

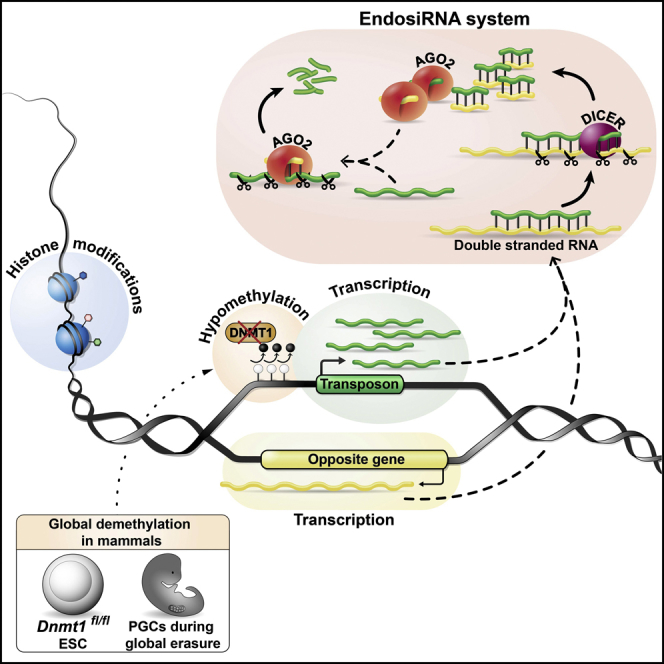
Summary
Erasure of DNA methylation and repressive chromatin marks in the mammalian germline leads to risk of transcriptional activation of transposable elements (TEs). Here, we used mouse embryonic stem cells (ESCs) to identify an endosiRNA-based mechanism involved in suppression of TE transcription. In ESCs with DNA demethylation induced by acute deletion of Dnmt1, we saw an increase in sense transcription at TEs, resulting in an abundance of sense/antisense transcripts leading to high levels of ARGONAUTE2 (AGO2)-bound small RNAs. Inhibition of Dicer or Ago2 expression revealed that small RNAs are involved in an immediate response to demethylation-induced transposon activation, while the deposition of repressive histone marks follows as a chronic response. In vivo, we also found TE-specific endosiRNAs present during primordial germ cell development. Our results suggest that antisense TE transcription is a “trap” that elicits an endosiRNA response to restrain acute transposon activity during epigenetic reprogramming in the mammalian germline.
Keywords: RNAi, DNMT1, germ line, primordial germ cell, transposable element, repeats, small RNAs, endogenous retroviruses, IAP elements
Graphical Abstract
Highlights
-
•
Global DNA demethylation in embryonic stem cells leads to transposon activation
-
•
Transposon activation increases the abundance of sense/antisense transcripts
-
•
ARGONAUTE2-bound endosiRNAs accumulate at high levels for acute repression
-
•
Longer-term transposon repression depends on repressive histone marks
In this issue of Cell Stem Cell, Berrens et al. report the control of transposable elements by endosiRNAs during global DNA demethylation induced in mouse embryonic stem cells. The study uncovered an “immediate” repression of transposons accomplished by endosiRNAs followed by their “chronic/long-term” silencing by repressive histone modifications.
Introduction
Epigenetic reprogramming in the mammalian germline is key for restoration of developmental potency and occurs at the preimplantation stage of embryonic development and during development of primordial germ cells (PGCs) (Reik and Surani, 2015). These events lead to global DNA methylation and H3K9me2 erasure together with the transient transcriptional activation of specific classes of transposable elements (TEs) (Hajkova et al., 2008, Rowe and Trono, 2011). This raises fundamental questions about the regulation of TE defense in the absence of repressive epigenetic marks.
TEs comprise ∼50% of the mammalian genome and can be categorized into two major classes: retrotransposons and DNA transposons (Lander et al., 2001). While most TEs in the genome are inactive due to mutations and/or truncations, around 1%–2% of long interspersed nuclear elements (LINEs) and endogenous retroviruses (ERVs) remain able to retrotranspose (Maksakova et al., 2006). Notably, the ERV family members intracisternal A particles (IAPs) and early transposons (ETns) are the most active TEs in the murine germline (Maksakova et al., 2006).
Due to their ability to retrotranspose, TEs are thought to play an important role in genome evolution, but can also cause genetic diseases (Goodier and Kazazian, 2008). In order to protect the genome from harmful mutations, regulatory mechanisms must be in place to limit their transcription.
TE activity is controlled by multiple epigenetic mechanisms including DNA methylation, repressive histone modifications, and small RNAs (Rowe and Trono, 2011). In somatic tissues, DNA methylation and H3K9me2/3 have been shown to be responsible for TE silencing (Walsh et al., 1998, Hutnick et al., 2010). However, in the germline, DNA methylation and H3K9me2 are globally erased, while H3K9me3 is maintained and H3K27me3 is redistributed (Iurlaro et al., 2017, Tang et al., 2016). Indeed, deletion of the H3K9me3 methyltransferase Setdb1 leads to activation of IAPs during PGC development as well as in mouse embryonic stem cells (ESCs) (Karimi et al., 2011, Maksakova et al., 2006). Further, global demethylation of naive ESCs results in transcriptional activation of TEs and subsequent resilencing by a redistribution of repressive histone marks (Walter et al., 2016).
A number of studies have demonstrated that small RNAs may also act post-transcriptionally as a second-tier defense against TEs, particularly in the germline. In mouse oocytes, microRNAs (miRNAs) and endogenous short interfering RNAs (endosiRNAs) that control TE expression have been identified (Tam et al., 2008, Flemr et al., 2013, Watanabe et al., 2006), and in the male germline PIWI-interacting small RNAs (piRNAs) can also control TE expression (Aravin et al., 2008). In ESCs, tRNA fragments have been recently described to play a role in ERV translational control (Schorn et al., 2017).
