

Abstract
Purpose:
Fuchs endothelial corneal dystrophy (FECD) is a progressive degenerative disease of the corneal endothelium. It is genetically heterogeneous and follows either an autosomal dominant or sporadic pattern of inheritance. Here, we have explored the association of four previously reported intronic single nucleotide polymorphisms and intronic CTG repeat expansions in TCF4 gene to FECD in an Indian cohort.
Methods:
The cohort consisting of 52 sporadic late-onset cases, 5 early-onset cases, and 148 controls was taken for the study. rs2286812 and rs613872 were genotyped by allele specific polymerase chain reaction (ASPCR) and PCR-based restriction digestion, respectively; rs17595731 and rs9954153 were genotyped by Taqman assay using real-time PCR. The quantitative assessment of the CTG repeat region was performed by PCR/Sanger DNA sequencing. The repeats were assessed qualitatively by short tandem repeat and triplet repeat primed PCR assays. The statistical analysis was performed using two-tailed Fisher's exact probability test.
Results:
SNPsrs613872 (G/T) for the ‘G’ allele (P value: 4.57 × 10−5) and rs17595731 (C/T) for the ‘C’ allele (P value: 1.87 × 10−5), respectively, showed a significant association to sporadic late-onset FECD. CTG repeat expansions were found to be associated with FECD with a P value = 2.4 × 10−3.
Conclusion:
rs613872, rs17595731, and CTG repeat expansions in intronic region of TCF4 are associated with increased risk of sporadic late-onset FECD in the Indian cohort studied.
Keywords: Fuchs endothelial corneal dystrophy, SNP, TCF4 gene, trinucleotide repeat
Fuchs endothelial corneal dystrophy (FECD: MIM136800; Mendelian Inheritance in Man) is a genetically heterogeneous, progressive degenerative disorder of the corneal endothelium with 50% of cases being autosomal dominant and the remaining, sporadic. FECD, broadly classified as early-onset (<40 years) and late-onset based on the age of presentation, is characterized by a gradual loss of corneal endothelial cells compromising its solute transport function. The hallmarks of FECD are localized thickening of Descemet's membrane due to abnormal accumulation of collagenous layer and formation of clinically visible deposits on the surface of the corneal endothelium called “guttae.” While COL8A2 gene mutations have been implicated in early-onset FECD,[1] mutations in SLC4A11 and TCF8 are reported to contribute to a small percentage of late-onset FECD.[2,3,4,5] Linkage analysis on multigenerational late-onset and small multiplex FECD families have identified many loci; FCD1(13pter-13q21.13), FCD2(18q21.2-q21.32), FCD3(5q33.1-q35.2), and FCD4(9p22.1-9p24.1), apart from other loci on chromosome 1,7,15,17,X.[6,7,8] Further, genome wide linkage analysis followed by either linkage region targeted next generation sequencing (NGS) or whole exome sequencing (WES) have identified LOXHD1 and AGBL1 as candidate genes at FCD2 locus and on chromosome 15, respectively.[9,10] Single nucleotide polymorphisms (rs17595731, rs9954153, rs613872, and rs2286812) and CTG trinucleotide repeat expansion in the intron of TCF4 gene are associated with an increased risk for sporadic late-onset FECD and have been significantly replicated in cohorts from different populations.[11,12,13,14,15,16,17,18,19,20,21] In this study, the association of these polymorphisms and CTG trinucleotide repeat expansion in the intronic region of TCF4 gene to late-onset FECD has been comprehensively assessed.
Methods
Subjects
A cohort consisting of 52 unrelated sporadic late-onset and 5 sporadic early-onset (<40 years) FECD cases were enrolled in the study. Patients were diagnosed with FECD based on complete ophthalmic examination and/or histopathological evidence following penetrating keratoplasty (PK) or endothelial transplantation.[5] A total of 148 unrelated age-matched subjects without any ocular abnormalities that were enrolled as part of an epidemiological study previously conducted were used as controls.[22,23] Ophthalmic examination for the cases and controls included slit lamp, fundus examination, and applanation tension. The cornea was assessed for thickness quantitatively by ultrasound corneal pachymetry (Tomey) and for endothelial morphology qualitatively by specular microscopy (Noncon, ROBO CA). The controls were ruled out for any corneal abnormalities, and clinical findings showed a compact cornea and the absence of “guttae.” The cases are a representation of patients across India with different sub-ethnic backgrounds.
