGCCUG repeats found in SCA36, can form more complex G-quadruplex structures in addition to hairpins (Mohan et. al. 2014, Walsh et. al. 2015). Such secondary structures lead to aggregation of expanded repeats in the nucleus, a characteristic shared by nearly all identified repeat expansion disorders mediated by RNA gain-of-function toxicity (Goodwin and Swanson 2013).
The finding that bidirectional transcription occurs across the repeat expansion in many of the repeat expansion disorders (Batra et. al. 2010) increases the complexity of disease mechanism. For repeat expansion disorders identified as proteotoxicity disorders, such as Huntington’s disease, identification of antisense transcripts may increase our understanding of the complexity of disease pathology. It is important to identify antisense transcription in other repeat disorders thought to be mediated by toxic proteins to determine if RNA-mediated toxicity is partially responsible for disease phenotypes.
Understanding the mechanisms by which decoy lncRNAs are regulated and their normal biological functions can facilitate deeper understanding of the mechanisms by which repeat expansions lead to disease. Alternatively, utilizing knowledge gained through extensive studies into repeat expansion disorders may aid in identifying additional functional decoy lncRNA, thereby increasing our understanding of this recently identified class of regulatory RNAs and unlocking another level of complexity encoded by the human genome.
Acknowledgments
The work in our lab is supported by National Institutes of Health grants R01AR045653, R01HL045565, and R01AR060733 (to T.A.C.), the Muscular Dystrophy Association (T.A.C) and the Myotonic Dystrophy Foundation (G.R.M).
Footnotes
Conflict of interest statement On behalf of all authors, the corresponding author states that there is no conflict of interest.
References
- Ash PE, Bierniek KF, Gendron TF, Caulfield T, Lin WL, DeJesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW, Rademakers R, Boylan KB, Dickson DW, Petrucelli L. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babitzke P, Romeo T. CsrB sRNA family: sequestration of RNA-binding regulatory proteins. Current Opinion in Microbiology. 2007;10:156–163. doi: 10.1016/j.mib.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Banez-Coronel M, Porta S, Kagerbauer B, Mateu-Huertas E, Pantano L, Ferrer I, Guzman M, Estivill X, Marti E. A pathogenic mechanism in Huntington’s disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genetics. 2012;8:e1002481. doi: 10.1371/journal.pgen.1002481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry G, Briggs JA, Vanichkina DP, Poth EM, Beveridge NJ, Ratnu VS, Nayler SP, Nones K, Hu J, Bredy TW, Nakagawa S, Rigo F, Taft RJ, Caims MJ, Blackshaw S, Wolvetang EJ, Mattick JS. The long non-coding RNA Gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Molecular Psychiatry. 2014;19:486–494. doi: 10.1038/mp.2013.45. [DOI] [PubMed] [Google Scholar]
- Batra R, Charizanis K, Manchanda M, Mohan A, Li M, Finn DJ, Goodwin M, Zhang C, Sobczak K, Thornton CA, Swanson MS. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Molecular Cell. 2014;56:311–322. doi: 10.1016/j.molcel.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra R, Charizanis K, Swanson MS. Partners in crime: bidirectional transcription in unstable microsatellite disease. Human Molecular Genetics. 2010;19:R77–82. doi: 10.1093/hmg/ddq132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomsztyk K, Denisenko O, Ostrowski J. hnRNP K: one protein multiple processes. Bioessays. 2004;26:629–638. doi: 10.1002/bies.20048. [DOI] [PubMed] [Google Scholar]
- Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- Brouwer JR, Willemsen R, Oostra BA. The FMR1 gene and fragile X-associated tremor/ataxia syndrome. American Journal of Medical Genetics, Part B. Neuropsychiatric Genetics. 1991;150B:782–798. doi: 10.1002/ajmg.b.30910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budworth H, McMurray CT. A brief history of triplet repeat diseases. Methods in Molecular Biology. 2013;1010:3–17. doi: 10.1007/978-1-62703-411-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen IC, Lin HY, Lee GC, Kao SHChen CM, Wu YR, Hsieh-Li HM, Su MT, Lee-Chen GJ. Spinocerebellar ataxia type 8 larger triplet expansion alters histone modification and induces RNA foci. BMC Molecular Biology. 2009;10:9. doi: 10.1186/1471-2199-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Carmichael GG. Altered nuclear retention of mRNAs containing inverted repeats in human embryonic stem cells: functional role of a nuclear noncoding RNA. Molecular Cell. 2009;35:467–478. doi: 10.1016/j.molcel.2009.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho DH, Thienes CP, Mahoney SE, Analau E, Filippova GN, Tapscott SJ. Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Molecular Cell. 2005;20:483–489. doi: 10.1016/j.molcel.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Cleary JD, Ranum LP. Repeat-associated non-ATG (RAN) translation in neurological disease. Human Molecular Genetics. 2013;22:R45–51. doi: 10.1093/hmg/ddt371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffee B, Zhang F, Warren ST, Reines D. Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nature Genetics. 1999;22:98–101. doi: 10.1038/8807. [DOI] [PubMed] [Google Scholar]
