

Abstract
We critically assess the proposal that succinate-fuelled reverse electron flow (REF) drives mitochondrial matrix superoxide production from Complex I early in reperfusion, thus acting as a key mediator of ischemia/reperfusion (IR) injury. Real-time surface fluorescence measurements of NAD(P)H and flavoprotein redox state suggest that conditions are unfavourable for REF during early reperfusion. Furthermore, rapid loss of succinate accumulated during ischemia can be explained by its efflux rather than oxidation. Moreover, succinate accumulation during ischemia is not attenuated by ischemic preconditioning (IP) despite powerful cardioprotection. In addition, measurement of intracellular reactive oxygen species (ROS) during reperfusion using surface fluorescence and mitochondrial aconitase activity detected major increases in ROS only after mitochondrial permeability transition pore (mPTP) opening was first detected. We conclude that mPTP opening is probably triggered initially by factors other than ROS, including increased mitochondrial [Ca2+]. However, IP only attenuates [Ca2+] increases later in reperfusion, again after initial mPTP opening, implying that IP regulates mPTP opening through additional mechanisms. One such is mitochondria-bound hexokinase 2 (HK2) which dissociates from mitochondria during ischemia in control hearts but not those subject to IP. Indeed, there is a strong correlation between the extent of HK2 loss from mitochondria during ischemia and infarct size on subsequent reperfusion. Mechanisms linking HK2 dissociation to mPTP sensitisation remain to be fully established but several related processes have been implicated including VDAC1 oligomerisation, the stability of contact sites between the inner and outer membranes, cristae morphology, Bcl-2 family members and mitochondrial fission proteins such as Drp1.
Abbreviations: 5-cH2DCFDA, 5-carboxy-2′,7′dichlorodihydrofluorescein diacetate; ANT, adenine nucleotide translocase; Bad, Bcl-2-associated death promoter; Bak, Bcl-2 homologous antagonist/killer; Bax, Bcl-2-like protein 4; Bcl-2, B cell lymphoma 2; BCECF, 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein; Bcl-xL, B-cell lymphoma-extra large; Bid, BH3 interacting-domain death agonist; CK, creatine kinase; CrP, creatine phosphate; CsA, cyclosporine A; Cyp-D, cyclophilin D; DHE, dihydroethidium; Drp1, dynamin-related protein 1; G-6-P, glucose-6-phosphate; GSK3β, glycogen synthase kinase 3β; HK, hexokinase; IF1, ATP synthase inhibitor factor 1; IMM, inner mitochondrial membrane; IP, ischemic preconditioning; IR, ischemia/reperfusion; Mdivi-1, mitochondrial division inhibitor 1; LS, light scattering; MCU, mitochondrial calcium uniporter; MitoPY1, Mitochondria Peroxy-Yellow 1; mPTP, mitochondrial permeability transition pore; NCLX, Na+/Ca2+ exchanger; OMM, outer mitochondrial membrane; PCr, phosphocreatine; PKCε, protein kinase Cε; pmf, proton motive force; PO1, Peroxy-Orange 1; REF, reverse electron flow; ROS, reactive oxygen species; SOD, superoxide dismutase; VDAC, voltage-dependent anion channel
Keywords: Succinate, Ischemia/reperfusion injury, Heart, Mitochondria, Reactive oxygen species, Calcium, Permeability transition pore, Hexokinase
Graphical abstract
Highlights
-
•
Mitochondrial ROS production during early reperfusion (RPF) occurs after mPTP opening.
-
•
Conditions do not favour ROS production by reverse electron flow during early RPF.
-
•
Ischemic preconditioning (IP) does not attenuate succinate accumulation in ischemia.
-
•
IP reduction of ROS and Ca2+ during RPF is secondary to attenuation of mPTP opening.
-
•
IP attenuates mPTP opening by mechanisms independent of ROS and Ca2+ such as HK2.
1. Introduction
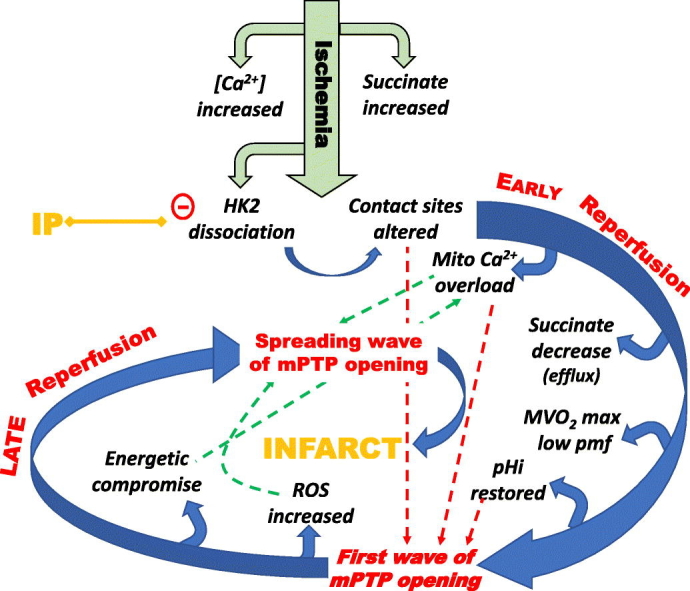
It is now widely accepted that the irreversible injury of the heart which occurs during reperfusion after a prolonged period of ischemia is mediated, at least in part, by opening of the mitochondrial permeability transition pore (mPTP) [1], [2]. This starts after about 2 min of reperfusion [3], [4] and coincides with the intracellular pH (pHi) returning to pre-ischemic values from the low values (< 6.5) that are reached after prolonged ischemia and which inhibit mPTP opening [5]. Opening of the mPTP not only compromises cellular bioenergetics, thus impairing the re-establishment of pre-ischemic levels of ions such as calcium, but also causes mitochondria to release reactive oxygen species (ROS) [6], [7]. The resulting increases in ROS and [Ca2+] induce further mPTP opening and bioenergetic compromise leading to a spreading wave of necrotic cell death that produces the infarct [1], [8]. Pharmacological inhibition of mPTP opening by cyclosporin A (CsA) [9], sanglifehrin A [10] or cinnamic anilides [11] can protect hearts from ischemia/reperfusion (IR) injury as can ischemic preconditioning (IP) and post-conditioning, both of which also inhibit mPTP opening [1], [8].
Although the precise molecular composition of the mPTP remains uncertain [2], [12], [13], it is well established that increases in matrix [Ca2 +] trigger mPTP opening and that its sensitivity to [Ca2+] is greatly enhanced by oxidative stress, elevated phosphate and decreased matrix adenine nucleotides [1]. These are all conditions associated with IR. Indeed, the combination of increased matrix [Ca2+] with oxidative stress is now widely regarded as the main trigger for mPTP opening during early reperfusion [1], [2]. Oxidative stress within the matrix is an especially attractive candidate since this overcomes the powerful inhibitory effect of adenine nucleotides on mPTP opening leading to a profound increase in Ca2+ sensitivity [14]. Furthermore, several studies, including our own, have shown oxidative stress to increase during reperfusion and to be greatly attenuated by a variety of protocols that induce cardioprotection including IP [15], [16], temperature preconditioning (TP) [17], urocortin [18], apomorphine [19], ischemic post-conditioning [20] and sequential exposure to isoproterenol and adenosine [21]. However, it is also well established that increased superoxide production can occur as a consequence of mPTP opening [6], [7], caused in part by the loss of cytochrome c and NADPH from the mitochondria, both of which are important for ROS scavenging [6], [22]. Thus, it is important to establish whether increased levels of ROS precede mPTP opening during early reperfusion or occur later as a consequence of mPTP opening.
Recently, Murphy, Krieg and colleagues have presented extensive data to implicate superoxide production from the matrix surface of Complex I early in reperfusion as a key player in IR injury [23], [24], [25], [26]. They propose that this superoxide production occurs because succinate accumulates in the heart during ischemia and is rapidly oxidised by reverse electron flow (REF) at the start of reperfusion. This induces a highly reduced state of the ubiquinone binding site on the matrix face of Complex I that drives superoxide production [23]. Here we critically assess the role of succinate-mediated superoxide production from Complex I in IR injury and conclude that it is unlikely to be the primary trigger of mPTP opening in the early phase of reperfusion and which is modulated by IP. Rather, we suggest that it is elevated [Ca2+] that initiates mPTP opens on reperfusion and that IP attenuates other factors that sensitise the mPTP to [Ca2+], including the well-established dissociation of hexokinase 2 (HK2) from its mitochondrial binding site that occurs during ischemia [27], [28], [29]. However, substantial ROS production does occur later in reperfusion as a consequence of initial mPTP opening, and this leads to further pore opening and an expanding area of necrotic cell death that forms the infarct. Cardioprotective protocols such as IP prevent HK2 loss from mitochondria during ischemia and so prevent both phases of mPTP opening.
2. Does mitochondrial superoxide production precede mPTP opening during reperfusion?
2.1. ROS measurements
The American Heart Association has recently published a “Scientific Statement” on the measurement of ROS species which provides a comprehensive review of the available methods, their limitations and what combined approaches are recommended for particular situations [30]. As this article makes abundantly clear, measurement of ROS species is not straight forward, and although many different methods can be used, each approach is fraught with potential pitfalls for the unwary. Some of these issues are noted in the discussion below, but the main focus of this section is to provide a critical review of the data relating the time course of ROS formation in the ischemic/reperfused heart to the time course of mPTP opening.
2.1.1. Studies using isolated cardiac myocytes
Studies using isolated adult cardiac myocytes subject to simulated ischemia and reperfusion have provided evidence that ROS production precedes mPTP opening and cell death [23], [31], [32], [33]. However, to simulate ischemia, these studies employed bicarbonate-free media and anoxia together with low pH, with or without the addition of l-lactate, followed by return to normal medium (still bicarbonate free) to mimic reperfusion. In such studies, the cardiomyocytes are usually quiescent or at best stimulated to beat at very low frequency and it is questionable whether these conditions adequately reproduce those occurring in the intact ischemic/reperfused heart. In the beating perfused heart there will be a much higher metabolic turnover and Ca2+ cycling rates than in isolated cardiac myocytes with the consequence that mitochondria will be in a different redox and bioenergetic state. This may reduce both their ability to accumulate Ca2+ and produce ROS. Furthermore, the concentration of myocytes in the heart, and their complex interactions with each other and endothelial cells, cannot be adequately reproduced when using isolated myocytes for fluorescence microscopy. Nor can the build-up and subsequent washout of metabolites that occurs in the ischemic reperfused heart, while the absence of bicarbonate will disrupt normal pH regulatory mechanisms. In addition, the studies with cardiac myocytes [32], [33] generally used much longer periods of simulated ischemia than are routinely employed in studies with the perfused heart and thus are difficult to compare with data obtained using the ischemic/reperfused heart. A more recent study using dihydroethidium (DHE) as the superoxide-sensitive dye has measured ROS production after 1–2 min of simulated reperfusion [23], but major concerns have been expressed over the use of DHE as a ROS probe, both in terms of whether the fluorescent species monitored really detects superoxide [34] and its sensitivity to changes in mitochondrial membrane potential [35] as discussed further below (Section 2.1.2). For these reasons, we suggest that any conclusions drawn from isolated cardiomyocyte experiments must be treated with caution, especially where they differ those obtained using the perfused heart.
2.1.2. Studies using the perfused heart
Measurements of ROS production/oxidative stress have been made in the ischemic/reperfused heart, but the majority of these studies lacked the time resolution to establish whether ROS increases before or after mPTP opening. Thus determinations of protein carbonylation [15], [16], [17], [19], [20], [21], [24], protein thiolation [36], lipid peroxidation [37], [38], [39] and aconitase activity [23] have all clearly demonstrated that oxidative stress does occur during reperfusion, but do not provide information on whether there is a significant increase in ROS within the first minute or two, prior to initial mPTP opening. The same is true for the measurement of mitochondrial superoxide using the mitochondrial targeted hydrogen peroxide probe, mito B, assayed using mass spectrometry of freeze clamped tissue. Here the earliest time point reported was at 15 min [23], [24]. More rapid detection of increased ROS production has been obtained using hearts perfused with the electron paramagnetic resonance (EPR) free radical probe 5,5-dimethyl-1-pyroline-n-oxide (DMPO) where peak concentrations of radical formation were detected after 10–20 s of reperfusion followed by a gradual decline over the next 1–2 min [40]. It was demonstrated that these radicals were derived from superoxide since reperfusion in the presence of enzymatically active recombinant human superoxide dismutase (SOD) markedly reduced the formation of the relevant EPR signals while inactive SOD had no effect. However, the effect of SOD implies that the location of the superoxide detected was extracellular since otherwise it would not be attenuated by membrane-impermeable SOD. In a separate set of experiments in which hearts were rapidly freeze-clamped and endogenous radicals measured using EPR, a similar time course of radical formation was detected [41] which is more likely to reflect intracellular superoxide formation over this time period. Indeed, measurements of the reversible glutathionylation of glyceraldehyde 3 phosphate dehydrogenase did detect an increase within 1 min of reperfusion after 30 min ischemia [42]. The luminescent probe lucigenin (N,N′-dimethyl-9,9′-biacridinium dinitrate) also detected increases in superoxide within a minute or so of reperfusion, but these only reached a peak at about 5 min [35] which is after mPTP opening [3], [4]. It has been suggested that the positive charge on this probe may lead to its preferential uptake and retention by mitochondria and some evidence for this has been presented [35]. However, others have argued that its permeability into cells is an issue and that a significant proportion of the signal may represent extracellular superoxide production [30].
