

Abstract
Human nonlysosomal glucosylceramidase (GBA2) is one of several enzymes that controls levels of glycolipids and whose activity is linked to several human disease states. There is a major need to design or discover selective GBA2 inhibitors both as chemical tools and as potential therapeutic agents. Here, we describe the development of a fluorescence polarization activity-based protein profiling (FluoPol-ABPP) assay for the rapid identification, from a 350+ library of iminosugars, of GBA2 inhibitors. A focused library is generated based on leads from the FluoPol-ABPP screen and assessed on GBA2 selectivity offset against the other glucosylceramide metabolizing enzymes, glucosylceramide synthase (GCS), lysosomal glucosylceramidase (GBA), and the cytosolic retaining β-glucosidase, GBA3. Our work, yielding potent and selective GBA2 inhibitors, also provides a roadmap for the development of high-throughput assays for identifying retaining glycosidase inhibitors by FluoPol-ABPP on cell extracts containing recombinant, overexpressed glycosidase as the easily accessible enzyme source.
Introduction
Human neutral glucosylceramidase (GBA2, nonlysosomal glucocerebrosidase), a member of the glycoside hydrolase (GH) family, GH116 (www.cazy.org), hydrolyzes glucosylceramide in the cytosol to produce glucose and ceramide.1−3 GBA2 is located at the cytosolic leaflet of the endoplasmic reticulum, Golgi apparatus, and endosomes.1,4 GBA2 controls levels of glucosylceramide, which is synthesized at the cytosolic leaflet of the Golgi apparatus prior to translocation to the lumen of the organelle for elongation to complex glycosphingolipids. GBA2 activity is complementary to GH30 human acid glucosylceramidase (GBA, glucocerebrosidase), which processes glucosylceramide in lysosomes.5 Genetic mutations in GBA are at the basis of the lysosomal storage disorder Gaucher disease. Gaucher macrophages are loaded with massive amounts of glucosylceramide.5 Other tissues in Gaucher, however, have remarkably normal glucosylceramide levels, and it is likely that GBA2 can compensate for reduced GBA activity in these tissues.5 Glucosylceramide has been linked to inflammation as well,6 underscoring the biomedical potential of compounds able to modulate glucosylceramide levels.
In addition to its interplay with GBA, GBA2 is itself implicated in many diseases. Mutations in the GBA2 gene cause hereditary spastic paraplegia and autosomal recessive cerebellar ataxia,7−9 indicating that compounds able to enhance GBA2 activity could have therapeutic potential. Conversely, compounds able to (partially) block GBA2 activity also hold therapeutic value; we recently found that lowering GBA2 activity in Niemann-Pick type C (NPC) mouse models led to elongated life-span and improved motor coordination.10 This finding we correlated to correction of GBA2 activity levels in Purkinje cells, which are among the most affected neuronal cells in NPC. Slowing-down of neuropathological processes in NPC mice was achieved by genetic ablation of the GBA2 gene and alternatively by pharmacological inhibition of the gene product, the latter with N-alkyldeoxynojirimycin derivatives such as N-butyldeoxynojirimycin or N-(5)-adamantane-1-ylmethoxypentyl deoxynojirimycin (respectively Zavesca (1) and AMP-DNM (2), Figure 1).10 Indeed, we and others have reported on the development of N-alkyldeoxynojirimycin derivatives as potent GBA2 inhibitors. The most potent GBA2 inhibitor at present is AMP-DNM (2), with a Ki of about 3 nM; this compound however also inhibits glucosylceramide synthase (GCS) and GBA at high nanomolar concentrations.11,12
Figure 1.
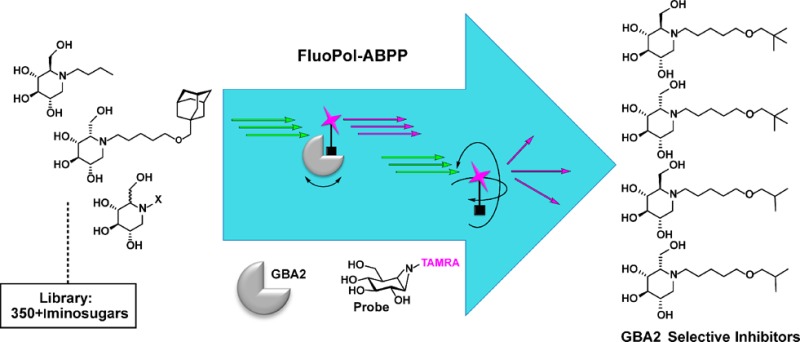
(A) Screening for GBA2 inhibitors (e.g., 1, 2, or 3) using activity-based probe 4 as a read-out in a FluoPol-ABPP assay. (B) 3-D structure (PDB 5NCX) of the TxGH116 β-glucosidase bound to cyclophellitol aziridine 5; the enzymatic nucleophile Glu441 and putative acid/base Asp593 are shown for reference. The map shown is a maximum-likelihood weighted 2Fobs – Fcalc at 0.4 e–/Å3.
