

Abstract
The heme enzyme chlorite dismutase (Cld) catalyzes the degradation of chlorite to chloride and dioxygen. Although structure and steady-state kinetics of Clds have been elucidated, many questions remain (e.g., the mechanism of chlorite cleavage and the pH dependence of the reaction). Here, we present high-resolution X-ray crystal structures of a dimeric Cld at pH 6.5 and 8.5, its fluoride and isothiocyanate complexes and the neutron structure at pH 9.0 together with the pH dependence of the Fe(III)/Fe(II) couple, and the UV–vis and resonance Raman spectral features. We demonstrate that the distal Arg127 cannot act as proton acceptor and is fully ionized even at pH 9.0 ruling out its proposed role in dictating the pH dependence of chlorite degradation. Stopped-flow studies show that (i) Compound I and hypochlorite do not recombine and (ii) Compound II is the immediately formed redox intermediate that dominates during turnover. Homolytic cleavage of chlorite is proposed.
Keywords: chlorite dismutase, heme enzyme, O2 generation, X-ray diffraction, neutron diffraction, stopped-flow spectroscopy, resonance Raman spectroscopy, spectroelectrochemistry
1. Introduction
Chlorite dismutases (Clds) are heme b-containing oxidoreductases (EC 1.13.11.49) found in prokaryotic organisms.1 They are able to efficiently decompose chlorite (ClO2– or OClO–) into harmless chloride (Cl–) and dioxygen (O2) with chlorite being the sole source of dioxygen.2 Thereby, a covalent oxygen–oxygen bond is formed, a biochemical reaction that was believed to be unique to the water-splitting manganese complex of photosystem II of oxygenic organisms.3
The ability to degrade chlorite was shown for several functional (i.e., efficiently chlorite degrading) Clds, all of them possessing a putatively flexible arginine in an otherwise hydrophobic distal heme pocket. Available X-ray crystal structures reveal two distinct conformations, either pointing toward the heme iron (“in”; e.g., chlorite dismutase from Dechloromonas aromatica (DaCld), PDB: 3Q08)4,5 or toward the substrate entry channel (“out”; e.g., chlorite dismutase from Nitrospira defluvii (NdCld), PDB: 3NN1).6
The mechanism of chlorite cleavage is still under discussion. Computational studies suggest homolytic cleavage of OClO– thereby producing chlorine monoxide (ClO•) and Compound II [Por···Fe(IV)=O], followed by a rebinding step and production of Fe(III)–peroxyhypochlorite that finally releases chloride and dioxygen (Reactions I and II).7 Binding of the anionic substrate chlorite (pKa = 1.728) to ferric Cld is exergonic7 and independent of the Cld-typical arginine.9
| I |
| II |
By contrast, biochemical studies on pentameric Clds suggest heterolytic cleavage of chlorite thereby forming Compound I [Por+•···Fe(IV)=O] and hypochlorite (HOCl/–OCl) (Reaction III).10−14 The following rebinding step produces Fe(III)–peroxyhypochlorite and, in consequence, chloride and dioxygen (Reaction IV).
| III |
| IV |
In contrast to KM, the turnover number (kcat) of Clds show a clear dependence on pH.11 The pH optimum of chlorite degradation is typically in the acidic pH range, and the turnover number decreases with rising pH. It has been postulated that the pH dependence of this reaction reflects the protonation state of the strictly conserved flexible distal arginine, which was reported to possess a pKa of ∼6.5 in DaCld14 and NdCld,11 both clade 1 members (see next paragraph). It was suggested that this basic amino acid supports both heterolytic cleavage of chlorite (Reaction III) and the recombination reaction between the postulated intermediate hypochlorite and Compound I (Reaction IV). Elimination of the distal Arg by mutation significantly decreases kcat and boosts the extent of irreversible inactivation. The latter event was proposed to be promoted by released hypochlorous acid, since scavengers of (HOCl/–OCl) are able to suppress inactivation and increase the extent of degraded chlorite.11 It should be noted that the exclusiveness of such scavengers for (HOCl/–OCl) is questionable, and it is not fully clear whether they would also detect chlorine monoxide or chlorine dioxide.
The known functional Clds can be divided into two phylogenetically distinct clades 1 and 2, which differ in subunit size and oligomerization. So far, biochemical and structural research has mainly focused on the pentameric and hexameric clade 1 proteins,6,10−17 whereas only few studies have addressed dimeric clade 2 Clds from Nitrobacter winogradskyi (NwCld),18Klebsiella pneumonia MGH 78578 (KpCld),19 and Cyanothece sp. PCC7425 (CCld).20 The present study provides the first comprehensive structural and biochemical investigation of a dimeric clade 2 chlorite dismutase (i.e., CCld) and focuses on comprehensive structural and mechanistic studies including the impact of pH on chlorite conversion.
In contrast to pentameric Clds, the spectral features of CCld in various redox states are shown to be better resolved and easier to assign. We report the pH dependence of (i) the pre-steady-state kinetics of the reaction of ferric CCld with both ClO2– and the putative reaction intermediate (HOCl/–OCl), (ii) the reduction potential of the Fe(III)/Fe(II) couple, and (iii) the UV–vis and resonance Raman spectra of the ferrous and ferric forms. We provide high-resolution X-ray structures of CCld at pH 6.5 (1.30 Å) and pH 8.5 (1.55 Å) as well as of its fluoride (1.18 Å) and isothiocyanate (1.28 Å) complexes at pH 6.5. Moreover, we present the first neutron structure of a ferric heme enzyme from this superfamily solved at pH 9.0 to a resolution of 2.35 Å together with the X-ray structure at room temperature. Our data demonstrate that the distal Arg is protonated at pH 9.0 and indicate that ClO• is the product of chlorite cleavage, at least in the O2 generative part of the reaction. This contradicts the hypothesis that the protonation state of the catalytic arginine modulates chlorite degradation.
On the basis of our data, we discuss the postulated mechanism(s) of chlorite conversion and provide a more detailed view of the mode of action including potential side reactions catalyzed by the enzyme.
2. Results
2.1. Impact of pH on the Spectral Properties of Dimeric Ferric and Ferrous CCld
The electronic absorption and resonance Raman (RR) spectra of ferric CCld are presented in Figure 1. The electronic absorption spectrum at acidic pH (i.e., 5.9) of the ferric protein (Figure 1a, spectrum (i), characterized by a Soret band at 406 nm, Q-bands at 503, 540, and 590 nm, and a charge-transfer (CT1) band at 636 nm, suggests a mixture of high-spin (HS) and low-spin (LS) species.
Figure 1.

(a) Electronic absorption and (b) high-frequency RR spectra of ferric CCld at pH 5.9 and pH 9.6. Experimental conditions: UV–vis: (a) 600 nm/min scan rate. The region between 450 and 700 nm has been expanded 8-fold. (b) RR: 413.1 nm exc., 5 mW laser power at the sample, and 10 min integration time. The region between 1450 and 1700 cm–1 has been expanded 2-fold. The intensity of the spectra is normalized to that of the ν4 band.
This is confirmed by the corresponding high-frequency RR spectrum (Figure 1b, bottom), which clearly indicates a mixture of three species; an aquo 6-coordinate (6c) HS (ν3 at 1485 cm–1, ν2 at 1562 cm–1), a 5-coordinate (5c) HS (ν3 at 1491 cm–1), and a 6cLS (ν3 at 1506 cm–1, ν10 at 1641 cm–1). Furthermore, ferric CCld is characterized by two coincident vinyl stretching modes at 1629 cm–1 as for myoglobin, which downshift to 1626 in the ferrous form (Figure S1b, spectrum a). The low-frequency RR spectrum (Figure S2, spectrum a) of ferric CCld at pH 5.8 is characterized by intense bands at 370 cm–1, due to the propionyl bending modes, and at 413 and 435 cm–1 due to the vinyl bending modes.
Figure S3a,b depicts the UV–vis spectral changes of CCld as a function of pH between pH 3.0 and 10.0, and pH 5.8 and 12.0, respectively. The pKa of the alkaline transition (i.e., deprotonation of the distal water) was calculated to be 8.1 ± 0.04.
At alkaline pH, the absorption spectrum (Figure 1a, top) becomes typical of a hydroxo-complex, characterized by a mixture of 6cHS and 6cLS species with a Soret band at 414 nm, Q bands at 540 and 575 nm, and a weak CT1 band at 610 nm. This is consistent with the high-frequency RR spectrum (Figure 1b, top) which displays a predominant 6cLS species (ν3 1506, ν2 1580, ν37 1605, ν10 1641 cm–1) and a weak 6cHS form (ν3 1478 cm–1). Accordingly, in the low-frequency region the ν(Fe–OH) stretching modes of the heme-hydroxide complexes have been identified by their sensitivity to 2H and 18O isotopic substitution (Figure S4a and Table S1) and on the basis of the difference spectra and curve fitting analysis (Figure S4b). The isotopic shifts observed for the 18O and 2H labeled species are in good agreement with those calculated using the diatomic harmonic oscillator model (Table S2). The LS and HS ν(Fe–OH) stretching modes have been assigned to the bands at the 510 and 485 cm–1, respectively (Figure 2a). It is noted that while the 6cLS is very similar to that found for DaCld and KpCld (514 and 509 cm–1, respectively), the 6cHS is more than 40 cm–1 higher (440 and 442 cm–1 in DaCld14 and KpCld,19 respectively).
Figure 2.

Low-frequency RR spectra of CCld. (a) ferric (pH 9.6), (b) ferrous, and (c) ferrous-CO at pH 5.8 showing the frequencies of the Fe-hydroxide LS, (red) and HS, (blue), Fe-Im, and the ν(Fe-CO) stretching and δ(FeCO) modes, respectively. Experimental: 413.1 nm (ferric and CO complex) and 441.6 nm (ferrous) exc.; (a) 5 mW laser power at the sample, average of 12 spectra with 120 min integration time; (b) 11 mW laser power at the sample, 2 spectra with 20 min integration time; (c) 5 mW laser power at the sample, average of 4 spectra with 40 min integration time.
