

Abstract
The VTA is necessary for reward behavior with dopamine cells critically involved in reward signaling. Dopamine cells in turn are innervated and regulated by neighboring inhibitory GABA cells. Using whole-cell electrophysiology in juvenile-adolescent GAD67-GFP male mice, we examined excitatory plasticity in fluorescent VTA GABA cells. A novel CB1-dependent LTD was induced in GABA cells that was dependent on metabotropic glutamate receptor 5, and cannabinoid receptor 1 (CB1). LTD was absent in CB1 knock-out mice but preserved in heterozygous littermates. Bath applied Δ9-tetrahydrocannabinol depressed GABA cell activity, therefore downstream dopamine cells will be disinhibited; and thus, this could potentially result in increased reward. Chronic injections of Δ9-tetrahydrocannabinol occluded LTD compared with vehicle injections; however, a single exposure was insufficient to do so. As synaptic modifications by drugs of abuse are often tied to addiction, these data suggest a possible mechanism for the addictive effects of Δ9-tetrahydrocannabinol in juvenile-adolescents, by potentially altering reward behavioral outcomes.
SIGNIFICANCE STATEMENT The present study identifies a novel form of glutamatergic synaptic plasticity in VTA GABA neurons, a currently understudied cell type that is critical for the brain's reward circuit, and how Δ9-tetrahydrocannabinol occludes this plasticity. This study specifically addresses a potential unifying mechanism whereby marijuana could exert rewarding and addictive/withdrawal effects. Marijuana use and legalization are a pressing issue for many states in the United States. Although marijuana is the most commonly abused illicit drug, the implications of legalized, widespread, or continued usage are speculative. This study in juvenile-adolescent aged mice identifies a novel form of synaptic plasticity in VTA GABA cells, and the synaptic remodeling that can occur after Δ9-tetrahydrocannabinol use.
Keywords: anandamide, endocannabinoid, mGluR5, THC, TRPV1, withdrawal
Introduction
Drug addiction represents a significant health and economic issue around the world. The United States alone spends >$193 billion dollars annually on costs related to illicit drug abuse. Although there is variation in the physiological mechanism of each drug of abuse, the common theme is that they alter VTA circuitry and function, usually to enhance DA release (Berridge and Robinson, 1998; Wise, 2008; Lüscher and Malenka, 2011). Dopaminergic cells are involved in motivation and learned reward behavior. Drugs of abuse enhance DA levels more than normal rewarding behaviors (McBride et al., 1999), and cause synaptic modifications leading to addiction (Lüscher and Malenka, 2011). Subsequent to the acute actions of drugs of abuse, synaptic and physiological changes to the reward circuit can persist long after clearance of these drugs. These synaptic changes in the VTA, or its projections, mediate some of the addictive components of drugs of abuse. All addictive psychoactive substances examined to date induce long-term changes of excitatory synapses of VTA DA neurons, whereas nonaddictive psychoactive substances do not (Mameli and Lüscher, 2011). For example, one exposure to cocaine induces LTP of excitatory input to VTA DA cells (Ungless et al., 2001). These synaptic changes are likely why addiction involves the compulsive drive to self-administer drugs even after mitigation of acute DA enhancements.
Endocannabinoids (eCBs) mediate many forms of plasticity, in particular LTD (Castillo et al., 2012). The eCBs are more recently characterized lipid-based signaling molecules that induce plasticity throughout the brain (Cristino et al., 2008), including the VTA (Mátyás et al., 2008). The eCBs include 2-arachidonylglycerol (2-AG) and anandamide that retrogradely activate receptors, such as cannabinoid receptor 1 (CB1) and transient receptor potential vanilloid 1 (TRPV1) (Di Marzo et al., 1994; Tanimura et al., 2010). These receptors have both been correlated to reward and addiction in the VTA (Marinelli et al., 2005; Rodriguez et al., 2011). The regulation of eCB production shares common mechanisms throughout the nervous system (Heifets and Castillo, 2009). Synthesis of eCBs 2-AG and anandamide often occurs in the postsynaptic cell, usually in response to Type I metabotropic glutamate receptor (mGluR) activation (Jung et al., 2005). When produced, these hydrophobic molecules escape from the postsynaptic cell and can bind to receptors on presynaptic neurons or adjacent cells. CB1 is particularly relevant to reward and addiction due to its ability to bind to the psychoactive ingredient in marijuana, Δ9-tetrahydrocannabinol (THC) (Gaoni and Mechoulam, 1964). Marijuana/THC use is especially negative for adolescents where it causes decreased cognition, abnormalities in the reward pathway, and increased probability of substance abuse (Quinn et al., 2008; Gilman et al., 2014; Hurd et al., 2014; Volkow et al., 2014). Indeed, a pathologic addiction state known as cannabis use disorder is on the rise and can lead to memory deficits (Smith et al., 2015). Cannabis use disorder, as defined in the DSM-V, is the most prevalent drug use disorder in the United States (Wu et al., 2016). The recent legalization of marijuana for medical and recreational use in several states will only increase the public issues associated with it and thus the demand for studies regarding its effects on reward, addiction, and cognition.
Last, as mentioned previously, while illicit drugs are known to cause synaptic modifications onto DA cells, data regarding drug modification of excitatory synapses onto VTA GABA neurons is conspicuously absent. Therefore, studies examining VTA GABA neurons are necessary, as drugs of abuse targeting these neurons, altering their activity and excitatory synaptic plasticity, could cause addictive behaviors as well. This is because decreasing GABA cell activity reduces inhibition to DA cells, thus enhancing DA release (Lassen et al., 2007). Our goal was to identify the mechanism of plasticity of excitatory inputs onto VTA GABA neurons and determine the influence of THC on this plasticity.
Materials and Methods
All experiments were performed in accordance with Institutional Animal Care and Use Committee protocols and followed National Institutes of Health Guide for the care and use of laboratory animals. Institutional Animal Care and Use Committee protocols for all experiments were approved by the Brigham Young University Institutional Animal Care and Use Committee, Animal Welfare Assurance Number A3783-01.
Animals.
Male juvenile/adolescent aged CD1 GAD67-GFP knock-in mice (P15–P40) produced by the Tamamaki laboratory were used so that GABA cells in the VTA could be positively identified using fluorescence (Tamamaki et al., 2003). Juvenile age in mice begins at ∼P14–P15 and adolescence begins at ∼P28–P35 (Iñiguez et al., 2016; Rovira and Geijo-Barrientos, 2016; Liu et al., 2017; Yohn and Blendy, 2017). In a prior report, we confirmed that extracted GFP-positive cells do indeed express GAD67 by PCR and performed immunohistochemistry experiments that confirm TH and GFP-positive cells are separate populations in the VTA (Merrill et al., 2015). All experiments and controls were interleaved among this age range. No differences in LTD were noted within this age range. Male CB1 knock-out mice (Dr. Ken Mackie's Laboratory, Department of Psychological and Brain Sciences, Indiana University, Bloomington, Indiana) were bred to female GAD67-GFP-positive/CB1 heterozygous mice to obtain knock-out and heterozygous littermates that also had positive GAD67-GFP labeling. GFP-positive mice were identified initially in screening using UV goggles, whereas CB1 knock-out, heterozygote, and littermate controls were identified by genotyping using PCR. Primer sequences for wild-type CB1 gene were as follows: forward, TACCACCATTTCACTGATTGTCT; and reverse, TGGCAAGAAAAGTCAGAGTGTAA. Sequences for knock-out were as follows: forward, AGGTCGGTCTTGACAAAAAGA; and reverse, GCCATTGAACAAGATGGATT.
Slice preparation.