Surface fluorescence of the Langendorff-perfused heart has also been used to monitor ROS production in real time during IR. There are significant problems associated with this approach including motion artefacts, internal filtering of the incident and emitted light by cytochromes, flavoproteins and myoglobin, and autofluorescence by NAD(P)H and flavoproteins. We have discussed these issues more fully elsewhere [7]. Of particular concern is that both autofluorescence and internal filtering change dramatically in the transition between normoxia and ischemia as myoglobin becomes deoxygenated and the cytochromes and flavoproteins become reduced. Although these effects rapidly reverse on reperfusion, it is only when this process is complete that reliable conclusions can be drawn from the crude fluorescent signals. This is a particular problem for dihydroethidium (DHE) which has been employed as a superoxide-specific fluorescent probe in several perfused heart studies [35], [43], [44], [45], [46], [47], [48], [49], [50]. DHE itself is not fluorescent but once oxidised by superoxide to 2-hydoxyethidium it fluoresces at > 600 nm when excited at ~ 535 nm. Unfortunately, this makes the probe especially sensitive to changes in myoglobin oxygenation and the redox state of cytochromes. Indeed, such effects probably account for the very rapid increase and decrease in fluorescence seen upon ischemia and reperfusion respectively [43]. This makes it very difficult to assess whether rapid changes in ROS are occurring early in reperfusion. Additional concerns have been expressed over the use of DHE as a ROS probe, both in terms of whether the fluorescent species monitored reflects superoxide [30], [34] and its sensitivity to changes in mitochondrial membrane potential [35]. Nevertheless, in the perfused heart slower increases in signal are seen after about 3–4 min of reperfusion that are prevented by IP [43] and would seem likely to reflect changes in ROS as revealed by our own studies with alternative probes described below.
We have recently published extensive data from experiments in which we continuously measured ROS production in the beating heart subject to IR using a bespoke surface fluorescence apparatus to monitor the surface fluorescence at multiple wavelengths with high time resolution [7]. Simultaneous measurements were made of hemodynamic function together with surface reflectances at both the excitation and emission wavelengths of all probes used. The latter measurements allowed monitoring of potential artefacts caused by changes in surface geometry (such as occurs during ischemic contracture), although in the optical configuration employed it was found that such artefacts were minimal. We also determined the fluorescence emission spectra for each dye loaded within the heart at various times of ischemia and reperfusion to assess the effects of internal filtering caused by myoglobin, cytochromes and flavoproteins. These measurements revealed that all fluorescent probes were likely to be subject to interference by internal filtering during the first 10–15 s of ischemia and reperfusion, although the use of the ROS-insensitive dye, calcein, monitored under the same conditions, allowed some estimate of these effects to be made. Measurements after 10–15 s of reperfusion were not subject to major artefacts and thus provided robust data. Three different ROS probes were employed to detect changes during reperfusion in control hearts and those subject to IP. These were 5-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (5-cH2DCFDA), a generic ROS probe whose exact specificity is uncertain [51], and two caged boronate probes that are specific for hydrogen peroxide: Peroxy-Orange 1 (PO1) which measures primarily cytosolic hydrogen peroxide and Mitochondria Peroxy-Yellow 1 (MitoPY1) that is targeted to mitochondria. In parallel we measured aconitase activity in mitochondria isolated after 90 s reperfusion as an additional indicator of mitochondrial ROS. As shown in Fig. 1, both 5-cH2DCFDA and PO1 revealed a similar pattern of ROS production; in both cases no significant increase in ROS was detected until after about 1.5–2 min of reperfusion. By measuring the loss of calcein entrapped within the mitochondria we confirmed that this was after mPTP opening had occurred [7]. The progressive increase in ROS observed after about 2 min of reperfusion was attenuated by both IP and CsA which also reduced infarct size and inhibited mPTP opening as detected previously using mitochondrial 2-deoxyglucose entrapment [52] and more recently with calcein entrapment [7]. Thus our data with both 5-cH2DCFDA and PO1 suggest that these probes only detected ROS production after initial mPTP opening. We were also unable to detect any increase in mitochondrial ROS at 90 s of reperfusion using either the mitochondria-targeted hydrogen peroxide probe MitoPY1 or aconitase activity measurements [7]. As noted before, both aconitase activity and the superoxide probe mitoB did detect an increase in mitochondrial ROS after 15 min reperfusion [23], but again this is after mPTP opening and thus may well be a consequence of mPTP opening rather than a primary cause.
Fig. 1.
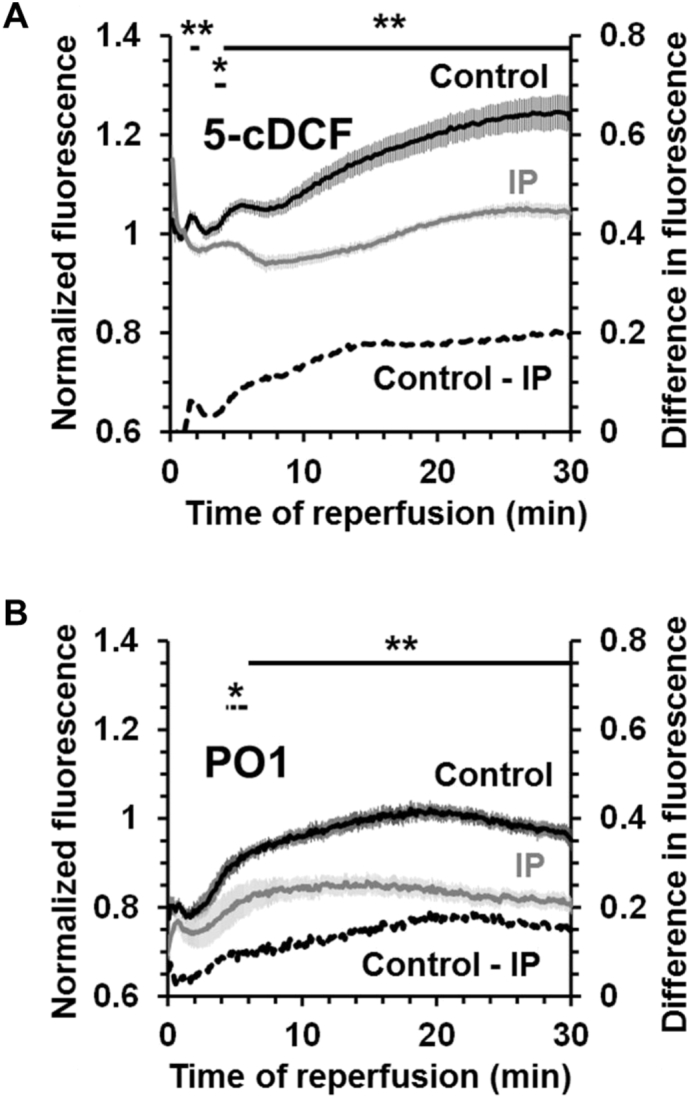
Time course of ROS production measured using surface fluorescence in the Langendorff-perfused heart subject to ischemia and reperfusion. Both control and IP hearts were pre-loaded with either 5-cH2DCFDA as its di-(acetoxymethyl ester) or PO1 and then subject to 30 min index ischemia followed by reperfusion for the time shown. Data are expressed as means ± SEM (error bars) for 6 (5-cH2DCFDA) or 9–11 (PO1) separate heart perfusions and are taken from [7] where further details may be found. Significant differences between control and IP hearts, calculated by unpaired t-test, are shown by horizontal lines (*p < 0.05; **p < 0.01).
Overall, we suggest that ROS probes such as DHE, DMPO and lucigenin that report increases in ROS levels within the first minute of reperfusion are probably not detecting ROS within the mitochondria. As such they provide no strong evidence for IP-sensitive increases in superoxide within the mitochondrial matrix early in reperfusion that could stimulate mPTP opening. It may well be that some of these probes are responding to either extracellular or cytosolic superoxide produced by enzymes such as NADPH oxidase, uncoupled nitric oxide synthase, monoamine oxidase and xanthine oxidase [53], [54], [55], [56]. Cytosolic superoxide is likely to be rapidly scavenged by SOD, glutathione peroxidase and glutathione reductase [57], [58] before it can enter the mitochondrial matrix as suggested by the rapid increase in glutathionylated proteins such as glyceraldehyde 3 phosphate dehydrogenase [42].
2.2. Effects of ROS scavengers
If an increase in mitochondrial superoxide production in the matrix is a key trigger for mPTP opening early in reperfusion, then intramitochondrial ROS scavengers (reviewed in [59]) should be cardioprotective. Mitochondria-targeted Szeto-Schiller peptides (also known as Bendavia) do reduce ROS production by isolated mitochondria [60] and are mildly cardioprotective in some models of IR injury [58], [60], [61], [62], [63], [64]. However, exactly how these agents work is unclear since they do not act as scavengers of defined reactive oxygen species produced in vitro [60] and thus are not acting as true ROS scavengers. The mitochondria-targeted ubiquinone derivative MitoQ is an established superoxide scavenger [65] and there is a single report of its ability to protect hearts from IR injury [66]. However, these data were obtained in a whole animal model in which rats were treated for 14 days with MitoQ in their drinking water before induction of myocardial ischemia. Thus, it cannot be ruled out that cardioprotection was secondary to longer term effects of MitoQ and not directly via mitochondrial ROS scavenging in the heart itself. Indeed, studies from this laboratory failed to demonstrate cardioprotection by MitoQ in the Langendorff-perfused heart [67]. Furthermore, in the many studies that have investigated the potential cardioprotective effects of a range of ROS scavengers during reperfusion, most have proved relatively ineffective at preventing IR injury in both the clinical setting and in vivo or ex vivo animal models of IR (reviewed in [68], [69]). Nevertheless, transgenic mice overexpressing either catalase or glutathione peroxidase are somewhat resistant to IR injury [70], [71].
The lack of effect of some ROS scavengers may be partially explained if ROS also play a role in cardioprotective signalling pathways as has been suggested [69], [72]. However, even in those cases where ROS scavengers do provide cardioprotection, it does not follow that this is the result of an inhibition of the initial phase of mPTP opening. Rather, it could be because of more efficient scavenging of the ROS produced after initial mPTP opening and thus the prevention of an escalating cascade of further ROS-sensitised mPTP opening and injury. This may also be true for the cardioprotection offered by pyruvate [73], [74], [75] and the anaesthetic propofol [76], [77], both of which are ROS scavengers. Pyruvate exerts especially powerful cardioprotection, but this is thought to be because it not only acts as a ROS scavenger but also maintains a more acid pH during ischemia and reperfusion [74], [75].
3. Assessing the evidence that succinate-driven reverse electron flow mediates superoxide production at Complex I during reperfusion
Complex I can generate large amounts of superoxide in the matrix by two mechanisms: first, at the flavin mononucleotide (FMN) site when it is in a highly reduced state such as under conditions of high proton motive force (pmf) and low rates of ATP synthesis (State 4); second, at another less well defined site when electrons are donated to the ubiquinone (CoQ) pool by REF. The latter occurs under conditions of very high pmf, especially when the pH gradient component is large, and is caused by the highly reduced state of the CoQ pool forcing electrons back from CoQH2 into Complex I, thus reducing NAD+ to NADH at the FMN site [78], [79]. Since the proportion of the FMN in the fully reduced state is set by the NADH/NAD+ ratio, the former mechanism produces more ROS in the presence of rotenone whereas the latter mechanism is blocked by rotenone. Importantly, superoxide production by either mechanism will be associated with a high matrix NADH/NAD+ ratio and this can be measured in the intact heart using surface fluorescence as can the redox state of flavoproteins which usually follows that of the NADH/NAD+ ratio [80], [81]. We have monitored the rapid changes in both the NADH/NAD+ and flavoprotein redox state as the heart is reperfused following ischemia using real time surface fluorescence and our data do not provide any support for a role of Complex I in superoxide production in the early phase of reperfusion [7].
3.1. Time course of Complex I oxidation
At the onset of ischemia, both NAD(P)H and flavoproteins rapidly become totally reduced as all oxygen is consumed, and they stay reduced until reperfusion is commenced when they rapidly re-oxidise as illustrated in Fig. 2.
Fig. 2.
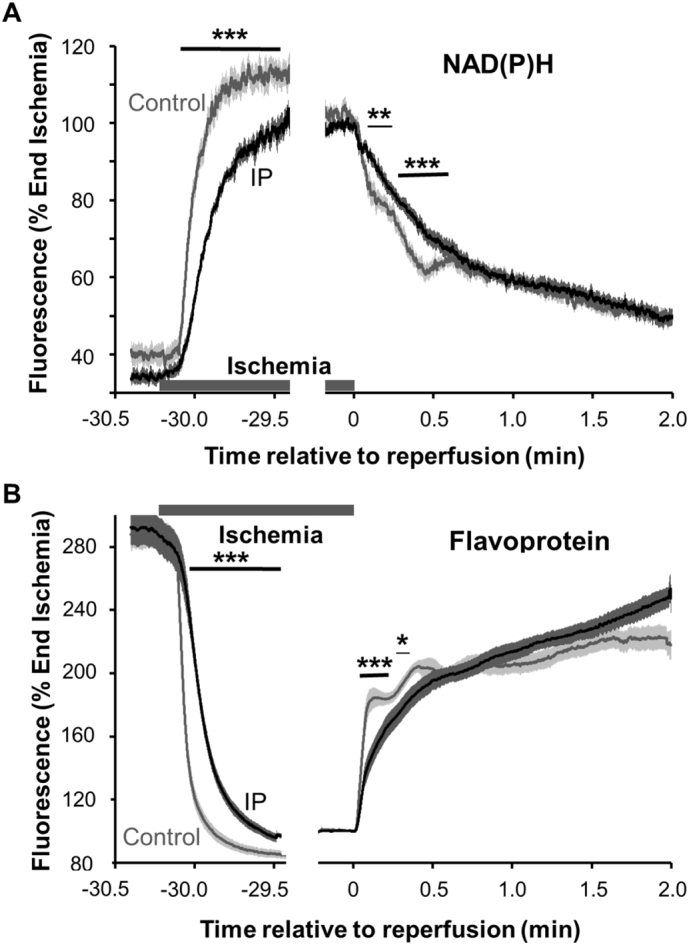
Time course of changes in flavoprotein and NAD(P)H surface fluorescence in the Langendorff-perfused heart subject to ischemia and reperfusion. Surface fluorescence data (normalised as % of values at the end of ischemia) are only shown for the start of ischemia (− 30 min) and reperfusion (0 min). Data are taken from [7] where further details may be found, and are presented as means ± SEM (error bars) of 7 control and IP hearts. Significant differences between control and IP hearts calculated by unpaired t-test are shown by horizontal lines (*p < 0.05; **p < 0.02, ***p < 0.01).