The electronic absorption and RR spectra of the reduced protein clearly indicate the presence of a pure 5cHS state (Figure S1a,b, spectra (i)). It is noted that the spectrum of ferrous CCld is invariant in the pH interval 5.8–9.8 (data not shown). The low-frequency RR spectrum of ferrous 5cHS hemes is of interest particularly due to the presence of a strong band resulting from the iron-imidazole stretching mode, ν(Fe-Im), in the range of 200–250 cm–1. The ν(Fe-Im) frequency is very sensitive to H-bonding interactions and, therefore, to the protein matrix surrounding the proximal His114 residue (CCld numbering).21,22 The intense band at 231 cm–1 observed for ferrous CCld at pH 5.8 (Figure 2b and Figure S2c) (higher than that observed for DaCld14 at 222 cm–1 but similar to that observed for KpCld19) is assigned to the ν(Fe-Im) mode, a frequency somewhat higher than that observed in myoglobin (220 cm–1), where the proximal His is hydrogen bonded to a weak H-bond acceptor.
2.2. Reactions of Ferric CCld with Hypochlorite
Hypochlorous acid is known to function as a two-electron oxidant that converts ferric peroxidases and catalases to the redox intermediate Compound I.23,24 In functional chlorite dismutases, the oxidation of ferric Cld by chlorite is proposed to transiently form hypochlorite (Reaction III) which is rapidly consumed in the subsequent recombination reaction (Reaction IV). Consequently, we asked whether ferric Cld can be oxidized by hypochlorite to Compound I followed by recombination of the latter with excess OCl–.
Thus, we followed the reaction of CCld with hypochlorite (HOCl/–OCl, pKa = 7.53) spectrophotometrically using the stopped-flow technique (Figure 3a–d) and polarographically using a Clark-type electrode (Figure 3e). At pH 5.0, effective Compound I formation can be observed (Figure 3a, red spectrum) with an apparent second-order rate constant of 2.5 × 106 M–1 s–1 (Figure 3c).
Figure 3.

Reaction of ferric CCld with hypochlorite (HOCl/-OCl). (a) Spectral changes of 1.5 μM CCld upon reaction with 13 μM hypochlorite at pH 5.0. Black: ferric CCld; red: Compound I; blue: Compound II-like species. For better visualization of the CT and Q bands, the 460–700 nm region is magnified 6-fold. Arrows illustrate the course of the reaction by both their colors and directions. (b) Corresponding time traces at 406 nm (ferric Soret maximum) and 418 nm (Soret maximum of Compound II-like species). Left: Time traces between 0.0 and 0.5 s. The single exponential fit of the time trace at 405 nm is shown in red. Right: Time traces over the total recording time. Recording time points of the intermediate spectra shown in (a) are labeled in corresponding colors. (c) Linear dependence of kobs from hypochlorite concentrations at pH 5.0. The rate constant kapp was obtained from the slope of the linear regression. (d) Spectral changes of 1.5 μM CCld upon reaction with 500 μM hypochlorite at pH 9.0. Black: ferric CCld; red and blue: intermediate spectra exhibiting ferric low-spin features. Peak maxima are labeled. (e) Reaction of 1.5 μM CCld with 20 μM chlorite at pH 5.0 (red) and with 40 μM hypochlorite at pH 5.0, 7.0, and 9.0 (black) followed by polarographic measurements using a Clark-type electrode. The negative control (no substrate) is shown in gray and reflects the amount of oxygen that is introduced to the O2-free system by triggering the reaction with enzyme solution.
For the depicted spectra and time traces at pH 5.0, 1.5 μM CCld and 13 μM hypochlorite were used. After 0.074 s, the Soret peak exhibits 50% hypochromicity, and a distinct band at 650 nm has emerged. Above a ∼6-fold excess of hypochlorite, a slow shift of Compound I to a Compound II-like species (Soret maximum at 418 nm and Q bands at 528 and 551 nm) is observed (Figure 3a, blue spectrum). Finally, the enzyme slowly returns to the ferric resting state (Figure 3b, time trace at 406 nm). The maximum abundance of the Compound II-like species is reached after ∼6.8 s. At all hypochlorite concentrations tested, heme bleaching could be observed to some extent.
At pH 7.0, using the same concentrations of enzyme and substrate, Compound I formation is about 4 times faster (9.6 × 106 M–1 s–1, data not shown) compared to pH 5.0. However, it overlaps to a greater extent with the evolution of the Compound II-like species (418, 528, and 551 nm) which is already fully developed after ∼0.3 s, approximately 23 times faster than at pH 5.0. At pH 9.0, the resting state of CCld exhibits a Soret band at 414 nm reflecting the presence of the OH–-complex described above. It does not directly react with hypochlorite. Only at higher concentration of oxidant, a very slow bathochromic shift is observed (Figure 3d). The final spectrum is similar to that of the CCld-cyanide complex. The nature of the ligand is unclear, one possibility is chlorate which can form upon disproportionation of hypochlorite.25 However, no spectral change could be observed upon incubation of CCld with chlorate at pH 9.0 (data not shown).
Importantly, at no pH studied (i.e., pH 5.0, 7.0, and 9.0), could any O2 generation be observed polarographically upon mixing 1.5 μM CCld with 40 μM hypochlorite (Figure 3e).
2.3. Chlorite Decomposition by Ferric CCld at Different pH Values
Recently we reported the steady-state kinetics of chlorite degradation of CCld and its pH dependence.20 The completeness of chlorite degradation as well as the initial reaction velocity increased with decreasing pH. At pH 5.0, the chlorite-degrading activity of CCld was found to have a kcat value of 1144 ± 23.8 s–1, a KM of 162 ± 10.0 μM, and a catalytic efficiency of (7.1 ± 0.6) × 106 M–1 s–1. Reactions below pH 4.0 could not be followed due to enzyme denaturation.
In the present work, we studied the pH-dependent degradation of chlorite by CCld using UV–vis stopped-flow spectroscopy. For comparison, we kept the substrate/enzyme ratio constant at all pH values and chose a 330-fold excess of chlorite, since this guaranteed full degradation of the oxidant over the whole range of pH. We observed a significantly prolonged phase of chlorite decomposition with increasing pH (time traces at 280 nm, Figure 4b). At pH 5.0, 500 μM chlorite are degraded within 160 ms by 1.5 μM CCld (Figure 4b, top). Within ∼40 ms, the spectrum shifts to a (not entirely developed) Compound II-like state (Figure 4a,b, top). At the very beginning of the reaction (∼4 ms after mixing), ∼25% hypochromicity and a shift of CT1 to 650 nm is observed, which might suggest Compound I formation to some extent. Higher substrate concentrations lead to immediate and full formation of Compound II (Soret maximum at 418 nm, typical double peak at 528 and 551 nm, Figure S5a). During chlorite degradation, this redox intermediate dominates and its spectrum is steadily converted to the ferric resting state. Interestingly, concomitant with chlorite degradation, an increase in absorbance at 360 nm is observed (Figure S5b,c). At this wavelength, chlorine dioxide exhibits its characteristic absorption maximum.26 From previous studies on horseradish peroxidase,24 it is known that this enzyme performs the classical peroxidase cycle upon reaction with chlorite, including Compound I and HOCl/–OCl formation. Concomitantly, chlorite is used as one-electron donor for Compound I and Compound II which leads to generation of chlorine dioxide, the latter being monitored UV–vis spectroscopically and being responsible for heme destruction when present in excess. However, in the case of CCld, after consumption of all the chlorite (0.160 s), the absorbance at 360 nm decreases. The latter event is accompanied by a retransition to a Compound II-like spectrum which then very slowly shifts back to the ferric resting state.
Figure 4.

Reaction of ferric CCld with chlorite followed by UV–vis stopped-flow spectroscopy (top to bottom: pH 5.0, 7.0, 9.0). (a) Spectral changes of 1.5 μM CCld upon reaction with 500 μM chlorite. Spectrum of ferric CCld is shown in red, the dominating Compound II-like intermediate spectrum is depicted in blue. Peak maxima are labeled. For better visualization of the CT and Q bands, the 460–700 nm region is magnified 6-fold. Arrows indicate the course of the reaction by both, their colors and directions. Spectra and corresponding recording time points are shown in corresponding colors. (b) Corresponding time traces at: 405 nm (pH 5.0 and 7.0) and 410 nm (pH 9.0), ferric Soret maximum, red; 418 nm, Soret maximum of the dominating Compound II-like intermediate species, blue; 280 nm, chlorite decomposition, black. To emphasize the pH-dependent differences between the rates of chlorite degradation (time trace at 280 nm), appropriate time frames are displayed in the left part of panel (b), and the respective time points of completed chlorite decomposition are labeled and marked by the continuous dashed line.
At pH 7.0, the degradation of 500 μM chlorite by 1.5 μM CCld takes 3.5 s (Figure 4a,b, middle). The spectral course of the reaction exhibits a direct and rapid transition of the resting state to fully developed Compound II (decrease in absorbance at 406 nm occurs simultaneously with red-shift of Soret maximum).
In contrast to pH 5.0, at pH 7.0 there is no indication of Compound I formation and the spectral features of Compound II remain fully established during chlorite degradation. A very slow conversion to the ferric state starts as soon as all chlorite is consumed (isosbestic point at 413 nm). The reaction at pH 9.0 is qualitatively invariant compared to pH 7.0, except that chlorite consumption proceeds over a longer time period (36 s) and no direct transition from Compound II to the resting state could be observed even after degradation of chlorite (Figure 4a,b, bottom). It is important to note that determination of apparent second-order rate constants for Compound II formation did not give reliable data. It was hampered by the fact that kobs values do not show any linear dependence on substrate concentrations below ∼66-fold excess.
2.4. Spectral Properties of Ligand-Bound Ferric and Ferrous CCld
The ligand binding behavior of pentameric Clds has been probed extensively,14,27,28 and it has been shown that the use of different buffer compositions can produce a pronounced heterogeneity of high- and low-spin species.14,27 Interestingly, binding of LS ligands like cyanide does not necessarily render the enzyme inactive or alter the kinetic parameters of chlorite degradation.29 This is also the case for ferric CCld, which we probed for binding to the exogenous HS-ligand fluoride and the LS-ligand thiocyanate. The dissociation constants for both ligands were determined by titration, followed by UV–vis spectroscopy, to be 12.6 mM for fluoride and 0.074 mM for thiocyanate (Figure S6).
Thiocyanate acts as a LS-ligand to the cofactor and binds in the form of isothiocyanate with its nitrogen atom oriented to the heme iron (q.v. Section 2.7). Accordingly, the presence of a low-spin heme is confirmed by a red-shifted Soret maximum at 414 nm and the intensity increase of Q-bands around 532 and 570 nm in the UV–vis spectrum (Figure S6a, dotted line). Fluoride acts as HS-ligand; accordingly in the UV–vis spectrum, the Soret maximum is blue-shifted to 404 nm, and strong bands at 503 and 611 nm are observed (Figure S6a, dashed line).