Mice were anesthetized with isoflurane and decapitated with a rodent guillotine. Brains were rapidly removed and sectioned horizontally on a vibratome at 300 μm using an ice-cold, sucrose-based cutting solution composed of 220 mm sucrose, 0.2 mm CaCl2, 3 mm KCl, 1.25 mm NaH2PO4, 25 mm NaHCO3, 12 mm MgSO4, and 10 mm glucose. After sectioning, the slices were placed in oxygenated ACSF composed of 119 mm NaCl, 26 mm NaHCO3, 2.5 mm KCl, 1 mm NaH2PO4, 2.5 mm CaCl2, 1.3 mm MgSO4, and 11 mm glucose in an incubator at 37°Celsius, where they remained until recording.
Recording protocol experimental design and statistical analyses.
Recordings began at least 1 h after cutting. Slices were placed in the recording chamber and bathed with oxygenated (95% O2, and 5% CO2) high divalent ACSF (4 mm CaCl2 and 4 mm MgCl2, replacing MgSO4), along with 100 μm picrotoxin to block GABAA currents. The VTA was visualized using an Olympus BX51W1 microscope with a 40× water-immersion objective. GAD67-GFP-positive cells were selected for recording from the same location in the VTA as our previous study (Merrill et al., 2015) at approximately the following coordinates from adult mouse bregma; anteroposterior −3.2 to 3.3, mediolateral ±0.6 to 0.7, dorsoventral −4.5 to 4.3. Patch pipette resistance was 2.5–5.5 mΩ. Cells were held at −65 mV and patched with a glass pipette filled with internal solution composed of 117 mm cesium gluconate, 2.8 mm NaCl, 20 mm HEPES, 5 mm MgCl2, and 1 mm QX-314 (Tocris Bioscience), pH 7.28 (275–285 mOsm). Current traces were recorded using Multiclamp 700B amplifier (Molecular Devices). Signals were filtered at 4 kHz and digitized with an Axon 1440A or 1550A digitizer (Molecular Devices) connected to a Dell personal computer with pClamp 10.2/10.5 Clampex software (Molecular Devices). The peak amplitude of the induced EPSC was calculated using Clampfit 10.5 software (Molecular Devices) and graphed using Origin 7.5. Within individual experiments, current amplitudes were averaged by minute (6 sweeps/min). Plasticity was induced using two stimulations at 100 Hz for 1 s, 20 s apart or 5 Hz for 3 min. Additional experiments include vehicle controls for ethanol and DMSO, or with AMPA and NMDA receptor antagonists to demonstrate that currents recorded from were glutamatergic (Fig. 1). Cells used for depolarization-induced suppression of excitation (DSE) experiments were initially held at −65 mV, then raised to 0 mV for 10 s before being returned to −65 mV. Wilcoxon matched pairs analysis was used to obtain p values for the paired-pulse ratios (PPRs) and coefficient of variance. All other p values were obtained using a two-way unequal variance t test and/or ANOVA comparing the 5 min before conditioning to a 5 min period at multiple time points after conditioning or after drug application, including 10–15, 15–20, and 20–25 min intervals, with 15–20 min being the norm. N values signify the number of cells used in each experiment, where 1–2 cells were acquired per animal. Statistics included two-way Student's t test to compare between two groups or ANOVA to compare within an individual experiment. Differences were considered significant at p < 0.05.
Figure 1.
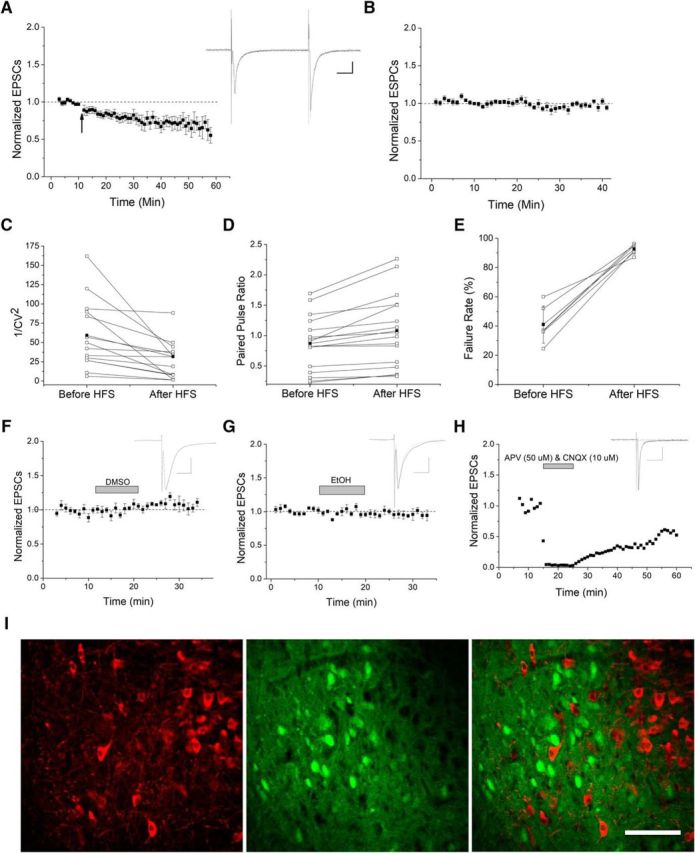
Presynaptic LTD in VTA GABA cells. A, Glutamate currents recorded from GAD67-GFP-positive VTA GABA cells. The 100 Hz HFS induces LTD as EPSC responses were reduced by 21 ± 5.5% at 10–15 min after HFS compared with baseline (n = 14, p ≪ 0.05, baseline compared with postconditioning at 10–15, 15–20, and 20–25 min) and eventually reached >30% depression. All plots are normalized EPSC amplitude means with standard error of the mean (SEM). Arrow indicates 100 Hz HFS conditioning induction. Insets, Here and throughout, Representative averaged EPSC traces (10–12 traces) taken from before (black) and 10–15 min after conditioning or drug application (gray). Calibration: Scale bars here and throughout are 100 pA, 10 ms. B, No treatment controls showed no change in EPSC amplitude, which demonstrates that HFS-LTD is not due to rundown effect (n = 8). C–E, Following HFS, the 1/CV2 decreased (n = 13 p < 0.05), PPR increased (n = 15, p < 0.05), and failure rates increased (n = 6, p < 0.05), suggesting a presynaptic mechanism for HFS-LTD. Connected open circles represent individual experiments before and 5–10 min after HFS. Connected closed circles represent averages from all experiments. F–H, Vehicle controls and glutamate transmission confirmation. F, Solvent DMSO vehicle controls demonstrate that DMSO does not alter basal neurotransmission of VTA GABA cells (n = 6, p > 0.5, post-DMSO application compared with pre-DMSO). G, EtOH vehicle controls also demonstrate no change in EPSCs compared with baseline (n = 7, p > 0.5). H, An individual experiment denotes that recorded EPSCs are indeed glutamatergic as they are blocked by a combination of AMPA receptor antagonist CNQX and NMDA receptor antagonist APV (n = 5). Washout occurs and recovers some of the glutamate response. I, Immunohistochemical image of the VTA. GAD67-GFP-positive mice were counterstained with TH antibody to label dopaminergic neurons. Anti-TH antibodies label cytosol of dopaminergic neurons as noted in the red channel, whereas GAD67-GFP-positive neurons are labeled in the green channel. In the merged image, no individual neuron is double-labeled, indicating that the inhibitory cells we recorded from are not dopaminergic, but that both are separate cell populations. Scale bar, 100 μm.
Immunohistochemistry.
Mouse GAD67-GFP brains were transcardially perfused with 0.1 m PBS followed by 4% PFA in 0.1 m PBS, pH 7.4. Brains were cryoprotected in 30% sucrose solution, frozen in OCT, sliced into 30 μm sections, and collected into 0.1 m PBS for a free-floating staining procedure. Slices were permeabilized with 0.2% Triton-X (Fisher Bioreagents) for 30 min, washed with 1% BSA in 0.1 m PBS for 2 h, and treated with primary antibody for sheep anti-TH (1:500, Novus Biologicals) in 5% normal goat serum and 1% BSA in PBS overnight at 4°C. Slices were then washed twice with 0.1 m PBS, followed by one wash of 0.2% Triton-X (Fisher Bioreagents) in 0.1 m PBS for 30 min, one wash of 1% BSA and 5% normal goat serum in 0.1 m PBS for 2 h, and a final application of anti-sheep (1:500, AlexaFluor546, Invitrogen) secondary antibody in 5% normal goat serum, 1% BSA in PBS for 2 h at room temperature. Slices were washed three times with Tris-buffered saline and mounted onto Superfrost Plus microscope slides (VWR). After drying overnight, slides were coverslipped with Fluoromount G (Southern Biotechnology) and imaged on an Olympus FluoView FV1000 laser scanning confocal microscope. Image capture was performed by sequential excitation of each fluorophore.