Sample collection
Genomic DNA was extracted from 10 mL whole blood for all cases and controls using NucleoSpin® Blood XL kit (Macherey-Nagel, GmbH, Germany) according to manufacturer's instructions. Approval for the study was obtained from the Institutional Review Board and Ethics Committee. The study adhered to the tenets of the Declaration of Helenski and all participants were recruited after written informed consent.
Genotyping of CTG repeats
Quantitative assessment by Sanger DNA sequencing
Primers (forward primer: AATGCCAGATGAGTTTGGTG; reverse primer: GCTGCCTGCCTAGGGCTAC) flanking the CTG repeats in the intron of TCF4 gene were designed using the Primer3 software (v. 0.4.0). Polymerase chain reaction (PCR) amplification was followed by EXO-SAP (Escherichia coli exonuclease I and fast alkaline phosphatase, Thermo Scientific, Massachusetts, USA) treatment. Sanger DNA sequencing was performed using Big Dye Terminator v.3.1 Ready Reaction kit (Applied Biosystems, Foster City, USA) in ABIPRISM® 3100-Avant Genetic Analyzer (Applied Biosystems, Foster City, USA). The number of repeats was manually counted using the Sequence Analysis v5.1.1 software (Applied Biosystems, Foster City, USA).
Triplet repeat primed polymerase chain reaction and short tandem repeat assay
The short tandem repeat (STR) and Triplet repeat primed PCR (TP-PCR) assays were performed based on protocol followed by Wieben et al.[14] followed by fragment analysis in ABIPRISM® 3100-Avant Genetic Analyzer (Applied Biosystems, Foster City, USA). Data analysis was performed using ABI Peak Scanner v1.0 software (Applied Biosystems, Foster City, USA). While STR assay suggests the homo/heterozygosity status of repeats, TP-PCR confirms the nature of homozygous calls in the STR assay, i.e., whether the sample is truly homozygous for the repeats or has an expanded heterozygous repeat which has not been captured.
Genotyping of SNPs rs17595731 and rs9954153 using Taq-Man assay
Allelic discrimination by TaqMan assay was performed using Applied Biosystems 7300 - real time PCR system (Applied Biosystems, Foster City, USA) for the SNPs rs17595731 and rs9954153. The PCR protocol was as per the TaqMan® SNP genotyping assays by Applied Biosystems.
Genotyping of SNPs rs613872 and rs2286812
rs613872 and rs2286812 were genotyped using allele specific PCR and PCR-based restriction digestion assays, respectively. Primers were designed using Primer3 software (v. 0.4.0) for both the SNPs.
Genotyping by restriction enzyme digestion of polymerase chain reaction product
PCR followed by digestion with restriction enzyme was performed for rs613872 where the ‘T’ nucleotide created a restriction site for enzyme ApoI. The presence of ‘T’ allele cut the 230 bp PCR amplified product into 117 bp and 113 bp fragments.
First, a 20 μL reaction was set up containing 10 mM Tris (pH 9.0), 50 mM KCl, 1.5 mM MgCl2 and 0.01% gelatin, 200 μM of dNTP blend (GeneAmpdNTP blend, Applied Biosystems, Foster City, CA, USA), 5 μM of each forward and reverse primer (Sigma-Aldrich, St. Louis, MO, USA) (forward primer-5’ CAGGCACTCCCCATTTACTG 3’, reverse primer-5’ ACCCCAGTAGGGTTGTGATG 3’), 1U of Taq DNA polymerase (GeNei, Bangalore, India), and 100 ng of genomic DNA. A touchdown PCR with annealing temperature of 61.3°C–54.3°C was performed.