These features, and the high-frequency RR spectra obtained with 441.6 nm excitation (ν3 at 1479 cm–1, ν2 at 1561 cm–1, ν10 at 1608 cm–1, data not shown), are characteristic of a 6cHS form, with fluoride coordinated as the sixth ligand of the iron ion. In the low-frequency RR spectra, the ν(Fe–F) stretching mode was assigned on the basis of its intensity enhancement upon excitation near the CT2 band,30,31 as shown by the comparison of the RR spectra obtained with excitation at 413.1 and 441.6 nm (Figure S7a,b). The ν(Fe–F) stretch is assigned to the band at 381 cm–1, which overlaps with the propionyl bending mode at 374 cm–1. The frequency is lower than the corresponding band of KpCld–F, and DaCld–F, complexes (385, 390 cm–1, respectively).28 This result agrees very well with the trans-effect of the proximal ligand on the Fe–F bond strength recently proposed for Clds. The higher the ν(Fe-Im) frequency, the weaker the ν(Fe–F) bond.28 CCld has the largest trans-effect among the different Clds.
To further probe the ligand binding properties of CCld, carbon monoxide binding to the ferrous form of the enzyme was studied. Complex formation gives rise to a 6cLS species (Figure S1a,b, spectra (ii)). The ν3 band at 1470 cm–1 in the RR spectrum indicates the presence of a small amount of 5cHS ferrous heme due to photolysis of the CO ligand under laser irradiation. Unlike DaCld,14 the spectrum of the carbon monoxide complex is invariant in the pH interval 5.8–9.8 (data not shown). Figure S8 depicts the RR spectra of the 12CO and 13CO complexes of CCld in the low- and high-frequency regions. Figure 2c shows the two isotope sensitive bands at 492 and 576 cm–1 in 12CO (which shift to 489 and 559 cm–1 for the CCld-13CO form, Figure S8a), assigned to the ν(Fe–C) stretching and δ(Fe–C–O) bending modes, respectively. Accordingly, a corresponding ν(CO) stretching mode is observed at 1952 cm–1, which shifts to 1908 cm–1 for the 13C-labeled form (Figure S8b). The ν(Fe–C)/ν(CO) frequencies, similar to those previously found for DaCld,14 are typical of heme-CO adducts that have weak interaction with the distal protein matrix.
Finally, we probed the pH dependence of the kinetics of cyanide binding to CCld. Cyanide acts as a strong LS-ligand for many heme enzymes. For CCld, the spectral properties of the complex are qualitatively pH-invariant and show a Soret maximum at 421 nm, broad Q-bands with a maximum at 538 nm, and a shoulder around 565 nm.20
In this work, binding rates have been determined by single exponential fitting of the decrease in absorbance at 405 nm (pH 5.0, 6.0 and 7.0) or double exponential fitting of the increase in absorbance at 410 or 423 nm (pH 8.0, 9.0 and 9.6) (Figure S9). At higher pH values, cyanide complexation is no longer the only reaction taking place since formation of the hydroxo-complex implies that OH– has to dissociate from the distal heme coordination site prior to CN– binding. Association constants increase with pH and range from 6.32 × 103 M–1 s–1 at pH 5.0 to 1.26 × 106 M–1 s–1 at pH 9.0 (Figure 5). Around pH 9.0, the rate reaches a maximum and starts to decrease as the pH is raised further.
Figure 5.

Binding of cyanide to ferric CCld. (a) Spectral changes during reaction of 1.5 μM ferric CCld (black spectrum) with 35 μM cyanide at pH 5.0. The red spectrum shows the emerging LS complex. The 460–700 nm region is magnified 6-fold. (b) Typical time trace at 405 nm. The single exponential fit of the curve is depicted in red. (c) Plot of kobs versus cyanide concentration. The apparent association constant kon was obtained from the slope of the regression line. (d) Apparent bimolecular rate constants of cyanide binding to ferric CCld at pH 5.0, 6.0, 7.0, 8.0, 9.0, and 9.6 determined by UV–vis stopped-flow spectroscopy. Plot of kon versus pH. The fit of the curve is depicted in red.
2.5. Impact of pH on the Standard Reduction Potential of the Fe(III)/Fe(II) Couple
Finally, we tested whether the pronounced pH dependence of the activity of CCld is related to pH-dependent changes in redox properties. In order to determine the standard reduction potential (E°′) of the Fe(III)/Fe(II) couple of CCld, spectroelectrochemical studies were performed. Figure 6 shows the fully oxidized and fully reduced (black lines, AλoxMax = 406 nm and AλredMax = 434 nm) as well as the equilibrium spectra of CCld (gray lines) at six different applied potentials in the optically transparent thin-layer spectroelectrochemical (OTTLE) cell (25 °C, pH 6.0). Two clear isosbestic points at 420 and 455 nm can be observed. From these data, we obtained a linear Nernst plot with a slope that is consistent with a one-electron reduction process (Figure 6b).32−34E°′ was calculated to be −94.4 ± 0.3 mV.
Figure 6.

Spectroelectrochemical titration of the Fe(III)/Fe(II) redox couple of the high-spin native form of CCld (a) Electronic absorption spectra of CCld at different applied potentials at pH 6.0. Black lines represent fully oxidized (AλoxMax at 406 nm) and fully reduced form (AλredMax at 434 nm). (b) Nernst plot with X representing (AλredMax – Aλred)/(AλoxMax – Aλox).
Almost identical spectral transitions were observed at pH 5.0 and 7.0 and from the corresponding Nernst plots E°′ values were calculated to be −82.5 ± 0.9 mV and −126.3 ± 1.9 mV, respectively (Figure S10a–d). Between pH 5.0 and 7.0, the reduction potential decreases by −18.5 mV/pH. At pH 10.0, the establishment of a redox equilibrium between the Fe(III)–hydroxo-complex (AλoxMax = 414 nm) and ferrous CCld (AλredMax = 434 nm) was observed with an isosbestic point at 421 nm. From the linear Nernst plot, a standard reduction potential of −172.7 ± 1.5 mV was obtained (Figure S10e,f).
2.6. X-ray Crystal Structure of CCld at pH 6.5 and 8.5
Homodimeric CCld crystallizes in space group P1 or P1211 and the asymmetric unit contains one or two dimers, depending on the crystallization conditions (Table 1). Each subunit binds one heme b and consists of a β-barrel that is made up of a three-stranded and a five-stranded antiparallel β-sheet. Eight α-helices complete the previously described ferredoxin-like fold (Figure 7a) which represents the most prominent common feature of all heme binding (i.e., functional chlorite dismutase, DyP-type peroxidase, aldoxime dehydratase, IsdG, and coproheme decarboxylase) and nonheme-dependent superfamily members. Interactions between the enzyme subunits include a salt bridge (Arg64-Glu103), hydrogen bonds (Gln3-Asp134, Ile59-Arg104, Ala62-Asn101), and π-stacking (Trp97-Phe141-His131-Phe55) (Figure S11). The enzyme was crystallized in the presence of the endogenous ligands (i) H2O (i.e., at pH 6.5) and (ii) OH– (i.e., pH 8.5), and the exogenous ligands (iii) thiocyanate (i.e., LS-ligand) and (iv) fluoride (i.e., HS-ligand). Crystals diffracted to high resolution [(i.e., 1.28 Å for SCN–, 1.18 Å for F– bound CCld, 1.55 Å for OH– bound CCld (pH 8.5) and 1.30 Å for H2O bound CCld (pH 6.5)] (Table 1). All of them are structurally very similar to a maximum rmsd value of 0.39 Å over 340 Cα atoms. Furthermore, the similarity to the only other solved dimeric chlorite dismutase structure of Nitrobacter winogradskyi is high with an rmsd of 0.81 Å over 337 equiv Cα atoms. For both, NwCld and CCld, a very flexible loop, encompassing the residues 40 to 49, could only be modeled in the OH– bound CCld structure (Figure S12c, the respective loops in both subunits are colored in red) due to special crystal packing which was the result of altered crystallization conditions (Figure S12a,b, pH 8.5 and 6.5, respectively).
Table 1. Crystallization Conditions, Data Collection, and Refinement Statistics for CClda.
| H2O | OH– | F– | SCN– | neutron | X-ray (room temperature) | |
|---|---|---|---|---|---|---|
| PDB accession code | 5MAU | 5K8Z | 5K91 | 5K90 | 5NKU | 5NKV |
| crystallization pH | 6.5 | 8.5 | 6.5 | 6.5 | 9.0 | 9.0 |
| exogenous ligand (mM) | - | - | 200 | 10 | - | - |
| Data Collection | ||||||
| beamline | ID30b (ESRFc) | ID23-2 (ESRF) | ID23-1 (ESRF) | ID02 (DLSd) | MaNDi (ORNLe) | in-house (ORNL) |
| temperature (K) | 100 | 100 | 100 | 100 | ambient | ambient |
| crystal modifications | - | - | - | - | deuterated | deuterated |
| resolution range (Å) | 46.38–1.30 (1.35–1.30) | 41.65–1.55 (1.61–1.55) | 46.34–1.18 (1.22–1.18) | 46.06–1.28 (1.33–1.28) | 17.22–2.35 (2.43–2.35) | 19.49–2.00 (2.07–2.00) |
| space group | P1 | P1211 | P1 | P1 | P1 | P1 |
| unit cell (Å, °) | a = 51.12 | a = 54.86 | a = 51.15 | a = 51.76 | a = 52.43 | a = 52.43 |
| b = 52.65 | b = 72.83 | b = 54.70 | b = 54.63 | b = 53.02 | b = 53.02 | |
| c = 54.73 | c = 112.49 | c = 94.27 | c = 94.10 | c = 55.34 | c = 55.34 | |
| α = 107.2 | α = 90.0 | α = 99.1 | α = 99.8 | α = 107.3 | α = 107.3 | |
| β= 99.2 | β = 94.6 | β = 94.8 | β = 94.8 | β = 98.5 | β = 98.5 | |
| γ = 108.9 | γ= 90.0 | γ = 99.0 | γ = 99.2 | γ = 109.9 | γ = 109.9 | |
| no. of dimers/AUb | 1 | 2 | 2 | 2 | 1 | 1 |
| multiplicity | 6.8 (7.0) | 4.3 (3.9) | 3.7 (3.6) | 1.8 (1.8) | 2.6 (1.6) | 2.6 (2.7) |
| completeness (%) | 95.0 (91.8) | 99.7 (99.8) | 90.3 (86.9) | 93.4 (91.4) | 66.7 (49.8) | 80.6 (42.1) |
| I/σ(I) | 10.6 (1.0) | 6.2 (0.9) | 6.7 (1.0) | 5.0 (0.3) | 7.7 (2.5) | 5.6 (2.1) |
| Wilson B-factor | 17.4 | 15.1 | 15.1 | 15.2 | 33.2 | 25.7 |
| Refinement | ||||||
| Rwork/Rfree | 0.147/0.176 | 0.198/0.246 | 0.190/0.222 | 0.210/0.236 | 0.239/0.253 | 0.140/0.186 |
| RMSD, bonds (Å) | 0.017 | 0.005 | 0.023 | 0.015 | 0.025 | 0.025 |
| RMSD, angles (deg) | 1.5 | 0.7 | 2.1 | 1.4 | 2.0 | 2.0 |
| Ramachandran outliers (%) | 0.00 | 0.28 | 0.00 | 0.00 | 0.29 | 0.29 |
| average B-factor (Å2) | 25.7 | 26.8 | 21.4 | 26.0 | 56.0 | 56.0 |
| macromolecules (Å2) | 23.5 | 26.5 | 19.4 | 25.1 | 56.6 | 56.6 |
| ligands (Å2) | 22.1 | 18.0 | 18.1 | 24.9 | 37.6 | 37.6 |
| solvent (Å2) | 39.5 | 30.9 | 34.1 | 32.3 | 55.7 | 55.7 |
Values in parentheses are for the highest-resolution shell.