GBA2 inhibitors thus have considerable potential both as cellular probes/tools and as therapeutic agents. Often, though, potent GBA2 inhibitors have significant off-targets; not only promiscuity toward GBA but also the enzyme responsible for the biosynthesis of glucosylceramide: GCS. Selective in vivo GBA2 inhibition is therefore complicated. For this reason, and considering the pharmaceutical potential of GBA2 as a drug target, selective GBA2 inhibitors are desirable commodities. Similarly, in order to access specific inhibitors, there is an urgent need for assays that rapidly identify potential GBA2 inhibitors. We here report on the development of a fluorescence polarization activity-based protein-profiling (FluoPol-ABPP) assay for fast identification of GBA2 inhibitors in extracts from cells overexpressing GBA2. Screening of our iminosugar library, which contains over 350 entries, provided lead structures bearing a relatively small (compared to existing nanomolar GBA2 inhibitors, including 2 and 3) apolar head groups. Building a focused library around this theme and assessment of the inhibitory potential of these against GBA2, GBA, and GCS yielded new leads for, and new insights into, future development of potent, selective GBA2 inhibitors. These leads, combined with our FluoPol-ABPP assay, are a useful addition to existing reagents and tools in unraveling GBA2 functioning in health and disease.
Results and Discussion
Our FluoPol-ABPP assay is based on our activity-based glycosidase profiling methodology that capitalizes on the two-step double displacement mechanism employed by retaining glycosidases. N-Alkylcyclophellitol aziridines, with the aziridine nitrogen bearing reporter groups (fluorophores, biotin), are in class, broad-spectrum retaining β-glucosidase activity-based probes (ABPs) reporting on GBA, GBA2, and the cytosolic retaining β-glucosidase GBA3 in human tissue (Figure 1A).13 In order to support specific reaction and covalency, we solved the structure of the Thermoanaerobacterium xylanolyticum TxGH116 β-glucosidase, a close homologue of human GBA2 with a conserved active site, in complex with cyclophellitol aziridine 5 (the precursor of ABP 4) to ascertain that cyclophellitol aziridines react with the active site nucleophile and thus that competitors of ABP 4 are true active site inhibitors, Figure 1B.14 The aziridine indeed binds to the invariant catalytic nucleophile Glu441, in an active site pocket on the enzyme surface. The active site pocket is freely accessible to solvent, with little steric hindrance for tagged, functionalized aziridines such as 4 (Supplementary Figure 4a). Binding of tagged ABP 4 to both GBA2 and TxGH116 can also be directly observed by fluorescent scanning of the respective proteins after ABP labeling (Figure 1A and Supplementary Figure 4B).
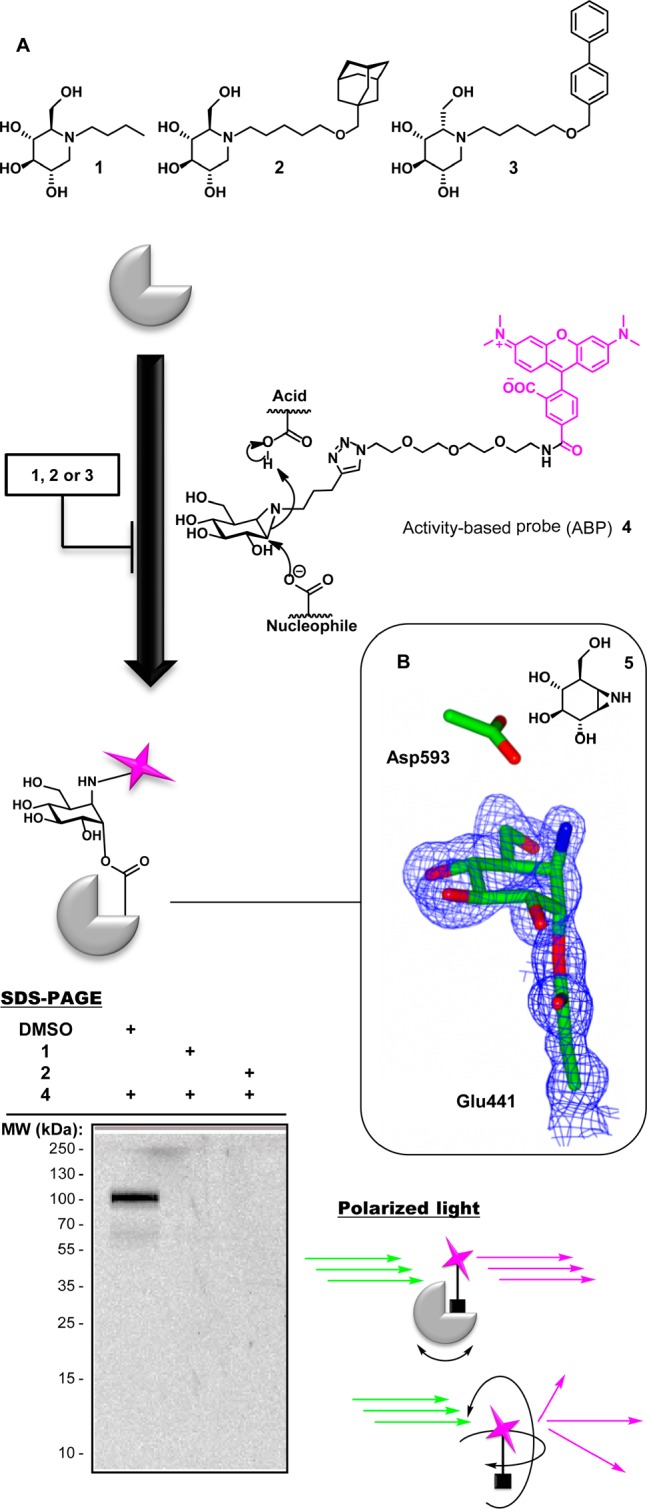
FluoPol-ABPP studies that have appeared in the literature make use of the tetramethylrhodamine (TMR) dye.15 In order to stay in tune with these studies, we therefore synthesized TMR-cyclophellitol aziridine 4 (for synthesis details see the Supporting Information, SI Schemes 1 and 2). GBA2 was stably overexpressed in HEK293 cells, constituting the majority of all β-glucosidase activity (the ratio of GBA2 levels over other targets labeled by 4 was established by prolonged imaging of the gel shown in Figure 1A; see the Supporting Information, SI Figure S1). Extracts of HEK293 overexpressing GBA2 were treated with ABP 4 at 500 nM at pH 7, and the protein content was resolved by SDS PAGE. Fluorescence scanning of the wet gel slab (λex 530 nm, λem 580 nm, Figure 1A) revealed a major band at about 102 kDa, which corresponds to the molecular weight of mature