In control and IP hearts re-oxidation of the flavoproteins was 50% complete within 5 and 15 s respectively and that of NAD(P)H at about 15 and 25 s respectively. The rapid re-oxidation is not unexpected since during ischemia ATP and creatine phosphate (CrP) are almost totally exhausted and are rapidly regenerated over the first few seconds of reperfusion [22], [82]. Indeed, oxygen consumption during the first 30 s of reperfusion is fast [7], consistent with the rapid oxidation of NADH required to fuel the increased rates of oxidative phosphorylation. However, several features of these data argue against significant superoxide production early in reperfusion. First, the highly reduced state of Complex I required for superoxide production is not maintained for more than a few seconds. Second, the pmf is unlikely to reach the high values associated with superoxide formation because of the rapid flow of protons back into the matrix through the F1Fo ATPase required for ATP re-synthesis. Third, the rate of re-oxidation of both flavoproteins and NAD(P)H is slower in IP hearts than control hearts. This does not readily fit with the proposal that IP reduces superoxide production from Complex I during early reperfusion as would be expected if superoxide is the key initial trigger for mPTP opening. This slower re-oxidation in IP hearts, and indeed the slower reduction during the onset of ischemia (Fig. 2), could reflect IP-mediated S-nitrosation of ND3 Cys39 and possibly other Complex I cysteines which inhibit its activity [24], [83], [84], [85], [86]. Another possible explanation would be that IP enhances inhibition of the mitochondrial ATP synthase by its inhibitor IF1 during ischemia, as has been observed in some studies (see [87]). This might lead to slower activation of oxidative phosphorylation and respiration during the first seconds of reperfusion. However, neither explanation would readily account for a reduction in matrix superoxide production by IP early in reperfusion.
3.2. Succinate measurements
Chouchani et al. [23] confirmed earlier observations [88], [89] that succinate concentrations increase several-fold in ischemic hearts and rapidly decrease again on reperfusion. They proposed that this loss of succinate early in reperfusion reflects its oxidation by REF that drives ROS production at Complex I. As shown in Fig. 3A, we were able to confirm that tissue succinate levels increase many-fold following 30 min ischemia of Langendorff-perfused rat hearts, but importantly we found that a similar accumulation of succinate occurs in IP hearts during ischemia [7]. Since IP hearts exhibit less oxidative stress and mPTP opening after IR than control hearts [7], [15], [16], [52], [90], [91], this is not what might be anticipated if succinate oxidation early in reperfusion drives REF and consequent ROS formation at Complex I. It should also be noted that a study published nearly twenty years ago [92] demonstrated that perfusion of hearts with succinate protected them from IR injury rather than exacerbating injury as might be predicted if succinate plays an important role in early ROS formation. Furthermore, treatment of hearts with fumarate is also cardioprotective [93], [94] as is genetic knockdown of fumarate hydratase that causes fumarate accumulation of the heart [95], and both conditions are associated with increased succinate levels. In ischemia, fumarate and thus succinate are formed during the catabolism of aspartate and glutamate and this pathway can produce some ATP via succinate thiokinase mediated substrate level phosphorylation. The reduction of fumarate to succinate, mediated via NADH oxidation through Complex 1, ubiquinone and Complex 2 may also help maintain the mitochondrial pmf during ischemia. These two effects are probably responsible for some of the cardioprotection [93], [94], [95], [96]. Fumarate may exert an additional longer term effect through stabilization of the transcriptional regulator Nrf2, perhaps involving succination of its binding partner Keap1, that enhances the transcription of protective antioxidant genes [95].
Fig. 3.

IP does not attenuate succinate accumulation in ischemia while dimethyl malonate impairs the hemodynamic performance of the Langendorff-perfused heart.
Panel A shows the succinate content (nmol per mg dry weight; mean ± SEM, n = 6) of freeze clamped control (CI) and IP (IPI) hearts following 30 min global ischemia; values in normoxic hearts were below our limit of detection (< 0.5 nmol per mg). Data are taken from [7]. Panel B shows that the Rate Pressure Product (RPP) of normoxic Langendorff-perfused rat hearts was decreased by the presence of increasing concentrations of dimethyl malonate (DMM). Data are expressed as a percentage of values in the absence of DMM (29,900 ± 2800 mmHg*beat/min) and are given as means ± SEM (error bars) of 7 hearts. Statistical significance was determined by paired Student's t-test (*p < 0.01).
It should also be noted that the decrease in succinate observed during early reperfusion [23] was shown previously to be associated with its rapid efflux from the heart within 1–2 min of reperfusion [88] and therefore might not reflect its rapid oxidation as proposed by Chouchani et al. [23]. It is likely that the proton-linked monocarboxylate carrier, MCT1, which is highly expressed in the heart [97], facilitates this efflux of succinate since the intracellular pH drops to < 6.5 in the ischemic heart [98] and this will convert > 10% of succinate (pKa2 = 5.64) to its monocarboxylate form. It is known that short-chain substituted monocarboxylates are good substrates for MCT1 [99], and we have confirmed this to be the case for the monocarboxylate form of succinate by studying its uptake into Xenopus laevis oocytes expressing MCT1 as shown in Fig. 4. Significant proton-linked succinate transport was observed at pH 6.0, where 30% of succinate is in its monocarboxylate form, and this was blocked by AR-C155858, a specific and potent MCT1 inhibitor [100], whereas no detectable transport was observed at pH 7.4 where < 2% of succinate is present in its monocarboxylate form.
Fig. 4.

Succinate is transported by MCT1 as the monocarboxylate anion.
MCT1 was expressed in Xenopus laevis oocytes proton-linked monocarboxylate transport measured in real time using the pH-sensitive fluorescent dye BCECF as described previously [100]. Panel A shows that 30 mmol/L l-lactate (L) is transported into and out of Xenopus laevis oocytes expressing MCT1 at both pH 7.4 and 6.0 but not control oocytes. By contrast, 30 mmol/L succinate (S) is only transported after the external pH has been decreased to 6.0 consistent with its transport as a monocarboxylate (i.e. in the monoprotonated form). Panel B shows that transport of both succinate and l-lactate at pH 6.0 is blocked by prior exposure of the oocyte to 1 μmol/L AR-C155858, a specific MCT1 inhibitor [100].
3.3. Cardioprotective effects of succinate dehydrogenase inhibition
Additional evidence provided by Chouchani et al. to support the importance of succinate oxidation for ROS production in early reperfusion was the demonstration that inhibition of succinate dehydrogenase (SDH) by malonate (generated from membrane-permeable dimethyl malonate – DMM) reduced ROS production in both isolated cardiomyocytes and hearts subject to IR [23]. In cardiomyocytes subjected to simulated IR the authors showed that DMM caused a significant decrease in ROS after 2 min “reperfusion”. However, they employed DHE as a ROS probe, which, as discussed earlier, is now recognised to be unreliable [30], [34], [35]. Moreover, as noted above, simulated IR in cardiomyocytes does not accurately reproduce the situation in the whole heart subject to IR. In an in vivo mouse model of reperfusion, Chouchani et al. showed that DMM decreased infarct size which confirms earlier studies that employed 3-nitropropionate [101], [102] and diazoxide [103], [104], [105], [106], [107] to inhibit succinate dehydrogenase. Although the authors used both mitoB and aconitase activity to show that DMM reduced matrix superoxide levels, these measurements were made after 15 min of reperfusion [23]. Thus, they may well reflect events occurring after mPTP opening and be secondary to cardioprotection mediated by DMM through other mechanisms. One such mechanism would be inhibition of flux through the citric acid cycle which utilises SDH as an essential component. This would lead to an impairment of both fatty acid and carbohydrate oxidation which might compromise the energy status of the heart. Indeed, as discussed more fully below, cardioprotection can be induced by a range of other agents and protocols that compromise the energy status of the heart, including a range of respiratory chain inhibitors [108], uncouplers [109] and transient ischemia on reperfusion (post-conditioning) [110]. Furthermore, as shown in Fig. 3B, we have demonstrated that in the Langendorff-perfused heart, treatment with DMM, at similar concentrations to those employed by Chouchani et al. [23], does lead to a progressive reduction in the rate pressure product (RPP) as the DMM concentration increased. This is entirely consistent with DMM mediating its cardioprotective effects through bioenergetic compromise.
3.4. Cardioprotective effects of Complex I inhibitors
Several studies have demonstrated that inhibiting Complex I during ischemia can reduce ROS formation and IR injury. This is the case for rotenone [111], amobarbital [108], [112], [113], [114], metformin [115], [116], [117], [118] and most recently by S-nitrosation of ND3 on Complex I mediated by the mitochondrial-targeted mitoSNO [24], [119]. It has been argued by Murphy, Kreig and colleagues that the cardioprotective effect of Complex I inhibition reflects inhibition of ROS production at Complex I [25], [26]. However, a range of other agents and protocols that compromise the energy status of the heart through diverse mechanisms have been shown to be cardioprotective. These include uncouplers of oxidative phosphorylation [109], [120], F1Fo ATP synthase inhibitors such as oligomycin and aurovertin [121], and the adenine nucleotide translocase (ANT) inhibitors atractyloside and bongkrekic acid [104], [105], [106], [107], [122], [123]. In addition, transient ischemia on reperfusion (known as post-conditioning) is a well-established cardioprotective protocol [110], [124] and is associated with impaired respiration and slower restoration of intracellular pH during reperfusion. In all these cases the heart is forced to generate ATP by glycolysis producing lactic acid which will result in a low intracellular pH being maintained for longer during the early stages of reperfusion. Opening of the mPTP is inhibited at pH values below 7 [5] and maintaining the mPTP in a closed state for longer during early reperfusion may allow restoration of ionic homeostasis, preventing calcium overload and thus an escalating cascade of mPTP opening, ROS production and injury so accounting for cardioprotection [1], [125].
4. Succinate stimulates ROS production following mPTP opening
As noted above, increased superoxide production can occur as a consequence of mPTP opening and this may lead to further mPTP opening in adjacent mitochondria [6], [7]. Thus an alternative explanation for the decreased ROS production caused by DMM treatment of both isolated cardiomyocytes subject to simulated IR and in the in vivo ischemic reperfused hearts [23] would be that succinate enhances ROS production after, rather than before, mPTP opening. In Fig. 5 we demonstrate that this is the case. Isolated heart mitochondria were energised with glutamate + malate in the presence (GMS) or absence (GM) of succinate and continuous measurements made of light scattering (LS), mitochondrial membrane potential (Δψ), extramitochondrial [Ca2 +] and production of hydrogen peroxide (rapidly formed from superoxide by SOD) as described previously [7]. As noted by others [126], [127], [128], hydrogen peroxide production before Ca2+ addition was greatly enhanced by succinate and this has been shown to involve superoxide production at both Complex I and SDH. Sequential additions of 100 μM Ca2+ were rapidly taken up by the mitochondria with a progressive reduction in both Δψ and the rate of hydrogen peroxide production until the mPTP opened (rapid and total loss of Δψ, release of accumulated Ca2+ and a large LS decrease). Hydrogen peroxide production then increased once more, confirming the increased superoxide production that occurs after mPTP opening. This may reflect the loss of cytochrome c and NADPH from the mitochondria, both of which are important for ROS scavenging [6], [22]. Importantly, even after mPTP opening, rates of hydrogen peroxide production were considerably greater when succinate was present than in its absence as shown in Fig. 5B. Very recent data from Weiss's laboratory have also confirmed that succinate increases ROS production in heart mitochondria following mPTP opening [129]. This ROS was shown to be produced primarily at Complex II but might also involve Complex III if this were damaged during ischemia [130].
Fig. 5.

The [Ca2+]-sensitivity of mPTP opening in isolated heart mitochondria is reduced by succinate despite greater ROS production both before and after pore opening. Panel A shows the effects of sequential additions of Ca2+ (100 μmol/L) on isolated rat heart mitochondria incubated with 5 mmol/L l-glutamate + 2 mmol/L l-malate (GM) or GM plus 5 mmol/L succinate (GMS) on mPTP opening and ROS production. Opening of the mPTP was detected as a loss of Δψ (Rhd-123 fluorescence increase) or accumulated Ca2+ (Fura-FF fluorescence increase) and a decrease in LS while ROS production was monitored using Amplex Red in a parallel experiment on the same mitochondria. Further details may be found elsewhere [7]. Panel B presents mean rates of ROS production (± SEM of 6 different mitochondrial preparations) before Ca2 + addition, after the penultimate Ca2+ addition and following pore opening.
Another important aspect of the data we present in Fig. 5A is that many more additions of Ca2+ were required to induce mPTP opening in the presence of succinate than its absence, despite succinate inducing much higher rates of hydrogen peroxide production. These data are in agreement with those obtained by others using skeletal muscle mitochondria [131] and suggest that succinate oxidation may actually protect against mPTP opening despite the greater ROS production. This casts further doubt on the proposal of Chouchani et al. [23] that succinate induced ROS production is a critical mediator of IR injury but is supportive of earlier studies in which succinate was found to be cardioprotective [92].
5. Alternative triggers for mPTP opening early in reperfusion
Our review of the available evidence leads us to conclude that it is unlikely that an increase in matrix superoxide production at Complex I, driven by REF from succinate, is the critical trigger for mPTP opening in the initial phase of reperfusion. Rather, we suggest that the majority of ROS are produced later in reperfusion and that this occurs as a consequence of mPTP opening. This helps to drive a progressive cascade of damage to the cardiomyocytes, including further mPTP opening and ROS production as well as damage to proteins involved in ionic homeostasis and calcium handling [132]. The result is an expanding area of necrotic cell death that forms the infarct. If this conclusion is correct then there must be other factors that trigger the initial mPTP opening and that can be attenuated by cardioprotective protocols such as IP. We consider some of these below.
5.1. The role of Ca2 +
It is well established that calcium acts as an essential trigger for mPTP opening and that many other factors that enhance pore opening do so by increasing its sensitivity to [Ca2+] [1], [12]. Increases in matrix [Ca2+] have been demonstrated previously in both cardiac myocytes [133] and perfused hearts [134] subject to ischemia and reperfusion. We have recently confirmed that increases in mitochondrial [Ca2+] can be detected in the Langendorff-perfused heart after 30 min ischemia and 90 s of reperfusion [7]. It seems probable that this increase in matrix [Ca2+] is important for mPTP opening since hearts of mice with an adult cardiomyocyte-specific deletion of the mitochondrial calcium uniporter (MCU) were shown to exhibit reduced IR injury [135], [136]. These data confirmed earlier reports that the MCU inhibitors ruthenium red and Ru360 are cardioprotective [137], [138]. Furthermore, mice with heart-specific deletion of the mitochondrial Na+/Ca2+ exchanger (NCLX), which is responsible for most Ca2+ efflux from the matrix of heart mitochondria, experienced substantial mitochondrial Ca2+ overload. This was accompanied by increased generation of superoxide and necrotic cell death in the heart and led to death of the mice within a few days. However, the mice survived considerably longer if mitochondrial permeability transition pore activation was attenuated by additional knockout of Cyp-D. Conversely, overexpression of NCLX in the mouse heart enhanced mitochondrial Ca2+ efflux, prevented opening of the mPTP and protected against ischemia-induced cardiomyocyte necrosis and heart failure [139].