AU, asymmetric unit.
ESRF, European Synchrotron Radiation Facility.
DLS, Diamond Light Source.
ORNL, Oak Ridge National Laboratory.
Figure 7.

Structure of CCld crystallized at pH 6.5 (i.e., highly active state). (a) The dimeric structure is shown as cartoon. Heme groups are depicted as gray sticks, the heme iron is shown as an orange sphere. (b) Left: Detailed view of the active site surrounding amino acid residues. Distal residues include Q74 and R127, proximal residues include H114, E167, K92, Y61, R104, and R105, the latter three as well as the former three generating two hydrogen bonding networks via the heme propionates. Waters (W) and heme iron are depicted as red and orange spheres, respectively. The heme is shown as gray sticks. Dashed lines indicate hydrogen bonding distances which are listed in Table 2. Right: Active site view rotated by 60° for better visualization of the separation of the proximal hydrogen bonding networks. (c) Left: Illustration of a computed intramolecular tunnel (shown in orange) that is filled with water molecules (shown as red spheres) and generates an active-site connection. The side chains of H131 of both subunits are pointing toward this tunnel. Right: Zoom on H131 which is hydrogen bonded to waters W503, W504, and W505, thereby interconnecting the two heme propionate groups.
Figure 7b shows the active site architecture of CCld at pH 6.5 (i.e., highly active state). His114 serves as the proximal fifth heme ligand, and its Nε2 is at a distance of 2.09 Å from the iron atom. The distal sixth heme ligand H2O binds to the heme iron at a distance of 2.60 Å. This distance is longer than expected for a water molecule bound to the heme iron. As previously observed for the ARP/CIP-SHA complex,35 the combined analysis of the spectroscopic data and X-ray crystal structures of CCld at pH 6.5 indicates that the Fe–H2O distance as measured by X-ray diffraction is an average resulting from the 5c and 6c species in equilibrium that, however, are directly revealed by RR spectroscopy. Very importantly, it has to be noted, that the extent of photoreduction in the crystal is strongly dependent on the applied radiation dose,36 and it has been shown that the redox sensitive metal centers get significantly reduced at already 10% of applied dose needed to collect the entire diffraction data set. Thus, the heme iron is very likely fully reduced, which is most probably at the basis of the unexpected increased distance to the distal ligand since water does not usually bind to the ferrous heme iron. The distal ligand is in hydrogen bonding distance (i.e., 2.87 Å) to a conserved H2O molecule (W502) found in every CCld X-ray data set, notably also in the neutron and X-ray data sets collected at room temperature (q.v. Section 2.8). W502 is in turn H-bonded (2.91 Å) to the Nε of Arg127 residue, which is strictly conserved among functional Clds.11,12 Interestingly, in all obtained CCld data sets, the Arg127 side chain is positioned away from the heme moiety. This out-conformation is putatively stabilized by hydrogen bonding between Nη1 of Arg127 and Oε1 of Gln74 (2.85 Å) (Figure 7b). For CCld, two separate hydrogen bonding networks on the proximal side of the heme can be identified. All involved residues (i.e., His114, Glu167, Lys92 and Tyr61, Arg104, Arg105) are conserved among functional chlorite dismutases with the exception of Arg105 which is characteristic for clade 2 representatives. As previously described, His114, Glu167, and Lys92 form an H-bonding network from the heme iron to the heme substituent propionate 6 (Figure 7b).12 A second network is generated by Tyr61, Arg104, and Arg105, which are interconnected via hydrogen bonds between their side chains. Tyr61 generates an additional hydrogen bond to propionate at position 7 via its backbone nitrogen (Figure 7b).
Further, two active site access channels per subunit can be identified. Since the subunits are rotated 180° with respect to each other, just one connection from the surface to the active site per subunit remains, whereas the other one disappears in the protein interface (Figure 7c). Using CAVER,37 it becomes evident that the latter channel interconnects both active sites (Figure 7c, left). CAVER reports an average bottleneck radius for the tunnel of 1.43 Å. The channel is approximately 25 Å in length (from propionate to propionate) and potentially enables a facilitated chemical cross-talk between CCld subunits.
Figure S12d shows the active site structure of CCld at pH 8.5 (i.e., moderately active state). Nε2 of His114 is at a distance of 2.30 Å from the coordinated iron atom of the heme group whereas the oxygen of OH– is at a distance of 2.51 Å. The aforementioned H2O (W502) is at 2.80 Å from Nε of Arg127. Nη1 of Arg127 is in hydrogen bonding distance (i.e., 2.84 Å) to Oε1 of Gln74.
2.7. X-ray Crystal Structures of Fluoride and Isothiocyanate Complexes of CCld
Figure S13a shows the active site of CCld with the heme bound to fluoride. The protein was crystallized at pH 6.5, and the electron density map was contoured at σ = 1.6. The distance of proximal histidine Nε2 to the iron atom is 2.07 Å, and the high-spin ligand fluoride is at a distance of 2.42 Å from the iron atom. W502 is at a distance of 2.91 Å from the ligand and at a distance of 2.95 Å from Nε of Arg127, whose Nη1 is 2.87 Å away from Oε1 of Gln74 (Table 2). Also in this case, the unexpected increased distance between the iron atom and the fluoride is most probably a consequence of the photoreduction since fluoride binds only ferric hemes.30
Table 2. Distance Analysis of Crystal Structures of Conserved Hydrogen Bonds in the Active Side of CClda.
| distance (Å) |
|||||||
|---|---|---|---|---|---|---|---|
| H2O (pH 6.5) | OH– (pH 8.5) | F– (pH 6.5) | SCN– (pH 6.5) | neutron/X-ray (pH 9.0) | X-ray room temperature (pH 9.0) | ||
| distalb | iron–ligand | 2.60 ± 0.01 | 2.51 ± 0.06 | 2.42 ± 0.20 | 2.04 ± 0.01 | 2.3 ± 0.1 | 2.5 ± 0.1 |
| ligand–W502 O | 2.87 ± 0.06 | 3.04 ± 0.08 | 2.91 ± 0.09 | 3.16 ± 0.16 | 3.0 ± 0.2 | 2.7 ± 0.1 | |
| W502 O–127 Nε | 2.91 ± 0.04 | 2.80 ± 0.13 | 2.95 ± 0.01 | 2.77 ± 0.01 | 2.6 ± 0.0 | 2.7 ± 0.2 | |
| 127 Nη1–74 Oε1 | 2.85 ± 0.01 | 2.84 ± 0.08 | 2.87 ± 0.03 | 2.85 ± 0.08 | 2.9 ± 0.2 | 3.1 ± 0.1 | |
| 127 Nη2–74 Oε1 | 2.98 ± 0.01 | 3.19 ± 0.19 | 3.02 ± 0.02 | 3.19 ± 0.04 | 3.5 ± 0.1 | 3.6 ± 0.2 | |
| proximal 1c | iron–114 Nε2 | 2.09 ± 0.03 | 2.30 ± 0.03 | 2.07 ± 0.03 | 2.11 ± 0.01 | 2.1 ± 0.1 | 2.4 ± 0.1 |
| 114 Nδ1–167 Oε1 | 2.77 ± 0.04 | 2.69 ± 0.04 | 2.77 ± 0.03 | 2.78 ± 0.05 | 2.8 ± 0.0 | 2.5 ± 0.1 | |
| 167 Oε2–92 Nζ | 2.74 ± 0.02 | 2.80 ± 0.06 | 2.73 ± 0.01 | 2.66 ± 0.01 | 2.6 ± 0.0 | 2.8 ± 0.0 | |
| 92 Nζ–500 O1/2D | 2.76 ± 0.01 | 2.75 ± 0.04 | 2.78 ± 0.01 | 2.85 ± 0.01 | 2.8 ± 0.1 | 2.8 ± 0.1 | |
| proximal 2d | 500 O2A–61 N | 2.80 ± 0.02 | 2.83 ± 0.04 | 2.81 ± 0.01 | 2.82 ± 0.06 | 2.8 ± 0.0 | 2.8 ± 0.0 |
| 61 O–105 Nη1 | 2.92 ± 0.02 | 2.92 ± 0.03 | 2.91 ± 0.04 | 2.85 ± 0.04 | 3.0 ± 0.0 | 3.0 ± 0.1 | |
| 104 Nη1–61 OH | 2.91 ± 0.01 | 2.93 ± 0.02 | 2.87 ± 0.02 | 2.90 ± 0.01 | 3.0 ± 0.1 | 2.9 ± 0.0 | |
| 500 O1D–W506 O | 2.65 ± 0.00 | 2.63 ± 0.09 | 2.63 ± 0.03 | 2.63 ± 0.05 | 2.6 ± 0.1 | 2.5 ± 0.0 | |
| W506 O–61 OH | 2.63 ± 0.04 | 2.64 ± 0.07 | 2.63 ± 0.04 | 2.72 ± 0.04 | 2.6 ± 0.1 | 2.6 ± 0.0 | |
[distance (Å) ± standard deviation]. Standard deviation was calculated from all subunits in the AU.
distal: heme iron–distal heme ligand −W502–Arg127–Gln74.
proximal 1: heme iron–His114–Glu167–Lys92–propionate 6 (500 O1/2D).
proximal 2: propionate 7 (500 O1/2A)–Tyr61–Arg105–Arg104–Tyr61–propionate 7; propionate 7–Tyr61–W506–propionate 6.