Solutions and pharmacological agents.
Picrotoxin (100 μm), and capsazepine (10 μm) were purchased from Abcam. APV (50 μm), AM-251 (2 μm), 2-AG (50 μm), R(+)methanandamide (50 μm), GDPβS (500 μm), DHPG (100 μm), Win55,212-2 (10 μm), THL (10 μm), 2-methyl-6(phenylethynyl)pyridine (MPEP), and RHC80267 (10 μm) were purchased from Tocris Bioscience. CNQX (10 μm) was purchased from Alomone Labs. THC was purchased from Sigma-Aldrich or provided by the National Institute on Drug Abuse Drug Supply Program. THC dissolved in ethanol was administered via intraperitoneal injection at 10 mg/kg, with a final injection of volume of 50 μl. Recordings began 24 h following the final injection. Vehicle controls included 0.1% EtOH and 0.1% DMSO. AM-251, 2-AG, and MPEP were diluted into DMSO in stock solutions of 20 mm, 125 μm, and 100 μm, respectively. All stock solutions were frozen at −20 C; the final concentration was attained by dilution in ACSF.
Results
Critical to the success of this study was to positively identify VTA GABA neurons for experiments. This was done by using a GAD67-GFP mouse model where GAD67-expressing GABA cells fluoresce green (Tamamaki et al., 2003). This was necessary as electrophysiological methods, such as Ih and sag potential that many prior studies used to discriminate the relatively fewer VTA GABA cells, are now considered fallible in their ability to discriminate GABA cells from DA cells (Margolis et al., 2006, 2012; Chieng et al., 2011). Therefore, this study is one of the few unequivocal examinations of VTA GABA cell plasticity. To determine the ability of VTA GABA cells to exhibit synaptic plasticity of their excitatory inputs, we applied either a 5 Hz or 100 Hz high-frequency stimulus (HFS) to voltage-clamped GAD67-GFP-positive GABA neurons in the mouse VTA. Whereas 5 Hz stimulation produced LTD of EPSC amplitude in ∼30% of cells (n = 7; data not shown), we observed a more consistent LTD following HFS in ∼85% of GABA cells (Fig. 1A). Therefore, HFS was used throughout these studies. As LTD had a slow onset, we performed a set of rundown experiments to ensure that LTD was not simply the result of depressed currents due to an experimental variable of whole-cell configuration that occurred in the absence of HFS. Control rundown experiments did not exhibit depression, and proved significantly different from HFS-LTD (Fig. 1B), suggesting that LTD was not an artifact of our experimental setup. As a note, a few cells slightly potentiated for 1–5 min before depressing, thus contributing to the slow onset appearance. To determine whether this plasticity was presynaptic or postsynaptic, coefficient of variance (1/CV2), PPRs, and failure rates were analyzed (Fig. 1C–E). 1/CV2 and PPRs are an analytical means to examine data comparing evoked EPSCs during baseline with those after HFS. 1/CV2 examines variation around the mean, which is decreased when LTD is presynaptic. PPRs examine the ratio of the second pulse divided by the first. As presynaptic LTD depresses the first pulse more than the second pulse, an increased PPR is indicative of presynaptic plasticity. Increased failure rates also suggest a presynaptic site of action, as it is more difficult to induce transmitter release after HFS. Whereas individually these results are suggestive of presynaptic LTD, collectively, these data strongly indicate a presynaptic mechanism for the observed plasticity. Additional controls demonstrate no effect of vehicle exposure on baseline and that currents recorded from are glutamatergic (Fig. 1F–H). EtOH (Fig. 1F) is a control of THC bath application and DMSO (Fig. 1G) for 2-AG, AM-251, and MPEP. Last, to confirm that VTA GFP cells are not dopaminergic, we performed immunohistochemistry on mouse VTA and note independent labeling of GFP (GAD67) and TH (Fig. 1I).
Presynaptic plasticity is often mediated by eCBs. Therefore, we examined the application of a nonhydrolysable form of the eCB anandamide, R(+)methanandamide, to see whether it would also induce depression of EPSCs, similar to that seen by HFS-induced LTD. Indeed similar to HFS, R(+)methanandamide induced significant depression (Fig. 2A). The usual mediator of eCB production is Type I mGluR activation. To test this, we applied the Type I mGluR agonist DHPG and once again observed significant depression (Fig. 2B). In the presence of the mGluR5 antagonist MPEP, HFS-LTD was blocked, suggesting that mGluR5 is required and sufficient for this plasticity (Fig. 2C). To further confirm that a postsynaptic GPCR such as mGluR was involved, a GDP analog, GDPβS, was added to the internal solution to block postsynaptic g-protein activation (Fig. 2D). After waiting at least 10 min for GDPβS to dialyze into the cell, HFS was applied. In the presence of GDPβS, HFS-LTD was inhibited, suggesting that postsynaptic GPCRs are necessary to initiate LTD and that the g-protein signal cascade involved is directly mediated by GABA cells. Collectively, these data support an eCB/mGluR5 pathway of LTD induction.
Figure 2.

eCB and mGluR5 involvement in VTA GABA cell plasticity. The eCBs are signaling molecules often involved in presynaptic plasticity mechanisms. A, A nonhydrolysable form of an eCB, R(+)methanandamide, induces depression of glutamatergic neurotransmission (n = 6, p ≪ 0.05). B, The Type 1 mGluR agonist DHPG also induced depression, similar to HFS-LTD (n = 7, p ≪ 0.05). C, The specific mGluR5 antagonist MPEP blocked HFS-LTD (n = 6; p < 0.05; compared to control LTD). D, To further test for the necessity of a postsynaptic GPCR (such as an mGluR) involvement in LTD, the GDP analog GDPβS, which inhibits G-proteins, was added to the internal solution and resulted in blocked HFS-LTD (n = 7; p > 0.05; compared to its baseline).
Among the candidate receptors known to bind eCBs and induce plasticity are TRPV1 and CB1. The TRPV1 antagonist capsazepine did not block HFS-induced LTD, suggesting that TRPV1 is not involved (Fig. 3A). However, application of the CB1 antagonist AM-251 effectively blocked LTD (Fig. 3B), suggesting that CB1 is required for HFS-LTD.
Figure 3.

CB1 mediates HFS-LTD. Two candidate receptors that could act as a presynaptic target for eCBs include TRPV1 and CB1. A, The TRPV1 antagonist capsazepine failed to block LTD, suggesting that TRPV1 is not involved in this HFS-LTD (n = 9; p > 0.05 compared with control LTD). B, The CB1 antagonist AM-251 significantly blocked LTD (n = 9, p < 0.05 compared with control LTD).
To verify CB1 involvement, we applied CB1 agonists without HFS to see whether agonist could induce a depression mimicking LTD. Win 55,212-5, an exogenous CB1 agonist, induced depression of ESPCs (Fig. 4A). To further confirm the involvement of eCBs and CB1 in HFS-LTD, occlusion experiments were done in which the cells were treated with Win55,212-2, then HFS was applied. Further depression by HFS was occluded, suggesting a single, common pathway of CB1 activation and HFS-induced LTD (Fig. 4A). The specificity of Win 55,212-2 for CB1 is noted as AM-251 blocked Win 55,212-2-induced depression (Fig. 4B). The endogenous agonist of CB1, 2-AG, also induced similar depression of EPSCs (Fig. 4C).