- Daughters RS, Tuttle DL, Gao W, Ikeda Y, Moseley ML, Ebner TJ, Swanson MS, Ranum LP. RNA gain-of-function in spinocerebellar ataxia type 8. PLoS Genetics. 2009;5:e1000600. doi: 10.1371/journal.pgen.1000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JW, Schut LJ, Moseley ML, Durand AC, Ranum LP. Spinocerebellar ataxia type 8: clinical features in a large family. Neurology. 2000;55:649–657. doi: 10.1212/wnl.55.5.649. [DOI] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AI, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkstra JM, Alexander DB. The “NF-κB interacting long noncoding RNA” (NKILA) transcript is antisense to cancer-associated gene PMEPA1. F1000Research. 2015;4:96. doi: 10.12688/f1000research.6400.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson-Stone C, Hallupp M, Bartley L, Shepherd CE, Halliday GM, Schofield PR, Hodges JR, Kwok JB. C9ORF72 repeat expansion in clinical and neuropathologic frontotemporal dementia cohorts. Neurology. 2012;79:995–1001. doi: 10.1212/WNL.0b013e3182684634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratta P, Mizielinska S, Nicoll JJ, Zloh M, Fisher EM, Parkinson G, Isaacs AM. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Scientific Reports. 2012;2:1016. doi: 10.1038/srep01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YH, Pizzuti A, Fenwick RG, King J, Rajnarayan S, Dunne PW, Dubel J, Nasser GA, Ashizawa T, de Jong P, et al. An unstable triplet repeat in a gene related to myotonic dystrophy. Science. 1992;255:1256–1258. doi: 10.1126/science.1546326. [DOI] [PubMed] [Google Scholar]
- Gendron TF, Bieniek KF, Zhang YJ, Jansen-West K, Ash PE, Caulfield T, Daughrity L, Dunmore JH, Castanedes-Casey M, Chew J, Cosio DM, van Blitterswijk M, Lee WC, Rademakers R, Boylan KB, Dickson DW, Petrucelli L. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathologica. 2013;126:829–844. doi: 10.1007/s00401-013-1192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordana MT, Ferrero P, Grifoni S, Pellerino A, Naldi A, Montuschi A. Dementia and cognitive impairment in amyotrophic lateral sclerosis: a review. Neurological Science. 2011;32:9–16. doi: 10.1007/s10072-010-0439-6. [DOI] [PubMed] [Google Scholar]
- Goodwin M, Swanson MS. RNA-binding protein mis-regulation in microsatellite expansion disorders. Advances in Experimental Medicine and Biology. 2013;825:353–388. doi: 10.1007/978-1-4939-1221-6_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff-Radford NR, Woodruff BK. Frontotemporal dementia. Seminars in Neurology. 2007;27:48–57. doi: 10.1055/s-2006-956755. [DOI] [PubMed] [Google Scholar]
- Gros-Louis F, Gaspar C, Rouleau GA. Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochemica Biophysica Acta. 2006;1762:956–972. doi: 10.1016/j.bbadis.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Gudde AEEG, van Heeringen SJ, de Oude AI, van Kessel IDG, Estabrook J, Wang ET, Wieringa B, Wansink DG. Antisense transcription of the myotonic dystrophy locus yields low-abundant RNAs with and without (CAG)n repeat. RNA Biology. 2017 doi: 10.1080/15476286.2017.1279787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha AD, Fung VS. Huntington’s disease. Current Opinion in Neurology. 2012;25:491–498. doi: 10.1097/WCO.0b013e3283550c97. [DOI] [PubMed] [Google Scholar]
- Hafner M, Lanthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jungkamp AC, Munschauer M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa VYeh HJ, McPhie P, Usdin K. The AUUCU repeats responsible for spinocerebellar ataxia type 10 form unusual RNA hairpins. Journal of Biological Chemistry. 2005;280:29340–29345. doi: 10.1074/jbc.M503495200. [DOI] [PubMed] [Google Scholar]
- Hersheson J, Haworth A, Houlden H. The inherited ataxias: genetic heterogeneity, mutation databases, and future directions in research and clinical diagnostics. Human Mutation. 2012;33:1324–1332. doi: 10.1002/humu.22132. [DOI] [PubMed] [Google Scholar]
- Hirose T, Virnicchi G, Tanigawa A, Naganuma T, Li R, Kimura H, Yokoi T, Nakagawa S, Benard M, Fox AH, Pierron G. NEAT1 long noncoding RNA regulates transcription via protein sequestration within subnuclear bodies. Molecular Biology of the Cell. 2014;25:169–183. doi: 10.1091/mbc.E13-09-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TH, Savkur RS, Poulos MG, Mancini MA, Swanson MS, Cooper TA. Colocalization of Muscleblind with RNA foci is separable from mis-regulation of alternative splicing in myotonic dystrophy. Journal of Cell Science. 2005;118:2923–2933. doi: 10.1242/jcs.02404. [DOI] [PubMed] [Google Scholar]
- Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes and Development. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- Holmes SE, O’Hearn E, Rosenblatt A, Callahan C, Hwang HS, Ingersoll-Ashworth RG, Fleisher A, Stevanin G, Brice A, Potter NT, Ross CA, Margolis RL. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nature Genetics. 2001;29:377–378. doi: 10.1038/ng760. [DOI] [PubMed] [Google Scholar]
- Hsu RJ, Hsiao KM, Lin MJ, Li CY, Wang LC, Chen LK, Pan H. Long tract of untranslated CAG repeats is deleterious in transgenic mice. PLoS One. 2011;6:e16417. doi: 10.1371/journal.pone.0016417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Cui X, Chen J, Feng Y, Song E, Li J, Liu Y. Long non-coding RNA NKILA inhibits migration and invasion of tongue squamous cell carcinoma cells via suppressing epithelial mesenchymal transition. Oncotarget. 2016;7:62520–62532. doi: 10.18632/oncotarget.11528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huarte M. The emerging role of lncRNAs in cancer. Nature Medicine. 2015;21:1253–1261. doi: 10.1038/nm.3981. [DOI] [PubMed] [Google Scholar]
- Hudson WH, Pickard MR, de Vera IMS, Kuiper EG, Mourtada-Maarabouni M, Conn GL, Kojetin DJ, Williams GT, Ortlund EA. Conserved sequence-specific lincRNA-steroid receptor interactions drive transcriptional repression and direct cell fate. Nature Communications. 2014;5:5395. doi: 10.1038/ncomms6395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguet A, Medja F, Nicole A, Vignaud A, Ferry A, Guiraud-Dogan C, Decostre V, Hogrel J-Y, Metzger F, Hoeflich A, Mouisel E, Gomes-Pereira M, Bassez G, Puymirat J, Furling D, Munnich A, Gourdon G. Molecular, physiological, and motor performance defects in DMSXL mice carrying >1000 CTG repeat form the human DM1 locus. PLoS Genetics. 8:e1003043. doi: 10.1371/journal.pgen.1003043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung T, Wang Y, Lin MF, Koegel AK, Kotake Y, Grant GD, Horlings HM, Shah N, Umbricht C, Wang P, Wang Y, Kong BLangerod A, Borresen-Dale A, Kim SK, van de Vijver M, Sukumar S, Whitfield ML, Kellis M, Xiong Y, Wong DJ, Chang HY. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nature Genetics. 2013;43:621–629. doi: 10.1038/ng.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y, Ohta Y, Kobayashi H, Okamoto M, Takamatsu K, Ota T, Manabe Y, Okamoto K, Koizumi A, Abe K. Clinical features of SCA36: a novel spinocerebellar ataxia with motor neuron involvement (Asidan) Neurology. 2012;79:333–341. doi: 10.1212/WNL.0b013e318260436f. [DOI] [PubMed] [Google Scholar]
- Imam H, Bano AS, Patel P, Holla P, Jameel S. The lncRNA NRON modulates HIV-1 replication in an NFAT-dependent manner and is differentially regulated by early and late viral proteins. Scientific Reports. 2015;5:8639. doi: 10.1038/srep08639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura K, Imamachi N, Akizuki G, Kumakura M, Kawaguchi A, Nagata K, Kato A, Kawaguchi Y, Sato H, Yoneda M, Kai C, Yada T, Suzuki Y, Yamada T, Ozawa T, Kaneki K, Inoue T, Kobayashi M, Kodama T, Wada Y, Sekimizu K, Akimitsu N. Long noncoding RNA NEAT1-dependent SFPQ relocation from promoter region to paraspeckle mediates IL8 expression upon immune stimuli. Molecular Cell. 2014;53:393–406. doi: 10.1016/j.molcel.2014.01.009. [DOI] [PubMed] [Google Scholar]
- Ip JY, Sone M, Nashiki C, Pan Q, Kitaichi K, Yanaka K, Abe T, Takao K, Miyakawa T, Blencowe BJ, Nakagawa S. Gomafu lncRNA knockout mice exhibit mild hyperactivity with enhanced responsiveness to the psychostimulant methamphetamine. Scientific Reports. 2016;6:27204. doi: 10.1038/srep27204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka A, Hasegawa Y, Ishida K, Yanaka K, Nakagawa S. Formation of nuclear bodies by the lncRNA Gomafu-associating proteins Celf3 and SF1. Genes to Cells. 2014;19:704–721. doi: 10.1111/gtc.12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazurek M, Ciesiolka A, Starega-Roslan J, Bilinska K, Krzyzosiak WJ. Identifying proteins that bind to specific–RNAs focus on simple repeat expansion diseases. Nucleic Acids Research. 2016;44:9050–9070. doi: 10.1093/nar/gkw803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Human Molecular Genetics. 2004;13:3079–3088. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- Jin P, Duan R, Qurashi A, Qin Y, Tian D, Rosser TC, Liu H, Feng Y, Warren ST. Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron. 2007;55:556–564. doi: 10.1016/j.neuron.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A, Singh RK, Gurha P, Ward AJ, Creighton CJ, Cooper TA. The Mef2 transcription network is disrupted in myotonic dystrophy heart tissue, dramatically altering miRNA and mRNA expression. Cell Reports. 2014;6:336–345. doi: 10.1016/j.celrep.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A, Xiao X, Ward AJ, Castle JC, Johnson JM, Burge CB, Cooper TA. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proceedings of the National Academy of Science. 2008;105:2033–20338. doi: 10.1073/pnas.0809045105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanadia RNJohnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- Kearse MG, Todd PK. Repeat-associated non-AUG translation and its impacts in neurodegenerative disease. Neurotherapeutics. 2014;11:721–731. doi: 10.1007/s13311-014-0292-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenneson A, Zhang F, Hagedorn CH, Warren ST. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and permutation carriers. Human Molecular Genetics. 2001;10:1449–1454. doi: 10.1093/hmg/10.14.1449. [DOI] [PubMed] [Google Scholar]