These data all confirm the importance of increased matrix [Ca2+] in triggering the mPTP opening that leads to increased ROS production and damage to the heart. However, we found no evidence that the cardioprotection afforded by IP was accompanied by any attenuation of this early rise in matrix [Ca2+], although IP did prevent the calcium overload that occurred later in reperfusion [7]. We confirmed that this later calcium overload was secondary to mPTP opening by demonstrating that it was prevented by pre-treatment of the hearts with CsA, a well-established mPTP inhibitor [7]. Others have come to a similar conclusion using an isolated myocyte model of IR [140]. It seems likely that the calcium overload induced by the initial phase of mPTP opening may act in conjunction with the parallel increases in ROS to cause additional mPTP opening in adjacent “unopened” mitochondria. This may mediate a progressive cascade of further ROS production and mPTP opening that will lead to a spreading area of necrotic cell death to form the infarct as discussed above. Indeed, waves of [Ca2+] and ROS that precede mPTP opening have been observed by confocal microscopy in the intact contraction-inhibited heart subject to hypoxia and re-oxygenation [141].
Overall, we conclude that, although IP attenuated the secondary calcium overload that follows mPTP opening, consistent with its ability to inhibit mPTP opening [7], [52], [90], [91], its inability to prevent the increase in matrix [Ca2+] that occurs in the first 90 s of reperfusion [7] implies that another factor must play a critical role in regulating this first phase of mPTP opening. One possible candidate is mitochondria-bound hexokinase 2 (HK2) whose extent of dissociation from mitochondria during ischemia correlates with infarct size and is prevented by IP [27], [28], [29].
5.2. The role of mitochondria-bound hexokinase 2
It has been known for many years that both hexokinase 1 (HK1) and hexokinase 2 (HK2) can bind to mitochondria and that such binding may play an important role in the regulation of hexokinase activity and thus of glucose metabolism [27], [142], [143]. However, more recently evidence has emerged that binding of these enzymes to the outer mitochondrial membrane (OMM) may also decrease apoptosis mediated by members of the Bcl2 family and antagonise mPTP opening [28]. Cardiac myocytes express both HK1 and HK2 and both bind to the OMM through an N-terminal hydrophobic domain but a significant proportion of HK2 is present in the cytosol [144], [145]. The extent of HK2 binding to mitochondria depends on the prevailing metabolic conditions with high concentrations of glucose-6-phosphate (G-6-P) favouring HK2 dissociation [143], [146]. Zuurbier and colleagues demonstrated a progressive loss of bound mitochondria-bound HK2 during ischemia of the heart [147] and we confirmed this, showing substantial loss of mitochondrial HK2, but not HK1, after 30 min global ischemia [148]. This was associated with a loss of cytochrome c from mitochondria suggesting enhanced permeability of the OMM. Furthermore, IP prevented both the dissociation of HK2 and the loss of cytochrome c [22] and also decreased the calcium sensitivity of mPTP by isolated mitochondria [16]. By manipulating the metabolism of the perfused heart prior to ischemia we were able to demonstrate [22] that there was a very strong inverse correlation between the amount of HK2 remaining bound to mitochondria at the end of ischemia and the infarct size after 120 min of reperfusion as shown in Fig. 6.
Fig. 6.
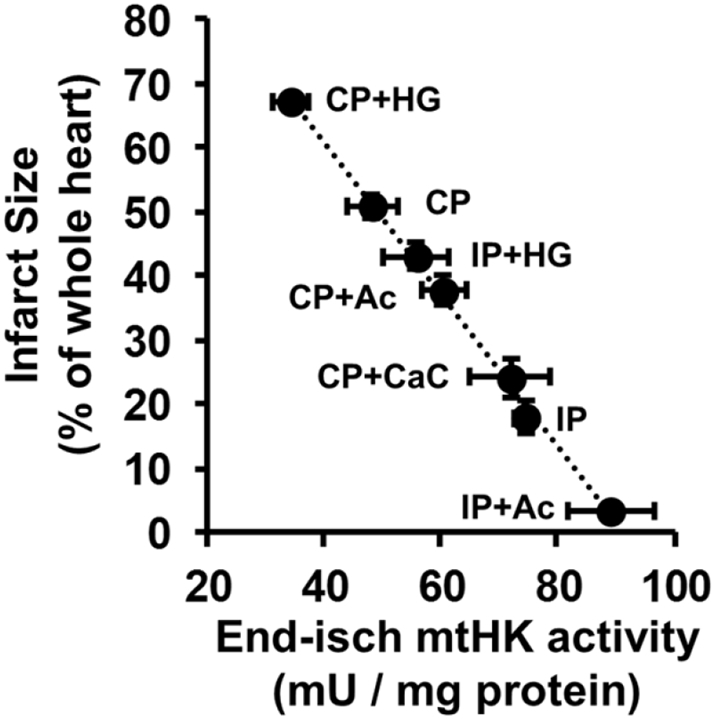
The extent of HK2 dissociation from mitochondria during ischemia is strongly correlated with infarct size following reperfusion. HK activity was measured in mitochondria isolated after 30 min ischemia and the infarct size after 2 h reperfusion. The changes in HK activity was confirmed to be due exclusively to HK2 dissociation by Western blotting. The pre-treatments used to modify HK2 binding at the end of ischemia were: Control (CP), ischemic preconditioned (IP) and perfusion with acetate (Ac), high [Ca2+] (CaC) or high glucose (HG) with or without IP. Data are taken from [22].
We have also shown that the increased susceptibility to IR of hearts from high fat fed mice is associated with less mitochondrial bound HK2 [149] while others have demonstrated that hearts from HK2+/− mice have reduced HK2 content and are more sensitive to IR injury [150]. As such HK2 binding to mitochondria behaves in a manner that would be expected of a key factor involved in determining the extent of mPTP opening and IR injury. Thus, HK2 dissociation from mitochondria during ischemia will enhance cytochrome c release and predispose the mPTP to open early in reperfusion under conditions of increased matrix [Ca2+]. By preventing this HK2 dissociation from mitochondria during ischemia, IP prevents both cytochrome c loss and mPTP sensitisation thus accounting, at least in part, for its cardioprotective effect. Very recent data from Zuurbier's laboratory have provided further evidence in support of this hypothesis. Rat hearts were perfused with low concentrations (200 nM) of a membrane permeable peptide (TAT-HKII) that dissociates bound HK2 from mitochondria and which was without significant effect on normoxic hearts. However, after 15 min ischemia, when reperfusion of control hearts caused only reversible injury (impaired hemodynamic function), the presence of the TAT-HKII peptide caused irreversible injury (lactate dehydrogenase release) [151].
Possible mechanisms by which IP may attenuate HK2 dissociation from mitochondria during ischemia have been comprehensively reviewed elsewhere [28] and are only briefly summarised here. Attenuation of the ischemia-induced increase in [G-6-P] and decrease intracellular pH are critical, and reflect decreased glycogen levels prior to ischemia [152], [153], [154]. Additional signalling pathways have also been proposed including translocation of PKCε to the mitochondria where it may phosphorylate VDAC1 to increase HK2 binding [155], although we were not able to confirm this [16]. GSK3β and Akt have also been implicated in IP [156], [157]. Indeed, it has been proposed that the mechanisms of many cardioprotective regimes converge to phosphorylate and inhibit GSK3β whose activity promotes mPTP opening [158], [159]. Interestingly, Akt has also been reported to phosphorylate Thr473 of HK2 directly with the result that its binding to mitochondria in cardiomycoytes is enhanced while its sensitivity to dissociation by increased concentrations of G-6-P is reduced [160], [161]. However, others have reported that neither Akt stimulation with insulin nor genetic ablation of VDAC isoforms 1 and 3 affect cell death induction by HK2 dissociation [144]. Furthermore, insulin, a potent activator of the Akt pathway, is not cardioprotective unless added during reperfusion [158]. Indeed, we found that pre-ischemic treatment of hearts with insulin in place of the IP protocol caused Akt and GSK3β phosphorylation to increase several-fold yet no cardioprotection was observed [16] while others have reported that such treatment may even suppress cardioprotection by IP [162]. Nor were we able to detect significant IP-mediated changes in phosphorylation of any mitochondrial proteins (including VDAC and HK2) [16]. Whatever the exact mechanisms(s) underlying the regulation of HK2 binding to mitochondria, the very strong inverse correlation between the extent of its binding at the end of ischemia with the extent of damage (infarct size) following reperfusion (Fig. 6) suggests that it may play a key role in cardioprotection. An attractive feature of this proposal is that it provides a plausible explanation of how, by converging on HK2 binding to mitochondria, a diverse range of known cardioprotective protocols with distinct signalling pathways can inhibit mPTP opening on reperfusion and so reduce IR injury.
5.3. The role of mitochondrial contact sites and cristae morphology
The mechanism(s) that link(s) HK2 binding to mPTP opening remain uncertain, but an attractive hypothesis for which there is increasing evidence is that HK2 binding modulates the interaction between the inner mitochondrial membrane (IMM) and the OMM. Dissociation of HK2 is proposed to alter the mitochondrial morphology and cristae structure in such a way as to increase OMM permeability to cytochrome c and sensitise mPTP opening to matrix [Ca2+] [28]. The presence of “contact sites” between the IMM and OMM, where the two membranes interact closely, was first revealed by freeze-fracture of mitochondria and were shown to be dynamic structures which are affected by the metabolic status of mitochondria [163], [164]. Several proteins have been implicated in their formation including VDAC, ANT and, when present, HK1, HK2 and creatine kinase (CK) [164], [165]. Originally thought to function primarily to enhance the transfer of ATP from the matrix to the cytosol [163], contact sites were subsequently implicated in the regulation of OMM permeability and mPTP opening [166]. We presented evidence that destabilisation of contact sites may facilitate opening of cristae junctions and thus access of cytochrome c within the cristae to the permeation pathways in the OMM, and also increase the sensitivity of the mPTP towards [Ca2+] [167]. HK2 binding stabilises contact sites [168], [169] and thus it would be predicted that the loss of HK2 during ischemia might lead to contact site breakage. In the perfused heart, there is good evidence that this is the case since operation of the creatine phosphate (CrP) shuttle, which involves the interaction of the ANT, CK and VDAC at contact sites, is impaired following ischemia [170] but less so following IP [22], [171]. Furthermore, using a variety of preconditioning stimuli to modify mitochondrial HK2 binding at the end of ischemia we confirmed that maintenance of contact sites during ischemia, as reflected in the recovery of CrP and hemodynamic function on reperfusion, was greatest in hearts with the highest mitochondrial HK2 binding [22]. The importance of contact sites in protecting mitochondria from mPTP opening is also consistent with the observation that knockout of CK, whose presence stabilises contact sites [172], makes hearts more sensitive to IR injury [173]. In addition, liver mitochondria from mice genetically modified to express mitochondrial CK (which is normally absent in liver) demonstrated a 3-fold increase in the number of contact sites as observed by EM [172], and were more resistant to mPTP opening by calcium [174].
Bcl-2 family members may also associate with the OMM at contact sites, possibly binding to VDAC [142], [175], [176], [177]. These proteins can influence both OMM permeability and mPTP activity (see [178], [179], [180]) and Bcl-xL has been shown to be lost from heart mitochondria during ischemia, perhaps leading to unmasking of pro-apoptotic members of the Bcl-2 family such as Bax and Bak that are already in the OMM [22]. Furthermore, adenovirus-mediated Bcl-xL gene transfer has been reported to protect hearts from IR injury [181] while hearts from mice in which both Bax and Bak have been knocked out are also protected from IR [182]. The activity of members of the Bcl-2 family can also be regulated by phosphorylation, as well as by proteolysis and translocation (see [178], [179], [180]) and this may provide yet further mechanisms by which preconditioning can attenuate OMM permeabilisation and mPTP opening in conjunction with its enhancement of HK2 binding. So too may the HK2-regulated oligomerisation of VDAC1 in the OMM that has recently been proposed as a pathway for cytochrome c release in apoptosis [176], [183]. Indeed, inhibitors of this process have been developed that prevent selenite-induced apoptosis and the associated cytochrome c release, ROS formation, mitochondrial depolarization and calcium overload [184]. Significantly this is accompanied by prevention of HK dissociation from the mitochondria and the authors propose that HK binding to VDAC1 prevents its oligomerization. This provides another plausible explanation for the relationship between mitochondrial HK binding and resistance to apoptosis and mPTP opening.
5.4. The role of mitochondrial fission
There may also be a link between contact site stability, mitochondrial cristae morphology and mitochondrial fission [28]. Thus the release of cytochrome c from the intermembrane space during apoptosis, which is associated with mitochondrial fission, involves changes in mitochondrial cristae morphology allowing the cytochrome c within the cristae to gain access to the permeabilised OMM [180]. A key player in mitochondrial fission is the GTPase Dynamin-related protein 1(Drp1) [180], [185] which must be recruited to the OMM. Importantly, mitochondria have been reported to undergo fission during ischemia of the perfused heart and this might be a critical factor in inducing the observed cytochrome c release [186]. Furthermore, inhibiting Drp1 activity with the inhibitor Mdiv-1 [186], [187], [188], [189] or transgenic expression of a dominant negative form of Drp1 [190] was found to reduce infarct size following IR. This was accompanied by preserved mitochondrial morphology, reduced mitochondrial ROS production and improved hemodynamic function [189]. Another attractive feature of Drp1 is the complex regulation of its translocation to and from mitochondria. Drp1 can be phosphorylated on either Ser616 or Ser637 by different protein kinases but only the former promotes its translocation to mitochondria [189]. Interestingly, the kinase responsible for Ser616 phosphorylation is PKCδ which becomes active during ischemia [191]. Similarly, Ser637 is phosphorylated by PKA that is active after pre-conditioning or TP [21] and promotes Drp1 dissociation from mitochondria. Thus, in a similar manner to HK2, it would seem possible that Drp1 can also act as a common locus that links different responses to IR and mitochondrial fate. Furthermore, Drp1 has been shown to associate with mitochondria during ischemia [187], [192], [193] and to facilitate calcium flux and cristae remodeling during apoptosis [194] in an Mid49/51-dependent and OPA1-independent fashion [195]. It is possible that the opposite translocation of Drp1 and HK2 during ischemia and cardioprotective regimens may reflect a shared but competing binding region on the OMM. Indeed, very recent studies in neonatal cardiac myocytes have demonstrated a reciprocal interaction between Drp1 and HK2 binding to mitochondria [196]. Treatment of the myocytes with 100 μM palmitic acid for 2 h enhanced Drp1 binding to mitochondria while reducing HK2 binding, and this was associated with increased mPTP opening and ROS and [Ca2+] levels. Treatment with 2-deoxy-glucose also reduced mitochondrial HK2 binding while promoting Drp1 translocation to mitochondria. Furthermore, knockdown of Drp1 using siRNA prevented the loss of HK2 induced by palmitic acid as did increasing Drp1 phosphorylation by Akt activation which also prevented the mPTP opening and the associated increases in [ROS] and [Ca2+].