Figure 4.

Postsynaptically produced 2-AG mediates LTD of VTA GABA cells. A, Application of the CB1 agonist Win55,212-2 depresses glutamate transmission at this synapse (n = 8). There is a significant depression between baseline and drug application (p < 0.05), but further HFS-induced depression was occluded (arrow; p > 0.5; WIN55,212-2 depression compared with post-HFS, ANOVA). B, When the CB1 antagonist AM-251 was present, Win55,212-2 caused no significant depression, demonstrating the specificity of Win 55,212-2 for CB1 (n = 5, p < 0.05 compared with Win 55,212-2 alone). C, Application of endogenous CB1 ligand 2-AG also induced significant depression (n = 9; p < 0.05 at both 15–20 and 20–25 min). D, When the DAGLα inhibitor RHC 80267 was applied to block postsynaptic production of 2-AG, HFS failed to produce a significant depression, suggesting that 2-AG is produced postsynaptically in the GABA neuron (n = 6, p > 0.5). E, To control for drug specificity, we applied a second DAGL antagonist, THL (10–20 μm) to the bath for at least 30–60 min before HFS. THL significantly blocked HFS-induced LTD (p < 0.05 compared with control LTD, n = 7). F, To further tie mGluR activation to CB1-induced LTD, we applied DHPG in the presence of CB1 antagonist AM-251 (2 μm). AM-251 significantly blocked DHPG-induced depression (n = 8; p < 0.05, DHPG-induced depression compared with DHPG-induced depression in the presence of AM-251 at 15–20 and 20–25 min after DHPG application; compare with Fig. 2B). G, H, DSE was used to further test for postsynaptic eCB production. VTA GABA neurons were depolarized to 0 mV for 10 s (arrowhead) to induce DSE. We observed a persistent depression following the DSE protocol (n = 7, p < 0.05), which was CB1-dependent, as it was blocked by AM-251 (n = 7; p < 0.05, DSE compared with DSE with AM-251).
To determine whether 2-AG was the endogenous eCB mediating LTD itself, and whether its production is postsynaptic, RHC 80267, a confirmed diacylglycerol lipase (DAGL) antagonist (Edwards et al., 2006) was applied to the intracellular solution to block this enzyme, which produces 2-AG. After whole-cell acquisition, RHC 80267 was allowed to dialyze into the cell for at least 10 min. When postsynaptic DAGL was blocked, no significant depression was seen following HFS (Fig. 4D). To confirm for drug specificity, an additional DAGL antagonist THL was applied. Bath application of THL (10 μm) for at least 30–60 min also resulted in a significant (p < 0.05) inhibition of LTD (Fig. 4E). Next, to tie Type I mGluR activation to CB1-induced LTD, we applied DHPG in the presence of CB1 antagonist AM-251. AM-251 blocks most of the DHPG-induced depression, which is significantly (p < 0.05) different from DHPG alone (Fig. 4F; DHPG-induced depression was 16.5% with AM-251 present and 47.1% in its absence at 10–15 min after DHPG application). Blocking HFS-LTD by postsynaptically inhibiting 2-AG synthesis (Fig. 4D) suggests that the 2-AG involved in HFS-LTD is made directly by the GABA cell. To further confirm that eCBs are being produced postsynaptically in GABA cells, we examined an additional type of plasticity unique to CB1: DSE. We observed a significant depression following a 10 s depolarization to 0 mV, which was surprisingly persistent throughout the duration of the recording (Fig. 4G). AM-251 blocked this depression (Fig. 4H), suggesting that VTA GABA cells can produce the eCBs that can bind to CB1 in this type of plasticity.
To further verify CB1 involvement using a genetic method, we used CB1 knock-out mice and heterozygous littermates. HFS continued to produce a significant depression in CB1+/− mice (Fig. 5A); however, LTD was not observed in CB1−/− (Fig. 5B). Collectively, our data strongly suggest that CB1 is required for HFS-LTD.
Figure 5.

HFS-LTD is blocked in CB1 knock-out mice. A, HFS induced a significant LTD in heterozygous mice compared with baseline (n = 7, p < 0.05). B, In CB1−/−, HFS failed to produce LTD, further demonstrating the importance of CB1 in this plasticity (n = 9, p < 0.05 compared with CB1+/−).
As CB1 can be activated by THC, this novel form of plasticity may have relevance to THC-induced reward or addictive phenomena, such as cannabis use disorder. Therefore, we next investigated whether the observed eCB-dependent HFS-LTD could be modified by drug exposure. Extracellular bath application of THC produced a significant depression of VTA GABA EPSCs (Fig. 6A), and this effect was blocked in the presence of AM-251 (Fig. 6B), demonstrating THC specificity for CB1. In CB1+/− mice, extracellular THC also produced depression (Fig. 6C), but not in CB1−/− mice (Fig. 6D), again confirming CB1 specificity for THC. We then determined whether acute THC bath application could occlude or alter HFS-LTD. Indeed, bath application with THC for at least 10 min occluded HFS-LTD (Fig. 6E). Therefore, THC acutely depresses GABA cell excitability, as well as occludes LTD induction.
Figure 6.

THC induces CB1-dependent depression. A, THC is the psychoactive component in marijuana, and a known CB1 agonist. When THC is applied to the extracellular bath, it produced a lasting depression (n = 6, p < 0.05). B, The CB1 antagonist AM-251 significantly blocked THC-induced depression (n = 6, p < 0.05, compared with THC without AM251). C, CB1 knock-out and heterozygous mice were also used to confirm CB1 involvement, where THC induced depression in heterozygous mice (n = 7, p < 0.05 compared with baseline). D, In CB1 KO mice, THC failed to produce significant depression (n = 9, p < 0.05; compared with THC in heterozygote mice). E, THC occluded HFS-LTD, suggesting that THC and HFS-LTD use the same pathway (p < 0.05 compared with control LTD).
As drugs of abuse often cause long-term synaptic modifications, we examined the role of acute and chronic THC injections on this LTD. To do this, juvenile-adolescent aged mice received either a single or chronic (7–10 consecutive days) THC intraperitoneal injections to study its effect on synaptic modification. We anticipated that a single THC injection might not have a long-term effect on plasticity, although chronic injections might, as long-term marijuana users are prone to THC-induced health issues. Indeed, a single injection failed to occlude LTD (Fig. 7A). However, chronic injections occluded LTD in slices (Fig. 7B), and in a few cells even tended to enhance EPSCs or reverse plasticity direction. Experiments from chronic vehicle ethanol control injections continued to exhibit LTD (Fig. 7C). This suggests that persistent, long-term THC exposure can modify plasticity at the glutamatergic inputs to VTA GABA cells of juvenile-adolescent age mice that may contribute to some of the long-term consequences of marijuana use.
Figure 7.