In contrast to somatic cells, increased pervasive transcription across TEs was reported in ESCs, suggesting that TEs may regulate transcription of long noncoding RNAs (lncRNAs) (Kelley and Rinn, 2012). Intriguingly, however, in yeast it was shown that genome-wide pervasive transcription antisense to transposons leads to an RNAi response as a defense mechanism against TEs (Cruz and Houseley, 2014). Sense/antisense transcription permits the production of double-stranded RNA (dsRNA) triggering RNAi (Fire et al., 1998), which has also been identified as a control mechanism of TEs (Robert et al., 2005).
Here we test the hypothesis that genic transcripts antisense to TEs serve as a trap for transcriptional activation of TEs during global demethylation in mammals. Generation of Dicer as well as Ago2 conditional and constitutive knockout ESC lines in the background of a Dnmt1 conditional knockout (cKO) line allowed us to define an “immediate” endosiRNA-dependent repressive response to TE activation and a subsequent “chronic” response, characterized by targeting of repressive histone modifications.
Results
Acute Dnmt1 Deletion Leads to TE Demethylation in ESCs
Our experimental system recapitulates epigenetic reprogramming of early embryos and PGCs in vitro. We used Cre-mediated conditional Dnmt1 deletion in ESCs (Dnmt1 cKO) (Sharif et al., 2016) and sampled DNA and RNA at several defined time points after Dnmt1 deletion for methylome, long and small transcriptome, and chromatin analyses (Figure 1A).
Figure 1.

Transcriptional Upregulation of Specific TE Classes upon Acute Dnmt1 Deletion
(A) Left: schematic overview of epigenetic reprogramming during preimplantation and male (blue) and female (red) germline development. Right: schematic of Dnmt1 cKO as an in vitro system for mechanistic study of TE regulation during epigenetic reprogramming.
(B) Violin plots showing the distribution of CpG methylation levels measured by WGBS-seq of WT (gray) and conditional Dnmt1 cKO ESCs induced for days depicted in the figure. The percentage of methylated cytosines was quantified in consecutive 50 CpG windows genome-wide. CGI, CpG island. For significance analysis, Wilcoxon rank-sum test with Bonferroni correction testing with a p value threshold of <0.05.
(C) Heatmap of unbiased hierarchical clustering of all TEs responsive to Dnmt1 cKO across the time course of KO induction. The relative expression (Z score) of TEs upon Dnmt1 cKO is shown; n = 2.
(D) Bar graph showing the percentage of genic antisense transcription upon Dnmt1 deletion in KO relative to WT samples; n = 2.
(E) Chromosome view of TE inserted antisense to gene. Position of TE is denoted (top panel) along with sense strand-specific RNA-seq reads (lower panels; sense transcription shown in blue; antisense transcription shown in red). Each read is depicted. Arrows indicate directionality of reads.
(F) Expression of TEs in conditional Dnmt1 cKO ESC. Shown are normalized RNA-seq read counts overlapping different TE classes in sense (filled bars) or antisense (hatched bars) orientation. The figure shows mean of n = 2.
See also Figures S1 and S4I and Data S1.
By whole-genome bisulfite sequencing (WGBS-seq), we confirmed that acute deletion of Dnmt1 led to genome-wide demethylation from an initial 85% CpG methylation to 35% at day 3 after deletion, and 20% at day 6 after deletion with no further demethylation thereafter (Figures 1B and S1A). The residual methylation can be attributed to the activity of the de novo DNA methyltransferases (Lei et al., 1996). Upon Dnmt1 cKO, loss of methylation was observed in genic and intergenic elements, CGIs, and non-CGI promoters (Figure 1B). Characteristic methylation profiles over gene bodies were reduced with the same kinetics as the rest of the genome upon Dnmt1 cKO (Figure S1B). Furthermore, low methylated regions (LMRs) (Stadler et al., 2011) and active enhancers became demethylated (Figure S1C). Thus, this in vitro model results in replication-dependent global demethylation of the genome, which closely resembles the dynamics of global reprogramming in early embryos and PGCs (von Meyenn et al., 2016).
To analyze TEs in WGBS-seq, RNA-seq, and chromatin immunoprecipitation (ChIP)-seq data, we only considered uniquely mapped reads and filtered out TEs overlapping the (± 2 kb) region surrounding genes. While unique mapping might not capture all information about young TEs (as they lack the increased sequence divergence of older TEs that makes unique mapping more efficient; Lerat et al., 2003), this conservative approach allows us to be confident that mapped reads can be definitively ascribed to specific TE subfamilies. Moreover, the filtering of the region (± 2 kb) surrounding genes avoids ambiguity about the origin of TE expression from promoters that are not their own (Figure S1D; Data S1).
Acute Dnmt1 deletion led to hypomethylation of TEs at the same rate as the rest of the genome (Figures 1B and S1E), with the exception of IAPs, RLTRs, and MMERVK10C, which preserved higher methylation levels (Figure S1F). Thus, our experimental system also closely recapitulates global demethylation dynamics of TEs in vivo, including the fact that IAPs are relatively resistant to global demethylation (Seisenberger et al., 2012, Kobayashi et al., 2013).
Increased Sense Transcription of TEs upon Hypomethylation Combines with Pervasive Antisense Transcription
Next, we performed total RNA-seq upon acute Dnmt1 deletion to examine if demethylation led to transcriptional activation of TEs. Transcriptional activation was limited to specific classes of ERVs (Figure 1C). We found TEs with increased transcription upon hypomethylation that remained active over the whole time course (MMERVK10C), as well as TEs initially activated but notably subsequently re-silenced (e.g., IAPs and MERVLs).