Twenty μL restriction digestion reaction was set up with 5 μL of amplified product, 2 μL of NEB buffer 3 (New England BioLabsInc, MA, USA.), 1 μL of 100X BSA, 1U of ApoI enzyme (New England BioLabs Inc., MA, USA). The reaction was incubated at 50°C for 16 h followed by heat inactivation of the enzyme at 80°C for 20 min. The amplified, digested products were analyzed on a 2% agarose gel. To eliminate the possibility of incomplete digestion, overnight restriction digestion protocol was performed, and samples were sequenced at random by Sanger DNA sequencing to confirm the genotype.
Genotyping by allele specific polymerase chain reaction
For SNP rs2286812, two allele specific reverse primers, for the ‘C’ and ‘T’ allele were designed additionally, apart from the forward and reverse primers (common forward primer-5’CCAGAGACATTTTGGAAATAACTT 3’, common reverse primer-5’ CAGTATGCTGGAGAGGGTCA 3’, allele specific reverse primer for ‘C’ allele-5’CAACTCTCAGTCTGTCATCATTAAG 3’, allele specific reverse primer for ‘T’ allele-5’CAACTCTCAGTCTGTCATCATTAAA 3’). Two PCRs were set up, each with the forward, reverse, and the allele specific reverse primers. Twenty μL reactions were set with 10 μM primer (Sigma-Aldrich, St. Louis, MO, USA), 10 mM Tris (pH 9.0), 50 mM KCl, 1.5 mM MgCl2, and 0.01% gelatin (GeNei, Bangalore, India), 100 μM of dNTP blend (GeneAmpdNTP blend, Applied Biosystems, Foster City, CA, USA), 1U of Taq DNA polymerase (GeNei, Bangalore, India) and 50 ng of genomic DNA. PCR amplification at annealing temperature of 61.4°C–54.4°C was followed by analysis on a 2% agarose gel.
Statistical analysis
Two-tailed Fisher's exact probability test was performed to study the association of CTG repeat expansion between cases and controls. Association of the SNPsrs17595731, rs9954153, rs613872, and rs2286812 and linkage disequilibrium (LD) between the SNPs to FECD was analyzed using the Haploview v4.2 software.[24]
Results
The 52 late-onset FECD cases included 23 males and 29 females with a mean age of 59.5 ± 10.9. The five early-onset cases were all females. A total of 148 controls were taken for the study with a mean age of 63.8 ± 7.9. The early onset cases in this cohort were previously screened for variations in the COL8A2 gene. No mutations were reported.[25] In the cases studied, 58% underwent a PK or Descemet Stripping Endothelial Keratoplasty (DSEK) in at least one eye and 42% had numerous guttae with polymegathism. Among the 42% who did not undergo corneal transplant, 9% of the cases showed corneal decompensation with corneal edema.
Assessment of CTG trinucleotide repeats in cases and controls
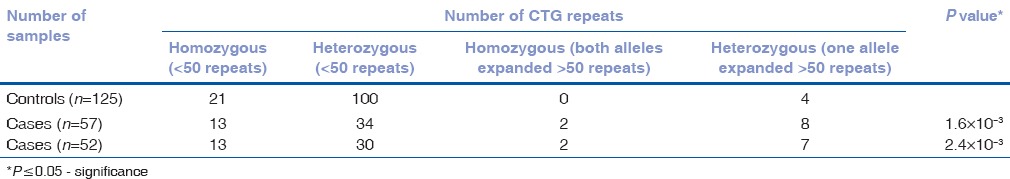
The CTG repeats were assessed quantitatively by manually counting the sequence electrophoretogram and qualitatively by TP-PCR and STR assays. We found 9/52 (17.3%) cases with repeat expansions >50 repeats when compared to 4/125 (3.2%) controls. Of the 9 cases with repeat expansions, 2 cases showed homozygous expansions >50 repeats in both alleles while the remaining 7 cases were heterozygous for the repeat expansion with >50 repeats in one allele. By Sanger DNA sequencing, we were able to count up to 75 repeats. The expansions in alleles were confirmed by TP-PCR assay. A significant association of expanded trinucleotide repeats to late-onset FECD was observed (P = 2.4 × 10−3). The odds of risk for late-onset FECD was found to be 6.3 times higher (odds ratio [OR] 6.3, 95% confidence interval [CI]: 1.8, 21.6), given the presence of expanded CTG trinucleotide repeats (>50 repeats) to baseline CTG repeats (<50 repeats) and was statistically significant. Association including the five early-onset cases was also done, and although we found a heterozygous expansion in one early-onset case, there was no notable change in P value [Table 1].