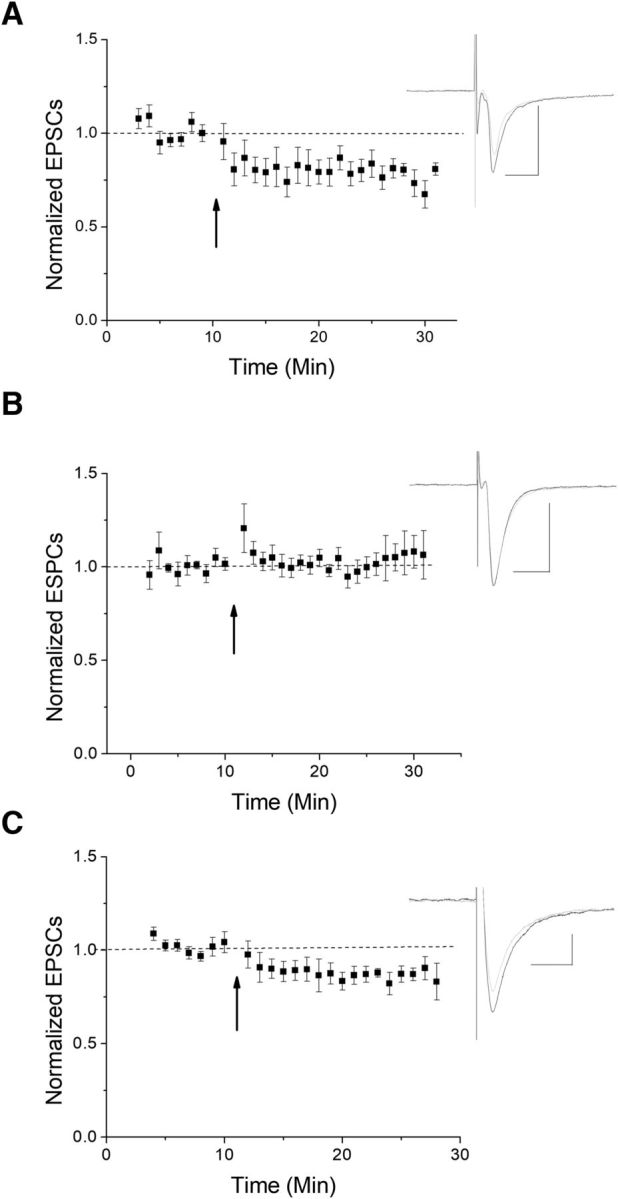
Chronic THC occludes HFS-LTD. We examined the effects of administering THC via intraperitoneal injections. A, A single THC injection failed to occlude LTD (n = 9 from 7 mice; p < 0.05 compared with baseline at 10–15 and 15–20 min). B, Chronic injections (7–10 consecutive days) of THC occluded LTD following HFS (n = 6 from 5 mice, p < 0.05, compared with vehicle only injections). C, Chronic injections of EtOH vehicle as a control demonstrate continued LTD (n = 6 from 5 mice, p < 0.05, compared with chronic THC).
Discussion
This study demonstrates a novel CB1-dependent LTD of excitatory inputs to VTA GABA cells, as summarized in Figure 8, which can be modified by intraperitoneally injected THC. HFS-LTD is induced by CB1 activation that is mediated by postsynaptically produced 2-AG, formed by DAGL via mGluR5 activation. THC induces synaptic depression of excitatory synapses onto VTA GABA cells when applied acutely and occludes it when injected chronically. Therefore, this novel form of plasticity has the potential to influence acute reward and long-term potential addictiveness following THC exposure.
Figure 8.
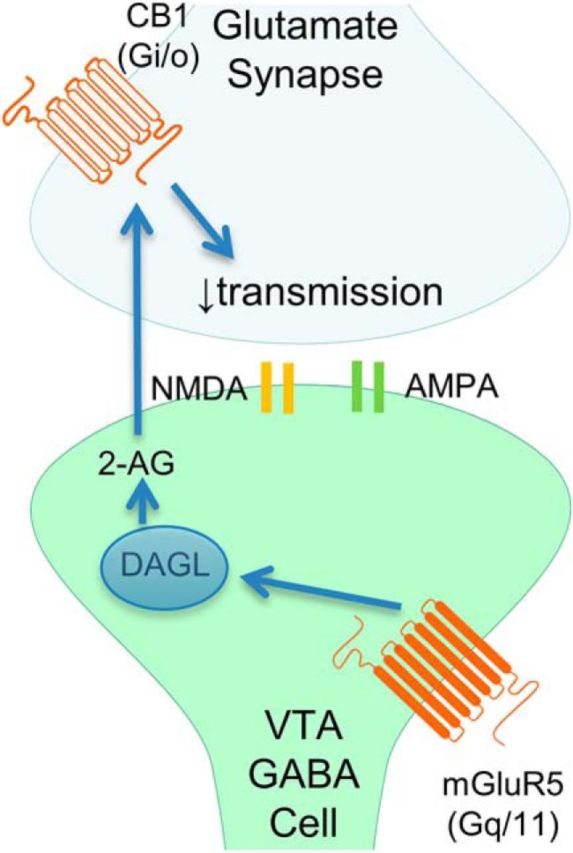
Current working model. HFS-LTD is induced by CB1 activation that is mediated by postsynaptically produced 2-AG, which is formed by DAGL and mGluR5 activation to reduce presynaptic neurotransmitter release.
CB1/mGluR-dependent GABA cell plasticity
To date, no one has fully studied excitatory plasticity of VTA GABA cells, despite the fact that GABA cells are involved in reward consumption (van Zessen et al., 2012), associative learning of reward (Brown et al., 2012), conditioned place preference (Bocklisch et al., 2013), and conditioned place aversion (Tan et al., 2012). The fact that GABA cells are critical to reward illustrates the importance of this study, which is the first to identify synaptic plasticity of excitatory inputs to VTA GABA cells that reduces GABA cell activity. Although one prior study examined plasticity of excitatory inputs to GABA cells, they used a pairing protocol (10 mV, 200 stimuli at 1 Hz) that failed to induce either LTP or LTD (Bonci and Malenka, 1999). However, this pairing protocol is normally used to examine NMDA-dependent plasticity. The plasticity we observed is mGluR-dependent, which accounts for the noted difference. We report that using a HFS protocol (2 trains of 100 Hz) depresses EPSCs of VTA GABA neurons in a CB1-dependent manner. This plasticity was presynaptic as 1/CV2 decreased (Del Castillo and Katz, 1954), PPR increased, and failure rate increased (Malinow and Tsien, 1990), which collectively strongly suggest presynaptic expression. Using failure rates examines changes specifically to evoked release versus spontaneous release. In addition, CB1-dependent plasticity is usually presynaptic (Castillo et al., 2012). As an important note, the slow LTD onset is real as control experiments in the absence of HFS were stable. Indeed, slow-onset LTD has been noted in mouse hippocampal GABAergic cells previously (Gibson et al., 2008) and may be a feature of inhibitory cells. The data also highlight that there are likely more eCB receptors at this synapse than just CB1 as R-(+) methanandamide and DHPG produce a larger effect than CB1 agonists alone, although CB1 is necessary for HFS-induced LTD. The former point was confirmed by the fact that AM-251 only partially blocked DHPG-induced depression, also suggesting that mGluR activation is indeed tied to CB1-induced depression. Forms of mGluR-dependent LTD are also a feature of VTA DA neurons. The mGluR1 induces LTD at excitatory synapses onto DA neurons, which reverses cocaine-induced strengthening of excitatory synapses (Bellone and Lüscher, 2006) mediated by exchanging GluR2-lacking AMPA receptors for those containing GluR2 (Mameli et al., 2007). However, the plasticity we examined is mGluR5 dependent. Whereas mGluR1 could still be involved in a postsynaptic or another plasticity form in VTA GABA cells, due to the large DPHG-induced depression noted, mGluR5 is required for presynaptic CB1-dependent LTD. DHPG-inducible eCB/CB1-dependent LTD at inhibitory inputs to DA neurons was demonstrated where cocaine enables a normally ineffective stimulus to evoke LTD of IPSCs (Pan et al., 2008, 2011; Yu et al., 2013). Therefore, whereas in VTA DA cells, CB1 induces IPSC-LTD; in VTA GABA cells, CB1 induces EPSC-LTD. Therefore, our data demonstrate that mGluR/CB1-dependent plasticity is a feature shared by both VTA GABA and DA cells.
A good cellular correlate to compare VTA GABA cells is the striatum (Lovinger and Mathur, 2012), where plasticity is strikingly similar. For example, LTD occurs at glutamatergic inputs onto striatal GABAergic medium spiny neurons, which is dependent on presynaptic CB1 and postsynaptic mGluR activation and eCB synthesis (Adermark et al., 2009; Lovinger, 2010), providing support for our findings. Finally, VTA DA cells exhibit CB1-dependent DSE where EPSCs are reduced for ∼1 min following depolarization to 0 mV (Melis et al., 2004). VTA GABA cells also exhibit DSE, which indicate that GABA cells can modulate their own activity by producing eCBs rather than relying on DA cells for their production. This DSE was surprisingly long-lasting; although we have no current rationale, it is not due to input resistance changes following a return in holding potential to −65 mV.