In addition to TEs, a small number of genes became activated upon loss of DNA methylation (Figures S1G and S1H), including the imprinted genes Xlr3a, Mirg, and Rian (Table S1), consistent with the known roles for methylation in regulation of these genes (Ferguson-Smith, 2011) (Figure S1I). DNA hypomethylation did not result in ESC differentiation, as indicated by the unaltered expression of the core pluripotency network (Figure S1J).
Interestingly, when quantifying reads overlapping with genes, we found upon global hypomethylation increased pervasive transcription in the antisense orientation to those genes (Figure 1D). These pervasive antisense transcripts are in fact produced by transcription of TEs that have integrated in an antisense orientation to the genes (Figure 1E). Consistent with previous studies, high numbers of TEs were found to be preferentially integrated in antisense orientation to genes (van de Lagemaat et al., 2006) (Figure S1K).
We next analyzed the total RNA-seq data to determine whether both sense and antisense transcription was detectable at sites of TE integration. Indeed, TE antisense transcription was found in all TE families, with sense transcripts of members of the ERVs being upregulated consistent with their activation in response to demethylation (Figure 1F). We also included TEs that were not activated by hypomethylation, but instead are regulated in a DICER-dependent manner (Figure 3E).
Figure 3.

TEs Are Repressed by a DICER Mechanism
(A) Knockdown (KD) of RNAi players. Left: schematic of siRNA KD in Dnmt1 cKO ESCs. The genome gets demethylated (5mC, orange) and IAPs get transcriptionally activated and resilenced (red) if small RNAs are present (gray); however, KD of the RNAi pathway will deplete small RNAs. Lower right: quantitative real-time PCR analysis showing KD efficiencies of Dicer, Ago2, and Dgcr8 upon treatment with siRNAs after Dnmt1 deletion. Upper right: expression of IAPs upon Dicer, Ago2, Dgcr8, or non-targeting siRNA transfection. The data are normalized to non-targeting control. Bars represent mean ± SD, n = 3. ∗p < 0.05, ∗∗p < 0.005, two-tailed Student’s t test.
(B) Small RNA-seq of Dicer/Dnmt1 cDKO and Dnmt1 cKO ESCs. Sense (orange) and antisense (blue) small RNAs are separated by size and were mapped to all TEs. Reads were normalized to non-induced WT (Dicerfl/fl/Dnmt1fl/fl) ESCs.
(C) Quantitative real-time PCR analysis of TE classes in ESCs following conditional Dnmt1 cKO or Dnmt1/Dicer cDKO by treatment with 4OHT or Dicer KO. Bars represent mean of two biological replicates with two technical replicates. Values were normalized to Atp5b and Hspcb, and major satellites were normalized to U1. ∗p < 0.05, ∗∗p < 0.005, two-tailed Student’s t test.
(D) Quantitative real-time PCR analysis of IAPEz in the indicated ESC lines. Conditional deletions were induced by treatment with 4OHT for the indicated days. Values were normalized to Atp5b and Hspcb and are relative to the respective WT sample for each KO line, indicated by dashed line. Error bars represent mean ± SD; n = 3 for Dnmt1 cKO, Dicer KO/Dnmt1 cKO, and Ago2 KO/Dnmt1 cKO; n = 2 for Dicer/Dnmt1 cDKO and Ago2/Dnmt1 cDKO. Ago2 KO/Dnmt1 cKO time points days 9 and 11 were not collected.
(E) Heatmap of unbiased hierarchical clustering of all TE classes responsive to Dicer KO. Heatmap depicts relative expression (Z score) of TEs upon Dicer KO.
See also Figures S3 and S4I and Tables S2 and S3.
Sense/Antisense Transcription of TEs Correlates with AGO2-Bound endosiRNAs
The production of sense and antisense transcripts across TEs is expected to lead to dsRNAs, which can subsequently induce an RNAi response and silence TEs post-transcriptionally. These results suggest that TE expression may be sensed by pervasive antisense transcription, thus constituting a TE “trap” (Figure 2A). To test this hypothesis, we performed small RNA-seq at defined time points after Dnmt1 deletion. The majority of small RNAs were miRNAs (Figures S2A–S2C) and were expressed independently of DNA methylation, with the exception of miRNAs from the imprinted Dlk and Xlr3 loci (Figures S2D and S2E). Small RNA quantitative real-time PCR of mature miRNAs confirmed their methylation-dependent regulation (Figure S2F). The Dlk locus serves as an example of the genome-wide response to acute Dnmt1 deletion with the imprint control region (ICR) becoming demethylated, leading to transcriptional upregulation of the imprinted locus and embedded miRNAs (Figure S2G).
Figure 2.

Generation of TE-Derived Small RNAs following Global Demethylation
(A) Schematic displaying the hypothesis of pervasive transcription overlapping TEs acting as a “trap” of transcriptional activation of TEs. This could work through the production of dsRNAs from sense and antisense transcripts that feed into the RNAi pathway, which subsequently silences the TEs.
(B) Small RNA-seq reads mapped to different classes of TEs from WT (gray) and conditional Dnmt1 cKO ESCs. ∗p < 0.05, ∗∗p < 0.005, two-tailed Student’s t test. Bars represent mean ± SD, n = 3. All reads of a size between 20 and 24 nt were mapped to TE consensus sequences.
(C) Small RNA-seq reads mapped to the consensus sequence of IAPEZ. All reads of a size between 20 and 36 nt were mapped to the IAPEZ consensus sequence.
(D) Schematic displaying AGO2 IP of small RNAs.
(E) Size distribution of AGO2-bound small RNAs after AGO2 IP of sense (black) and antisense (gray) small RNAs mapping to repeatmasker consensus sequences using the piPipes small RNA-seq pipeline (Han et al., 2015).