GBA2. Levels of active GBA2 in the overexpressing cells are such that endogenous GBA and GBA3, which are also targets of cyclophellitol aziridine ABPs, are hardly visible, even though labeling of GBA and GBA3 is saturating at the probe concentrations used. GBA2 labeling with 4 can be abolished by preincubation with the known GBA2 inhibitors Zavesca (1) (100 μM final concentration) and AMP-DNM (2) (10 μM final concentration) in a competitive ABPP experiment (Figure 1A, lanes 2 and 3, respectively). In fluorescence polarization assays, a fluorophore is excited at a specific wavelength with linear polarized light. The orientation of emitted light and thereby the degree of polarized light depend on the movement the excited fluorophore has undergone, which depends on its size: large molecules (such as GBA2-bound 4) move and rotate slower than small molecules (here: free 4). Samples are irradiated with linear polarized light (λex 530 nm), and the degree of loss of polarized light emitted at λem 580 nm represents the degree of inhibition of GBA2 labeling with 4. Initial experiments using fluorescence polarization as a read-out were performed in 96-well plates. The pH optimum of GBA2 is reported1 to be at pH 5.8, and contemporary assays for measuring GBA2 inhibitor activities are performed at or close to this pH. We (Figure 2A) established that, in a range from pH 4 to pH 8 (the range at which GBA2 possesses measurable activity), maximal return of polarized light occurs at pH 7. The optimal final probe concentration in the assay conditions used (protein concentration 0.5 mg/mL, Vfinal 75 μL), which intrinsically depends on the amount of active GBA2 present in the extracts (which we keep constant), was found to be 25–50 nM (Figure 2B); in this setting a suitable difference in polarized light emission (Δ about 180 mPol) was achieved between samples derived from GBA2-overexpressing cells and wild-type cells. IC50 values of a set of known GBA2 inhibitors, both competitive (Zavesca (1), AMP-DNM (2), l-ido-AMP-DNM (6), DNM (7)) and mechanism-based (cyclophellitol, 8), were next established using the FluoPol-ABPP format using 50 nM ABP 4. Values obtained (Figure 2D) were in line with literature values on the same compound, for both the potent (2, 3, 6) and less potent (1, 7, 8), which were all established using fluorogenic substrate assays, thereby validating our FluoPol-ABPP assay.16
Figure 2.
Optimization and validation of FluoPol-ABPP for GBA2. (A) Effect of pH. (B) Effect of probe concentration. (C) Structures of additional established inhibitors. (D) Inhibition by established inhibitors (1, 2, 6, 7, and 8). Error bars represent standard error of the mean (SEM).
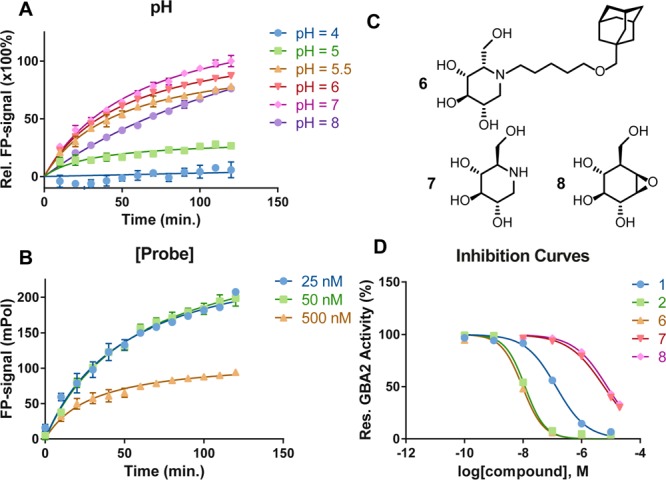
We next transferred our assay to a 384-well plate format (Vfinal 15 μL) and screened our complete iminosugar library, containing both previously published and until now unpublished compounds (the structures of all 358 compounds and the synthesis of new compounds are in the Supporting Information). All compounds were assessed at 100 nM final concentration, and the percentage of GBA2 inhibition calculated from the relative loss of fluorescence polarization was observed. Besides N-alkyldeoxynojirimycin, the corresponding d-galacto- and l-ido-configured iminosugars are known to be potent GBA2 inhibitors, and library entries adhering to this general description are picked up in our FluoPol-ABPP assay as well (Figure 3).16 Alternatively configured piperidines, partially deoxygenated deoxynojirimycin derivatives, and hydroxylated pyrrolidines show up only rarely, if at all, as inhibitors in our assays (for a more detailed grouping of compound classes and GBA2 inhibition see the Supporting Information).