The importance of GABA cell involvement in reward, either directly through axonal projects to regions outside the VTA or indirectly with local GABA cells regulating VTA DA cell activity, highlights the critical nature in understanding GABA cell modification by synaptic plasticity, a necessary element in understanding VTA reward mechanisms. Directly, GABA cells in the VTA project to the NAc where they enhance associated reward learning (Brown et al., 2012) and to the tegmental pedunculopontine nucleus where they mediate the DA-independent reinforcing properties of opioids (Ting-A-Kee and van der Kooy, 2012). Indirectly, decreased local GABA cell activity results in increased VTA DA cell activity via disinhibition (Bocklisch et al., 2013), and conversely increased GABA cell activity inhibits DA cell activity and DA release (Tan et al., 2012; van Zessen et al., 2012), altering reward behavior. These prior studies demonstrate that local VTA GABA cells do indeed innervate and regulate VTA DA neuron activity. Therefore, local GABA neurons can regulate DA cell activity; thus, our studies could directly correlate to changes in DA levels. It is also important to note that other GABAergic sources, including rostromedial tegmental nucleus (Matsui and Williams, 2011) and NAc (Brown et al., 2012; Edwards et al., 2017) innervate and regulate DA cell activity and should be considered. Collectively, this study fills a missing piece of our understanding of reward physiology and suggests that this novel LTD could potentially modulate reward behavior. Further confirming this possibility is that VTA DA cells are under tonic inhibition from VTA GABAergic cells, including inputs from the recently identified rostromedial tegmental nucleus (Barrot et al., 2012), making any change in GABA function pertinent to DA cell function, as only phasic, or brief inhibition or excitation of DA cells mimics negative reward (Chang et al., 2016) or positive behavioral reward conditioning (Tsai et al., 2009), respectively. Clearly, GABA cells projecting outside the VTA or within the VTA play crucial roles in mesolimbic circuitry, similar to direct excitatory inputs to DA neurons.
Reward
Regarding THC-induced reward, acute exposure directly modifies dopaminergic cells by increasing firing rate. This occurs via presynaptic CB1 receptors on GABA inputs to DA cells, which disinhibit DA cells (Johnson et al., 1992; Tan et al., 2012). Our current study proposes a novel mechanism, one synapse upstream in the circuit, namely, that THC reduces excitatory input to GABA cells and thus decreases inhibition of VTA GABA cells to VTA DA cells. Therefore, exogenous THC could simultaneously alter both synapses synergistically, resulting in increased DA release and reward.
Addiction
To date, all addictive psychoactive substances examined induce long-term changes of excitatory synapses onto VTA DA neurons, whereas nonaddictive psychoactive substances do not (Mameli and Lüscher, 2011). This finding portends the importance of understanding forms of glutamatergic VTA GABA plasticity and their potential role in addiction. Drug-evoked or drug-inhibited plasticity alters VTA circuits leading to drug addiction by creating drug-adaptive behaviors (Lüscher and Malenka, 2011). Although the addictive nature of THC is debatable, alterations in the mesolimbic pathway following chronic THC exposure are particularly relevant for adolescents. Indeed, human adolescent marijuana use correlates to decreased IQ and cognitive impairment (Meier et al., 2012), cortex development issues, schizophrenia (Volkow et al., 2014), alterations in NAc (Gilman et al., 2014), and increased probability for long-term substance abuse (Hurd et al., 2014; Verweij et al., 2016). Therefore, studies examining THC impact on adolescent brain are critical. The mice age range used here correlates to juvenile-adolescent ages, making this study of potential importance as to THC effects on immature brain, which could have implications in adolescent-related increases in substance abuse.
Regarding acute and chronic THC use, while a single THC injection was sufficient to alter plasticity in the NAc (Mato et al., 2004), we noted that a single THC injection had no effect on CB1-dependent LTD of VTA GABA cells. However, chronic THC did occlude LTD, suggesting this occlusion mechanism responds to chronic or repeated usage. As during cannabis withdrawal, where there is a decrease in mesolimbic DA (Oleson and Cheer, 2012), it is possible that THC-occluded LTD prevents GABA cells from reducing their activity, potentially contributing to some of decreased DA levels in withdrawal. Chronic THC use outcomes were also recently reviewed (Colizzi et al., 2016). The plasticity described here is a similar mechanism to previously reported acute/chronic synaptic modifications; however, ours is the first study to identify this in VTA GABA neurons.
Significance
Marijuana use and legalization is a current “hot button” issue. Although marijuana is the most commonly used illicit drug, the implications of legalized, widespread, or continued usage are speculative. After alcohol and opiate-related substance abuse, marijuana surprisingly ranks third for those entering substance abuse treatment programs (SAMHSA survey; Substance Abuse and Mental Health Services Administration, 2010), portending the importance of understanding THC's effect on the brain's reward system. In addition, it appears that THC has a more aversive impact on adolescents than adults, and marijuana use is becoming increasingly prevalent among adolescents in the United States. Therefore, an understanding of how THC effects the adolescent immature brain is essential. Current state trends of marijuana legalization may increase usage and associated sequelae (Cerdá et al., 2012), thus increasing demand for studies regarding its effects on reward, addiction, and cognition. Even THC use for medical purposes is important to understand, particularly when prescribed for adolescents. This study in juvenile-adolescent aged mice emphasizes the vital role VTA GABA cells play in reward circuitry, and the synaptic remodeling that can occur after THC use.
In conclusion, the present study demonstrates a novel form of plasticity on VTA GABA neurons, an understudied cell population in the brain's reward circuit. This study examines a novel and unifying mechanism whereby marijuana could exert some of its rewarding or addictive/withdrawal effects. This is important as, although marijuana abuse has a high use prevalence among illicit drugs (Compton and Baler, 2016), conditions, such as cannabis use disorder, remain difficult to treat partly because its cognitive altering mechanisms are poorly understood (Elkashef et al., 2008). Therefore, as those attempting to overcome addiction often relapse, novel targets to treat addiction are essential. Indeed, GABA neurons make for promising targets for novel addiction treatments (Bocklisch et al., 2013), as their excitatory transmission can be remodeled, similar to DA neurons.
Note Added in Proof: The last author's name was accidentally incorrectly listed in the Early Release version published October 16, 2017. The author's name has now been corrected.
Footnotes
This work was supported by National Institute of Neurological Diseases and Stroke Grant R15NS078645 and National Institute of Drug Abuse Grant R15DA038092. This work was also supported by institutional Mentoring Environment Grants to J.G.E. and Brigham Young University Graduate Fellowship Awards to L.F. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Neurological Disorders and Stroke, National Institute of Drug Abuse, or the National Institutes of Health. We thank National Institute on Drug Abuse Drug Supply Program for some of the THC used in this study; Dr. Scott Steffensen for the use of GAD67-GFP mice; and Dr. Ken Mackie for providing the CB1 knock-out mice.
The authors declare no competing financial interests.