- Kino T, Hurt DE, IT, Nader N, Chrousos GP. Noncoding RNA Gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Science Signaling. 2010;3(107):ra8. doi: 10.1126/scisignal.2000568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y, Habu T, Liu W, Okuda H, Koizumi A. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. American Journal of Human Genetics. 2011;89:121–130. doi: 10.1016/j.ajhg.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob MD, Moseley ML, Schut LJ, Benzow KA, Bird TD, Day JW, Ranum LP. An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8) Nature Genetics. 1999;21:379–384. doi: 10.1038/7710. [DOI] [PubMed] [Google Scholar]
- Krans A, Kearse MG, Todd PK. Repeat-associated non-AUG translation from antisense CCG repeats in Fragile X tremor/ataxia syndrome. Annals of Neurology. 2016;80:871–881. doi: 10.1002/ana.24800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk M, Emerson BM. p50-associated COX-2 extragenic RNA (PACER) activates COX-2 gene expression by occluding repressive NF-κB complexes. eLife. 2014;3:e01776. doi: 10.7554/eLife.01776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzyzosiak WJ, Sobczak KWajciechowska M, Riszer A, Mykowska A, Kozlowski P. Triplet repeat RNA structure and its role as a pathogenic agent and therapeutic target. Nucleic Acids Research. 2012;40:11–26. doi: 10.1093/nar/gkr729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung JTY, Colognori D, Lee JT. Long noncoding RNAs: past, present, and future. Genetics. 2013;193:651–669. doi: 10.1534/genetics.112.146704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Molecular Cell. 2007;38:68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D–Y, McMurray CT. Trinucleotide expansion in disease: why is there a length threshold? Current Opinion in Genetics and Development. 2014;26:131–140. doi: 10.1016/j.gde.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kopp F, Chang T, Sataluri A, Chen B, Sivakumar S, Yu H, Xie Y, Mendell JT. Noncoding RNA NORAD regulates genomic stability by sequestering PUMILIO proteins. Cell. 2016;164:69–80. doi: 10.1016/j.cell.2015.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LB, Yu Z, Teng X, Bonini NM. RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature. 2008;453:1107–1111. doi: 10.1038/nature06909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, Swanson MS, Thornton CA. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Human Molecular Genetics. 2006;15:2087–2097. doi: 10.1093/hmg/ddl132. [DOI] [PubMed] [Google Scholar]
- Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- Liu B, Sun L, Liu Q, Gong C, Yao Y, Lv X, Lin L, Yao H, Su F, Li D, Zeng M, Song E. A cytoplasmic NF-κB interacting long noncoding RNA blocks IκB phosphorylation and suppresses breast cancer metastasis. Cancer Cell. 2015;27:370–381. doi: 10.1016/j.ccell.2015.02.004. [DOI] [PubMed] [Google Scholar]
- Liu MY, Gui G, Wei B, Preston JF, Oakford L, Yuksel U, Giedroc DP, Romeo T. The RNA molecule CsrB binds to the global regulatory protein CsrA and antagonizes its activity in Escherichia coli. Journal of Biological Chemistry. 1997;272:17502–17510. doi: 10.1074/jbc.272.28.17502. [DOI] [PubMed] [Google Scholar]
- Liu W, Ikeda Y, Hishikawa N, Yamashita T, Deguchi K, Abe K. Characteristic RNA foci of the abnormal hexanucleotide GGCCUG repeat expansion in spinocerebellar ataxia type 36 (Asidan) European Journal of Neurology. 2014;21:1377–1386. doi: 10.1111/ene.12491. [DOI] [PubMed] [Google Scholar]
- Liu X, Li D, Zhang W, Guo M, Zhan Q. Long non-coding RNA gadd7 interacts with TDP-43 and regulates Cdk6 mRNA decay. EMBO Journal. 2012;31:4415–4427. doi: 10.1038/emboj.2012.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Cui Y, Mukherjee A, Chattergee AK. Characterization of a novel RNA regulator of Ewinia carotovora ssp. Carotovora that controls production of extracellular enzymes and secondary metabolites. Molecular Microbiology. 1998;29:219–234. doi: 10.1046/j.1365-2958.1998.00924.x. [DOI] [PubMed] [Google Scholar]
- Longman D, Johnstone IL, Caceres JF. Functional characterization of SR and SR-related genes in Caenorhabditis elegans. EMBO Journal. 2000;19:1625–1637. doi: 10.1093/emboj/19.7.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankodi A, Urbinati CR, Yuan QP, Moxley RT, Sansone V, Krym M, Henderson D, Schalling M, Swanson MS, Thornton CA. Muscleblind localized to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Human Molecular Genetics. 2001;10:2165–2170. doi: 10.1093/hmg/10.19.2165. [DOI] [PubMed] [Google Scholar]