Table 1.
CTG repeat expansion in late onset cohort alone (n=52) and with early onset cases (n=57)
Association of rs613872 and rs17595731 to late-onset Fuchs endothelial corneal dystrophy
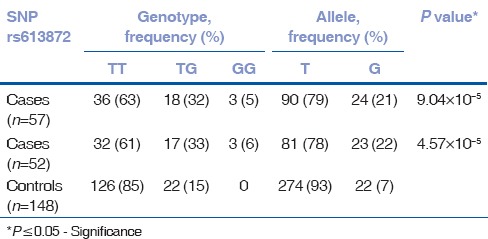
Of the four SNPs (rs17595731, rs9954153, rs613872, and rs2286812), rs613872 (P = 4.57 × 10−5, OR 3.6, 95% CI: 1.7, 7.3), and rs17595731 (P = 1.87 × 10−5, OR 15.3, 95% CI: 3.1, 73.4) showed a significant association to late-onset FECD. Although the minor allele frequency of SNP rs17595731 was 0.028, reports on the inclusion of SNPs with minor allele frequency >0.01 and the proven association of rs17595731 to late-onset FECD indicate an association of the SNP to the disease.[26,27,28] The P value for rs2286812 indicated significance; however, the Hardy–Weinberg Equilibrium was considerably deviated. SNP rs9954153 did not show any association. There was no substantial change in the P value of the 4 SNPs with the inclusion of 5 early-onset cases in the study [Tables 2–5]. The four SNPs are not in LD.
Table 2.
Genotype data of SNP rs9954153 for late-onset cohort alone (n=52) and with early-onset cases (n=57)
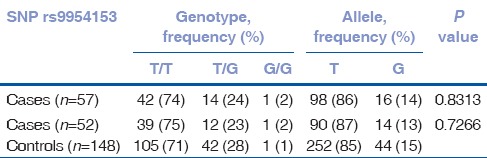
Table 5.
Genotype data of SNP rs17595731 for late onset cohort alone (n=52) and with early onset cases (n=57)

Table 3.
Genotype data of SNP rs2286812 for late-onset cohort alone (n=52) and with early-onset cases (n=57)
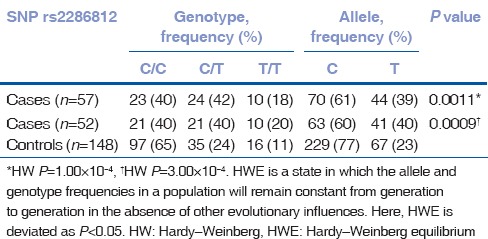
Table 4.
Genotype data of SNP rs613872 for late-onset cohort alone (n=52) and with early-onset cases (n=57)
Discussion
Late-onset FECD is a complex disease involving genetic and environmental factors. A vast proportion of PK has been indicated by FECD in North America (28%) and Europe (20.6%).[29] In the Indian population, a study has shown that about 5.3% of PK cases came to clinic with FECD as the cause.[30]
Apart from the identification of various candidate genes contributing to FECD, association studies gained importance when Baratz et al. identified genetic risk variants by Genome-wide association studies for the first time in 2010. Four SNPs in the intron of TCF4 were found to be independently associated with FECD.[31] Further, replication studies on these risk variants in various populations identified a significant association of rs613872.[19,20] This SNP was studied in two Indian cohorts and association was reported in only one of the two studies which involved 82 sporadic late-onset FECD cases.[16,32] We have genotyped these four risk variants and observed that the ‘G’ allele of rs613872, and additionally, the ‘C’ allele of rs17595731 was associated with sporadic late-onset FECD cases in our cohort. Our data adds value to the significance of association of this SNP to late-onset FECD and for the first time shows an association of rs17595731 to FECD in this Indian cohort.