(F) Small RNA-seq of AGO2-bound small RNAs mapped to TE classes of WT (gray) and conditional Dnmt1 cKO ESCs induced for 9 days (light blue). ∗p < 0.05, ∗∗p < 0.005, two-tailed Student’s t test. Bars represent mean ± SD, n = 4.
See also Figures S2 and S4I and Data S1.
Due to the short reads in small RNA-seq, we used TE consensus sequence mapping to analyze global TE-derived small RNAs. This method allows unambiguous alignment to unique TE classes. Notably, we observed a substantial increase of small RNAs mapping to IAP, MERVL, and ETn upon Dnmt1 deletion (Figure 2B), which in the case of IAPs mapped across the whole length of the element (Figure 2C). Small RNAs mapping to L1MdGf and MMERVK10C were detected both in wild-type (WT) and Dnmt1 cKO ESCs, respectively (Figure 2B).
The mammalian ARGONAUTE proteins (AGO) are critical components of the RNA-induced silencing complex (RISC). AGO2 can bind miRNAs as well as endosiRNAs and has the ability to “slice” its targets (Doi et al., 2003). We performed AGO2 IP from Dnmt1 cKO ESCs at day 9 after deletion and analyzed the pull-down by small RNA-seq (Figure 2D). The AGO2 IP small RNA-seq libraries of both WT and Dnmt1 cKO ESCs were composed 90% of known miRNAs, while 40% of the remaining small RNAs mapped to TEs (Figure S2H, Dnmt1 cKO shown). This subset of AGO2-bound small RNAs was 22 nt long and mapped to sense and antisense strands of TEs (Figure 2E); the small RNAs had 5′ U overhangs (Figure S2I) and formed characteristic 5′-5′ overlaps at nucleotide 20, identifying them as bona fide endosiRNAs (Figure S2J) (Ghildiyal and Zamore, 2009). AGO2-bound endosiRNAs mapping to MERVL and RLTR45 were expressed throughout the time course while endosiRNAs mapping to L1, IAP, and ETn or MMERVK10C were significantly enriched upon Dnmt1 deletion (Figure 2F), suggesting that functional endosiRNAs against specific TE classes are generated during global demethylation.
We also generated small RNA-seq libraries of male and female PGCs from embryonic day (E)13.5 and E14.5 embryos and found that ∼10% of all 20–24 nt small RNAs mapped to TEs in both male and female E13.5 and E14.5 PGCs, with small RNAs mapping to IAPEZ and L1MdGf particularly enriched in E14.5 PGCs (Figures S2K and S2L). These small RNAs had the defining properties of endosiRNAs (Figures S2M–S2O), suggesting that a similar response to the one we discovered in ESCs exists during global demethylation in the germline in vivo.
Key RNAi Components Are Involved in the Repression of Specific TE Classes
To investigate whether the observed endosiRNAs were involved in restraining TE expression, we knocked down key components of the endosiRNA and miRNA pathways in Dnmt1 cKO and monitored IAP expression by quantitative real-time PCR. Upon knockdown of Dicer or Ago2, both essential components of the RNAi pathway, IAP transcription was strongly upregulated, while knockdown of Dgcr8 (dispensable for endosiRNA function) had no effect on IAP expression (Figure 3A). This suggests that TEs are controlled by functional endosiRNAs.
To examine the role of the RNAi pathway during global hypomethylation in more detail, we generated conditional Dicer/Dnmt1 cDKO (conditional double-knockout) ESCs (Figure S3A) and carried out a number of quality controls. Loss of Dicer activity was confirmed by loss of expression of mmu-miR-93, while Dicer-independent small nucleolar RNAs (snoRNAs) were still expressed (Figure S3A). We generated total RNA-seq data from Dicer/Dnmt1 cDKO ESCs and found increased antisense transcripts in these cells, as seen earlier in the Dnmt1 cKO ESCs (Figure S3B). Furthermore, small RNA-seq of Dicer/Dnmt1 cDKO ESCs showed a depletion of all miRNAs (Figure S3C) and a loss of 21–24 nt small RNAs mapping to all TEs as well as specifically to L1MdGf and IAPEz (Figures 3B and S3D), which proves that the described small RNAs are DICER-dependent products.
Acute conditional deletion of both Dicer and Dnmt1 together resulted in significantly higher levels of transcription of IAPs by day 10 in comparison to those in Dnmt1 cKO ESCs (Figure 3C). Importantly, there was no notable resilencing of IAP transcripts in Dicer/Dnmt1 cDKO. This demonstrates that DICER plays a role in the re-repression of IAPs upon global hypomethylation. LINEs and major satellites (non-TE pericentric repeats), while not upregulated upon Dnmt1 deletion, were also dramatically upregulated following Dicer deletion (Figure 3C). Dicer/Dnmt1 cDKO ESCs started to show signs of cell death from day 12 after deletion, potentially as a result of TE mobilization, as has been shown in constitutive Dicer KO (Bodak et al., 2017).
We next asked whether deletion of RNAi components downstream of DICER would lead to a similar response and generated conditional Ago2/Dnmt1 cDKO ESCs (Figure S3E). While we initially expected that Ago2/Dnmt1 cDKO might show comparable results to the Dicer/Dnmt1 cDKO, we found that the deletion kinetics of Ago2 KO were substantially slower than those of Dicer KO (Figures S3F and S3G). Surprisingly, however, we found that transcriptional upregulation of TEs in the Ago2/Dnmt1 cDKO was considerably blunted (Figure 3D).
To gain deeper insights into the blunted TE expression, we constitutively deleted Ago2 or Dicer using CRISPR/Cas9 genome editing in the background of Dnmt1 cKO ESCs (Figures S3H–S3J). We first determined the effect of Dicer KO on genic and transposon transcription and were able to identify TEs that were solely dependent on DICER for their silencing, such as L1MdGf (Figures 3E and S3K–S3O).