Figure 3.
FluoPol-ABPP assay with an iminosugar library (358 entries). (A) Competition at 100 nM. (B) Examples of second-generation analogues of compounds 9 and 10.
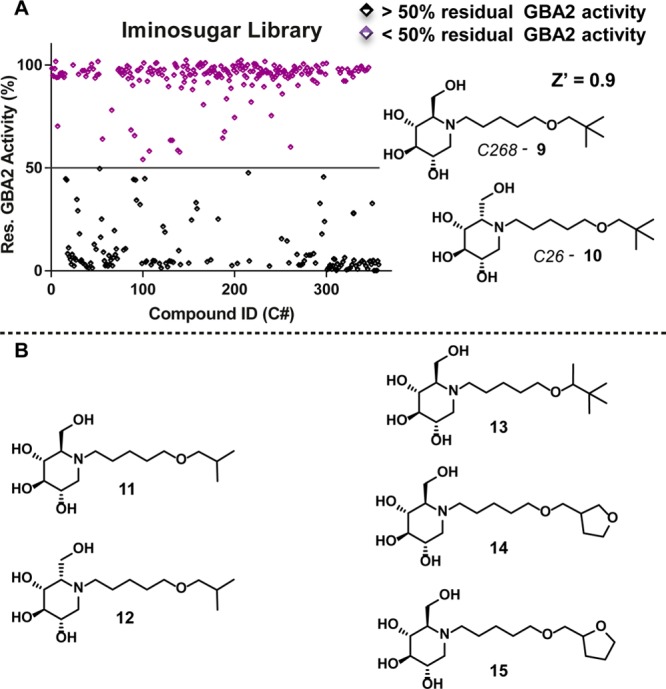
Among the most potent GBA2 inhibitors we identified neopentyloxypentyl-deoxynojirimycin 9 and its l-ido-congener 10 (Figure 3). Intriguingly, for these compounds the hydrophobic N-alkyl group is relatively small compared to that of 2 and 3. We previously found that large (as in 2) and preferably aromatic (as in 3) alkoxy substituents are preferred by GCS and reasoned that a reduced size of this part of the molecule may be favorable for achieving GBA2 selectivity over the ceramide glucosyltransferase. Accordingly, we synthesized a set of close analogues of compounds 9 and 10 and assessed their inhibitory potency against GBA2, GBA, GBA3, and GCS (examples of second-generation library 11–15 in Figure 3; for synthesis and analytical details see the Supporting Information).
As can be seen in Table 1, compounds 9 and 10 are highly potent and rather selective GBA2 inhibitors. Removal of one methyl group, as in isopropyl derivatives 11 and 12, led to an increase of inhibitory potency against GBA2 with concomitant (slight) decrease of inhibitory potency against GCS. l-ido-configured compounds 10 and 12 are over 100 000-fold selective for GBA2 over GBA1, which is a significant improvement compared to previously reported inhibitors showing a maximal 10 000-fold selectivity (for selectivity ratios of the compounds in Table 1 for GBA2 offset against GBA, GBA3, and GCS, respectively, see the Supporting Information, SI 8). As mentioned earlier, the activity against GCS is much reduced when comparing to our previously reported adamantyl and biphenyl compounds, respectively 2 and 3. Compounds 9, 13, and 14 are about 1000-fold more potent on GBA2 over GCS, while previously reported compounds are at best 150-fold more selective. While substitution of the neopentyl moiety for tetrahydrofuranylmethyl groups, as in 14 and 15, had a detrimental effect on inhibitory potency toward GBA, GBA2, and GCS, it has no significant effect on the selectivity. Based on both potency and selectivity, neopentyl and isopropyl derivatives 9–12 are perhaps the most potent and selective GBA2 inhibitors reported to date.