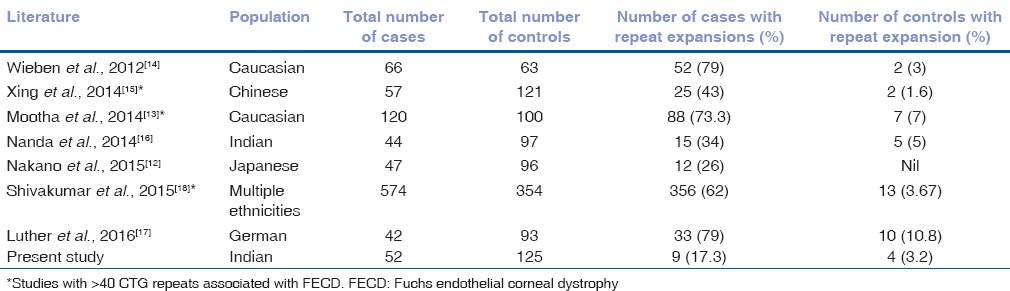
Breschel et al. reported novel polymorphic unstable trinucleotide repeats (CTG) in the intron of TCF4 gene in the population while scanning chromosome 18 specific library in a study on bipolar disorder. However, the CTG repeat expansions did not show association to the disorder.[33] As intronic region of TCF4 gene is a hotspot for SNPs associated with late-onset FECD, Wieben et al. studied the significance of these repeats and reported an association of >50 expanded CTG repeats to late-onset FECD[14] and the same has been replicated across different ethnicities.[12,13,15,16,17,18] Some studies have considered >40 repeats as criteria for expansion and have found a significant association to the disease.[13,15,18] Our study confirmed a strong association of >50 repeat expansions with late-onset FECD. We did not observe intermediate repeat expansions between 40 and 50 repeats in our cohort. However, compared to the seven other studies on various ethnicities including that of another Indian cohort, we observed a lower percentage of cases with expanded CTG repeats [Table 6].
Table 6.
Comparison of expanded CTG repeats in cases and controls across ethnicities based on literature
TCF4 gene coding for the E2-2 protein is expressed in the corneal endothelium and plays a vital role in the regulation of cell adhesion protein, E-cadherin and is thus hypothesized to promote endothelial-mesenchymal transition (EMT) by increased expression of ZEB1. When there is a loss of corneal endothelial cells, the stem cells from the limbus, migrate to the site of loss or damage and replenish the endothelial cells by the mechanism of EMT. The exact sequence of events in FECD remain unknown, and it is speculated that the dysregulation of E2-2 expression leading to reduced ZEB1 expression may affect migration of progenitor cells to replenish the lost or damaged endothelial cells. Due to loss of cells, subsequent degeneration and progressive vision impairment could occur.[34]
Du et al. observed RNA foci in the corneal endothelial cells of late-onset FECD patients with CTG repeat expansions. When compared with RNA foci in myotonic dystrophy (DM1), the foci were similar in morphology. RNA Seq data revealed differential splicing of five genes including MBNL1 in the corneal endothelium of FECD patients which was also observed in DM1. Splicing of MBNL1 is implicated in EMT thus providing evidence for a possible role in FECD pathogenesis.[35] Further characterization studies may provide insight into the mechanisms involved in the pathogenesis of FECD due to repeat expansion and its relevance to EMT.