We next performed a time course of Dnmt1 deletion in Ago2 KO/Dnmt1 cKO and in Dicer KO/Dnmt1 cKO and measured IAP expression by quantitative real-time PCR. Notably, we found substantially attenuated upregulation of IAPs upon Dnmt1 deletion in both ESC lines, which was confirmed by total RNA-seq (Figures 3D and S3O). These results indicate that, in addition to DNA methylation and RNAi, alternative TE silencing mechanisms can be recruited. While DICER-dependent mechanisms restrict the expression of specific TE classes upon deletion of Dnmt1, ablation of the RNAi pathway prior to demethylation triggers the engagement of another silencing mechanism. Since repressive histone marks have been shown to contribute to TE repression in somatic tissues and in ESCs (Karimi et al., 2011, Maksakova et al., 2006, Walter et al., 2016), we asked whether these might constitute the additional repressive mechanism observed here.
TE Silencing by Repressive Histone Marks
To study the involvement of chromatin in TE regulation upon global hypomethylation, we carried out ChIP-seq analyses of the repressive histone marks H3K9me2, H3K9me3, and H3K27me3 at 4 and 8 days after deletion of Dnmt1, i.e., before and after transcriptional upregulation of the relevant TE classes. Genome-wide distribution of the repressive histone marks—H3K27me3, H3K9me2, and H3K9me3—confirmed earlier studies (Iurlaro et al., 2017, Tang et al., 2016) with H3K27me3 enrichment in gene bodies and H3K9me2/3 enrichment in TEs (Figure S4A). Additionally, H3K27me3 was enriched in promoter regions but depleted at transcription start sites (TSSs) (Figures S4B and S4C). Upon Dnmt1 deletion, neither of these repressive histone marks were redistributed genome-wide (Figure S4D).
However, DICER-independent MERVLs showed increased H3K27me3 deposition upon Dnmt1 deletion, recapitulating what has been reported in naive hypomethylated ESCs (Walter et al., 2016) (Figure 4A). We found H3K9me3 enrichment across IAPs independent of DNA methylation levels, confirming previous results (Figures S4E and S4F) (Walter et al., 2016, Sharif et al., 2016). Importantly, H3K27me3 and H3K9me2 deposition was found on IAPs 9 days after Dnmt1 deletion, explaining why early, but not late, depletion of Dicer or Ago2 results in sustained TE expression. These results show that two repressive pathways are in place to control TE expression in ESCs (Figure S4I), and importantly, that they are staggered in time, with an “immediate” RNAi response and a subsequent “chronic” chromatin response.
Figure 4.

Repressive Histone Modifications Enriched at TEs upon Global Demethylation
(A) Heatmap showing relative enrichment (Z score) of repressive histone marks (H3K9me3, H3K27me3, and H3K9me2) at TE classes differentially regulated upon both Dicer KO (Figure 3A) and Dnmt1 cKO (Figure 1C) and normalized to enrichment in WT ESCs upon acute deletion of Dnmt1.
(B) H3K27me3, H3K9me3, and H3K9me2 enrichment over TEs dependent on Dicer and Dnmt1. Heatmap depicts ChIP-seq data of H3K27me3 mapped to TE families at depicted days after Dnmt1 cKO, Dicer KO, and Dnmt1/Dicer cDKO in comparison to WT ESCs.
(C) Schematic of the two levels of TE control upon global demethylation. Upon deletion of Dnmt1, DNA methylation (5mC; orange)-mediated repression is lost, and transposon expression increases (as an example, IAP expression is shown in green). Subsequently, small RNAs (red; “immediate” response) and repressive histone marks (chromatin, blue; “chronic” response) establish a new repressive environment. Also indicated are the time points at which the different experimental manipulations interfere with the system.
To obtain insights into the attenuated IAP expression in Dicer KO/Dnmt1 cKO, we performed ChIP-seq of the same repressive histone marks. While we did not observe a genome-wide redistribution of H3K27me3, H3K9me2, and H3K9me3 in the Dicer KO or the Dicer KO/Dnmt1 cKO (Figures S4G and S4H), we observed a clear redistribution of repressive histone marks over TEs in Dicer KO and in particular an enrichment of H3K27me3 and of H3K9me2 at IAPs. This was even further increased upon Dnmt1 deletion (Figure 4B). Hence, acute deletion of Dicer during global demethylation abrogates re-silencing of IAPs while constitutive deletion of Dicer instigates a repressive chromatin response in IAPs that suppresses reactivation upon hypomethylation (Figure 4C).
Discussion
How TEs are controlled during global epigenetic reprogramming in the mammalian germline is a highly relevant question. The present study provides, to our knowledge, the first evidence of AGO2-bound endosiRNAs in ESCs during global DNA hypomethylation, which restrict TE expression as judged by acute depletion of Dicer or Ago2. As we also detect DICER-dependent endosiRNAs in PGCs, it is likely that the described mechanism also operates in vivo. This mechanism constitutes a first line of TE defense during epigenetic reprogramming. A second line of defense is provided by chromatin targeting and retargeting, presumably through the evolution of sequence-specific recognition modules of TEs such as zinc-finger proteins (Rowe and Trono, 2011). Our work also indicates a link between these systems; they are staggered in time and thus potentially connected.
Many TE families are associated with transcribed genes or lncRNAs in ESCs (Kelley and Rinn, 2012). This provides the potential for sense/antisense transcription to occur when TEs become demethylated, as observed here (Figure 1F). In oocytes, pseudogenes provide the antisense strand to TEs to feed into an RNAi pathway (Tam et al., 2008) and TEs have been shown to give rise to dsRNA in preimplantation embryos due to their bidirectional promoters (Svoboda et al., 2004). Indeed, we found intragenic active TEs preferentially integrated in antisense direction to the gene (Figure S1K). Previous studies had concluded that this could prevent disruption of normal gene expression (van de Lagemaat et al., 2006). We suggest an additional reason why this direction of insertion is evolutionarily favored: it produces a trapping system (“trap”) for transposon activation during epigenetic reprogramming, in order to tame newly invading TEs (Figure 2A).