Table 1. Inhibition Values and Selectivity Ratios of Deoxynojirimycin Derivatives on GCS, GBA, GBA2, and GBA3.
| compound | GCSa | GBA1b | GBA2b | GBA3b | GCS/GBA2c | GBA1/GBA2c | GBA3/GBA2c |
|---|---|---|---|---|---|---|---|
| 1 | 50 | 675 | 0.326 | >1000 | 153 | 2071 | >3067 |
| 2 | 0.15 | 0.28 | 0.0035 | 27.6 | 43 | 80 | 7886 |
| 3 | 0.008 | 25.3 | 0.0027 | 27.8 | 3 | 9370 | 10296 |
| 6 | 0.15 | 12.4 | 0.0083 | 66.1 | 18 | 1494 | 7964 |
| 9 | 5.05 | 3.8 | 0.0051 | 27.6 | 990 | 745 | 5412 |
| 10 | 2.77 | 120 | 0.0077 | 119 | 360 | 15584 | 15455 |
| 11 | 3.87 | 10.8 | 0.0075 | 128 | 516 | 1440 | 17067 |
| 12 | 0.729 | 1223 | 0.0095 | 177 | 77 | 128737 | 18632 |
| 13 | 6 | 7.3 | 0.0059 | 116 | 1017 | 1237 | 19661 |
| 14 | 53 | 92 | 0.0536 | 180 | 989 | 1716 | 3358 |
| 15 | >50 | 122 | 0.1771 | 145 | >282 | 689 | 819 |
Inhibition value for in situ assay is given as IC50 (μM).
Inhibition value for in vitro assay is given as Ki (μM).
Relative GBA2 offset (ratio)
In a final in situ target engagement experiment we performed competitive ABPP on live cells. Cells overexpressing GBA2 or GBA3 and containing endogenous GBA levels were treated with compounds 9–12 at various final concentrations, lysed, and then treated with ABP 4. As can be seen from the images of the SDS PAGE gels (Figure 4), all compounds are cell permeable and selectively block GBA2 over GBA and GBA3 at the concentrations tested.
Figure 4.

Competitive ABPP experiment of HEK293T cells overexpressing GBA2 or GBA3 (and expressing endogenous GBA) treated with compounds 9–12 at various final concentrations prior to cell lysis and ABPP profiling of remaining enzyme activity.
Conclusions
Collectively, this work describes the development of an effective FluoPol-ABPP assay for the rapid screening of GBA2 inhibitors. Fluorescence polarization (FluoPol) is often used for the analysis of the potency of small molecules to inhibit enzymes17 but has to the best of our knowledge not been used in the context of glycosidase inhibitor assays. This certainly holds true for the combination of FluoPol and ABPP,18 as we show here. FluoPol-ABPP can be conducted on target enzymes in complex mixtures, as long as the ABP used modifies with considerable selectivity the target enzyme. This can be achieved even with broad-spectrum ABPs such as the one used here, simply by bringing the target enzyme to overexpression. Target selectivity is easily established by running an SDS PAGE on the ABP-labeled sample. One potential caveat of FluoPol-ABPP is the covalent and irreversible nature of the ABP used. Most studies on FluoPol-ABPP inhibitor screening entail the assessment of covalent inhibitors.18 Here, we show that competitive inhibitors can also be identified using FluoPol-ABPP as an initial screen, after which more in-depth kinetic studies can be executed on selected compounds using conventional assays using fluorogenic substrates. Here, we identified a few compounds from our 358-compound collection that possess considerable GBA2 selectivity, and on the basis of these lead structures we prepared a small focused library to add to our growing library of iminosugar glycosidase inhibitors. In-depth analysis revealed that, indeed, modulating the size of the hydrophobic nitrogen substituent yields inhibitors more potent, and crucially also more selective, for GBA2 compared to existing inhibitors. From a technical point of view, the FluoPol-ABPP assay can be adapted to allow high-throughput screening of large compound selections on GBA2 inhibition. Conceptually, with this study we show that FluoPol-ABPP, as a recently proposed strategy for high-throughput screening of various enzymes, is also eminently feasible for searching for glycosidase inhibitors, especially considering that the list of activity-based glycosidase probes targeting a variety of retaining glycosidases is growing.
Experimental Section
Cell Culture
Human embryonic kidney 293T (HEK293T) cells (Sigma) were cultured in DMEM high glucose (Gibco) supplemented with 10% NBS and 100 units/mL penicillin/streptomycin (Gibco) at 37 °C and 5% CO2. RAW 264.7 cells (American Type Culture Collection) were cultured in RPMI (Gibco) supplemented with 10% fetal calf serum, 1 mM Glutamax, and 100 units/mL penicillin/streptomycin (Gibco) at 37 °C and 5% CO2.
Transient and Stable Overexpression of Human GBA2 and GBA3
Primers used for transient GBA2-expressing HEK293T were designed based on NCBI reference sequence NM_020944.2. The full-length coding sequences were cloned into pcDNA3.1/Myc-His (Invitrogen, Life Technologies, Carlsbad, CA, USA). Subconfluent HEK293T cells were transfected with empty pcDNA3.1 or GBA2pcDNA3.1 (plasmid:PEI ratio 1:3). Media was refreshed 24 h later, and cells were collected 72 h after transfection in phosphate-buffered saline (PBS) buffer. Cells were centrifuged at 1000 rpm for 10 min, after which the supernatant was removed. Cell pellets were snap-frozen with liquid nitrogen and stored at −80 °C.