References
- Adermark L, Talani G, Lovinger DM (2009) Endocannabinoid-dependent plasticity at GABAergic and glutamatergic synapses in the striatum is regulated by synaptic activity. Eur J Neurosci 29:32–41. 10.1111/j.1460-9568.2008.06551.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrot M, Sesack SR, Georges F, Pistis M, Hong S, Jhou TC (2012) Braking dopamine systems: a new GABA master structure for mesolimbic and nigrostriatal functions. J Neurosci 32:14094–14101. 10.1523/JNEUROSCI.3370-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone C, Lüscher C (2006) Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci 9:636–641. 10.1038/nn1682 [DOI] [PubMed] [Google Scholar]
- Berridge KC, Robinson TE (1998) What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Brain Res Rev 28:309–369. 10.1016/S0165-0173(98)00019-8 [DOI] [PubMed] [Google Scholar]
- Bocklisch C, Pascoli V, Wong JC, House DR, Yvon C, de Roo M, Tan KR, Lüscher C (2013) Cocaine disinhibits dopamine neurons by potentiation of GABA transmission in the ventral tegmental area. Science 341:1521–1525. 10.1126/science.1237059 [DOI] [PubMed] [Google Scholar]
- Bonci A, Malenka RC (1999) Properties and plasticity of excitatory synapses on dopaminergic and GABAergic cells in the ventral tegmental area. J Neurosci 19:3723–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MT, Tan KR, O'Connor EC, Nikonenko I, Muller D, Lüscher C (2012) Ventral tegmental area GABA projections pause accumbal cholinergic interneurons to enhance associative learning. Nature 492:452–456. 10.1038/nature11657 [DOI] [PubMed] [Google Scholar]
- Castillo PE, Younts TJ, Chávez AE, Hashimotodani Y (2012) Endocannabinoid signaling and synaptic function. Neuron 76:70–81. 10.1016/j.neuron.2012.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerdá M, Wall M, Keyes KM, Galea S, Hasin D (2012) Medical marijuana laws in 50 states: investigating the relationship between state legalization of medical marijuana and marijuana use, abuse and dependence. Drug Alcohol Depend 120:22–27. 10.1016/j.drugalcdep.2011.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CY, Esber GR, Marrero-Garcia Y, Yau HJ, Bonci A, Schoenbaum G (2016) Brief optogenetic inhibition of dopamine neurons mimics endogenous negative reward prediction errors. Nat Neurosci 19:111–116. 10.1038/nn.4191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chieng B, Azriel Y, Mohammadi S, Christie MJ (2011) Distinct cellular properties of identified dopaminergic and GABAergic neurons in the mouse ventral tegmental area. J Physiol 589:3775–3787. 10.1113/jphsiol.2011.210807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colizzi M, McGuire P, Pertwee RG, Bhattacharyya S (2016) Effect of cannabis on glutamate signalling in the brain: a systematic review of human and animal evidence. Neurosci Biobehav Rev 64:359–381. 10.1016/j.neubiorev.2016.03.010 [DOI] [PubMed] [Google Scholar]
- Compton WM, Baler R (2016) The epidemiology of DSM-5 cannabis use disorders among U.S. adults: science to inform clinicians working in a shifting social landscape. Am J Psychiatry 173:551–553. 10.1176/appi.ajp.2016.16030314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristino L, Starowicz K, De Petrocellis L, Morishita J, Ueda N, Guglielmotti V, Di Marzo V (2008) Immunohistochemical localization of anabolic and catabolic enzymes for anandamide and other putative endovanilloids in the hippocampus and cerebellar cortex of the mouse brain. Neuroscience 151:955–968. 10.1016/j.neuroscience.2007.11.047 [DOI] [PubMed] [Google Scholar]
- Del Castillo J, Katz B (1954) Quantal components of the end-plate potential. J Physiol 124:560–573. 10.1113/jphysiol.1954.sp005129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D (1994) Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 372:686–691. 10.1038/372686a0 [DOI] [PubMed] [Google Scholar]
- Edwards DA, Kim J, Alger BE (2006) Multiple mechanisms of endocannabinoid response initiation in hippocampus. J Neurophysiol 95:67–75. 10.1152/jn.00813.2005 [DOI] [PubMed] [Google Scholar]
- Edwards NJ, Tejeda HA, Pignatelli M, Zhang S, McDevitt RA, Wu J, Bass CE, Bettler B, Morales M, Bonci A (2017) Circuit specificity in the inhibitory architecture of the VTA regulates cocaine-induced behavior. Nat Neurosci 20:438–448. 10.1038/nn.4482 [DOI] [PubMed] [Google Scholar]
- Elkashef A, Vocci F, Huestis M, Haney M, Budney A, Gruber A, el-Guebaly N (2008) Marijuana neurobiology and treatment. Subst Abus 29:17–29. 10.1080/08897070802218166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaoni Y, Mechoulam R (1964) Isolation, structure, and partial synthesis of an active constituent of hashish. J Am Chem Soc 86:1646 10.1021/ja01062a046 [DOI] [Google Scholar]
- Gibson HE, Edwards JG, Page RS, Van Hook MJ, Kauer JA (2008) TRPV1 channels mediate long-term depression at synapses on hippocampal interneurons. Neuron 57:746–759. 10.1016/j.neuron.2007.12.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman JM, Kuster JK, Lee S, Lee MJ, Kim BW, Makris N, van der Kouwe A, Blood AJ, Breiter HC (2014) Cannabis use is quantitatively associated with nucleus accumbens and amygdala abnormalities in young adult recreational users. J Neurosci 34:5529–5538. 10.1523/JNEUROSCI.4745-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heifets BD, Castillo PE (2009) Endocannabinoid signaling and long-term synaptic plasticity. Annu Rev Physiol 71:283–306. 10.1146/annurev.physiol.010908.163149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd YL, Michaelides M, Miller ML, Jutras-Aswad D (2014) Trajectory of adolescent cannabis use on addiction vulnerability. Neuropharmacology 76:416–424. 10.1016/j.neuropharm.2013.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iñiguez SD, Aubry A, Riggs LM, Alipio JB, Zanca RM, Flores-Ramirez FJ, Hernandez MA, Nieto SJ, Musheyev D, Serrano PA (2016) Social defeat stress induces depression-like behavior and alters spine morphology in the hippocampus of adolescent male C57BL/6 mice. Neurobiol Stress 5:54–64. 10.1016/j.ynstr.2016.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, Mercuri NB, North RA (1992) 5-Hydroxytryptamine1B receptors block the GABAB synaptic potential in rat dopamine neurons. J Neurosci 12:2000–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung KM, Mangieri R, Stapleton C, Kim J, Fegley D, Wallace M, Mackie K, Piomelli D (2005) Stimulation of endocannabinoid formation in brain slice cultures through activation of group I metabotropic glutamate receptors. Mol Pharmacol 68:1196–1202. 10.1124/mol.105.013961 [DOI] [PubMed] [Google Scholar]
- Lassen MB, Brown JE, Stobbs SH, Gunderson SH, Maes L, Valenzuela CF, Ray AP, Henriksen SJ, Steffensen SC (2007) Brain stimulation reward is integrated by a network of electrically coupled GABA neurons. Brain Res 1156:46–58. 10.1016/j.brainres.2007.04.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Zhao Y, Yang J, Zhang X, Zhang W, Wang P (2017) Neonatal repeated exposure to isoflurane not sevoflurane in mice reversibly impaired spatial cognition at juvenile-age. Neurochem Res 42:595–605. 10.1007/s11064-016-2114-7 [DOI] [PubMed] [Google Scholar]
- Lovinger DM. (2010) Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology 58:951–961. 10.1016/j.neuropharm.2010.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, Mathur BN (2012) Endocannabinoids in striatal plasticity. Parkinsonism Relat Disord 18 [Suppl 1]:S132–S134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Malenka RC (2011) Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron 69:650–663. 10.1016/j.neuron.2011.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Tsien RW (1990) Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature 346:177–180. 10.1038/346177a0 [DOI] [PubMed] [Google Scholar]
- Mameli M, Lüscher C (2011) Synaptic plasticity and addiction: learning mechanisms gone awry. Neuropharmacology 61:1052–1059. 10.1016/j.neuropharm.2011.01.036 [DOI] [PubMed] [Google Scholar]
- Mameli M, Balland B, Luján R, Lüscher C (2007) Rapid synthesis and synaptic insertion of GluR2 for mGluR-LTD in the ventral tegmental area. Science 317:530–533. 10.1126/science.1142365 [DOI] [PubMed] [Google Scholar]