6. Conclusions
Our own data together with a critical analysis of the published data of others lead us to conclude that it is unlikely that significant superoxide production in the mitochondrial matrix, driven by succinate-fuelled REF, occurs in the early phase of reperfusion (i.e. the first 2 min). Hence we suggest that this mechanism is unlikely to be a critical mediator of IR injury. Rather we would argue that the evidence favours the initial phase of mPTP opening being triggered by factors other than matrix ROS while the large increase in ROS seen later in reperfusion occurs as a consequence of mPTP opening. This secondary rise in ROS may play an important role in causing an escalating cascade of mPTP opening that leads to a spreading wave of necrotic cell death and the formation of the infarct. An important stimulus for the first phase of mPTP opening is likely to be the observed increase in mitochondrial matrix [Ca2+] during ischemia and early reperfusion. However, it cannot be the only trigger because the increase in [Ca2+] is not attenuated by IP, yet IP inhibits mPTP opening and provides cardioprotection. A promising additional candidate for regulating the initial phase of mPTP opening is mitochondrial bound HK2 which dissociates from the mitochondria during ischemia, but not following IP. Dissociation of bound HK2 is associated with an increase loss of cytochrome c from the mitochondria during ischemia and an enhanced sensitivity to mPTP opening. This provides an explanation of the very close correlation between the extent of HK2 loss from mitochondria during ischemia and the extent of injury (infarct size) on subsequent reperfusion. The mechanisms linking HK2 dissociation to OMM permeabilisation, cytochrome c release and mPTP sensitisation remain to be fully established but the available evidence implicates several related processes: the stability of contact sites between the inner and outer membranes, cristae morphology and VDAC1 oligomerisation. This is illustrated diagrammatically in Fig. 7. The involvement of these interacting processes provides potential loci for the established effects on IR injury of mitochondrial fission proteins such as Drp1 (knockout/inhibition is cardioprotective), members of the Bcl-2 family such as Bcl-xL, whose overexpression is cardioprotective, and Bax and Bak whose knockout is cardioprotective, and proteins enhancing contact site formation such as HK2 and mitochondrial CK whose knockout enhances damage. However, further work will be required to test this hypothesis and elucidate the underlying mechanisms that link these processes to the regulation of mPTP opening and the development of IR injury.
Fig. 7.

Proposed sequence of events in ischemia and reperfusion that lead to mPTP opening and reperfusion injury and their attenuation by IP. This scheme is a slightly modified version of those presented previously [7], [28].
Sources of funding
Medical Research Council G0801237.
British Heart Foundation RG/08/001/24717 and PG/12/40/29634.
Disclosures
None.
Acknowledgements
We thank Emily Saunderson for her assistance with measuring succinate transport in MCT1-expressing oocytes.
References
- 1.Halestrap A.P. A pore way to die: the role of mitochondria in reperfusion injury and cardioprotection. Biochem. Soc. Trans. 2010;38:841–860. doi: 10.1042/BST0380841. [DOI] [PubMed] [Google Scholar]
- 2.Bernardi P., Di Lisa F. The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J. Mol. Cell. Cardiol. 2015;78:100–106. doi: 10.1016/j.yjmcc.2014.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Griffiths E.J., Halestrap A.P. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem. J. 1995;307:93–98. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Halestrap A.P., Connern C.P., Griffiths E.J., Kerr P.M. Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol. Cell. Biochem. 1997;174:167–172. [PubMed] [Google Scholar]
- 5.Halestrap A.P. Calcium-dependent opening of a non-specific pore in the mitochondrial inner membrane is inhibited at pH values below 7- implications for the protective effect of low pH against chemical and hypoxic cell damage. Biochem. J. 1991;278:715–719. doi: 10.1042/bj2780715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zorov D.B., Juhaszova M., Sollott S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014;94:909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andrienko T., Pasdois P., Rossbach A., Halestrap A.P. Real-Time Fluorescence Measurements of ROS and [Ca2 +] in ischemic/reperfused rat hearts: detectable increases occur only after mitochondrial pore opening and are attenuated by ischemic preconditioning. PLoS One. 2016;11 doi: 10.1371/journal.pone.0167300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ong S.B., Samangouei P., Kalkhoran S.B., Hausenloy D.J. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J. Mol. Cell. Cardiol. 2015;78:23–34. doi: 10.1016/j.yjmcc.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 9.Griffiths E.J., Halestrap A.P. Protection by cyclosporin A of ischemia reperfusion-induced damage in isolated rat hearts. J. Mol. Cell. Cardiol. 1993;25:1461–1469. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- 10.Clarke S.J., McStay G.P., Halestrap A.P. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J. Biol. Chem. 2002;277:34793–34799. doi: 10.1074/jbc.M202191200. [DOI] [PubMed] [Google Scholar]
- 11.Fancelli D., Abate A., Amici R., Bernardi P., Ballarini M., Cappa A. Cinnamic anilides as new mitochondrial permeability transition pore inhibitors endowed with ischemia-reperfusion injury protective effect in vivo. J. Med. Chem. 2014;57:5333–5347. doi: 10.1021/jm500547c. [DOI] [PubMed] [Google Scholar]
- 12.Halestrap A.P., Richardson A.P. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 2015;78:129–141. doi: 10.1016/j.yjmcc.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 13.Karch J., Molkentin J.D. Identifying the components of the elusive mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. U. S. A. 2014;111:10396–10397. doi: 10.1073/pnas.1410104111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Halestrap A.P., Woodfield K.Y., Connern C.P. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J. Biol. Chem. 1997;272:3346–3354. doi: 10.1074/jbc.272.6.3346. [DOI] [PubMed] [Google Scholar]
- 15.Khaliulin I., Schwalb H., Wang P., Houminer E., Grinberg L., Katzeff H. Preconditioning improves post-ischemic mitochondrial function and diminishes oxidation of mitochondrial proteins. Free Radic. Biol. Med. 2004;37:1–9. doi: 10.1016/j.freeradbiomed.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 16.Clarke S.J., Khaliulin I., Das M., Parker J.E., Heesom K.J., Halestrap A.P. Inhibition of mitochondrial permeability transition pore opening by ischemic preconditioning is probably mediated by reduction of oxidative stress rather than mitochondrial protein phosphorylation. Circ. Res. 2008;102:1082–1090. doi: 10.1161/CIRCRESAHA.107.167072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khaliulin I., Clarke S.J., Lin H., Parker J., Suleiman M.-S., Halestrap A.P. Temperature preconditioning of isolated rat hearts - a potent cardioprotective mechanism involving a reduction in oxidative stress and inhibition of the mitochondrial permeability transition pore. J. Physiol. 2007;581:1147–1161. doi: 10.1113/jphysiol.2007.130369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Townsend P.A., Davidson S.M., Clarke S.J., Khaliulin I., Carroll C.J., Scarabelli T.M. Urocortin prevents mitochondrial permeability transition in response to reperfusion injury indirectly, by reducing oxidative stress. Am. J. Phys. 2007;293:H928–H938. doi: 10.1152/ajpheart.01135.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khaliulin I., Schneider A., Houminer E., Borman J.B., Schwalb H. Apomorphine prevents myocardial ischemia/reperfusion-induced oxidative stress in the rat heart. Free Radic. Biol. Med. 2004;37:969–976. doi: 10.1016/j.freeradbiomed.2004.06.029. [DOI] [PubMed] [Google Scholar]
- 20.Paillard M., Gomez L., Augeul L., Loufouat J., Lesnefsky E.J., Ovize M. Postconditioning inhibits mPTP opening independent of oxidative phosphorylation and membrane potential. J. Mol. Cell. Cardiol. 2009;46:902–909. doi: 10.1016/j.yjmcc.2009.02.017. [DOI] [PubMed] [Google Scholar]
- 21.Khaliulin I., Parker J.E., Halestrap A.P. Consecutive pharmacological activation of PKA and PKC mimics the potent cardioprotection of temperature preconditioning. Cardiovasc. Res. 2010;88:324–333. doi: 10.1093/cvr/cvq190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pasdois P., Parker J.E., Halestrap A.P. Extent of mitochondrial hexokinase II dissociation during ischemia correlates with mitochondrial cytochrome c release, reactive oxygen species production, and infarct size on reperfusion. J. Am. Heart Assoc. 2012;2 doi: 10.1161/JAHA.112.005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chouchani E.T., Pell V.R., Gaude E., Aksentijevic D., Sundier S.Y., Robb E.L. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chouchani E.T., Methner C., Nadtochiy S.M., Logan A., Pell V.R., Ding S. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 2013;19:753–759. doi: 10.1038/nm.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pell V.R., Chouchani E.T., Frezza C., Murphy M.P., Krieg T. Succinate metabolism: a new therapeutic target for myocardial reperfusion injury. Cardiovasc. Res. 2016;111:134–141. doi: 10.1093/cvr/cvw100. [DOI] [PubMed] [Google Scholar]
- 26.Pell V.R., Chouchani E.T., Murphy M.P., Brookes P.S., Krieg T. Moving forwards by blocking back-flow: the yin and yang of MI therapy. Circ. Res. 2016;118:898–906. doi: 10.1161/CIRCRESAHA.115.306569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nederlof R., Eerbeek O., Hollmann M.W., Southworth R., Zuurbier C.J. Targeting hexokinase II to mitochondria to modulate energy metabolism and reduce ischaemia-reperfusion injury in heart. Br. J. Pharmacol. 2014;171:2067–2079. doi: 10.1111/bph.12363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Halestrap A.P., Pereira G.C., Pasdois P. The role of hexokinase in cardioprotection - mechanism and potential for translation. Br. J. Pharmacol. 2015;172:2085–2100. doi: 10.1111/bph.12899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calmettes G., Ribalet B., John S., Korge P., Ping P., Weiss J.N. Hexokinases and cardioprotection. J. Mol. Cell. Cardiol. 2015;78:107–115. doi: 10.1016/j.yjmcc.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Griendling K.K., Touyz R.M., Zweier J.L., Dikalov S., Chilian W., Chen Y.R. Measurement of reactive oxygen species, reactive nitrogen species, and redox-dependent signaling in the cardiovascular system: a scientific statement from the American Heart Association. Circ. Res. 2016;119:e39–e75. doi: 10.1161/RES.0000000000000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vanden Hoek T.L., Li C., Shao Z., Schumacker P.T., Becker L.B. Significant levels of oxidants are generated by isolated cardiomyocytes during ischemia prior to reperfusion. J. Mol. Cell. Cardiol. 1997;29:2571–2583. doi: 10.1006/jmcc.1997.0497. [DOI] [PubMed] [Google Scholar]
- 32.Kim J.S., Jin Y.G., Lemasters J.J. Reactive oxygen species, but not Ca2 + overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am. J. Phys. 2006;290:H2024–H2034. doi: 10.1152/ajpheart.00683.2005. [DOI] [PubMed] [Google Scholar]
- 33.Assaly R., de Tassigny A.D., Paradis S., Jacquin S., Berdeaux A., Morin D. Oxidative stress, mitochondrial permeability transition pore opening and cell death during hypoxia-reoxygenation in adult cardiomyocytes. Eur. J. Pharmacol. 2012;675:6–14. doi: 10.1016/j.ejphar.2011.11.036. [DOI] [PubMed] [Google Scholar]
- 34.Zhao H., Joseph J., Fales H.M., Sokoloski E.A., Levine R.L., Vasquez-Vivar J. Detection and characterization of the product of hydroethidine and intracellular superoxide by HPLC and limitations of fluorescence. Proc. Natl. Acad. Sci. U. S. A. 2005;102:5727–5732. doi: 10.1073/pnas.0501719102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Näpänkangasa J.P., Liimatta E.V., Joensuuc P., Bergmann U., Ylitalo K., Hassinen I.E. Superoxide production during ischemia-reperfusion in the perfused rat heart: a comparison of two methods of measurement. J. Mol. Cell. Cardiol. 2012;53:906–915. doi: 10.1016/j.yjmcc.2012.09.011. [DOI] [PubMed] [Google Scholar]
- 36.Eaton P., Byers H.L., Leeds N., Ward M.A., Shattock M.J. Detection, quantitation, purification, and identification of cardiac proteins S-thiolated during ischemia and reperfusion. J. Biol. Chem. 2002;277:9806–9811. doi: 10.1074/jbc.M111454200. [DOI] [PubMed] [Google Scholar]
- 37.Paradies G., Petrosillo G., Pistolese M., DiVenosa N., Federici A., Ruggiero F.M. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart - involvement of reactive oxygen species and cardiolipin. Circ. Res. 2004;94:53–59. doi: 10.1161/01.RES.0000109416.56608.64. [DOI] [PubMed] [Google Scholar]