Overlapping sense/antisense transcription feeds into an endosiRNA pathway regulated by DICER and AGO2 to silence TEs. The generation of the two constitutive and conditional KO ESCs in the background of the Dnmt1 cKO allowed us to dissect the dynamics of TE control during global hypomethylation, revealing an “immediate” response that is characterized by endosiRNAs and affected by acute depletion of Dicer or Ago2. This is followed by a “chronic” response, which is defined by targeting of repressive histone modifications (particularly H3K27me3 and H3K9me2) and occurs subsequent to the endosiRNA response in Dnmt1 cKO and Dnmt1/Dicer cDKO ESCs (Figure 4C). Intriguingly, non-acute depletion of Dicer also instigates deposition of H3K27me3 and H3K9me2 independently of DNA demethylation, suggesting that the two systems are linked. We suggest a mechanism of TE control by which the “immediate” endosiRNA response to global methylation erasure is followed by a “chronic” repressive chromatin response. Interestingly, the “chronic” response is initiated by deletion of Dnmt1 as well as by abrogation of the “immediate” defense. Therefore, the “immediate” and “chronic” responses are not only staggered in time, but also appear mechanistically linked. Unravelling the molecular underpinnings of this link will be an important topic of future work.
The specific response of IAPs and LINEs to loss of DICER may be explained by the fact that they embody the most active retrotransposition competent TE copies in the mouse germline (Maksakova et al., 2006) and are primarily guarded by endosiRNAs, with chromatin playing a secondary role in their transcriptional restriction. Other TEs, in contrast, are primarily controlled by chromatin redistribution upon global demethylation. The present study highlights the exquisite variety and interplay of epigenetic modifications by which the transcription of different TE families is regulated. Future work in this area, particularly with high-coverage long-read sequencing, will hopefully allow the characterization of transcriptional and epigenetic regulation of individual TE copies in the genome.
We identified DICER as an important factor in small RNA-dependent silencing of TEs. Nonetheless, DICER-independent AGO2-bound small RNAs may also play a role in TE silencing (Babiarz et al., 2008, Murchison et al., 2005). DICER-independent small RNAs might also explain the repression of ETns, to which increasing amounts of AGO2-bound small RNAs mapped, but which were not responsive to Dicer KO.
TEs benefit from transcriptional activation in the germline, but not in somatic cells (Haig, 2016). Hence, one might speculate that they may regulate aspects of epigenetic reprogramming in germ cells to their benefit. In this respect, TEs may not be the sole benefactors of their own mobilization, but it also impacts the creation of novelty in the host genome. Nevertheless, unrestrained activation and transposition would presumably be detrimental to the host genome, and hence a sophisticated balance of regulatory mechanisms for TEs has evolved in the germline, including the chromatin retargeting and the endosiRNA pathway we report here.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-CD4 microbead | Miltenyl Biotec | Cat #: 130-045-101 |
| Alexa Fluor 647, goat anti-mouse IgG antibody | Thermo Fisher Scientific | Cat# A-21236; RRID: AB_141725 |
| Alexa Fluor 568 donkey anti - rabbit IgG antibody | Thermo Fisher Scientific | Cat# A10042; RRID: AB_2534017 |
| Rabbit Anti-Nanog Polyclonal Antibody, Unconjugated | Abcam | Cat# ab80892; RRID: AB_2150114 |
| AGO2 antibody | Dr. O’Carrolls lab | N/A |
| Histone H3K9me3 antibody | Active Motif | Cat #: 61013; RRID: AB_2687870 |
| H3K27me3-mouse antibody | Active Motif | Cat #: 39155; RRID: AB_2561020 |
| Histone H3K9me2 antibody | Abcam | Cat #: ab1220; RRID: AB_449854 |
| Bacterial and Virus Strains | ||
| E.coli: One Shot TOP10 chemically competent cells | Thermo Fisher Scientific | Cat #: K450001 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tamoxifen | Sigma-Aldrich | Cat #: T5648-1G |
| mouse LIF | Stem Cell Institute, Cambridge | N/A |
| Na/Deoxycholate | Sigma-Aldrich | Cat #: D6750-10G |
| N-lauroylsarcosine | Sigma-Aldrich | Cat #: 61739-5G |
| Vanadyl ribonucleoside complex | New England Biolabs | Cat #: S1402S |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat #: 11668027 |
| Protein G-coupled Dynabeads | Thermo Fisher Scientific | Cat #: 10003D |
| HiFi Uracil+ ReadyMix | KAPABiosystems | Cat #: KK2801 |
| T4 RNA Ligase 2, truncated | New England Biolabs | Cat #: M0242S |
| Tri-Reagent | Sigma-Aldrich | Cat #: T9424-200ML |
| Phenol/chloroform/isoamylalcohol (25:24:1) | Life Technologies | Cat #: 15593031 |