- Masuda A, Andersen HS, Doktor TK, Okamoto T, Ito M, Andresen BS, Ohno K. CUGBP1 and MBNL1 preferentially bind to the 3’ UTRs and facilitate mRNA decay. Scientific Reports. 2012;2:209. doi: 10.1038/srep00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matilla-Duenas A, Corral-Juan M, Volpini V, Sanchez I. The spinocerebellar ataxias: clinical aspects and molecular genetics. Advances in Experimental Medicine and Biology. 2012;724:351–374. doi: 10.1007/978-1-4614-0653-2_27. [DOI] [PubMed] [Google Scholar]
- Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, Khajavi M, McCall AE, Davis CF, Zu L, Achari M, Pulst SM, Alonso E, Noebels JL, Nelson DL, Zoghbi HY, Ashizawa T. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nature Genetics. 2000;26:191–194. doi: 10.1038/79911. [DOI] [PubMed] [Google Scholar]
- Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nature Reviews: Genetics. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- Michalowski S, Miller JW, Urbinati CR, Paliouras M, Swanson MS, Griffith J. Visualization of double-stranded RNAs from the myotonic dystrophy protein kinase gene and interactions with CUG-binding protein. Nucleic Acids Research. 1999;27:3534–3542. doi: 10.1093/nar/27.17.3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel L, Huguet-Lachon A, Gourdon G. Sense and antisense DMPK RNA foci accumulate in DM1 tissues during development. PLoS One. 2015;10:e0137620. doi: 10.1371/journal.pone.0137620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS. Recruitment of human Muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO Journal. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan A, Goodwin M, Swanson MS. RNA-protein interactions in unstable microsatellite diseases. Brain Research. 2014;1584:3–14. doi: 10.1016/j.brainres.2014.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, Haass C, Edbauer D. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- Moseley ML, Zu T, Ikeda Y, Gao W, Mosemiller AK, Daughters RS, Chen G, Weatherspoon MR, Clark HB, Ebner TJ, Day JW, Ranum LP. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nature Genetics. 2006;38:758–769. doi: 10.1038/ng1827. [DOI] [PubMed] [Google Scholar]
- Mooers BH, Logue JS, Berglund JA. The structural basis of myotonic dystrophy from the crystal structure of CUG repeats. Proceedings of the National Academy of Science. 2005;102:16626–16631. doi: 10.1073/pnas.0505873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muslimov IA, Patel MV, Rose A, Tiedge H. Spatial code recognition in neuronal RNA targeting: role of RNA-hnRNP A2 interactions. Journal of Cell Biology. 2011;194:441–457. doi: 10.1083/jcb.201010027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mykowska A, Sobczak K, Wojciechowska M, Kozlowski P, Krzyzosiak WJ. CAG repeats mimic CUG repeats in the misregulation of alternative splicing. Nucleic Acids Research. 2011;39:8938–8951. doi: 10.1093/nar/gkr608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napierala M, Krzyzosiak WJ. CUG repeats present in myotonin kinase RNA form metastable ‘slippery’ hairpins. Journal of Biological Chemistry. 1997;272:31079–31085. doi: 10.1074/jbc.272.49.31079. [DOI] [PubMed] [Google Scholar]
- Nie M, Deng Z, Liu J, Wang D. Noncoding RNAs, emerging regulators of skeletal muscle development and disease. BioMed Research International. 2015;2015:1–17. doi: 10.1155/2015/676575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimi Y, Takahashi M, Sugawara E, Umeda S, Obayashi M, Sato N, Ishiguro T, Higashi M, Eishi Y, Mizusawa H, Ishikawa K. Abnormal RNA structures (RNA foci) containing penta-nucleotide repeat (UGGAA)n in the Purkinje cell nucleus is associated with spinocerebellar ataxia type 31 pathogenesis. Neuropathology. 2013;33:600–611. doi: 10.1111/neup.12032. [DOI] [PubMed] [Google Scholar]
- Orr HT. Cell biology of spinocerebellar ataxia. Journal of Cell Biology. 2012;197:167–177. doi: 10.1083/jcb.201105092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annual Review Neuroscience. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- Park H, Gonzalez AL, Yildirim I, Tran T, Lohman JR, Fang P, Guo M, Disney MD. Crystallographic and computational analyses of AUUCU repeating RNA that causes spinocerebellar ataxia type 10 (SCA10) Biochemistry. 2015;54:3851–3859. doi: 10.1021/acs.biochem.5b00551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson HL, Perez MK, Trottier Y, Trojanowski JQ, Subramony SH, Das SS, Vig P, Mandel JL, Fischbeck KH, Pittman RN. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron. 1997;19:333–344. doi: 10.1016/s0896-6273(00)80943-5. [DOI] [PubMed] [Google Scholar]