Stable GBA2-expressing HEK293T cells were generated as follows. The PCR-amplified human GBA2 (GBA2, acc. no. NM_020944.2) coding sequence (using the following primers: sense 5′-GGGGACAAGTTTGTACA AAAAAGCAGGCTTAACCACCATGGGGACCCAGGAT CCAG-3′ and antisense 5′- GGGGACCACTTTGTAC AAGAAAGCTGGGTTTCACTCTGGGCTCAGGTTTG-3′) was cloned into pDNOR-221 and subcloned in pLenti6.3/TO/V5-DEST using the Gateway system (Invitrogen). Correctness of the construct was verified by sequencing. HEK293T cells were transfected with pLenti6.3-GBA2 in combination with the envelope and packaging plasmids pMD2G, pRRE, and pRSV. Subsequently, culture supernatant containing viral particles was collected and used for infection of HEK293T cells. Selection using blasticidin for several weeks rendered cells stably expressing human GBA2 as determined by activity assays. For stable expression of GBA3 in HEK293T cells, the PCR-amplified GBA3 (GBA3, acc. no. NM_020973.4) coding sequence (using the following oligonucleotides: sense 5′-GAATTCGCCGCACCATGGCTTTCCCTGCAGGATTTG-3′ and antisense 5′-GCGGCCGCAGATGTGCTTCAA GGCCATTG-3′) was cloned into pcDNA3.1/Zeo and transfected into HEK293T cells using FuGene 6 Transfection Reagent (Promega Benelux, Leiden, The Netherlands). Selection using Zeocin for several weeks rendered cells stably expressing human GBA3 as determined by activity assays.
Preparation of Lysates
Cell pellets were resuspended in lysis buffer (20 mM hepes, 2 mM DTT, 0.25 M sucrose, 1 mM MgCl2, 2.5 U/mL benzonase, pH 7.0). The homogenate was incubated on ice for 30 min after lysis and homogenization using SilentCrusher (Heidolph). Ultracentrifugation was performed at 32.000 rpm for 30 min at 4 °C. Supernatant fractions were aliquoted in appropriate volumes, after the total protein concentration was determined via a Bradford assay, using BSA (Sigma) for standards and BioRad Quickstart Bradford Reagents. Aliquots were snap-frozen with liquid nitrogen and stored at −80 °C.
Gel-Activity-Based Protein Profiling Experiments
Prepared lysate was diluted in assay buffer (20 mM Hepes, 2 mM DTT at pH 7.0) until appropriate final protein concentration was reached. Samples were incubated with 500 nM probe at 37 °C for 1 h (Vfinal = 20 μL). Protein content was denatured using Laemli buffer (4×) at 100 °C for 3 min. Reactions were resolved by 12.5% SDS-PAGE electrophoresis, and wet slabs were scanned for fluorescence (Molecular Imager Gel Doc XR, Biorad). For the in situ competitive ABPP experiments stable GBA2- and GBA3-overexpressing HEK293T cells were incubated for 1 h at 37 °C with various concentrations of the selected inhibitors. Cells were washed two times with PBS and then harvested by scraping in 25 mM potassium phosphate buffer (pH 6.5) containing 0.1% Triton X-100 (v/v). Cells were lysed via snap-freezing in liquid nitrogen. Protein concentration was determined via BCA assay (Thermo Fischer). Labeling of the glucosidases was conducted using 500 nM ABP 4 for 30 min at 37 °C on 20 μg of protein. Protein content was denatured using Laemli buffer (5×) at 100 °C for 5 min. Reactions were resolved by 10% SDS-PAGE electrophoresis, and wet slabs were scanned as described above.
Optimization of the FluoPol-ABPP Assay
Optimal probe concentration on the FluoPol signal was determined by varying probe concentrations from 25 to 500 nM probe at a constant lysate concentration (0.5 mg/mL) and neutral pH. FluoPol-ABPP assays were also performed at different pH values. Hepes was replaced by 20 mM citric acid–citrate buffer for reactions at pH ≤ 6. The pH experiments were performed at optimal probe concentration (50 nM). Competition experiments were conducted by 30 min of preincubation of compounds in the lysates at 37 °C (2.5% DMSO). All reactions (Vfinal 75 μL) containing bovine gamma-globulin (Sigma) and Chaps (Sigma), respectively 0.5 and 1 mg/mL, were carried out in 96-well plates (flat-black bottom, Greiner). FluoPol signals were monitored on an Infinite M1000Pro (Tecan) using λex 530 nm and λem 580 nm. Mock-containing lysates were used as reference samples, samples without probe as blanks to correct for background polarization, and GBA2-containing lysates without inhibitor as controls. All samples were corrected for background polarization, and the residual enzyme activity was calculated based on the polarization signal from the controls. Polarization signals were plotted against time or inhibitor concentration and processed in GraphPad Prism 6.0. IC50 values were calculated via nonlinear regression using the mentioned software (N = 2, n = 3).
FluoPol-ABPP Screen of the Iminosugar Library
The screen, using the optimized conditions as described above, was conducted in a 384-well black-bottom plate (Greiner) with reaction volumes set at 15 μL. Concentration of the iminosugars during the screen was 100 nM. The FluoPol signal was measured on a PHERAstar (BMG Labtech). Resulting polarization signals were processed as described above. Residual enzyme activities were plotted against the corresponding iminosugar.