| Triton X-100 | Sigma-Aldrich | Cat #: RES9690T |
| Dimethylsulfoxide (DMSO) | Thermo Fisher Scientific | Cat #: TS-20684 |
| Ampicillin | Sigma-Aldrich | Cat #: A9518-5G |
| Penicillin/Streptomycin | Thermo Fisher Scientific | Cat #: 15140122 |
| L-glutamine | Thermo Fisher Scientific | Cat #: 25030081 |
| Non-essential amino acids | Thermo Fisher Scientific | Cat #: 11140050 |
| 2-Mercaptoethanol (50mM) | Life technologies | Cat #: 31350-010 |
| RNase A | Thermo Fisher Scientific | Cat #: EN0531 |
| cOmplete Protease Inhibitor Cocktail | Sigma-Aldrich | Cat #: 00000001169 7498001 |
| Proteinase K | Thermo Fisher Scientific | Cat #: EO0491 |
| Paraformaldehyde 16% Solution | Agar Scientific | Cat #: AGR1026 |
| Gelatine | Sigma-Aldrich | Cat #: G9391 |
| DTT | Sigma-Aldrich | Cat #: D0632-1G |
| Fetal Bovine Serum (FBS) | Stem Cell Institute, Cambridge | N/A |
| DMEM (High Glucose) w/L-Glutamine andamp; Na Pyr | Life Technologies | Cat #: 41966-052 |
| NEBuffer 2 | New England Biolabs | Cat #: B7002S |
| Trypsin EDTA (1x) 100ml | Life technologies | Cat #: 25300-054 |
| HyperLadder 1kb, 100bp | Bioline | Cat #: BIO-33053, BIO-33029 |
| SYBR Safe | Invitrogen | Cat #: S33102 |
| SYBR Gold | Life Technologies | Cat #: S11494 |
| PvuI | New England Biolabs | Cat #: R0150S |
| EcoRI HF | New England Biolabs | Cat #: R3101L |
| T4 Polynucleotide Kinase | New England Biolabs | Cat #: M0201L |
| T4 Ligase | New England Biolabs | Cat #: M0202T |
| Ampure XP beads | Beckman Coulter | Cat #: A63880 |
| T5 Exonuclease | New England Biolabs | Cat #: M0363S |
| Exonuclease I | New England Biolabs | Cat #: M0293S |
| Klenow exo- | New England Biolabs | Cat #: M0212L |
| Glycoblue | Ambion | Cat #: AM9516 |
| Optimem | GIBCO | Cat #: 31985062 |
| DAPI | Thermo Fisher Scientific | Cat #: 62248 |
| MyTaq Redmix | Bioline | Cat #: BIO-25043 |
| Orange G dye | Sigma-Aldrich | Cat #: 861286-25G |
| Critical Commercial Assays | ||
| TruSeq Small RNA Library Prep Kit -Set A (24 rxns) (Set A-c: indexes 1-36) | Illumina | Cat #:RS-200-0012, RS-200-0024, RS-200-0036 |
| NEBNext DNA Library Prep Master Mix Set for Illumina | New England Biolabs | Cat #: E6040S |
| Imprint DNA Modification Kit | Sigma-Aldrich | Cat #: MOD50-1KT |
| TruSeq RNA library preparation kit v2 | Illumina | Cat #: RS-122-2001 |
| MicroPlex Library Preparation kit | Diagenode | Cat #: C05010012 |
| SmallRNA qRTPCR miRNA kit: mmu_miR93 | Taqman | Cat #: TM001090 |
| SmallRNA qRTPCR miRNA kit: mmu_miR7081_mat | Taqman | Cat #: TM467052_mat |
| SmallRNA qRTPCR miRNA kit: snoRNA202 | Taqman | Cat #: 001232 |
| Dharmacon siGENOME SMARTpool, mouse Dicer | Dharmacon | Cat #: MU-040892-01-0005 |
| Dharmacon siGENOME SMARTpool, mouse Dgcr8 | Dharmacon | Cat #: MU-051365-00-0002 |
| Dharmacon siGENOME SMARTpool, mouse Ago2 | Dharmacon | Cat #: MU-058989-01-0005 |
| Dharmacon siGENOME SMARTpool, mouse Dicer | Dharmacon | Cat #: D-001210-02-05 |
| Miniprep kit | QIAGEN | Cat #: 27106 |
| Gel extraction kit | GeneJET | Cat #: K0691 |
| PCR Purification kit | GeneJET | Cat #: K0701 |
| Qiaamp DNA micro kit | QIAGEN | Cat #: 56304 |
| TURBO DNA-free kit | Life Technologies | Cat #: AM1907 |
| Quant-iT PicoGreen dsDNA Assay kit | Life Technologies | Cat #: P11496 |
| Platinum SYBR Green qPCR SuperMix-UDG w/ROX | Life Technologies | Cat #: 11744100 |
| QuickExtract | Epicenter | Cat #: QE09050 |
| Kapa Library Quantification kit | Kapa Biosystems | Cat #: KK4847 |
| High Sensitivity DNA kit | Agilent | Cat #: 5067-4626 |
| High Sensitivity total RNA kit | Agilent | Cat #: 5067-1513 |
| Deposited Data | ||
| Raw and analyzed data | This study | GEO: GSE89698 |
| Mouse reference genome NCBI build 37, NCBIM37 | Mouse Genome Sequencing Consortium | http://may2012.archive.ensembl.org/Mus_musculus/Info/Index |
| Mouse repeats | repeatmasker v4.0.3, library version 20130422 | http://www.repeatmasker.org/ |
| Mouse ESCs enhancer annotation track | Chen et al., 2012, Creyghton et al., 2010 | N/A |
| CpG island promoters | Illingworth and Bird, 2009 | N/A |
| Promoters: regions −1kb to the transcription start site | Ensemble, NCBIM37 version 67 | N/A |
| Experimental Models: Cell Lines | ||
| Dnmt1 cKO: Passage 12 Dnmt1loxP/loxP (C57BL/6) ESCs | Sharif et al., 2016 | N/A |
| Dicer/Dnmt1 cDKO: Passage 21 Dicer loxP/loxP/Dnmt1 loxP/loxP ESCs | This study | See STAR Methods section CRISPR cKO and KO |
| Ago2/Dnmt1 cDKO: Passage 21 Ago2 loxP/loxP /Dnmt1 loxP/loxP ES cells | This study | See STAR Methods section CRISPR cKO and KO |
| Dicer KO: Passage 17 Dicer KO/Dnmt1loxP/loxP ES cells | This study | See STAR Methods section CRISPR cKO and KO |