- Pettersson OJ, Aagaard L, Andrejeva D, Thomsen R, Jensen TG, Damgaard CK. DDX6 regulates sequestered nuclear CUG-expanded DMPK-mRNA in dystrophia myotonica type 1. Nucleic Acids Research. 2014;42:7186–7200. doi: 10.1093/nar/gku352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickard MR, Williams GT. The hormone response element mimic sequence of Gas5 lncRNA is sufficient to induce apoptosis in breast cancer cells. Oncotarget. 2016;7:10104–10116. doi: 10.18632/oncotarget.7173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puvvula PK, Rohini DD, Pineau P, Marchio A, Moon A, Dejean A, Bischof O. Long noncoding RNA PANDA and scaffold-attachment-factor SAFA control senescence entry and exit. Nature Communications. 2014;5:5323. doi: 10.1038/ncomms6323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian M, Yang X, Li Z, Jiang C, Song D, Yan W, Liu T, Wu Z, Kong J, Wei H, Xiao J. P50-associated COX-2 extragenic RNA (PACER) overexpression promotes proliferation and metastasis of osteosarcoma cells by activating COX-2 gene. Tumor Biology. 37:3879–3886. doi: 10.1007/s13277-015-3838-8. [DOI] [PubMed] [Google Scholar]
- Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nature Reviews Neurology. 2012;8:423–434. doi: 10.1038/nrneurol.2012.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapicavoli NA, Qu K, Zhang J, Mikhail M, Laberge R, Chang HY. A mammalian pseudogene lncRNA at the interface of inflammation and anti-inflammatory therapeutics. eLife. 2013;2:e00762. doi: 10.7554/eLife.00762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau F, Freyermuth F, Fugier C, Villemin J-P, Fischer M-C, Jost B, Dembele D, Gourdon G, Nicole A, Duboc D, et al. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nature Structural Molecular and Biology. 2011;18:840–845. doi: 10.1038/nsmb.2067. [DOI] [PubMed] [Google Scholar]
- Reddy K, Zamiri B, Stanley SY, Macgregor RB, Pearson CE. The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms a tract length-dependent uni- and multimolecular RNA G-quadruplex structures. Journal of Biological Chemistry. 2013;288:9860–9866. doi: 10.1074/jbc.C113.452532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AI, Holtta-Vuori M, Ikonen E, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p12-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring HZ, Lis JT. The SR protein B52/SRp55 is essential for Drosophila development. Molecular and Cell Biology. 1994;14:7499–7506. doi: 10.1128/mcb.14.11.7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nature Reviews Neuroscience. 2013;14:248–264. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- Roberts TC, Morris KV, Wood MJA. The role of long non-coding RNAs in neurodevelopment, brain function and neurological disease. Philosophical Transactions of the Royal Society B. 2014;369:20130507. doi: 10.1098/rstb.2013.0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer JD, Guerreiro R, Vandrovcova J, Uphill J, Reiman D, Beck J, Isaacs AM, Authier A, Ferrari R, Fox NC, Mackenzie IR, Warren JD, de Silva R, Holton J, Revesz T, Hardy J, Mead S, Rossor MN. The heritability and genetics of frontotemporal lobar degeneration. Neurology. 2009;73:1451–1456. doi: 10.1212/WNL.0b013e3181bf997a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnicki DD, Holmes SE, Lin MW, Thornton CA, Ross CA, Margolis RL. Huntington’s disease-like 2 is associated with CUG repeat-containing RNA foci. Annals of Neurology. 2007;61:272–282. doi: 10.1002/ana.21081. [DOI] [PubMed] [Google Scholar]
- Rudnicki DD, Pletnikova O, Vonsattel JP, Ross CA, Margolis RL. A comparison of Huntington disease and Huntington disease-like 2 neuropathology. Journal of Neuropathology and Experimental Neurology. 2008;67:366–374. doi: 10.1097/NEN.0b013e31816b4aee. [DOI] [PubMed] [Google Scholar]
- Sato N, Amino T, Kabayashi K, Asakawa S, Ishiguro T, Tsunemi T, Takahashi M, Matsuura T, Flanigan KM, Iwasaki S, Ishino F, Saito Y, Murayama S, Yoshida M, Hashizume Y, Takahashi Y, Tsuji S, Shimizu N, Toda T, Ishikawa K, Mizusawa H. Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n. American Journal of Human Genetics. 2009;85:544–557. doi: 10.1016/j.ajhg.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt MHM, Pearson CE. Disease-associated repeat instability and mismatch repair. DNA Repair. 2016;38:117–126. doi: 10.1016/j.dnarep.2015.11.008. [DOI] [PubMed] [Google Scholar]
- Schmitt AM, Chang HY. Long noncoding RNAs in Cancer Pathways. Cancer Cell. 2016;29:452–463. doi: 10.1016/j.ccell.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seixas AI, Holmes SE, Takeshima H, Pavlovish A, Sachs N, Pruitt JL, Silveira I, Ross CA, Margolis RL, Rudnicki DD. Loss of junctophilin-3 contributes to Huntington disease-like 2 pathogenesis. Annals of Neurology. 2012;71:245–257. doi: 10.1002/ana.22598. [DOI] [PubMed] [Google Scholar]