Analysis of Selected Compounds as Inhibitors of Enzymatic Activity of GCS, GBA, GBA2, and GBA3
Enzyme assays of GCS, GBA1, GBA2, and GBA3 were carried out as previously described (refs (16) and (19)), and inhibition by selected compounds was assessed. Briefly, for GBA1, pure recombinant human enzyme (Cerezyme from Genzyme) was used. Activity was measured with 3.7 mM 4-methylumbeliferone (4MU)-β-d-glucopyranoside (Sigma) in 150 mM McIlvaine buffer pH 5.2 supplemented with 0.2% sodium taurocholate (w/v), 0.1% Triton X-100 (v/v), and 0.1% bovine serum albumin (BSA) (w/v) for 30 min. The reaction was stopped with excess 1 M NaOH-glycine (pH 10.3), and liberated 4MU fluorescence was measured with an LS55 fluorimeter (PerkinElmer) using λEx 366 nm and λEm 445 nm. For GBA2 measurements, cellular homogenates of a stable HEK293T-overexpressing GBA2 cell line preincubated for 30 min with an inhibitor of GBA1 (1 mM conduritol β epoxide CBE from Sigma) were used. Activity was measured with 3.7 mM 4MU-β-d-glucopyranoside in 150 mM McIlvaine pH 5.8 and 0.1% BSA (w/v) for 1 h. Reactions were stopped as described above for GBA1. For GBA3 measurements, cellular homogenates of a stable HEK293T-overexpressing GBA3 cell line also preincubated for 30 min with an inhibitor of GBA1 (1 mM conduritol β epoxide CBE from Sigma) were used. Reactions were conducted in 100 mM hepes pH 7.0 and 0.1% BSA (w/v) for 1 h. All IC50 values were determined in triplicate, and the inhibitors tested were preincubated for 30 min at 37 °C. Ki values were determined in duplicate using a range from 0.05 to 5 mM 4MU-β-d-glucopyranoside in appropriate buffer containing inhibitor. Incubation time and temperature were the same as described above, but without the preincubation step. Observed fluorescence was curve-fitted against inhibitor or substrate concentrations using GraphPad Prism 6.0 in order to obtain IC50 or Ki values.
IC50 values for GCS were determined in situ with 6-[N-methyl-N-(7-nitrobenz-2-oxa-1,3-diazol-4-ylaminododecanoyl]sphingosine (NBD-ceramide) as substrate. RAW 264.7 cells were grown to confluence in six-well plates and preincubated for 1 h with an inhibitor of GBA1 activity (300 μM CBE), followed by 1 h incubation at 37 °C with 1 μM C6-NBD-ceramide and in the presence of a range of inhibitor concentrations. The cells were washed 3× with PBS and harvested by scraping. After lipid extraction (described by Bligh and Dyer20), the C6-NBD lipids were separated and detected by high-performance liquid chromatography (λEx 470 nm and λEm 530 nm). IC50 values were determined in duplicate from the titration curves of observed formed C6-NBD-glucosylceramide, and data were curve-fitted via GraphPad Prism 6.0.
Acknowledgments
We thank The Netherlands Organization for Scientific Research (NWO-CW, TOP-PUNT grant to H.S.O.), the China Scholarship Council (CSC, Ph.D. fellowship to B.L.), and the European Research Council (ERC-2011-AdG-290836 “Chembiosphing”, to H.S.O., ERC-2012-AdG-32294 “Glycopoise”, to G.J.D.; G.J.D. holds the Royal Society Ken Murray Research Professorship) for financial support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b07352.
Synthetic procedures for ABP 4 and (unpublished) deoxynojirimycin analogues, 1H and 13C NMR spectra, IR and LC-MS data; figures validating the FluoPol-ABPP method and additional figures for the screen showing a preference for gluco- and ido-configured iminosugars; compound structures and inhibitory potencies on GCS, GBA1, and GBA2 (PDF)
Author Contributions
# D. Lahav and B. Liu contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Van Weely S.; Brandsma M.; Strijland A.; Tager J. M.; Aerts J. M. F. G. Biochim. Biophys. Acta, Mol. Basis Dis. 1993, 1181, 55–62. 10.1016/0925-4439(93)90090-N. [DOI] [PubMed] [Google Scholar]
- Yildiz Y.; Matern H.; Thompson B.; Allegood J. C.; Warren R. L.; Ramirez D. M. O.; Hammer R. E.; Hamra F. K.; Matern S.; Russell D. W. J. Clin. Invest. 2006, 116, 2985–2994. 10.1172/JCI29224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boot R. G.; Verhoek M.; Donker-Koopman W.; Strijland A.; Van Marle J.; Overkleeft H. S.; Wennekes T.; Aerts J. M. F. G. J. Biol. Chem. 2007, 282, 1305–1312. 10.1074/jbc.M610544200. [DOI] [PubMed] [Google Scholar]