Conclusion
SNP rs613872 and CTG repeat expansions have consistently shown association to late-onset FECD across populations of various ethnicities. As the age of onset and progression of the disease was highly variable in this cohort, no genotype-phenotype correlation was done. Genotyping of the associated SNPs in sporadic cases of late-onset FECD may be useful in counseling the patient for regular follow-up and timely clinical management, and the same may be offered to the at risk family members such as siblings and offspring. A larger sample size with a higher representation of different sub-ethnic groups of the Indian population will strengthen our finding. This is the first study to report a significant association of rs17595731 to late-onset FECD in the Indian population.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
The authors would like to thank the patients for their participation. The study was supported by a financial grant from the Indian Council for Medical Research, India, project no: 53/10/2011-BMS.
References
- 1.Biswas S, Munier FL, Yardley J, Hart-Holden N, Perveen R, Cousin P, et al. Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum Mol Genet. 2001;10:2415–23. doi: 10.1093/hmg/10.21.2415. [DOI] [PubMed] [Google Scholar]
- 2.Riazuddin SA, Vithana EN, Seet LF, Liu Y, Al-Saif A, Koh LW, et al. Missense mutations in the sodium borate cotransporter SLC4A11 cause late-onset Fuchs corneal dystrophy. Hum Mutat. 2010;31:1261–8. doi: 10.1002/humu.21356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riazuddin SA, Zaghloul NA, Al-Saif A, Davey L, Diplas BH, Meadows DN, et al. Missense mutations in TCF8 cause late-onset Fuchs corneal dystrophy and interact with FCD4 on chromosome 9p. Am J Hum Genet. 2010;86:45–53. doi: 10.1016/j.ajhg.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mehta JS, Vithana EN, Tan DT, Yong VH, Yam GH, Law RW, et al. Analysis of the posterior polymorphous corneal dystrophy 3 gene, TCF8, in late-onset Fuchs endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2008;49:184–8. doi: 10.1167/iovs.07-0847. [DOI] [PubMed] [Google Scholar]
- 5.Vithana EN, Morgan PE, Ramprasad V, Tan DT, Yong VH, Venkataraman D, et al. SLC4A11 mutations in Fuchs endothelial corneal dystrophy. Hum Mol Genet. 2008;17:656–66. doi: 10.1093/hmg/ddm337. [DOI] [PubMed] [Google Scholar]
- 6.Riazuddin SA, Eghrari AO, Al-Saif A, Davey L, Meadows DN, Katsanis N, et al. Linkage of a mild late-onset phenotype of Fuchs corneal dystrophy to a novel locus at 5q33.1-q35. Invest Ophthalmol Vis Sci. 2009;50:5667–71. doi: 10.1167/iovs.09-3764. [DOI] [PubMed] [Google Scholar]
- 7.Sundin OH, Broman KW, Chang HH, Vito EC, Stark WJ, Gottsch JD. A common locus for late-onset Fuchs corneal dystrophy maps to 18q21.2-q21. Invest Ophthalmol Vis Sci. 2006;47:3919–26. doi: 10.1167/iovs.05-1619. [DOI] [PubMed] [Google Scholar]
- 8.Sundin OH, Jun AS, Broman KW, Liu SH, Sheehan SE, Vito EC, et al. Linkage of late-onset Fuchs corneal dystrophy to a novel locus at 13pTel-13q12.13. Invest Ophthalmol Vis Sci. 2006;47:140–5. doi: 10.1167/iovs.05-0578. [DOI] [PubMed] [Google Scholar]