- 38.Petrosillo G., DiVenosa N., Ruggiero F.M., Pistolese M., DAgostino D., Tiravanti E. Mitochondrial dysfunction associated with cardiac ischemia/reperfusion can be attenuated by oxygen tension control. Role of oxygen-free radicals and cardiolipin. Biochim. Biophys. Acta. 2005;1710:78–86. doi: 10.1016/j.bbabio.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 39.Paradies G., Petrosillo G., Paradies V., Ruggiero F.M. Role of cardiolipin peroxidation and Ca2 + in mitochondrial dysfunction and disease. Cell Calcium. 2009;45:643–650. doi: 10.1016/j.ceca.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 40.Zweier J.L. Measurement of superoxide-derived free radicals in the reperfused heart. Evidence for a free radical mechanism of reperfusion injury. J. Biol. Chem. 1988;263:1353–1357. [PubMed] [Google Scholar]
- 41.Zweier J.L., Kuppusamy P., Williams R., Rayburn B.K., Smith D., Weisfeldt M.L. Measurement and characterization of postischemic free radical generation in the isolated perfused heart. J. Biol. Chem. 1989;264:18890–18895. [PubMed] [Google Scholar]
- 42.Eaton P., Wright N., Hearse D.J., Shattock M.J. Glyceraldehyde phosphate dehydrogenase oxidation during cardiac ischemia and reperfusion. J. Mol. Cell. Cardiol. 2002;34:1549–1560. doi: 10.1006/jmcc.2002.2108. [DOI] [PubMed] [Google Scholar]
- 43.Kevin L.G., Camara A.K., Riess M.L., Novalija E., Stowe D.F. Ischemic preconditioning alters real-time measure of O-2 radicals in intact hearts with ischemia and reperfusion. Am. J. Phys. 2003;284:H566–H574. doi: 10.1152/ajpheart.00711.2002. [DOI] [PubMed] [Google Scholar]
- 44.Novalija E., Kevin L.G., Eells J.T., Henry M.M., Stowe D.F. Anesthetic preconditioning improves adenosine triphosphate synthesis and reduces reactive oxygen species formation in mitochondria after ischemia by a redox dependent mechanism. Anesthesiology. 2003;98:1155–1163. doi: 10.1097/00000542-200305000-00018. [DOI] [PubMed] [Google Scholar]
- 45.Stowe D.F., Aldakkak M., Camara A.K.S., Riess M.L., Heinen A., Varadarajan S.G. Cardiac mitochondrial preconditioning by Big Ca2 +-sensitive K + channel opening requires superoxide radical generation. Am. J. Phys. 2006;290:H434–H440. doi: 10.1152/ajpheart.00763.2005. [DOI] [PubMed] [Google Scholar]
- 46.Lu L.S., Liu Y.B., Sun C.W., Lin L.C., Su M.J., Wu C.C. Optical mapping of myocardial reactive oxygen species production throughout the reperfusion of global ischemia. J. Biomed. Opt. 2006;11 doi: 10.1117/1.2186321. [DOI] [PubMed] [Google Scholar]
- 47.Camara A.K., Riess M.L., Kevin L.G., Novalija E., Stowe D.F. Hypothermia augments reactive oxygen species detected in the guinea pig isolated perfused heart. Am. J. Physiol. Heart. 2004;286:H1289–H1299. doi: 10.1152/ajpheart.00811.2003. [DOI] [PubMed] [Google Scholar]
- 48.Riess M.L., Camara A.K., Kevin L.G., An J., Stowe D.F. Reduced reactive O2 species formation and preserved mitochondrial NADH and [Ca2 +] levels during short-term 17 degrees C ischemia in intact hearts. Cardiovasc. Res. 2004;61:580–590. doi: 10.1016/j.cardiores.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 49.Biary N., Xie C., Kauffman J., Akar F.G. Biophysical properties and functional consequences of reactive oxygen species (ROS)-induced ROS release in intact myocardium. J. Physiol. 2011;589:5167–5179. doi: 10.1113/jphysiol.2011.214239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Novalija E., Kevin L.G., Camara A.K., Bosnjak Z.J., Kampine J.P., Stowe D.F. Reactive oxygen species precede the epsilon isoform of protein kinase C in the anesthetic preconditioning signaling cascade. Anesthesiology. 2003;99:421–428. doi: 10.1097/00000542-200308000-00024. [DOI] [PubMed] [Google Scholar]
- 51.Karlsson M., Kurz T., Brunk U.T., Nilsson S.E., Frennesson C.I. What does the commonly used DCF test for oxidative stress really show? Biochem. J. 2010;428:183–190. doi: 10.1042/BJ20100208. [DOI] [PubMed] [Google Scholar]
- 52.Javadov S.A., Clarke S., Das M., Griffiths E.J., Lim K.H.H., Halestrap A.P. Ischaemic preconditioning inhibits opening of mitochondrial permeability transition pores in the reperfused rat heart. J. Physiol. 2003;549:513–524. doi: 10.1113/jphysiol.2003.034231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Granger D.N., Kvietys P.R. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol. 2015;6:524–551. doi: 10.1016/j.redox.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matsushima S., Tsutsui H., Sadoshima J. Physiological and pathological functions of NADPH oxidases during myocardial ischemia-reperfusion. Trends Cardiovasc. Med. 2014;24:202–205. doi: 10.1016/j.tcm.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andreyev A.Y., Kushnareva Y.E., Starkov A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry. 2005;70:200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 56.Andreyev A.Y., Kushnareva Y.E., Murphy A.N., Starkov A.A. Mitochondrial ROS metabolism: 10 years later. Biochemistry. 2015;80:517–531. doi: 10.1134/S0006297915050028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dey S., Sidor A., O'Rourke B. Compartment-specific control of reactive oxygen species scavenging by antioxidant pathway enzymes. J. Biol. Chem. 2016;291:11185–11197. doi: 10.1074/jbc.M116.726968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kezic A., Spasojevic I., Lezaic V., Bajcetic M. Mitochondria-targeted antioxidants: future perspectives in kidney ischemia reperfusion injury. Oxidative Med. Cell. Longev. 2016;2016 doi: 10.1155/2016/2950503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smith R.A., Murphy M.P. Mitochondria-targeted antioxidants as therapies. Discov. Med. 2011;11:106–114. [PubMed] [Google Scholar]
- 60.Brown D.A., Hale S.L., Baines C.P., del Rio C.L., Hamlin R.L., Yueyama Y. Reduction of early reperfusion injury with the mitochondria-targeting peptide bendavia. J. Cardiovasc. Pharmacol. Ther. 2014;19:121–132. doi: 10.1177/1074248413508003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao K.S., Zhao G.M., DL Wu, Soong Y., Birk A.V., Schiller P.W. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J. Biol. Chem. 2004;279:34682–34690. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- 62.Dai W., Cheung E., Alleman R.J., Perry J.B., Allen M.E., Brown D.A. Cardioprotective effects of mitochondria-targeted peptide SBT-20 in two different models of rat ischemia/reperfusion. Cardiovasc. Drugs Ther. 2016;30:559–566. doi: 10.1007/s10557-016-6695-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kloner R.A., Hale S.L., Dai W., Gorman R.C., Shuto T., Koomalsingh K.J. Reduction of ischemia/reperfusion injury with bendavia, a mitochondria-targeting cytoprotective peptide. J. Am. Heart Assoc. 2012;1 doi: 10.1161/JAHA.112.001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Szeto H.H. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid. Redox Signal. 2008;10:601–619. doi: 10.1089/ars.2007.1892. [DOI] [PubMed] [Google Scholar]
- 65.Smith R.A., Murphy M.P. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann. N. Y. Acad. Sci. 2010;1201:96–103. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]
- 66.Adlam V.J., Harrison J.C., Porteous C.M., James A.M., Smith R.A.J., Murphy M.P. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 67.Hansson M.J., Llwyd O., Morin D., de Paulis D., Arnoux T., Gouarne C. Differences in the profile of protection afforded by TRO40303 and mild hypothermia in models of cardiac ischemia/reperfusion injury. Eur. J. Pharmacol. 2015;760:7–19. doi: 10.1016/j.ejphar.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 68.Becker L.B. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc. Res. 2004;61:461–470. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 69.Pagliaro P., Penna C. Redox signalling and cardioprotection: translatability and mechanism. Br. J. Pharmacol. 2015;172:1974–1995. doi: 10.1111/bph.12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yoshida T., Watanabe M., Engelman D.T., Engelman R.M., Schley J.A., Maulik N. Transgenic mice overexpressing glutathione peroxidase are resistant to myocardial ischemia reperfusion injury. J. Mol. Cell. Cardiol. 1996;28:1759–1767. doi: 10.1006/jmcc.1996.0165. [DOI] [PubMed] [Google Scholar]
- 71.Li G.Q., Chen Y., Saari J.T., Kang Y.J. Catalase-overexpressing transgenic mouse heart is resistant to ischemia-reperfusion injury. Am. J. Phys. 1997;273:H1090–H1095. doi: 10.1152/ajpheart.1997.273.3.H1090. [DOI] [PubMed] [Google Scholar]
- 72.Heusch G. Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ. Res. 2015;116:674–699. doi: 10.1161/CIRCRESAHA.116.305348. [DOI] [PubMed] [Google Scholar]
- 73.Deboer L.W.V., Bekx P.A., Han L.H., Steinke L. Pyruvate enhances recovery of rat hearts after ischemia and reperfusion by preventing free radical generation. Am. J. Phys. 1993;265:H1571–H1576. doi: 10.1152/ajpheart.1993.265.5.H1571. [DOI] [PubMed] [Google Scholar]
- 74.Kerr P.M., Suleiman M.S., Halestrap A.P. Reversal of permeability transition during recovery of hearts from ischemia and its enhancement by pyruvate. Am. J. Phys. 1999;276:H496–H502. doi: 10.1152/ajpheart.1999.276.2.H496. [DOI] [PubMed] [Google Scholar]
- 75.Mallet R.T., Sun J., Knott E.M., Sharma A.B., Olivencia-Yurvati A.H. Metabolic cardioprotection by pyruvate: recent progress. Exp. Biol. Med. 2005;230:435–443. doi: 10.1177/153537020523000701. [DOI] [PubMed] [Google Scholar]
- 76.Javadov S.A., Lim K.H.H., Kerr P.M., Suleiman M.S., Angelini G.D., Halestrap A.P. Protection of hearts from reperfusion injury by propofol is associated with inhibition of the mitochondrial permeability transition. Cardiovasc. Res. 2000;45:360–369. doi: 10.1016/s0008-6363(99)00365-x. [DOI] [PubMed] [Google Scholar]
- 77.Lim K.H.H., Halestrap A.P., Angelini G.D., Suleiman M.S. Propofol is cardioprotective in a clinically relevant model of normothermic blood cardioplegic arrest and cardiopulmonary bypass. Exp. Biol. Med. 2005;230:413–420. doi: 10.1177/15353702-0323006-09. [DOI] [PubMed] [Google Scholar]
- 78.Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brand M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016;100:14–31. doi: 10.1016/j.freeradbiomed.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 80.Hassinen I.E. Reflectance spectrophotometric and surface fluorometric methods for measuring the redox state of nicotinamide nucleotides and flavins in intact tissues. Methods Enzymol. 1986;123:311–320. doi: 10.1016/s0076-6879(86)23036-0. [DOI] [PubMed] [Google Scholar]
- 81.Mayevsky A., Chance B. Intracellular oxidation-reduction state measured in situ by a multichannel fiber-optic surface fluorometer. Science. 1982;217:537–540. doi: 10.1126/science.7201167. [DOI] [PubMed] [Google Scholar]
- 82.Vuorinen K., Ylitalo K., Peuhkurinen K., Raatikainen P., Alarami A., Hassinen I.E. Mechanisms of ischemic preconditioning in rat myocardium: roles of adenosine, cellular energy state, and mitochondrial F1F0-ATPase. Circulation. 1995;91:2810–2818. doi: 10.1161/01.cir.91.11.2810. [DOI] [PubMed] [Google Scholar]
- 83.Sun J., Murphy E. Protein S-nitrosylation and cardioprotection. Circ. Res. 2010;106:285–296. doi: 10.1161/CIRCRESAHA.109.209452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Murphy E., Kohr M., Sun J., Nguyen T., Steenbergen C. S-nitrosylation: a radical way to protect the heart. J. Mol. Cell. Cardiol. 2012;52:568–577. doi: 10.1016/j.yjmcc.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sun J., Morgan M., Shen R.F., Steenbergen C., Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ. Res. 2007;101:1155–1163. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- 86.Burwell L.S., Nadtochiy S.M., Tompkins A.J., Young S., Brookes P.S. Direct evidence for S-nitrosation of mitochondrial complex I. Biochem. J. 2006;394:627–634. doi: 10.1042/BJ20051435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Murphy E., Steenbergen C. Did a classic preconditioning study provide a clue to the identity of the mitochondrial permeability transition pore? Circ. Res. 2013;113:852–855. doi: 10.1161/CIRCRESAHA.113.301950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pisarenko O., Studneva I., Khlopkov V., Solomatina E., Ruuge E. An assessment of anaerobic metabolism during ischemia and reperfusion in isolated guinea pig heart. Biochim. Biophys. Acta. 1988;934:55–63. doi: 10.1016/0005-2728(88)90119-3. [DOI] [PubMed] [Google Scholar]
- 89.Peuhkurinen K.J., Takala T.E., Nuutinen E.M., Hassinen I.E. Tricarboxylic acid cycle metabolites during ischemia in isolated perfused rat heart. Am. J. Phys. 1983;244:H281–H288. doi: 10.1152/ajpheart.1983.244.2.H281. [DOI] [PubMed] [Google Scholar]
- 90.Argaud L., GateauRoesch O., Chalabreysse L., Gomez L., Loufouat J., ThivoletBejui F. Preconditioning delays Ca2 +-induced mitochondrial permeability transition. Cardiovasc. Res. 2004;61:115–122. doi: 10.1016/j.cardiores.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 91.Hausenloy D.J., Yellon D.M., Mani-Babu S., Duchen M.R. Preconditioning protects by inhibiting the mitochondrial permeability transition. Am. J. Phys. 2004;287:H841–H849. doi: 10.1152/ajpheart.00678.2003. [DOI] [PubMed] [Google Scholar]
- 92.Sakamoto M., Takeshige K., Yasui H., Tokunaga K. Cardioprotective effect of succinate against ischemia/reperfusion injury. Surg. Today. 1998;28:522–528. doi: 10.1007/s005950050177. [DOI] [PubMed] [Google Scholar]
- 93.Hochachka P.W., Storey K.B. Metabolic consequences of diving in animals and man. Science. 1975;187:613–621. doi: 10.1126/science.163485. [DOI] [PubMed] [Google Scholar]