- Körschen H. G.; Yildiz Y.; Raju D. N.; Schonauer S.; Bönigk W.; Jansen V.; Kremmer E.; Kaupp U. B.; Wachten D. J. Biol. Chem. 2013, 288, 3381–3393. 10.1074/jbc.M112.414714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraz M. J.; Kallemeijn W. W.; Mirzaian M.; Herrera Moro D.; Marques A.; Wisse P.; Boot R. G.; Willems L. I.; Overkleeft H. S.; Aerts J. M. F. G. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2014, 1841, 811–825. 10.1016/j.bbalip.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Nagata M.; Izumi Y.; Ishikawa E.; Kiyotake R.; Doi R.; Iwai S.; Zakaria O.; Yamaji T.; Miyamoto T.; Bamba T.; Yamasaki S. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 3285–3294. 10.1073/pnas.1618133114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer M. B.; Eleuch-Fayache G.; Schottlaender L. V.; Nehdi H.; Gibbs J. R.; Arepalli S. K.; Chong S. B.; Hernandez D. G.; Sailer A.; Liu G.; Mistry P. K.; Cai H.; Shrader G.; Sassi C.; Bouhlal Y.; Houlden H.; Hentati F.; Amouri R.; Singleton A. B. Am. J. Hum. Genet. 2013, 92, 245–251. 10.1016/j.ajhg.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E.; Schüle R.; Smets K.; Rastetter A.; Boukhris A.; Loureiro J. L.; Gonzalez M. A.; Mundwiller E.; Deconinck T.; Wessner M.; Jornea L.; Oteyza A. C.; Durr A.; Martin J. J.; Schöls L.; Mhiri C.; Lamari F.; Züchner S.; De Jonghe P.; Kabashi E.; Brice A.; Stevanin G. Am. J. Hum. Genet. 2013, 92, 238–244. 10.1016/j.ajhg.2012.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana S.; Truong N. Y.; Vieira D. B.; Wigger J. G. D.; Forrester A. M.; Veinotte C. J.; Berman J. N.; van der Spoel A. C. Zebrafish 2016, 13, 177–187. 10.1089/zeb.2015.1152. [DOI] [PubMed] [Google Scholar]
- Marques A. R. A.; Aten J.; Ottenhoff R.; Van Roomen C. P. A. A.; Moro D. H.; Claessen N.; Veloz M. F. V.; Zhou K.; Lin Z.; Mirzaian M.; Boot R. G.; De Zeeuw C. I.; Overkleeft H. S.; Yildiz Y.; Aerts J. M. F. G. PLoS One 2015, 10, 1–18. 10.1371/journal.pone.0135889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overkleeft H. S.; Renkema G. H.; Neele J.; Vianello P.; Hung I. O.; Strijland A.; van der Burg A. M.; Koomen G.; Pandit U. K.; Aerts J. M. F. G. J. Biol. Chem. 1998, 273, 26522–26527. 10.1074/jbc.273.41.26522. [DOI] [PubMed] [Google Scholar]
- Wennekes T.; van den Berg R. J. B. H. N.; Boot R. G.; van der Marel G. A.; Overkleeft H. S.; Aerts J. M. F. G. Angew. Chem., Int. Ed. 2009, 48, 8848–8869. 10.1002/anie.200902620. [DOI] [PubMed] [Google Scholar]
- Kallemeijn W. W.; Li K.-Y.; Witte M. D.; Marques A. R. A.; Aten J.; Scheij S.; Jiang J.; Willems L. I.; Voorn-Brouwer T. M.; van Roomen C. P. A. A.; Ottenhoff R.; Boot R. G.; van den Elst H.; Walvoort M. T. C.; Florea B. I.; Codée J. D. C.; van der Marel G. A.; Aerts J. M. F. G.; Overkleeft H. S. Angew. Chem. 2012, 124, 12697–12701. 10.1002/ange.201207771. [DOI] [PubMed] [Google Scholar]
- Charoenwattanasatien R.; Pengthaisong S.; Breen I.; Mutoh R.; Sansenya S.; Hua Y.; Tankrathok A.; Wu L.; Songsiriritthigul C.; Tanaka H.; Williams S. J.; Davies G. J.; Kurisu G.; Cairns J. R. K. ACS Chem. Biol. 2016, 11, 1891–1900. 10.1021/acschembio.6b00192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachovchin D. A.; Brown S. J.; Rosen H.; Cravatt B. F. Nat. Biotechnol. 2009, 27, 387–394. 10.1038/nbt.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennekes T.; Meijer A. J.; Groen A. K.; Boot R. G.; Groener J. E.; van Eijk M.; Ottenhoff R.; Bijl N.; Ghauharali K.; Song H.; Shea T. J. O.; Liu H.; Yew N.; Copeland D.; van den Berg R. J.; van der Marel G. A.; Overkleeft H. S.; Aerts J. M. F. G. J. Med. Chem. 2010, 53, 689–698. 10.1021/jm901281m. [DOI] [PubMed] [Google Scholar]
- Lea W. A.; Simeonov A. Expert Opin. Drug Discovery 2011, 6, 17–32. 10.1517/17460441.2011.537322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niphakis M. J.; Cravatt B. F. Annu. Rev. Biochem. 2014, 83, 341–377. 10.1146/annurev-biochem-060713-035708. [DOI] [PubMed] [Google Scholar]
- Dekker N.; Voorn-Brouwer T.; Verhoek M.; Wennekes T.; Narayan R. S.; Speijer D.; Hollak C. E. M.; Overkleeft H. S.; Boot R. G.; Aerts J. M. F. G. Blood Cells, Mol., Dis. 2011, 46, 19–26. 10.1016/j.bcmd.2010.07.009. [DOI] [PubMed] [Google Scholar]
- Bligh E. G.; Dyer W. J. Can. J. Biochem. Physiol. 1959, 37, 911–917. 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.