- Sellier C, Buijsen RAM, He F, Natla S, Jung L, Tropel P, Gaucherot A, Jacobs H, Meziane H, Vincent A, Champy M-F, Sorg T, Pavlovic G, Wattenhofer-Donze M, Birling M-C, Oulad-Abdelghani M, Eberling P, Ruffenach F, Joint M, Anheim M, Martinez-Cerdeno V, Tassone F, Willemsen R, Hukema RK, Viville S, Martinat C, Todd PK, Charlet-Berguerand N. Translation of expanded CGG repeats into FMRpolyG is pathogenic and may contribute to Fragile X tremor ataxia syndrome. Neuron. 2017;93:331–347. doi: 10.1016/j.neuron.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, Schneider A, Richard S, Willemsen R, Elliott DJ, Hagerman PJ, Charlet-Berguerand N. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO Journal. 2010;29:1248–1261. doi: 10.1038/emboj.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Findlay GM, Bandukwala HS, Oberdoerffer S, Baust B, Li Z, Schmidt V, Hogan PG, Sacks DB, Rao A. Dephosphorylation of the nuclear factor of activated T cells (NFAT) transcription factor is regulated by an RNA-protein scaffold complex. Proceedings of the National Academy of Science. 2011;108:11381–11386. doi: 10.1073/pnas.1019711108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh SY, Bonini NM. Genes and pathways affected by CAG-repeat RNA-based toxicity in Drosophila. Human Molecular Genetics. 2011;20:4810–4821. doi: 10.1093/hmg/ddr420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicot G, Gomes-Pereira M. RNA toxicity in human disease and animal models: from the uncovering of a new mechanism to the development of promising therapies. Biochemica et Biophysica Acta. 2013;1832:1390–1409. doi: 10.1016/j.bbadis.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Sigdel KR, Cheng A, Wang Y, Duan L, Zhang Y. The emerging functions of long noncoding RNA in immune cells: autoimmune diseases. Journal of Immunology Research. 2015;2015:1–9. doi: 10.1155/2015/848790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simionescu-Bankston A, Kumar A. Noncoding RNAs in the regulation of skeletal muscle biology in health and disease. Journal of Molecular Medicine. 2016;94:853–866. doi: 10.1007/s00109-016-1443-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobczak K, de Mezer M, Michlewski G, Krol J, Krzyzosiak WJ. RNA structure of trinucleotide repeats associated with human neurological diseases. Nucleic Acids Research. 2003;31:5469–5482. doi: 10.1093/nar/gkg766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobczak K, Michlewski G, de Mezer M, Kierzek E, Krol J, Olejniczak M, Kierzek R, Krzyzosiak WJ. Structural diversity of triplet repeat RNAs. Journal of Biological Chemistry. 2010;285:12755–12764. doi: 10.1074/jbc.M109.078790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofola OA, Jin P, Qin Y, Duan R, Liu H, de Haro M, Nelson DL, Botas J. RNA-binding proteins hnRNP A1/B1 and CUGBP1 suppress fragile X CGG permutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron. 2007;55:565–571. doi: 10.1016/j.neuron.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcliffe JS, Nelson DL, Zhang F, Pieretti M, Caskey CT, Saxe D, Warren ST. DNA methylation represses FMR-1 transcription in fragile X syndrome. Human Molecular Genetics. 1992;1:397–400. doi: 10.1093/hmg/1.6.397. [DOI] [PubMed] [Google Scholar]
- Tassone F, Iwahashi C, Hagerman PJ. FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS) RNA Biology. 2004;1:103–105. doi: 10.4161/rna.1.2.1035. [DOI] [PubMed] [Google Scholar]
- Taylor JP. A PR plug for the nuclear pore in amyotrophic lateral sclerosis. Proceedings of the National Academy of Science. 2017;114:1445–1447. doi: 10.1073/pnas.1621085114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teive HA, Munhoz RP, Arruda WO, Raskin S, Werneck LC, Ashizawa T. Spinocerebellar ataxia type 10 – A review. Parkinsonism Related Disorders. 2011;17:655–661. doi: 10.1016/j.parkreldis.2011.04.001. [DOI] [PubMed] [Google Scholar]
- Thornton CA. Myotonic Dystrophy. Neurologic Clinics. 2014;32:705–719. doi: 10.1016/j.ncl.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, White RJ, Xia T, Welle S, Turner DH, Mathews MB, Thornton CA. Expanded CUG repeat RNAs from hairpins that activate the double-stranded RNA-dependent protein kinase PKR. RNA. 2000;6:79–87. doi: 10.1017/s1355838200991544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tichon A, Gil N, Lubelsky Y, Solomon TH, Lemze D, Itzkovitz S, Stern-Ginossar N, Ulitsky I. A conserved abundant cytoplasmic long noncoding RNA modulates repression by Pumilio proteins in human cells. Nature Communications. 2016;7:12209. doi: 10.1038/ncomms12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, Renoux AJ, Chen K-C, Scaglione M, Basrur V, Elenitoba-Johnson K, Vonsattel JP, Louis ED, Sutton MA, Taylor JP, Mills RE, Charlet-Berguerand N, Paulson HL. CGG repeat-associated translation mediated neurodegeneration in Fragile X tremor ataxia syndrome. Neuron. 2013;78:440–455. doi: 10.1016/j.neuron.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, Freier SM, Bennett CF, Sharma A, Bubulya PA, Blencowe BJ, Prasanth SG, Prasanth KV. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Molecular Cell. 2010;39:925–938. doi: 10.1016/j.molcel.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi H, Chan HYE. Expression of expanded CAG transcripts triggers nucleolar stress in Huntington’s disease. Cerebellum. 2013;12:310–312. doi: 10.1007/s12311-012-0447-6. [DOI] [PubMed] [Google Scholar]