Figure S13b shows the active site of CCld in complex with SCN– which was crystallized at pH 6.5. The distance of Nε2 of His114 to the iron atom is 2.11 Å. The LS-ligand SCN– binds with its nitrogen atom facing the heme iron at a distance of 2.04 Å. W502 is at 3.16 Å from the ligand nitrogen atom and 2.77 Å distant from Nε of Arg127. Once again, hydrogen bonding can be observed between Nη1 of Arg127 and Oε1 of Gln74 at a distance of 2.85 Å. To confirm the depicted orientation of the ligand, a data set was collected at a wavelength of 1.9 Å (Figure S13c) at which the anomalous scattering of nitrogen and carbon is negligible and the strong difference anomalous electron density peak corresponds to the sulfur atom, corroborating the orientation of the ligand. The data enable us to conclude that the sulfur does not directly interact with the iron atom.
2.8. Neutron and X-ray Crystal Structure of CCld at pH 9.0 and Room Temperature
Two of the most substantial limiting factors of using X-rays for protein structure determination are that (i) positions of hydrogens and protons, which contribute up to 50% or more to the total number of atoms in a protein, often cannot be determined from X-ray scattering data and (ii) photoreduction occurs, which is especially an issue for metal containing enzyme cofactors like heme.38 Neutrons are able to overcome both issues since neutrons (i) are scattered by hydrogen atoms to the same extent as as carbons, nitrogen or oxygen atoms and they (ii) are nonionizing. Consequently, photoreduction does not occur at all.39 Here, we used neutron crystallography as a complementary technique to X-ray diffraction studies to obtain comprehensive structural information on CCld in its active ferric state, including protonation states of important amino acid residues. In particular, the protonation state of this Arg is the subject of ongoing debate as it is believed that an untypically low pKa value (i.e., around 6.5 for DaCld) of the amino acid side chain dictates activity profiles and inactivation behavior of Clds.10,11,14
Crystal growth of hydrogenated CCld in quartz capillaries was facilitated by applying the method of microseeding. Crystals formed within 5–10 days. Hydrogen/deuterium exchange and change of pH from 6.5 to pD 9.4 was achieved by repeated crystallization buffer exchange. The dimensions of the investigated diamond shaped crystal were approximately 2.0 × 1.0 × 0.4 mm. The ratio of crystal volume to asymmetric unit volume was calculated to be 60, a value underlining the challenging character of the experiment.40 The characterization of the heme ligand and determination of the protonation states of amino acid residues (i.e., Lys92, Arg104, Arg105, His114, Arg127, His131, Glu167) were expected to assist a rationalization of the inactivity of the enzyme at this pH. This experiment yielded the 54th protein structure solved by neutron crystallography (Protein Data Bank, May 2017) and the first oxidized chlorite dismutase structure collected at room temperature. Using neutrons, the crystal diffracted to a maximum resolution of 2.35 Å, whereas with X-rays it diffracted to 2.0 Å. The CCld model was refined against X-ray data alone, followed by joint X-ray and neutron refinement.
Figure 8a shows that Nδ1 of His114 is protonated and in hydrogen bonding distance (i.e., 2.75 Å) to Oε1 of Glu167. The deprotonated Nε2 of His114 is at 2.12 Å from the heme iron and serves as proximal ligand. Comparing the His114 Nε2 to iron distances of all structures in complex with endogenous ligands H2O or OH–, it can be summarized that they increase in the order X-ray crystallography (H2O, 100 K, synchrotron) < Neutron crystallography (OH–) < X-ray crystallography (OH–, 100 K, synchrotron) < X-ray crystallography (OH–, room temperature, in-house source) (Table 2).
Figure 8.

Comparison of neutron (yellow, σ = 1.4) and X-ray (purple, σ = 1.5) 2F0 – Fc density maps in the active site of CCld at pH 9.0. (a) Proximal H-bonding players His114, Glu167, and Lys92. (b) H-bonding network between Tyr61, a conserved deuterium atom (W506) and the heme propionate 6. (c) Catalytically important Arg127 with the side chain oriented toward the substrate entrance channel (out-conformation). The distal heme ligand OD– is depicted as sticks, the heme iron is shown as an orange sphere. Nitrogen: blue, oxygen: red, deuterium: turquoise.
Further, Oε2 of Glu167 is hydrogen bonded to Nζ of Lys92 which in turn makes hydrogen bonding contacts with the heme propionate at position 6. At the backbone nitrogen of Tyr61, there is no nuclear density observable indicating that the hydrogen is either not easily accessible or that it is involved in a hydrogen bond with the heme propionate at position 7 (Figure 8b). Both situations could hinder its exchange for deuterium. The hydroxyl group of Tyr61 is in hydrogen bonding distance to the guanidinium group of Arg104; however, nuclear density clearly reveals an involvement of Tyr61 in a hydrogen bond to W506, which is coordinated by O1D of propionate 6 (Figure 8b). Protonated Nη1 of Arg105 is 2.99 Å away from the backbone oxygen of Tyr61.
Most importantly, at pD 9.4 Arg127 is fully protonated and Nε of this residue is at a distance of 2.59 Å from W502, engaged in a hydrogen bond. Supported by spectroscopic measurements, the nature of the distal heme ligand was characterized to be an OH– at the studied pH, with an average distance of the oxygen to the iron atom of 2.33 Å, longer than expected, therefore, either due to the coexistence of both 6cHS and LS hydroxo-complexes observed at room temperature or to the photoreduction since also OH– is not a specific ligand of ferrous heme. Comparing all CCld structures in complex with OH–, the ligand-to-iron distance increases in the order neutron crystallography < X-ray crystallography (in-house source) < X-ray crystallography (synchrotron), reflecting the correlation between ligand distance and the reducing power of the applied radiation.
His131 is another critical amino acid residue as it is strictly conserved among functional chlorite dismutases, and its side chain is found at an important position toward the intramolecular channel described above (Figure 7c, left). Three water molecules (W503, W504, W505) form a hydrogen bonding network that spans the His131 side chain and both heme propionates (Figure 7c, right). All three waters can be clearly identified in all obtained density maps (neutron and X-ray). Notably, the signals for the room temperature measurement with neutrons are distinct, with B-factors comparable to protein atoms, suggesting that these water molecules are tightly bound. This in turn emphasizes their important functional role in properly positioning the heme propionate groups.
3. Discussion
Chlorite dismutase from Cyanothece sp. PCC7425 is a dimeric representative of clade 2 functional Clds and represents the first protein of this clade which has been comprehensively characterized regarding structure–function relationships. Similar to its characterized homologues (e.g., NdCld, DaCld, Cld from GR-1, KpCld), it is capable of efficiently converting chlorite into chloride and molecular oxygen, the reaction being strongly dictated by pH. The (increasingly incomplete) substrate turnover is significantly slowed down with rising pH. In this work, we have focused on the elucidation of the impact of pH on structure and the molecular mechanism of chlorite degradation by CCld.
3.1. Impact of pH on the Active Site Architecture of CCld
Spectroelectrochemical studies revealed that the reduction potential E°′ of the [Fe(III)/Fe(II] couple of CCld at pH 5.0 is −82.5 mV, which is significantly higher compared to plant type heme b peroxidases like horseradish peroxidase (HRP) (∼ −260 mV). This reflects the more hydrophilic distal heme pocket of HRP that stabilizes the ferric state. In CCld, E°′ undergoes a decrease of −90 mV between pH 5.0 (−82.5 mV) and pH 10.0 (−172.7 mV). A main contribution to this modulation results from the deprotonation of the distal heme ligand H2O with increasing pH giving rise to a predominant hydroxo-LS ligand [ν(Fe–OH) stretching mode at 510 cm–1], which selectively stabilizes Fe(III)–heme over Fe(II)-heme for electrostatic as well as coordinative reasons.41 In the case of CCld, the pKa of this alkaline transition was found to be 8.1, a value slightly lower than those reported for pentameric chlorite dismutases (DaCld: 8.7;14 IdCld: 8.5;15 chlorite dismutase from the chlorate respiring strain GR-1:8.242).
The redox properties of a heme protein are determined by (i) the proximal heme ligand and its noncovalent interactions with other active site residues, (ii) the conformations and interactions of the heme propionates, and (iii) the nature of distal (catalytic) residues and their interactions with both the protein matrix and the solvent. In Clds, a His serves as proximal heme ligand. Studying ferrous CCld, resonance Raman spectroscopy revealed a high ν(Fe-Im) frequency (231 cm–1) that is in agreement with the distance (2.09 ± 0.03 Å) between the heme iron and the His114 Nε2 found in the X-ray structure obtained at pH 6.5. It shows that the proximal His114 is H-bonded to the nearby Glu167 residue and thereby obtains a significant imidazolate character, similar to that observed in heme peroxidases.43 However, unlike the heme containing peroxidases, the ν(Fe-Im) frequency does not vary at basic pH. In fact, for peroxidases at alkaline pH, a downshift of the ν(Fe–His) frequency is observed due to a weakening of the H-bond interaction.44
The neutron structure of CCld nicely demonstrates the Cld-typical conserved and rigid proximal H-bonding network (Figure 8). It shows that the deprotonated Nε2 of His114 coordinates the ferric heme iron whereas Nδ1 of His114 is protonated and hydrogen bonded to Oε1 of Glu167. Further, Oε2 of Glu167 is H-bonded to Nζ of Lys92, which in turn makes hydrogen bonding contacts with the heme propionate at position 6. Except for the distance between the heme iron and Nε2 of His114, the lengths of these conserved hydrogen bonds are only very slightly modulated by pH and the presence of distal ligands (Table 2).
Participation of heme propionates in this network is also underlined by the intense propionate bending band at 370 cm–1 (361 cm–1 for the ferrous form), suggesting a moderate hydrogen bond interaction between the propionate substituents and nearby residues.45 Besides interaction of propionate 6 with Lys92, the neutron structure additionally shows that Tyr61, which is connected to propionate 7 via its backbone nitrogen, makes strong H-bonding contacts to propionate 6 via a conserved water molecule (W506). Moreover, the electronic state of the heme cofactor is further influenced by the hydrogen bonding network built by His131 and three conserved waters (W503, W504, and W505), spanning from one propionate to the other (Figure 7c).