- 9.Riazuddin SA, Parker DS, McGlumphy EJ, Oh EC, Iliff BW, Schmedt T, et al. Mutations in LOXHD1, a recessive-deafness locus, cause dominant late-onset Fuchs corneal dystrophy. Am J Hum Genet. 2012;90:533–9. doi: 10.1016/j.ajhg.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riazuddin SA, Vasanth S, Katsanis N, Gottsch JD. Mutations in AGBL1 cause dominant late-onset Fuchs corneal dystrophy and alter protein-protein interaction with TCF4. Am J Hum Genet. 2013;93:758–64. doi: 10.1016/j.ajhg.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li D, Peng X, Sun H. Association of TCF4 polymorphisms and Fuchs’ endothelial dystrophy: A meta-analysis. BMC Ophthalmol. 2015;15:61. doi: 10.1186/s12886-015-0055-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakano M, Okumura N, Nakagawa H, Koizumi N, Ikeda Y, Ueno M, et al. Trinucleotide repeat expansion in the TCF4 gene in Fuchs’ endothelial corneal dystrophy in Japanese. Invest Ophthalmol Vis Sci. 2015;56:4865–9. doi: 10.1167/iovs.15-17082. [DOI] [PubMed] [Google Scholar]
- 13.Mootha VV, Gong X, Ku HC, Xing C. Association and familial segregation of CTG18.1 trinucleotide repeat expansion of TCF4 gene in Fuchs’ endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2014;55:33–42. doi: 10.1167/iovs.13-12611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wieben ED, Aleff RA, Tosakulwong N, Butz ML, Highsmith WE, Edwards AO, et al. A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2-2) gene predicts Fuchs corneal dystrophy. PLoS One. 2012;7:e49083. doi: 10.1371/journal.pone.0049083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xing C, Gong X, Hussain I, Khor CC, Tan DT, Aung T, et al. Transethnic replication of association of CTG18.1 repeat expansion of TCF4 gene with Fuchs’ corneal dystrophy in Chinese implies common causal variant. Invest Ophthalmol Vis Sci. 2014;55:7073–8. doi: 10.1167/iovs.14-15390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nanda GG, Padhy B, Samal S, Das S, Alone DP. Genetic association of TCF4 intronic polymorphisms, CTG18.1 and rs17089887, with Fuchs’ endothelial corneal dystrophy in an Indian population. InvestOphthalmol Vis Sci. 2014;55:7674–80. doi: 10.1167/iovs.14-15297. [DOI] [PubMed] [Google Scholar]
- 17.Luther M, Grünauer-Kloevekorn C, Weidle E, Passarge E, Rupprecht A, Hoffmann K, et al. TGC repeats in intron 2 of the TCF4 gene have a good predictive power regarding to fuchs endothelial corneal dystrophy. Klin Monbl Augenheilkd. 2016;233:187–94. doi: 10.1055/s-0035-1546138. [DOI] [PubMed] [Google Scholar]
- 18.Vasanth S, Eghrari AO, Gapsis BC, Wang J, Haller NF, Stark WJ, et al. Expansion of CTG18.1 Trinucleotide repeat in TCF4 is a potent driver of Fuchs’ corneal dystrophy. Invest Ophthalmol Vis Sci. 2015;56:4531–6. doi: 10.1167/iovs.14-16122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li YJ, Minear MA, Rimmler J, Zhao B, Balajonda E, Hauser MA, et al. Replication of TCF4 through association and linkage studies in late-onset Fuchs endothelial corneal dystrophy. PLoS One. 2011;6:e18044. doi: 10.1371/journal.pone.0018044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riazuddin SA, McGlumphy EJ, Yeo WS, Wang J, Katsanis N, Gottsch JD. Replication of the TCF4 intronic variant in late-onset Fuchs corneal dystrophy and evidence of independence from the FCD2 locus. Invest Ophthalmol Vis Sci. 2011;52:2825–9. doi: 10.1167/iovs.10-6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Igo RP, Jr, Kopplin LJ, Joseph P, Truitt B, Fondran J, Bardenstein D, et al. Differing roles for TCF4 and COL8A2 in central corneal thickness and fuchs endothelial corneal dystrophy. PLoS One. 2012;7:e46742. doi: 10.1371/journal.pone.0046742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agarwal S, Raman R, Paul PG, Rani PK, Uthra S, Gayathree R, et al. Sankara Nethralaya-Diabetic Retinopathy Epidemiology and Molecular Genetic Study (SN-DREAMS 1): Study design and research methodology. Ophthalmic Epidemiol. 2005;12:143–53. doi: 10.1080/09286580590932734. [DOI] [PubMed] [Google Scholar]