- Tsuiji H, Yoshimoto R, Hasegawa Y, Furuno M, Yoshida M, Nakagawa S. Competition between a noncoding exon and introns: Gomafu contains tandem UACUAAC repeats and associates with splicing factor-1. Genes to Cells. 2011;16:479–490. doi: 10.1111/j.1365-2443.2011.01502.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Robberecht W. Clinical implications of recent breakthroughs in amyotrophic lateral sclerosis. Current Opinion in Neurology. 2013 doi: 10.1097/WCO.0b013e328364c063. [DOI] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, Eussen BE, van Ommen GB, Blonden LAJ, Riggins GJ, Chastain JL, Kunst CB, Galjaard H, Caskey CT, Nelson DL, Oostra BA, Warren ST. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- Walsh MJ, Cooper-Knock J, Dodd JE, Stopford MJ, Mihaylov SR, Kirby J, Shaw PJ, Hautbergue GM. Decoding the pathophysiological mechanisms that underlie RNA dysregulation in neurodegenerative disorders: a review of the current state of the art. Neuropathology and Applied Neurobiology. 2015;41:109–134. doi: 10.1111/nan.12187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan P, Su W, Zhuo Y. The role of long noncoding RNAs in neurodegenerative diseases. Molecular Neurobiology. 2016 doi: 10.1007/s12035-016-9793-6. [DOI] [PubMed] [Google Scholar]
- Wang ET, Cody NAL, Jog S, Biancolella M, Wang TT, Treacy DJ, Luo S, Schroth GP, Housman DE, Reddy S, et al. Transcriptome-wide regulation of pre-mRNA localization by muscleblind proteins. Cell. 2012;150:710–724. doi: 10.1016/j.cell.2012.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LC, Chen KY, Pan H, Wu CC, Chen PH, Liao YT, Li C, Huang ML, Hsiao KM. Muscleblind participates in RNA toxicity of expanded CAG and CUG repeats in Caenorhabditis elegans. Cell Molecular Life Sciences. 2010;68:1255–1267. doi: 10.1007/s00018-010-0522-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White M, Xia G, Gao R, Wakamiya M, Sarkar PS, McFarland K, Ashizawa T. Transgenic mice with SCA10 pentanucleotide repeats show motor phenotype and susceptibility to seizure: a toxic RNA gain-of-function model. Journal of Neuroscience Research. 2012;90:706–714. doi: 10.1002/jnr.22786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MC, Gao R, Xu W, Mandal SM, Lim JG, Hazra TK, Wakamiya M, Edwards SF, Raskin S, Teive HA, Zoghbi HY, Sarkar PS, Ashizawa T. Inactivation of hnRNP K by expanded intronic AUUCU repeat induces apoptosis via translocation of PKCdelta to mitochondria in spinocerebellar ataxia 10. PLoS Genetics. 2010;6:e1000984. doi: 10.1371/journal.pgen.1000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilburn B, Rudnicki DD, Zhao J, Weitz TM, Cheng Y, Gu X, Greiner E, Park CS, Wang N, Sopher BL, La Spada AR, Osmand A, Margolis RL, Sun YE, Yang XW. An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington’s disease-like 2 mice. Neuron. 2011;70:427–440. doi: 10.1016/j.neuron.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willingham AT, Orth AP, Batalov S, Peters EC, Wen BG, Aza-Blanc P, Hogenesch JB, Schultz PG. A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science. 2005;309:1570–1573. doi: 10.1126/science.1115901. [DOI] [PubMed] [Google Scholar]
- Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes and Development. 2009;23:1494–1504. doi: 10.1101/gad.1800909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Yin Q, Luo Z, Yao R, Zheng C, Zhang J, Xiang J, Yang L, Chen L. Unusual processing generates SPA lncRNAs that sequester multiple RNA binding proteins. Molecular Cell. 2016;64:534–548. doi: 10.1016/j.molcel.2016.10.007. [DOI] [PubMed] [Google Scholar]
- Xu X, Yang D, Ding JH, Wang W, Chu PH, Dalton ND, Wang HY, Bermingham JR, Jr, Ye Z, Liu F, Rosenfeld MG, Manley JL, Ross J, Jr, Chen J, Xiao RP, Cheng H, Fu XD. ASF/SF2-regulated CaMKIIdelta alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell. 2005;120:59–72. doi: 10.1016/j.cell.2004.11.036. [DOI] [PubMed] [Google Scholar]
- Yang F, Zhang H, Mei Y, Wu M. Reciprocal regulation of HIF-1α and lincRNA-p21 modulates the Warburg effect. Molecular Cell. 2014;53:88–100. doi: 10.1016/j.molcel.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Yang Y, Wen L, Zhu H. Unveiling the hidden function of long non-coding RNA by identifying its major partner-protein. Cell and Bioscience. 2015;5:59–68. doi: 10.1186/s13578-015-0050-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu AD, Wang Z, Morris KV. Long noncoding RNAs: a potent source of regulation in immunity and disease. Immunology and Cell Biology. 2015;93:277–283. doi: 10.1038/icb.2015.2. [DOI] [PubMed] [Google Scholar]
- Yum K, Wang ET, Kalsotra A. Myotonic dystrophy: disease repeat range, penetrance, age of onset, and relationship between repeat size and phenotypes. Curr Opin Genet Dev. 2017;44:30–37. doi: 10.1016/j.gde.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Donnely CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, Gupta S, Thomas MA, Hong I, Chiu SL, Huganir RL, Ostrow LW, Matunis MJ, Wang J, Sattler R, Lloyd TE, Rothstein JD. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525:56–61. doi: 10.1038/nature14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, Ranum LP. Non-ATG-initiated translation directed by microsatellite expansions. Proceedings of the National Academy of Science. 2011;108:260–265. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]