These H-bonding interactions stabilize CCld in the ferric state at all pH values studied, although this stabilizing effect is more pronounced at alkaline pH (see above). Around the (acidic) pH optimum, the resting ferric heme iron participates in a distal H-bonding network that includes the metal ion, W501, W502, conserved Arg127 in “out” conformation and Gln74 (Figure 7b). Distance analyses based on both the X-ray structure at pH 8.5 and the neutron structure at pH 9.0, indicate that deprotonation of W501 at alkaline pH shortens the distance to the heme iron in agreement with the formation of the Fe–OH ligation (highlighted by RR at room temperature). Deprotonation of W501 also significantly weakens or even breaks the interaction between OH– and W502 and simultaneously strengthens the interaction between W502 and Arg127 (Table 2). The interaction between Arg127 and Gln74 is not affected by the alkaline transition.
One of the main questions of the present study was the role of conserved Arg127 in catalysis and whether its protonation status modulates the rate and extent of chlorite degradation. In this work, we could obtain neutron and X-ray diffraction data from the same H/D exchanged crystal at pH 9.0. Therefore, both data sets could be simultaneously refined (joint neutron/X-ray refinement), which can compensate for the increased number of parameters that must be refined in the neutron structure. This is due to the addition of hydrogens atoms into the structure essentially doubling the number of atoms that are present in refinement compared to a typical X-ray structure.
For H/D exchanged samples, phenix.refine automatically determines such sites and performs constrained occupancy refinement, constraining the sum of occupancies of H and D to one. For Arg127, phenix.refine calculates occupancies of exchangeable H/D in the side chain for chain A of 1 and for chain B of 0.97. Control refinements setting the Dη22 and Hη22 simultaneously to zero, thereby mimicking a deprotonated state, did not result in a positive density peak at this position. These data clearly suggest that Arg127 is protonated at pH 9.0 and rule out that it may serve as distal base and dictate the activity profile of CCld. This is also underlined by recently conducted studies about the pKa value of Arg which conclude that the guanidinium moiety is virtually always charged in a protein, even in hydrophobic core regions and pockets.46,47
3.2. Impact of Ligand Binding on the Structure of the Heme Cavity
Elucidation of the crystal structures of HS and LS complexes studied at pH 6.5 demonstrate that the addition of a ligand, irrespective of its nature, eliminates W501 but not W502 from the heme cavity and keeps Arg127 in the “out” conformation (Figure S13). The binding of the HS ligand fluoride does not induce obvious rearrangements of the distal H-bonding network (Figure S13a, Table 2). By contrast, binding of the LS ligand isothiocyanate (Figure S13b) results in (i) a significantly reduced distance between the nitrogen atom of the ligand and the heme iron with respect to the water or hydroxo-ligands, and in (ii) weakening of the H-bond between the nitrogen atom and W502 which in the complex is closer to Arg127 (Table 2). For DFT calculations, the isothiocyanate complex [Por···Fe(III)––NCS] structure of pentameric AoCld (PDB: 2VXH) was used to construct a model of the enzyme–substrate complex [Por···Fe(III)––OClO] before O–Cl bond cleavage.48 However, any conclusions drawn from these studies should carefully consider that in the AoCld X-ray crystal structure the distal Arg is found in the “in” conformation, thereby displacing a potential bridging water (W502 in CCld) between ligand and distal Arg.
The interaction between ligands, the solvent and the distal protein matrix was also probed by RR spectroscopy. Principally, the absorption maximum of the CT1 band is a sensitive probe of the interaction of fluoride with distal amino acid residues. It red-shifts when the ligand acts as an H-bond acceptor, since the interaction between the p orbitals of the ligand and the iron dπ orbitals decreases the energy of the latter.30,31 In CCld, the low energy of the CT1 band, together with the low frequency of the ν(Fe–F–) stretch, indicate the presence of strong H-bonding interactions supporting the X-ray crystal structure depicted in Figure S13a. Fluoride serves as H-bond acceptor for W502, which in turn is H-bonded to Arg127. This finding is supported by RR spectra of the Fe(II)-CO complex, which reveal very moderate polar interactions of carbon monoxide with the distal protein matrix. CO is not able to interact with water and the Arg127 in the “out” conformation is positioned too far to enable direct contact.
The hypothesis that Arg127 may serve as distal base could also be ruled out by determination of association rate constants of cyanide. The pKa of HCN is 9.14 and, typically, the anionic form (CN–) binds to the ferric heme protein. In heme peroxidases, a basic amino acid, typically histidine, acts as a proton acceptor forming the anionic ligand that binds to the ferric state. Thus, formation of the LS complex depends on the corresponding pKa of the distal histidine.49 In the case of functional Clds, the observed pH dependence clearly underlines that these enzymes lack a distal base that supports deprotonation of HCN. Between pH 5.0 and 8.0, the apparent bimolecular rate constant of cyanide binding to ferric CCld strictly increased with increasing pH and the ionization of HCN (Figure 5). Accordingly, both the obtained neutron structure of CCld combined with kinetic information on cyanide binding clearly refute the hypothesis that Arg127 acts as a base and dictates the pH dependence of chlorite degradation.
3.3. Molecular Mechanism of Chlorite Degration
It has been demonstrated previously by isotope-labeling experiments that both oxygen atoms in the evolved O2 derive from chlorite and that the reaction stoichiometry (chlorite:oxygen) is 1:1.2 Finally, based on experimental studies on pentameric Clds6,10−17 and theoretical studies,48 two mechanisms were proposed that both involve a (heterolytic or homolytic) O–Cl bond cleavage in the [Por···Fe(III)––OClO] complex leading to a ClO fragment (hypochlorous acid or chlorine monoxide) and a high–valent oxoiron(IV) intermediate (Compound I or Compound II).
Studying pre-steady-state kinetics of NdCld11,12 and DaCld2,14 was so far hampered by the fact that (i) the reaction with chlorite proceeds too fast to be tracked by the stopped-flow technique and that (ii) the spectral transitions were ambiguous and thus difficult to interpret. By contrast, the spectral features of dimeric CCld in its various oxidation states are much more pronounced, thus allowing their interconversions during chlorite degradation to be followed.
From studies on heme peroxidases and catalases it is known that (HOCl/–OCl) functions as a two-electron oxidant of heme peroxidases thereby rapidly producing Compound I.23,24 At biologically relevant pH, hypochlorous acid is a strong two-electron oxidant [E°′(HOCl, H+/Cl–, H2O) = 1280 mV at pH 7.0 and 1490 mV at pH 0.0].50 As a consequence, it mediates the two-electron oxidation of lactoperoxidase (LPO) or myeloperoxidase (MPO)51 to the corresponding Compound I state. This applies also to peracetic acid [E°′(CH3COOOH, H+/CH3COO–, H2O) = 1636 mV at pH 6.0].10,52 Here, we could demonstrate that CCld follows the same reaction. Hypochlorite rapidly oxidizes the ferric enzyme to Compound I which is characterized by about 50% hypochromicity at the Soret maximum and a distinct band at 650 nm (Scheme 1, (4)). Compound I slowly shifts to an intermediate with spectral features similar to Compound II which is better defined as Compound I* having the porphyryl radical quenched by electrons spent by the protein matrix (Scheme 1, (5)). This internal (unspecific) electron transfer is increased with increasing pH which was also seen with Compound I of pentameric DaCld produced by peracetic acid.13 For DaCld, freeze-quench EPR confirmed the presence of an uncoupled protein-based radical formed upon decay of Compound I to Compound I*.13
Scheme 1. Proposed Main (Highlighted in Green) and Side Reactions (Highlighted in Blue) of Ferric CCld with Chlorite.
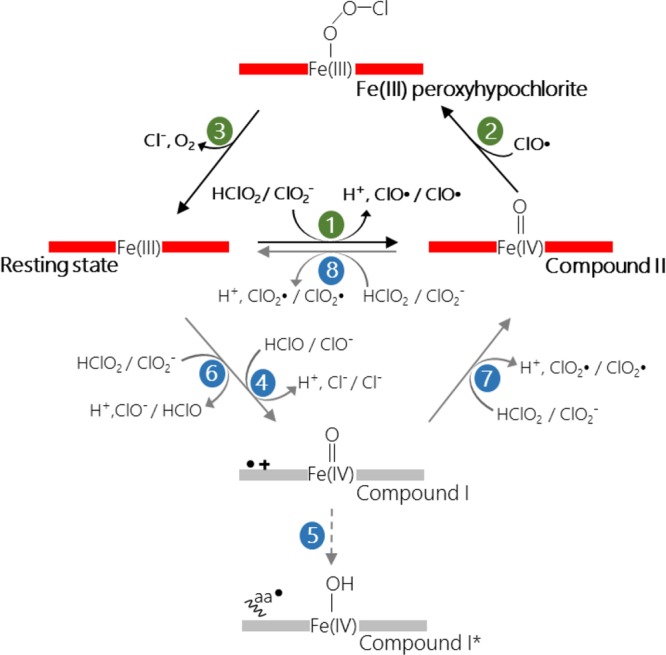
Importantly, even in the presence of excess (HOCl/–OCl), neither with CCld nor with DaCld could O2 formation be measured polarographically. This clearly contradicts an efficient rebound mechanism between Compound I and hypochlorite as described by Reaction IV. This is also underlined by the fact that Compound I formation mediated by chlorite (which must be accompanied by generation of HOCl) has never been detected with pentameric chlorite dismutases by using UV–vis stopped-flow spectroscopy. Moreover, heterolytic cleavage of chlorite was ruled out by DFT calculations due to the absence of spin localized at the porphyrin macrocycle and an unfavorable high energy barrier.48
In contrast, mixing of dimeric CCld with chlorite in the stopped-flow apparatus leads to the formation of Compound II (Soret maximum at 418 nm, two prominent bands at 528 and 551 nm) within milliseconds (Scheme 1, (1)). Chlorite is a weaker two-electron oxidant compared to HOCl/–OCl. Its standard reduction potential for the redox couple (HClO2, 2H+/HOCl, H2O) is reported to be 1080 mV at pH 7.0, and it increases with 88 mV per pH unit with decreasing pH (being 1256 at pH 5.0 and 1645 mV at pH 0.0).53 Chlorite is well-known to mediate Compound I formation in horseradish peroxidase (HRP) [E°′(Fe(III)/Fe(II)) = −310 mV; E°′ (Compound I/ferric HRP) = 920 mV at pH 7.0].24,54 Under acidic conditions, oxidation of HRP by chlorite becomes more favorable because of the divergent pH dependencies of both redox couples. The potential of the couple (Compound I/ferric HRP) increases with 59 mV per decreasing pH unit.54 In contrast to HRP, chlorite is unable to mediate Compound I formation of MPO [E°′(Fe(III)/Fe(II)) = +5 mV; E°′ (Compound I/ferric MPO) = 1160 mV at pH 7.0]55,56 or bovine LPO [E°′(Fe(III)/Fe(II)) = −183 mV; E°′ (Compound I/ferric HRP) = 1090 mV at pH 7.0].57,58 Although we do not know E°′ (Compound I/ferric) of CCld, it is reasonable to assume that the two-electron oxidation of CCld by chlorite is thermodynamically unfavorable. As already discussed above, due to the hydrophobic heme cavity, E°′[Fe(III)/Fe(II)] of CCld is high (in between E°′ of LPO and MPO), which suggests that also E°′ (Compound I/ferric CCld) exhibits values comparable to LPO and MPO (>1100 mV). By contrast, the one-electron oxidation of ferric CCld by chlorite should be thermodynamically feasible since (in analogy to heme peroxidases) E°′ (Compound II/ferric CCld) < E°′ (Compound I/ferric CCld) and Compound II will be more stable in a hydrophobic cavity compared to Compound I. This is also indicated by computational studies.48
Due to increasing E°′ values of the redox couple (HClO2, 2H+/HOCl, H2O) with decreasing pH, the two-electron oxidation of ferric CCld might become feasible in the acidic region (Scheme 1, (6)). Indeed, upon mixing ferric CCld with chlorite at pH 5.0, we monitored some hypochromicity in the Soret absorbance within the first milliseconds before Compound II is formed (Figure 4a). This might indicate formation of Compound I to a certain extent. Nevertheless, at pH > 5.0 ferric CCld is converted directly to Compound II.