- Margolis EB, Lock H, Hjelmstad GO, Fields HL (2006) The ventral tegmental area revisited: is there an electrophysiological marker for dopaminergic neurons? J Physiol 577:907–924. 10.1113/jphysiol.2006.117069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Toy B, Himmels P, Morales M, Fields HL (2012) Identification of rat ventral tegmental area GABAergic neurons. PLoS One 7:e42365. 10.1371/journal.pone.0042365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli S, Pascucci T, Bernardi G, Puglisi-Allegra S, Mercuri NB (2005) Activation of TRPV1 in the VTA excites dopaminergic neurons and increases chemical-and noxious-induced dopamine release in the nucleus accumbens. Neuropsychopharmacology 30:864–870. 10.1038/sj.npp.1300615 [DOI] [PubMed] [Google Scholar]
- Mato S, Chevaleyre V, Robbe D, Pazos A, Castillo PE, Manzoni OJ (2004) A single in-vivo exposure to delta 9THC blocks endocannabinoid-mediated synaptic plasticity. Nat Neurosci 7:585–586. 10.1038/nn1251 [DOI] [PubMed] [Google Scholar]
- Matsui A, Williams JT (2011) Opioid-sensitive GABA inputs from rostromedial tegmental nucleus synapse onto midbrain dopamine neurons. J Neurosci 31:17729–17735. 10.1523/JNEUROSCI.4570-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mátyás F, Urbán GM, Watanabe M, Mackie K, Zimmer A, Freund TF, Katona I (2008) Identification of the sites of 2-arachidonoylglycerol synthesis and action imply retrograde endocannabinoid signaling at both GABAergic and glutamatergic synapses in the ventral tegmental area. Neuropharmacology 54:95–107. 10.1016/j.neuropharm.2007.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride WJ, Murphy JM, Ikemoto S (1999) Localization of brain reinforcement mechanisms: intracranial self-administration and intracranial place-conditioning studies. Behav Brain Res 101:129–152. 10.1016/S0166-4328(99)00022-4 [DOI] [PubMed] [Google Scholar]
- Meier MH, Caspi A, Ambler A, Harrington H, Houts R, Keefe RS, McDonald K, Ward A, Poulton R, Moffitt TE (2012) Persistent cannabis users show neuropsychological decline from childhood to midlife. Proc Natl Acad Sci U S A 109:E2657–E2664. 10.1073/pnas.1206820109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL (2004) Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci 24:53–62. 10.1523/JNEUROSCI.4503-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill CB, Friend LN, Newton ST, Hopkins ZH, Edwards JG (2015) Ventral tegmental area dopamine and GABA neurons: physiological properties and expression of mRNA for endocannabinoid biosynthetic elements. Sci Rep 5:16176. 10.1038/srep16176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleson EB, Cheer JF (2012) A brain on cannabinoids: the role of dopamine release in reward seeking. Cold Spring Harb Perspect Med 2:a012229. 10.1101/cshperspect.a012229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Hillard CJ, Liu QS (2008) Endocannabinoid signaling mediates cocaine-induced inhibitory synaptic plasticity in midbrain dopamine neurons. J Neurosci 28:1385–1397. 10.1523/JNEUROSCI.4033-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Zhong P, Sun D, Liu QS (2011) Extracellular signal-regulated kinase signaling in the ventral tegmental area mediates cocaine-induced synaptic plasticity and rewarding effects. J Neurosci 31:11244–11255. 10.1523/JNEUROSCI.1040-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn HR, Matsumoto I, Callaghan PD, Long LE, Arnold JC, Gunasekaran N, Thompson MR, Dawson B, Mallet PE, Kashem MA, Matsuda-Matsumoto H, Iwazaki T, McGregor IS (2008) Adolescent rats find repeated Delta(9)-THC less aversive than adult rats but display greater residual cognitive deficits and changes in hippocampal protein expression following exposure. Neuropsychopharmacology 33:1113–1126. 10.1038/sj.npp.1301475 [DOI] [PubMed] [Google Scholar]
- Rodriguez JS, Boctor SY, Flores LC, Phelix CF, Martinez JL Jr (2011) Local pretreatment with the cannabinoid CB1 receptor antagonist AM251 attenuates methamphetamine intra-accumbens self-administration. Neurosci Lett 489:187–191. 10.1016/j.neulet.2010.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovira V, Geijo-Barrientos E (2016) Intra- and interhemispheric propagation of electrophysiological synchronous activity and its modulation by serotonin in the cingulate cortex of juvenile mice. PLoS One 11:e0150092. 10.1371/journal.pone.0150092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MJ, Cobia DJ, Reilly JL, Gilman JM, Roberts AG, Alpert KI, Wang L, Breiter HC, Csernansky JG (2015) Cannabis-related episodic memory deficits and hippocampal morphological differences in healthy individuals and schizophrenia subjects. Hippocampus 25:1042–1051. 10.1002/hipo.22427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Substance Abuse and Mental Health Services Administration (2010) Results from the 2010 National Survey on Drug Use and Health: Summary of national findings. Office of Applied Statistics, NSDUH Series H-41, HHS Publication No. (SMA) 11-4658, Rockville, MD. [Google Scholar]
- Tamamaki N, Yanagawa Y, Tomioka R, Miyazaki J, Obata K, Kaneko T (2003) Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol 467:60–79. 10.1002/cne.10905 [DOI] [PubMed] [Google Scholar]
- Tan KR, Yvon C, Turiault M, Mirzabekov JJ, Doehner J, Labouèbe G, Deisseroth K, Tye KM, Lüscher C (2012) GABA neurons of the VTA drive conditioned place aversion. Neuron 73:1173–1183. 10.1016/j.neuron.2012.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimura A, Yamazaki M, Hashimotodani Y, Uchigashima M, Kawata S, Abe M, Kita Y, Hashimoto K, Shimizu T, Watanabe M, Sakimura K, Kano M (2010) The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase alpha mediates retrograde suppression of synaptic transmission. Neuron 65:320–327. 10.1016/j.neuron.2010.01.021 [DOI] [PubMed] [Google Scholar]
- Ting-A-Kee R, van der Kooy D (2012) The neurobiology of opiate motivation. Cold Spring Harb Perspect Med 2:a012096. 10.1101/cshperspect.a012096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HC, Zhang F, Adamantidis A, Stuber GD, Bonci A, de Lecea L, Deisseroth K (2009) Phasic firing in dopaminergic neurons is sufficient for behavioral conditioning. Science 324:1080–1084. 10.1126/science.1168878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A (2001) Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 411:583–587. 10.1038/35079077 [DOI] [PubMed] [Google Scholar]
- van Zessen R, Phillips JL, Budygin EA, Stuber GD (2012) Activation of VTA GABA neurons disrupts reward consumption. Neuron 73:1184–1194. 10.1016/j.neuron.2012.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verweij KJ, Creemers HE, Korhonen T, Latvala A, Dick DM, Rose RJ, Huizink AC, Kaprio J (2016) Role of overlapping genetic and environmental factors in the relationship between early adolescent conduct problems and substance use in young adulthood. Addiction 111:1036–1045. 10.1111/add.13303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Baler RD, Compton WM, Weiss SR (2014) Adverse health effects of marijuana use. N Engl J Med 370:2219–2227. 10.1056/NEJMra1402309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RA. (2008) Dopamine and reward: the anhedonia hypothesis 30 years on. Neurotox Res 14:169–183. 10.1007/BF03033808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LT, Zhu H, Swartz MS (2016) Trends in cannabis use disorders among racial/ethnic population groups in the United States. Drug Alcohol Depend 165:181–190. 10.1016/j.drugalcdep.2016.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yohn NL, Blendy JA (2017) Adolescent chronic unpredictable stress exposure is a sensitive window for long-term changes in adult behavior in mice. Neuropsychopharmacology 42:1670–1678. 10.1038/npp.2017.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Zhong P, Liu X, Sun D, Gao HQ, Liu QS (2013) Metabotropic glutamate receptor I (mGluR1) antagonism impairs cocaine-induced conditioned place preference via inhibition of protein synthesis. Neuropsychopharmacology 38:1308–1321. 10.1038/npp.2013.29 [DOI] [PMC free article] [PubMed] [Google Scholar]