- 94.Laplante A., Vincent G., Poirier M., Des Rosiers C. Effects and metabolism of fumarate in the perfused rat heart. A 13C mass isotopomer study. Am. J. Phys. 1997;272:E74–E82. doi: 10.1152/ajpendo.1997.272.1.E74. [DOI] [PubMed] [Google Scholar]
- 95.Ashrafian H., Czibik G., Bellahcene M., Aksentijevic D., Smith A.C., Mitchell S.J. Fumarate is cardioprotective via activation of the Nrf2 antioxidant pathway. Cell Metab. 2012;15:361–371. doi: 10.1016/j.cmet.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Taegtmeyer H. Metabolic responses to cardiac hypoxia. Increased production of succinate by rabbit papillary muscles. Circ. Res. 1978;43:808–815. doi: 10.1161/01.res.43.5.808. [DOI] [PubMed] [Google Scholar]
- 97.Halestrap A.P., Wang X.M., Poole R.C., Jackson V.N., Price N.T. Lactate transport in heart in relation to myocardial ischemia. Am. J. Cardiol. 1997;80:A17–A25. doi: 10.1016/s0002-9149(97)00454-2. [DOI] [PubMed] [Google Scholar]
- 98.Gabel S.A., Cross H.R., London R.E., Steenbergen C., Murphy E. Decreased intracellular pH is not due to increased H + extrusion in preconditioned rat hearts. Am. J. Phys. 1997;42:H2257–H2262. doi: 10.1152/ajpheart.1997.273.5.H2257. [DOI] [PubMed] [Google Scholar]
- 99.Halestrap A.P. Monocarboxylic acid transport. Compr Physiol. 2013;3:1611–1643. doi: 10.1002/cphy.c130008. [DOI] [PubMed] [Google Scholar]
- 100.Ovens M.J., Davies A.J., Wilson M.C., Murray C.M., Halestrap A.P. AR-C155858 is a potent inhibitor of monocarboxylate transporters MCT1 and MCT2 that binds to an intracellular site involving transmembrane helices 7-10. Biochem. J. 2010;425:523–530. doi: 10.1042/BJ20091515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Turan N., Csonka C., Csont T., Giricz Z., Fodor G., Bencsik P. The role of peroxynitrite in chemical preconditioning with 3-nitropropionic acid in rat hearts. Cardiovasc. Res. 2006;70:384–390. doi: 10.1016/j.cardiores.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 102.Hu Z., Yang Y., Zhang K., Sun Z. Chemical preconditioning by 3-nitropropionic acid reduces ischemia-reperfusion injury in rat heart. J. Huazhong Univ. Sci. Technolog. Med. Sci. 2005;25:439–441. doi: 10.1007/BF02828217. [DOI] [PubMed] [Google Scholar]
- 103.Coetzee W.A. Multiplicity of effectors of the cardioprotective agent, diazoxide. Pharmacol. Ther. 2013;140:167–175. doi: 10.1016/j.pharmthera.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hanley P.J., Daut J. K-ATP channels and preconditioning: a re-examination of the role of mitochondrial KATpchannels and an overview of alternative mechanisms. J. Mol. Cell. Cardiol. 2005;39:17–50. doi: 10.1016/j.yjmcc.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 105.Minners J., Lacerda L., Yellon D.M., Opie L.H., McLeod C.J., Sack M.N. Diazoxide-induced respiratory inhibition - a putative mitochondrial K(ATP) channel independent mechanism of pharmacological preconditioning. Mol. Cell. Biochem. 2007;294:11–18. doi: 10.1007/s11010-005-9066-6. [DOI] [PubMed] [Google Scholar]
- 106.Bednarczyk P., Barker G.D., Halestrap A.P. Determination of the rate of K(+) movement through potassium channels in isolated rat heart and liver mitochondria. Biochim. Biophys. Acta. 2008;1777:540–548. doi: 10.1016/j.bbabio.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 107.Drose S., Hanley P.J., Brandt U. Ambivalent effects of diazoxide on mitochondrial ROS production at respiratory chain complexes I and III. Biochim. Biophys. Acta. 2009;1790:558–565. doi: 10.1016/j.bbagen.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 108.Chen Q., Camara A.K., Stowe D.F., Hoppel C.L., Lesnefsky E.J. Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion. Am. J. Phys. 2007;292:C137–C147. doi: 10.1152/ajpcell.00270.2006. [DOI] [PubMed] [Google Scholar]
- 109.Brennan J.P., Southworth R., Medina R.A., Davidson S.M., Duchen M.R., Shattock M.J. Mitochondrial uncoupling, with low concentration FCCP, induces ROS-dependent cardioprotection independent of KATP channel activation. Cardiovasc. Res. 2006;72:313–321. doi: 10.1016/j.cardiores.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 110.Ovize M., Baxter G.F., Di Lisa F., Ferdinandy P., Garcia-Dorado D., Hausenloy D.J. Postconditioning and protection from reperfusion injury: where do we stand?: Position Paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc. Res. 2010;87:406–423. doi: 10.1093/cvr/cvq129. [DOI] [PubMed] [Google Scholar]
- 111.Lesnefsky E.J., Chen Q., Moghaddas S., Hassan M.O., Tandler B., Hoppel C.L. Blockade of electron transport during ischemia protects cardiac mitochondria. J. Biol. Chem. 2004;279:47961–47967. doi: 10.1074/jbc.M409720200. [DOI] [PubMed] [Google Scholar]
- 112.Chen Q., Hoppel C.L., Lesnefsky E.J. Blockade of electron transport before cardiac ischemia with the reversible inhibitor amobarbital protects rat heart mitochondria. J. Pharmacol. Exp. Ther. 2006;316:200–207. doi: 10.1124/jpet.105.091702. [DOI] [PubMed] [Google Scholar]
- 113.Chen Q., Moghaddas S., Hoppel C.L., Lesnefsky E.J. Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion. J. Pharmacol. Exp. Ther. 2006;319:1405–1412. doi: 10.1124/jpet.106.110262. [DOI] [PubMed] [Google Scholar]
- 114.Aldakkak M., Stowe D.F., Chen Q., Lesnefsky E.J., Camara A.K. Inhibited mitochondrial respiration by amobarbital during cardiac ischaemia improves redox state and reduces matrix Ca2 + overload and ROS release. Cardiovasc. Res. 2008;77:406–415. doi: 10.1016/j.cardiores.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 115.Paiva M., Riksen N.P., Davidson S.M., Hausenloy D.J., Monteiro P., Goncalves L. Metformin prevents myocardial reperfusion injury by activating the adenosine receptor. J. Cardiovasc. Pharmacol. 2009;53:373–378. doi: 10.1097/FJC.0b013e31819fd4e7. [DOI] [PubMed] [Google Scholar]
- 116.Bhamra G.S., Hausenloy D.J., Davidson S.M., Carr R.D., Paiva M., Wynne A.M. Metformin protects the ischemic heart by the Akt-mediated inhibition of mitochondrial permeability transition pore opening. Basic Res. Cardiol. 2008;103:274–284. doi: 10.1007/s00395-007-0691-y. [DOI] [PubMed] [Google Scholar]
- 117.Calvert J.W., Gundewar S., Jha S., Greer J.J., Bestermann W.H., Tian R. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes. 2008;57:696–705. doi: 10.2337/db07-1098. [DOI] [PubMed] [Google Scholar]
- 118.Batandier C., Guigas B., Detaille D., El-Mir M.Y., Fontaine E., Rigoulet M. The ROS production induced by a reverse-electron flux at respiratory-chain complex 1 is hampered by metformin. J. Bioenerg. Biomembr. 2006;38:33–42. doi: 10.1007/s10863-006-9003-8. [DOI] [PubMed] [Google Scholar]
- 119.Prime T.A., Blaikie F.H., Evans C., Nadtochiy S.M., James A.M., Dahm C.C. A mitochondria-targeted S-nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia-reperfusion injury. Proc. Natl. Acad. Sci. U. S. A. 2009;106:10764–10769. doi: 10.1073/pnas.0903250106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hausenloy D., Wynne A., Duchen M., Yellon D. Transient mitochondrial permeability transition pore opening mediates preconditioning-induced protection. Circulation. 2004;109:1714–1717. doi: 10.1161/01.CIR.0000126294.81407.7D. [DOI] [PubMed] [Google Scholar]
- 121.Grover G.J., Atwal K.S., Sleph P.G., Wang F.L., Monshizadegan H., Monticello T. Excessive ATP hydrolysis in ischemic myocardium by mitochondrial F1F0-ATPase: effect of selective pharmacological inhibition of mitochondrial ATPase hydrolase activity. Am. J. Phys. 2004;287:H1747–H1755. doi: 10.1152/ajpheart.01019.2003. [DOI] [PubMed] [Google Scholar]
- 122.Husainy M.A., Dickenson J.M., Galinanes M. The MPTP status during early reoxygenation is critical for cardioprotection. J. Surg. Res. 2012;174:62–72. doi: 10.1016/j.jss.2010.11.879. [DOI] [PubMed] [Google Scholar]
- 123.Lim K.H.H., Javadov S.A., Das M., Clarke S.J., Suleiman M.S., Halestrap A.P. The effects of ischaemic preconditioning, diazoxide and 5-hydroxydecanoate on rat heart mitochondrial volume and respiration. J. Physiol. 2002;545:961–974. doi: 10.1113/jphysiol.2002.031484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhao Z.Q., VintenJohansen J. Postconditioning: reduction of reperfusion-induced injury. Cardiovasc. Res. 2006;70:200–211. doi: 10.1016/j.cardiores.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 125.Heusch G. Postconditioning: old wine in a new bottle? J. Am. Coll. Cardiol. 2004;44:1111–1112. doi: 10.1016/j.jacc.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 126.Quinlan C.L., Orr A.L., Perevoshchikova I.V., Treberg J.R., Ackrell B.A., Brand M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012;287:27255–27264. doi: 10.1074/jbc.M112.374629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Moreno-Sanchez R., Hernandez-Esquivel L., Rivero-Segura N.A., Marin-Hernandez A., Neuzil J., Ralph S.J. Reactive oxygen species are generated by the respiratory complex II—evidence for lack of contribution of the reverse electron flow in complex I. FEBS J. 2013;280:927–938. doi: 10.1111/febs.12086. [DOI] [PubMed] [Google Scholar]
- 128.Quinlan C.L., Perevoshchikova I.V., Hey-Mogensen M., Orr A.L., Brand M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013;1:304–312. doi: 10.1016/j.redox.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Korge P., John S.A., Calmettes G., Weiss J.N. Reactive oxygen species production induced by pore opening in cardiac mitochondria: the role of Complex II. J. Biol. Chem. 2017 doi: 10.1074/jbc.M116.768325. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Korge P., Calmettes G., John S.A., Weiss J.N. Reactive oxygen species production induced by pore opening in cardiac mitochondria: the role of Complex III. J. Biol. Chem. 2017 doi: 10.1074/jbc.M116.768325. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Fontaine E., Eriksson O., Ichas F., Bernardi P. Regulation of the permeability transition pore in skeletal muscle mitochondria—modulation by electron flow through the respiratory chain complex. J. Biol. Chem. 1998;273:12662–12668. doi: 10.1074/jbc.273.20.12662. [DOI] [PubMed] [Google Scholar]
- 132.Wagner S., Rokita A.G., Anderson M.E., Maier L.S. Redox regulation of sodium and calcium handling. Antioxid. Redox Signal. 2013;18:1063–1077. doi: 10.1089/ars.2012.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Silverman H.S., Stern M.D. Ionic basis of ischaemic cardiac injury: insights from cellular studies. Cardiovasc. Res. 1994;28:581–597. doi: 10.1093/cvr/28.5.581. [DOI] [PubMed] [Google Scholar]
- 134.Varadarajan S.G., An J., Novalija E., Smart S.C., Stowe D.F. Changes in [Na(+)](i), compartmental [Ca(2 +)], and NADH with dysfunction after global ischemia in intact hearts. Am. J. Phys. 2001;280:H280–H293. doi: 10.1152/ajpheart.2001.280.1.H280. [DOI] [PubMed] [Google Scholar]
- 135.Kwong J.Q., Lu X., Correll R.N., Schwanekamp J.A., Vagnozzi R.J., Sargent M.A. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 2015;12:15–22. doi: 10.1016/j.celrep.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Luongo T.S., Lambert J.P., Yuan A., Zhang X., Gross P., Song J. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep. 2015;12:23–34. doi: 10.1016/j.celrep.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Stone D., Darley-Usmar V., Smith D.R., Oleary V. Hypoxia-reoxygenation induced increase in cellular Ca-2 + in myocytes and perfused hearts - the role of mitochondria. J. Mol. Cell. Cardiol. 1989;21:963–973. doi: 10.1016/0022-2828(89)90795-5. [DOI] [PubMed] [Google Scholar]
- 138.Garcia-Rivas Gde J., Carvajal K., Correa F., Zazueta C. Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post-ischaemic functional recovery in rats in vivo. Br. J. Pharmacol. 2006;149:829–837. doi: 10.1038/sj.bjp.0706932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Luongo T.S., Lambert J.P., Gross P., Nwokedi M., Lombardi A.A., Shanmughapriya S. The mitochondrial Na +/Ca2 + exchanger is essential for Ca2 + homeostasis and viability. Nature. 2017;545:93–97. doi: 10.1038/nature22082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lemasters J.J., Theruvath T.P., Zhong Z., Nieminen A.L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta. 2009;1787:1395–1401. doi: 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Davidson S.M., Yellon D.M., Murphy M.P., Duchen M.R. Slow calcium waves and redox changes precede mitochondrial permeability transition pore opening in the intact heart during hypoxia and reoxygenation. Cardiovasc. Res. 2012;93:445–453. doi: 10.1093/cvr/cvr349. [DOI] [PubMed] [Google Scholar]
- 142.Pastorino J.G., Hoek J.B. Hexokinase II: the integration of energy metabolism and control of apoptosis. Curr. Med. Chem. 2003;10:1535–1551. doi: 10.2174/0929867033457269. [DOI] [PubMed] [Google Scholar]
- 143.Wilson J.E. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J. Exp. Biol. 2003;206:2049–2057. doi: 10.1242/jeb.00241. [DOI] [PubMed] [Google Scholar]
- 144.Chiara F., Castellaro D., Marin O., Petronilli V., Brusilow W.S., Juhaszova M. Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS One. 2008;3 doi: 10.1371/journal.pone.0001852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Xie G.C., Wilson J.E. Rat brain hexokinase: the hydrophobic N-terminus of the mitochondrially bound enzyme is inserted in the lipid bilayer. Arch. Biochem. Biophys. 1988;267:803–810. doi: 10.1016/0003-9861(88)90090-2. [DOI] [PubMed] [Google Scholar]