In the time course of chlorite degradation, Compound II is the dominating redox intermediate at all pH values. It should be mentioned that the spectral properties of Compound II are completely different from those of the LS (cyanide, isothiocyanate, hydroxide) complexes (421–538 nm, 414–532 nm, 414–540 nm, respectively) ruling out that the observed dominating intermediate during turnover represents [Por···Fe(III)–OOCl–], i.e. the reaction product of the rebinding step (Scheme 1, (2)). According to computational studies, this intermediate can be considered as a transient (short-lived) LS complex of ferric Cld.48
At all pH values, Compound II is formed immediately upon mixing ferric CCld with chlorite, whereas the total time for chlorite degradation (followed at 280 nm) significantly increases with increasing pH. A mechanistic explanation of this behavior is yet to be found since the presented structural data disprove the hypothesis that the distal Cld-typical Arg dictates the pH dependence. As outlined above, it is protonated at all studied pH values (even in the presence of anionic ligands) and its orientation in crystal structures is always “out”. The dominating “out” conformation is also supported by RR data that suggest weak interactions of the CO ligand with the protein matrix. However, we do not want to dispute the hypothesis that Arg127 is flexible and can adopt the “in” conformation in solution during enzyme turnover.
Based on these findings we propose the following reaction mechanism. The reaction cycle of dimeric CCld is initiated by binding of the anionic substrate (pKa = 1.72) to the Fe(III) center which occurs spontaneously and independent of Arg.11 At this step, Arg is in the “out” conformation and seems to be relevant for chlorite recognition only.9 The resulting enzyme–substrate complex [Por···Fe(III)––OClO] is either hydrogen bonded to W502 as suggested by the CCld-isothiocyanate complex [Por···Fe(III)––NCS] or directly to Arg127, which in the “in” conformation would displace W502. As suggested by computational studies, in the “in” conformation both oxygen atoms of chlorite act as H-bond acceptors for the Arg. By homolytic cleavage chlorine monoxide and Compound II are formed (Scheme 1, (1)). Our stopped-flow studies clearly show that Compound II formation is extremely fast between pH 5.0 and 9.0 suggesting that the rate of homolytic bond cleavage is independent of pH. This might suggest that the following rebound step (Scheme 1, (2)) is responsible for the observed pH dependence. These findings also rule out that there is any mechanistic difference in the replacement of either W501 (at acidic pH) or its deprotonated version (at alkaline pH) (pKa = 8.1) that could lead to the observed pH dependence.
With increasing pH, the O–O bond formation apparently becomes inefficient and the rate of chlorite degradation decreases significantly. Rebinding of Compound II and chlorine monoxide needs rotation of the latter and rearrangements of H-bonds with Arg127 to support O–O bond formation which–according to computational studies–is a very fast process.48 Finally, to facilitate the departure of the chloride anion from the resulting LS complex [Por···Fe(III)–OOCl–], the H-bonds between the Arg and the two oxygen atoms are required to rearrange again to the terminal Cl atom, an event which might provide the stabilization for the leaving negatively charged chloride anion (Scheme 1, (3)).
Besides the decrease of the rate of chlorite degradation, Clds become inactivated at alkaline pH. As a consequence, the degradation of higher amounts of chlorite is incomplete under these conditions. This is also the case for CCld.20 Based on studies on pentameric Clds and a proposed heterolytic cleavage of chlorite, it was hypothesized that hypochlorite is responsible for the irreversible inactivation of the enzyme.11 Inefficient rebinding of hypochlorite to Compound I was proposed to lead to the release of (HOCl/–OCl) which oxidatively modifies both the heme cofactor and the protein. This was demonstrated by addition of hypochlorite traps. However, it is not possible to exclude that other chlorine species (chlorine monoxide or the observed byproduct chlorine dioxide, see below) also react with the trapping agents that were applied in the cited study. It is reasonable to assume that under suboptimal conditions for the rebound reaction the postulated intermediate chlorine monoxide escapes from the reaction sphere and promotes enzyme inactivation.
The present study also shows that chlorine dioxide is a byproduct of chlorite degradation. Typically, it derives from the one-electron oxidation of chlorite by redox intermediates like Compound I and Compound II of heme peroxidases.24 The standard reduction potential of the couple (ClO2/ClO2–) has a value of 934 mV and is independent of pH above pH 2.0.59 Both Compound I and II of CCld could be able to mediate the oxidation of chlorite to chlorine dioxide. As mentioned above, at acidic pH, some Compound I formation might occur as a side reaction. Its reduction to Compound II would generate chlorine dioxide (Scheme 1, (7)). Moreover, at excess of chlorite and/or under nonoptimal conditions for the rebound reaction, chlorite can compete with chlorine monoxide and act as electron donor for Compound II. Thereby, chlorine monoxide is eliminated from the reaction sphere and chlorine dioxide is produced (Scheme 1, (8)). These reactions and the release of the generated chlorine species might contribute to the pH dependence and enzyme inactivation at increasing pH values.
However, further investigations are definitely necessary to elucidate the exact mechanism of the pH dependence of chlorite degradation. The present study unequivocally demonstrates that CCld is an excellent model enzyme to study these reactions. It is possible to grow high-quality crystals at various pH to obtain high-resolution X-ray and neutron structures. Most importantly, it allows assignment of all relevant redox intermediates according to their spectral features and study of the kinetics of their interconversion by multimixing stopped-flow spectroscopy.
Acknowledgments
This project was supported by the Austrian Science Fund, FWF [Doctoral program BioToP—Molecular Technology of Proteins (W1224) and the projects P25270 and P22276] and the Federal Ministry of Economy, Family and Youth through the initiative “Laura Bassi Centres of Expertise”, funding the Center of Optimized Structural Studies, No. 253275. Research at the Spallation Neutron Source (SNS) at ORNL was sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy. We thank Elisabeth Lobner, Christa Jakopitsch, and Rupert Tscheließnig for lab assistance, discussions and assistance in data analysis.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.7b01749.