- 23.George R, Arvind H, Baskaran M, Ramesh SV, Raju P, Vijaya L. The Chennai glaucoma study: Prevalence and risk factors for glaucoma in cataract operated eyes in urban Chennai. Indian J Ophthalmol. 2010;58:243–5. doi: 10.4103/0301-4738.62655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 25.Soumittra N, Loganathan SK, Madhavan D, Ramprasad VL, Arokiasamy T, Sumathi S, et al. Biosynthetic and functional defects in newly identified SLC4A11 mutants and absence of COL8A2 mutations in Fuchs endothelial corneal dystrophy. J Hum Genet. 2014;59:444–53. doi: 10.1038/jhg.2014.55. [DOI] [PubMed] [Google Scholar]
- 26.Gorlov IP, Gorlova OY, Sunyaev SR, Spitz MR, Amos CI. Shifting paradigm of association studies: Value of rare single-nucleotide polymorphisms. Am J Hum Genet. 2008;82:100–12. doi: 10.1016/j.ajhg.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koller DL, Ichikawa S, Lai D, Padgett LR, Doheny KF, Pugh E, et al. Genome-wide association study of bone mineral density in premenopausal European-American women and replication in African-American women. J Clin Endocrinol Metab. 2010;95:1802–9. doi: 10.1210/jc.2009-1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tabangin ME, Woo JG, Martin LJ. The effect of minor allele frequency on the likelihood of obtaining false positives. BMC Proc. 2009;3(Suppl 7):S41. doi: 10.1186/1753-6561-3-S7-S41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matthaei M, Sandhaeger H, Hermel M, Adler W, Jun AS, Cursiefen C, et al. Changing indications in penetrating keratoplasty: A systematic review of 34 years of global reporting. Transplantation. 2017;101:1387–99. doi: 10.1097/TP.0000000000001281. [DOI] [PubMed] [Google Scholar]
- 30.Dandona L, Ragu K, Janarthanan M, Naduvilath TJ, Shenoy R, Rao GN. Indications for penetrating keratoplasty in India. Indian J Ophthalmol. 1997;45:163–8. [PubMed] [Google Scholar]
- 31.Baratz KH, Tosakulwong N, Ryu E, Brown WL, Branham K, Chen W, et al. E2-2 protein and Fuchs's corneal dystrophy. N Engl J Med. 2010;363:1016–24. doi: 10.1056/NEJMoa1007064. [DOI] [PubMed] [Google Scholar]
- 32.Gupta R, Kumawat BL, Paliwal P, Tandon R, Sharma N, Sen S, et al. Association of ZEB1 and TCF4 rs613872 changes with late onset Fuchs endothelial corneal dystrophy in patients from northern India. Mol Vis. 2015;21:1252–60. [PMC free article] [PubMed] [Google Scholar]
- 33.Breschel TS, McInnis MG, Margolis RL, Sirugo G, Corneliussen B, Simpson SG, et al. A novel, heritable, expanding CTG repeat in an intron of the SEF2-1 gene on chromosome 18q21.1. Hum Mol Genet. 1997;6:1855–63. doi: 10.1093/hmg/6.11.1855. [DOI] [PubMed] [Google Scholar]
- 34.Wright AF, Dhillon B. Major progress in Fuchs's corneal dystrophy. N Engl J Med. 2010;363:1072–5. doi: 10.1056/NEJMe1007495. [DOI] [PubMed] [Google Scholar]
- 35.Du J, Aleff RA, Soragni E, Kalari K, Nie J, Tang X, et al. RNA toxicity and missplicing in the common eye disease fuchs endothelial corneal dystrophy. J Biol Chem. 2015;290:5979–90. doi: 10.1074/jbc.M114.621607. [DOI] [PMC free article] [PubMed] [Google Scholar]