- 146.Pastorino J., Hoek J. Regulation of hexokinase binding to VDAC. J. Bioenerg. Biomembr. 2008;40:171–182. doi: 10.1007/s10863-008-9148-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gurel E., Smeele K.M., Eerbeek O., Koeman A., Demirci C., Hollmann M.W. Ischemic preconditioning affects hexokinase activity and HKII in different subcellular compartments throughout cardiac ischemia-reperfusion. J. Appl. Physiol. 2009;106:1909–1916. doi: 10.1152/japplphysiol.90537.2008. [DOI] [PubMed] [Google Scholar]
- 148.Pasdois P., Parker J.E., Griffiths E.J., Halestrap A.P. The role of oxidized cytochrome c in regulating mitochondrial reactive oxygen species production and its perturbation in ischaemia. Biochem. J. 2011;436:493–505. doi: 10.1042/BJ20101957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Littlejohns B., Pasdois P., Duggan S., Bond A.R., Heesom K., Jackson C.L. Hearts from mice fed a non-obesogenic high-fat diet exhibit changes in their oxidative state, calcium and mitochondria in parallel with increased susceptibility to reperfusion injury. PLoS One. 2014;9 doi: 10.1371/journal.pone.0100579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wu R., Smeele K.M., Wyatt E., Ichikawa Y., Eerbeek O., Sun L. Reduction in hexokinase II levels results in decreased cardiac function and altered remodeling after ischemia/reperfusion injury. Circ. Res. 2011;108:60–69. doi: 10.1161/CIRCRESAHA.110.223115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Nederlof R., Gurel-Gurevin E., Eerbeek O., Xie C., Deijs G.S., Konkel M. Reducing mitochondrial bound hexokinase II mediates transition from non-injurious into injurious ischemia/reperfusion of the intact heart. J. Physiol. Biochem. 2017 doi: 10.1007/s13105-017-0555-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Finegan B.A., Lopaschuk G.D., Gandhi M., Clanachan A.S. Ischemic preconditioning inhibits glycolysis and proton production in isolated working rat hearts. Am. J. Phys. 1995;269:H1767–H1775. doi: 10.1152/ajpheart.1995.269.5.H1767. [DOI] [PubMed] [Google Scholar]
- 153.McNulty P.H., Darling A., Whiting J.M. Glycogen depletion contributes to ischemic preconditioning in the rat heart in vivo. Am. J. Phys. 1996;271:H2283–H2289. doi: 10.1152/ajpheart.1996.271.6.H2283. [DOI] [PubMed] [Google Scholar]
- 154.King L.M., Opie H. Does preconditioning act by glycogen depletion in the isolated rat heart? J. Mol. Cell. Cardiol. 1996;28:2305–2321. doi: 10.1006/jmcc.1996.0224. [DOI] [PubMed] [Google Scholar]
- 155.Baines C.P., Song C.X., Zheng Y.T., Wang G.W., Zhang J., Wang O.L. Protein kinase Cepsilon interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ. Res. 2003;92:873–880. doi: 10.1161/01.RES.0000069215.36389.8D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hausenloy D.J., Yellon D.M. Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc. Res. 2006;70:240–253. doi: 10.1016/j.cardiores.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 157.Yang X., Cohen M.V., Downey J.M. Mechanism of cardioprotection by early ischemic preconditioning. Cardiovasc. Drugs Ther. 2010;24:225–234. doi: 10.1007/s10557-010-6236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Juhaszova M., Zorov D.B., Kim S.H., Pepe S., Fu Q., Fishbein K.W. Glycogen synthase kinase-3 beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Invest. 2004;113:1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Juhaszova M., Zorov D.B., Yaniv Y., Nuss H.B., Wang S., Sollott S.J. Role of glycogen synthase kinase-3beta in cardioprotection. Circ. Res. 2009;104:1240–1252. doi: 10.1161/CIRCRESAHA.109.197996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Miyamoto S., Murphy A.N., Brown J.H. Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 2008;15:521–529. doi: 10.1038/sj.cdd.4402285. [DOI] [PubMed] [Google Scholar]
- 161.Roberts D.J., Tan-Sah V.P., Smith J.M., Miyamoto S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J. Biol. Chem. 2013;288:23798–23806. doi: 10.1074/jbc.M113.482026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Fullmer T.M., Pei S., Zhu Y., Sloan C., Manzanares R., Henrie B. Insulin suppresses ischemic preconditioning-mediated cardioprotection through Akt-dependent mechanisms. J. Mol. Cell. Cardiol. 2013;64:20–29. doi: 10.1016/j.yjmcc.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Brdiczka D. Contact sites between mitochondrial envelope membranes - structure and function in energy-transfer and protein-transfer. Biochim. Biophys. Acta. 1991;1071:291–312. doi: 10.1016/0304-4157(91)90018-r. [DOI] [PubMed] [Google Scholar]
- 164.Knoll G., Brdiczka D. Changes in freeze-fractured mitochondrial membranes correlated to their energetic state dynamic interactions of the boundary membranes. Biochim. Biophys. Acta. 1983;733:102–110. doi: 10.1016/0005-2736(83)90095-0. [DOI] [PubMed] [Google Scholar]
- 165.Beutner G., Ruck A., Riede B., Brdiczka D. Complexes between porin, hexokinase, mitochondrial creatine kinase and adenylate translocator display properties of the permeability transition pore. Implication for regulation of permeability transition by the kinases. Biochim. Biophys. Acta. 1998;1368:7–18. doi: 10.1016/s0005-2736(97)00175-2. [DOI] [PubMed] [Google Scholar]
- 166.Brdiczka D.G., Zorov D.B., Sheu S.S. Mitochondrial contact sites: their role in energy metabolism and apoptosis. Biochim. Biophys. 2006;1762:148–163. doi: 10.1016/j.bbadis.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 167.Doran E., Halestrap A.P. Cytochrome c release from isolated rat liver mitochondria can occur independently of outer-membrane rupture: possible role of contact sites. Biochem. J. 2000;348:343–350. [PMC free article] [PubMed] [Google Scholar]
- 168.Beutner G., Ruck A., Riede B., Brdiczka D. Complexes between hexokinase, mitochondrial porin and adenylate translocator in brain: regulation of hexokinase, oxidative phosphorylation and permeability transition pore. Biochem. Soc. Trans. 1997;25:151–157. doi: 10.1042/bst0250151. [DOI] [PubMed] [Google Scholar]
- 169.Kottke M., Adams V., Wallimann T., Nalam V.K., Brdiczka D. Location and regulation of octameric mitochondrial creatine kinase in the contact sites. Biochim. Biophys. Acta. 1991;1061:215–225. doi: 10.1016/0005-2736(91)90287-i. [DOI] [PubMed] [Google Scholar]
- 170.Rauch U., Schulze K., Witzenbichler B., Schultheiss H.P. Alteration of the cytosolic-mitochondrial distribution of high-energy phosphates during global myocardial ischemia may contribute to early contractile failure. Circ. Res. 1994;75:760–769. doi: 10.1161/01.res.75.4.760. [DOI] [PubMed] [Google Scholar]
- 171.Laclau M.N., Boudina S., Thambo J.B., Tariosse L., Gouverneur G., Bonoron-Adele S. Cardioprotection by ischemic preconditioning preserves mitochondrial function and functional coupling between adenine nucleotide translocase and creatine kinase. J. Mol. Cell. Cardiol. 2001;33:947–956. doi: 10.1006/jmcc.2001.1357. [DOI] [PubMed] [Google Scholar]
- 172.Speer O., Back N., Buerklen T., Brdiczka D., Koretsky A., Wallimann T. Octameric mitochondrial creatine kinase induces and stabilizes contact sites between the inner and outer membrane. Biochem. J. 2005;385:445–450. doi: 10.1042/BJ20040386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Spindler M., Meyer K., Stromer H., Leupold A., Boehm E., Wagner H. Creatine kinase-deficient hearts exhibit increased susceptibility to ischemia-reperfusion injury and impaired calcium homeostasis. Am. J. Phys. 2004;287:H1039–H1045. doi: 10.1152/ajpheart.01016.2003. [DOI] [PubMed] [Google Scholar]
- 174.Dolder M., Walzel B., Speer O., Schlattner U., Wallimann T. Inhibition of the mitochondrial permeability transition by creatine kinase substrates—requirement for microcompartmentation. J. Biol. Chem. 2003;278:17760–17766. doi: 10.1074/jbc.M208705200. [DOI] [PubMed] [Google Scholar]
- 175.Shoshan-Barmatz V., Mizrachi D. VDAC1: from structure to cancer therapy. Front. Oncol. 2012;2:164. doi: 10.3389/fonc.2012.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Shoshan-Barmatz V., Ben-Hail D., Admoni L., Krelin Y., Tripathi S.S. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim. Biophys. Acta. 2015;1848:2547–2575. doi: 10.1016/j.bbamem.2014.10.040. [DOI] [PubMed] [Google Scholar]
- 177.Adams V., Bosch W., Schlegel J., Wallimann T., Brdiczka D. Further characterization of contact sites from mitochondria of different tissues - topology of peripheral kinases. Biochim. Biophys. Acta. 1989;981:213–225. doi: 10.1016/0005-2736(89)90031-x. [DOI] [PubMed] [Google Scholar]
- 178.Soriano M.E., Scorrano L. The interplay between BCL-2 family proteins and mitochondrial morphology in the regulation of apoptosis. Adv. Exp. Med. Biol. 2010;687:97–114. doi: 10.1007/978-1-4419-6706-0_6. [DOI] [PubMed] [Google Scholar]
- 179.Tait S.W., Green D.R. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 180.Martinou J.C., Youle R.J. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev. Cell. 2011;21:92–101. doi: 10.1016/j.devcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Huang J.H., Ito Y., Morikawa M., Uchida H., Kobune M., Sasaki K. Bcl-xL gene transfer protects the heart against ischemia/reperfusion injury. Biochem. Biophys. Res. Commun. 2003;311:64–70. doi: 10.1016/j.bbrc.2003.09.160. [DOI] [PubMed] [Google Scholar]
- 182.Whelan R.S., Konstantinidis K., Wei A.C., Chen Y., Reyna D.E., Jha S. Bax regulates primary necrosis through mitochondrial dynamics. Proc. Natl. Acad. Sci. U. S. A. 2012;109:6566–6571. doi: 10.1073/pnas.1201608109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Keinan N., Tyomkin D., Shoshan-Barmatz V. Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol. Cell. Biol. 2010;30:5698–5709. doi: 10.1128/MCB.00165-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ben-Hail D., Begas-Shvartz R., Shalev M., Shteinfer-Kuzmine A., Gruzman A., Reina S. Novel compounds targeting the mitochondrial protein VDAC1 inhibit apoptosis and protect against mitochondria dysfunction. J. Biol. Chem. 2016;291:24986–25003. doi: 10.1074/jbc.M116.744284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hall A.R., Burke N., Dongworth R.K., Hausenloy D.J. Mitochondrial fusion and fission proteins: novel therapeutic targets for combating cardiovascular disease. Br. J. Pharmacol. 2014;171:1890–1906. doi: 10.1111/bph.12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ong S.-B., Subrayan S., Lim S.Y., Yellon D.M., Davidson S.M., Hausenloy D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- 187.Disatnik M.H., Ferreira J.C., Campos J.C., Gomes K.S., Dourado P.M., Qi X. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J. Am. Heart Assoc. 2013;2 doi: 10.1161/JAHA.113.000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Gao D., Zhang L., Dhillon R., Hong T.T., Shaw R.M., Zhu J. Dynasore protects mitochondria and improves cardiac lusitropy in Langendorff perfused mouse heart. PLoS One. 2013;8 doi: 10.1371/journal.pone.0060967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Sharp W.W., Fang Y.H., Han M., Zhang H.J., Hong Z., Banathy A. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. 2014;28:316–326. doi: 10.1096/fj.12-226225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zepeda R., Kuzmicic J., Parra V., Troncoso R., Pennanen C., Riquelme J.A. Drp1 loss-of-function reduces cardiomyocyte oxygen dependence protecting the heart from ischemia-reperfusion injury. J. Cardiovasc. Pharmacol. 2014;63:477–487. doi: 10.1097/FJC.0000000000000071. [DOI] [PubMed] [Google Scholar]
- 191.Murriel C.L., Churchill E., Inagaki K., Szweda L.I., Mochly Rosen D. Protein kinase C delta activation induces apoptosis in response to cardiac ischemia and reperfusion damage - a mechanism involving bad and the mitochondria. J. Biol. Chem. 2004;279:47985–47991. doi: 10.1074/jbc.M405071200. [DOI] [PubMed] [Google Scholar]
- 192.Din S., Mason M., Volkers M., Johnson B., Cottage C.T., Wang Z. Pim-1 preserves mitochondrial morphology by inhibiting dynamin-related protein 1 translocation. Proc. Natl. Acad. Sci. U. S. A. 2013;110:5969–5974. doi: 10.1073/pnas.1213294110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Dong Y., Undyala V.V., Przyklenk K. Inhibition of mitochondrial fission as a molecular target for cardioprotection: critical importance of the timing of treatment. Basic Res. Cardiol. 2016;111:59. doi: 10.1007/s00395-016-0578-x. [DOI] [PubMed] [Google Scholar]
- 194.Prudent J., Zunino R., Sugiura A., Mattie S., Shore G.C., McBride H.M. MAPL SUMOylation of Drp1 stabilizes an ER/mitochondrial platform required for cell death. Mol. Cell. 2015;59:941–955. doi: 10.1016/j.molcel.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 195.Otera H., Miyata N., Kuge O., Mihara K. Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J. Cell Biol. 2016;212:531–544. doi: 10.1083/jcb.201508099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Yang Y.-L., Li J., Liu K., Zhang L., Liu Q., Liu B. Ginsenoside Rg5 increases cardiomyocyte resistance to ischemic injury through regulation of mitochondrial hexokinase-II and dynamin-related protein 1. Cell Death Dis. 2017;8:e2625. doi: 10.1038/cddis.2017.43. [DOI] [PMC free article] [PubMed] [Google Scholar]