Author Contributions
◆ (I.S. and G.M.) These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Hofbauer S.; Schaffner I.; Furtmüller P. G.; Obinger C. Biotechnol. J. 2014, 9, 461–473. 10.1002/biot.201300210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A. Q.; Streit B. R.; Zdilla M. J.; Abu-Omar M. M.; DuBois J. L. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 15654–15659. 10.1073/pnas.0804279105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J. R. Annu. Rev. Plant Biol. 2015, 66, 23–48. 10.1146/annurev-arplant-050312-120129. [DOI] [PubMed] [Google Scholar]
- Goblirsch B. R.; Streit B. R.; DuBois J. L.; Wilmot C. M. JBIC, J. Biol. Inorg. Chem. 2010, 15, 879–888. 10.1007/s00775-010-0651-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goblirsch B. R.; Streit B. R.; DuBois J. L.; Wilmot C. M. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2009, 65, 818–821. 10.1107/S1744309109026785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostan J.; Sjöblom B.; Maixner F.; Mlynek G.; Furtmüller P. G.; Obinger C.; Wagner M.; Daims H.; Djinović-Carugo K. J. Struct. Biol. 2010, 172, 331–342. 10.1016/j.jsb.2010.06.014. [DOI] [PubMed] [Google Scholar]
- Keith J. M.; Abu-Omar M. M.; Hall M. B. Inorg. Chem. 2011, 50, 7928–7930. 10.1021/ic2009732. [DOI] [PubMed] [Google Scholar]
- Fabian I.; Gordon G. Inorg. Chem. 1991, 30, 3785–3787. 10.1021/ic00019a045. [DOI] [Google Scholar]
- Sündermann A.; Reif M. M.; Hofbauer S.; Obinger C.; Oostenbrink C. Biochemistry 2014, 53, 4869–4879. 10.1021/bi500467h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc B.; Mayfield J. A.; McDonald C. A.; Lukat-Rodgers G. S.; Rodgers K. R.; DuBois J. L. Biochemistry 2012, 51, 1895–1910. 10.1021/bi2017377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofbauer S.; Gruber C.; Pirker K. F.; Sündermann A.; Schaffner I.; Jakopitsch C.; Oostenbrink C.; Furtmüller P. G.; Obinger C. Biochemistry 2014, 53, 3145–3157. 10.1021/bi500401k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofbauer S.; Gysel K.; Bellei M.; Hagmuller A.; Schaffner I.; Mlynek G.; Kostan J.; Pirker K. F.; Daims H.; Furtmüller P. G.; Battistuzzi G.; Djinovic-Carugo K.; Obinger C. Biochemistry 2014, 53, 77–89. 10.1021/bi401042z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayfield J. A.; Hammer N. D.; Kurker R. C.; Chen T. K.; Ojha S.; Skaar E. P.; DuBois J. L. J. Biol. Chem. 2013, 288, 23488–23504. 10.1074/jbc.M112.442335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit B. R.; Blanc B.; Lukat-Rodgers G. S.; Rodgers K. R.; DuBois J. L. J. Am. Chem. Soc. 2010, 132, 5711–5724. 10.1021/ja9082182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenklo K.; Danielsson Thorell H.; Bergius H.; Aasa R.; Nilsson T. JBIC, J. Biol. Inorg. Chem. 2001, 6, 601–607. 10.1007/s007750100237. [DOI] [PubMed] [Google Scholar]
- Streit B. R.; DuBois J. L. Biochemistry 2008, 47, 5271–5280. 10.1021/bi800163x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ginkel C. G.; Rikken G. B.; Kroon A. G.; Kengen S. W. Arch. Microbiol. 1996, 166, 321–326. 10.1007/s002030050390. [DOI] [PubMed] [Google Scholar]
- Mlynek G.; Sjöblom B.; Kostan J.; Füreder S.; Maixner F.; Gysel K.; Furtmüller P. G.; Obinger C.; Wagner M.; Daims H.; Djinović-Carugo K. J. Bacteriol. 2011, 193, 2408–2417. 10.1128/JB.01262-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celis A. I.; Geeraerts Z.; Ngmenterebo D.; Machovina M. M.; Kurker R. C.; Rajakumar K.; Ivancich A.; Rodgers K. R.; Lukat-Rodgers G. S.; DuBois J. L. Biochemistry 2015, 54, 434–446. 10.1021/bi501184c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffner I.; Hofbauer S.; Krutzler M.; Pirker K. F.; Bellei M.; Stadlmayr G.; Mlynek G.; Djinovic-Carugo K.; Battistuzzi G.; Furtmüller P. G.; Daims H.; Obinger C. Mol. Microbiol. 2015, 96, 1053–1068. 10.1111/mmi.12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori H.; Kitagawa T. J. Am. Chem. Soc. 1980, 102, 3608–3613. 10.1021/ja00530a049. [DOI] [Google Scholar]
- Stein P.; Mitchell M.; Spiro T. G. J. Am. Chem. Soc. 1980, 102, 7795–7797. 10.1021/ja00546a035. [DOI] [Google Scholar]
- Floris R.; Wever R. Eur. J. Biochem. 1992, 207, 697–702. 10.1111/j.1432-1033.1992.tb17097.x. [DOI] [PubMed] [Google Scholar]
- Jakopitsch C.; Spalteholz H.; Furtmüller P. G.; Arnhold J.; Obinger C. J. Inorg. Biochem. 2008, 102, 293–302. 10.1016/j.jinorgbio.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Prince L. A. Anal. Chem. 1964, 36, 613–616. 10.1021/ac60209a017. [DOI] [Google Scholar]
- Chen T.-H. Anal. Chem. 1967, 39, 804–813. 10.1021/ac60251a023. [DOI] [Google Scholar]
- De Schutter A.; Correia H. D.; Freire D. M.; Rivas M. G.; Rizzi A.; Santos-Silva T.; Gonzalez P. J.; Van Doorslaer S. J. Phys. Chem. B 2015, 119, 13859–13869. 10.1021/acs.jpcb.5b04141. [DOI] [PubMed] [Google Scholar]
- Geeraerts Z.; Rodgers K. R.; DuBois J. L.; Lukat-Rodgers G. S. Biochemistry 2017, 56, 4509–4524. 10.1021/acs.biochem.7b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire D. M.; Rivas M. G.; Dias A. M.; Lopes A. T.; Costa C.; Santos-Silva T.; Van Doorslaer S.; Gonzalez P. J. J. Inorg. Biochem. 2015, 151, 1–9. 10.1016/j.jinorgbio.2015.07.006. [DOI] [PubMed] [Google Scholar]
- Droghetti E.; Nicoletti F. P.; Bonamore A.; Sciamanna N.; Boffi A.; Feis A.; Smulevich G. J. Inorg. Biochem. 2011, 105, 1338–1343. 10.1016/j.jinorgbio.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Nicoletti F. P.; Droghetti E.; Boechi L.; Bonamore A.; Sciamanna N.; Estrin D. A.; Feis A.; Boffi A.; Smulevich G. J. Am. Chem. Soc. 2011, 133, 20970–20980. 10.1021/ja209312k. [DOI] [PubMed] [Google Scholar]
- Battistuzzi G.; Bellei M.; Bortolotti C. A.; Rocco G. D.; Leonardi A.; Sola M. Arch. Biochem. Biophys. 2004, 423, 317–331. 10.1016/j.abb.2003.12.036. [DOI] [PubMed] [Google Scholar]
- Dong S.; Niu J.; Cotton T. M.. Ultraviolet/visible spectroelectrochemistry of redox proteins. In Methods in Enzymology; Kenneth S., Ed.; Academic Press: San Diego, 1995; Vol. 246, pp 701–732. [DOI] [PubMed] [Google Scholar]
- Millis C. D.; Cai D. Y.; Stankovich M. D.; Tien M. Biochemistry 1989, 28, 8484–8489. 10.1021/bi00447a032. [DOI] [PubMed] [Google Scholar]
- Indiani C.; Santoni E.; Becucci M.; Boffi A.; Fukuyama K.; Smulevich G. Biochemistry 2003, 42, 14066–14074. 10.1021/bi035290l. [DOI] [PubMed] [Google Scholar]
- Meharenna Y. T.; Doukov T.; Li H.; Soltis S. M.; Poulos T. L. Biochemistry 2010, 49, 2984–2986. 10.1021/bi100238r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petřek M.; Otyepka M.; Banáš P.; Košinová P.; Koča J.; Damborský J. BMC Bioinf. 2006, 7, 316. 10.1186/1471-2105-7-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macedo S.; Pechlaner M.; Schmid W.; Weik M.; Sato K.; Dennison C.; Djinović-Carugo K. J. Synchrotron Radiat. 2009, 16, 191–204. 10.1107/S0909049509003331. [DOI] [PubMed] [Google Scholar]
- Kwon H.; Smith O.; Raven E.; Moody P. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2017, 73, 141–147. 10.1107/S2059798316016314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakeley M. P.; Hasnain S. S.; Antonyuk S. V. IUCrJ 2015, 2, 464–474. 10.1107/S2052252515011239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casalini S.; Battistuzzi G.; Borsari M.; Ranieri A.; Sola M. J. Am. Chem. Soc. 2008, 130, 15099–15104. 10.1021/ja8040724. [DOI] [PubMed] [Google Scholar]
- Hagedoorn P. L.; de Geus D. C.; Hagen W. R. Eur. J. Biochem. 2002, 269, 4905–4911. 10.1046/j.1432-1033.2002.03208.x. [DOI] [PubMed] [Google Scholar]
- Zamocky M.; Hofbauer S.; Schaffner I.; Gasselhuber B.; Nicolussi A.; Soudi M.; Pirker K. F.; Furtmüller P. G.; Obinger C. Arch. Biochem. Biophys. 2015, 574, 108–119. 10.1016/j.abb.2014.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smulevich G., Howes B. D., Droghetti E.. Structural and functional properties of heme-containing peroxidases: a resonance Raman perspective for the superfamily of plant, fungal and bacterial peroxidases In Heme Peroxidases; Raven E., Dunford B., Eds.; The Royal Society of Chemistry: Cambridge, U.K., 2015; pp 61–98. [Google Scholar]
- Cerda-Colón J. F.; Silfa E.; López-Garriga J. J. Am. Chem. Soc. 1998, 120, 9312–9317. 10.1021/ja972654m. [DOI] [Google Scholar]
- Fitch C. A.; Platzer G.; Okon M.; Garcia-Moreno E. B.; McIntosh L. P. Protein Sci. 2015, 24, 752–761. 10.1002/pro.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms M. J.; Schlessman J. L.; Sue G. R.; Garcia-Moreno E. E. B. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 18954–18959. 10.1073/pnas.1104808108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S.; Li Z. S.; Chen S. L. Dalton Trans. 2014, 43, 973–981. 10.1039/C3DT52171K. [DOI] [PubMed] [Google Scholar]
- Araiso T.; Dunford H. B. J. Biol. Chem. 1981, 256, 10099–10104. [PubMed] [Google Scholar]
- Arnhold J.; Monzani E.; Furtmüller P. G.; Zederbauer M.; Casella L.; Obinger C. Eur. J. Inorg. Chem. 2006, 2006, 3801–3811. 10.1002/ejic.200600436. [DOI] [Google Scholar]
- Furtmüller P. G.; Burner U.; Jantschko W.; Regelsberger G.; Obinger C. Redox Rep. 2000, 5, 173–178. 10.1179/135100000101535717. [DOI] [PubMed] [Google Scholar]
- Awad M. I.; Denggerile A.; Ohsaka T. J. Electrochem. Soc. 2004, 151, E358–E363. 10.1149/1.1812733. [DOI] [Google Scholar]
- Epstein I. R.; Kustin K. J. Phys. Chem. 1985, 89, 2275–2282. 10.1021/j100257a025. [DOI] [Google Scholar]
- Hayashi Y.; Yamazaki I. J. Biol. Chem. 1979, 254, 9101–9106. [PubMed] [Google Scholar]
- Arnhold J.; Furtmüller P. G.; Obinger C. Redox Rep. 2003, 8, 179–186. 10.1179/135100003225002664. [DOI] [PubMed] [Google Scholar]
- Arnhold J.; Furtmüller P. G.; Regelsberger G.; Obinger C. Eur. J. Biochem. 2001, 268, 5142–5148. 10.1046/j.0014-2956.2001.02449.x. [DOI] [PubMed] [Google Scholar]
- Furtmüller P. G.; Arnhold J.; Jantschko W.; Zederbauer M.; Jakopitsch C.; Obinger C. J. Inorg. Biochem. 2005, 99, 1220–1229. 10.1016/j.jinorgbio.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Jakopitsch C.; Pirker K. F.; Flemmig J.; Hofbauer S.; Schlorke D.; Furtmüller P. G.; Arnhold J.; Obinger C. J. Inorg. Biochem. 2014, 135, 10–19. 10.1016/j.jinorgbio.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaning U. K.; Sehested K.; Holcman J. J. Phys. Chem. 1985, 89, 760–763. 10.1021/j100251a008. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.