| Ago2 KO: Passage 17 Ago2 KO/Dnmt1loxP/loxP ES cells | This study | See STAR Methods section CRISPR cKO and KO |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J female mice carrying the Oct4-GFP transgene in the developing gonad: B6.Cg-Tg(GOF18/EGFP)11Ymat/Rbrc | Yoshimizu et al., 1999 | RRID: IMSR_RBRC00868 |
| Oligonucleotides | ||
| Primers for CRISPR clone generation, see Table S3 | This paper | N/A |
| Primers for RTqPCR clone generation, see Table S2 | This paper | N/A |
| Recombinant DNA | ||
| Cas9 plasmid: pSpCas9(BB)-2A-GFP | Ran et al., 2013 | Addgene Plasmid #48138 |
| pSpCas9(BB)-2A-hCD4 | This study | N/A |
| Software and Algorithms | ||
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Bismark | Krueger and Andrews, 2011 | https://www.bioinformatics.babraham.ac.uk/projects/bismark/, version 0.14.4 |
| TopHat | Trapnell et al., 2009 | http://ccb.jhu.edu/software/tophat/index.shtml |
| piPipes | Han et al., 2015 | https://github.com/bowhan/piPipes/wiki |
| Trim Galore | N/A | http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/, Version 0.4.1 |
| SeqMonk software | N/A | http://www.bioinformatics.babraham.ac.uk/projects/seqmonk/ |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html, version 3.5 |
| Transposon analysis | this study | STAR Methods section Transposon analysis |
| R | Data analysis | https://www.r-project.org/, version 3.2.5 |
| Adobe Illustrator | Figures | http://www.adobe.com/de/products/illustrator.html, version CC 2015.3 |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Rebecca Berrens (rebecca.berrens@gmail.com). The AGO2 antibody was obtained from EMBL, after establishing an MTA with the laboratory of Prof. Donal O’Carroll at University of Edinburgh.
Experimental Model and Subject Details
Cell lines
Mouse embryonic stem cell (ESC) lines were used in this study. Dnmt1loxP/loxP ESCs (strain C57BL/6) were obtained from Haruhiko Koseki, RIKEN Center for Integrative Medical Sciences, Yokohama City, Japan (Sharif et al., 2016). Dicer/Dnmt1 cDKO, Ago2/Dnmt1 cDKO, Dicer KO and Ago2 KO ESC lines were generated using Dnmt1loxP/loxP ESCs using the CRISPR/Cas9 targeting and screening primers mentioned in Table S3.
Mice
All in vivo PGC samples were collected from timed matings of C57Bl/6J male with C57BL/6J female mice carrying the Oct4-GFP transgene expressed in the developing gonad (Yoshimizu et al., 1999). Primordial germ cells from male and female embryos at E13.5 and E14.5 were collected. All procedures were covered by a project license (to WR) under the Animal (Scientific Procedures) Act 1986, and are locally regulated by the Babraham Institute Animal Welfare, Experimentation, and Ethics Committee.
Method Details
DNA/RNA Extraction
Genomic DNA was prepared using QIAmp DNA Micro Kit (QIAGEN). RNA was extracted using TriReagent (Sigma) and Phase Lock tubes (5Prime) following manufacturers’ instructions and subjected to DNase treatment using the DNA-free kit (Ambion DNA-free DNA Cat #1311027) according to the manufacturers’ instructions.
Small RNA Quantitative Real-Time PCR
For small RNA qPCR Taqman miRNA kits were used according to the manufacturer’s’ instructions for mmu_miR93 (Taqman, Cat. No. TM001090), mmu_miR7081_mat (Taqman, Cat. No. TM467052_mat) and snoRNA202 (Taqman, Cat. No. 001232) was used as a positive control. Quantitative real-time PCR primers are listed in Table S2.
AGO2 IP
ESCs were cultured on 15 cm dishes and harvested in 1 x PBS. Pellets were frozen at −80°C until further processing. ESC were resuspended in 300 μl Lysis buffer (50 mM Tris, pH8, 150 mM NaCl, 5 mM MgCl2, 15% Glycerol, 1 mM DTT, 0.5% Sodium deoxycholate, 0.5% Triton X-100, Protease inhibitor cocktail (Roche), 50μg/ml yeast tRNA, 2mM Vanadyl ribonucleoside complex) and cells were pelleted at 10,000 rpm, 10 min, 4°C. The supernatant was used as whole ESC extract. 25 μL beads (protein G Sepharose) were washed 3 times with 1 mL of Wash Buffer (10 mM Tris pH 8, 150 mM NaCl, 1 mM MgCl2, 0,01% NP-40). 50 μl of purified AGO2 antibody (O’Carroll lab) was added, filled up to 1mL with Wash Buffer and incubated O/N at 4°C in a rotating wheel. On the next day, the beads were washed 3 times with Wash Buffer and the negative control (beads with extract but without serum) was prepared. The ESC extract was pre-spun to remove precipitated proteins and 200μL extract was added to the beads and filled up to 600μL with Lysis buffer. The mix was incubated for 2-4h at 4°C in a rotating wheel and subsequently washed 5 times with wash buffer and the IP was eluted with 300μL Proteinase K buffer (10 mM Tris pH 7,5, 0,5% SDS, 5 mM EDTA, 1 μL Proteinase K/reaction) after 30 min for 50°C incubation on the thermomixer, at 850 rpm. RNA was isolated by phenol extraction and eluted in 8 μl H2O.
RNAi knockdown of Ago2, Dicer1, Dgcr8 in Dnmt1fl/fl ES cells
RNA interference experiments were performed according to manufacturers’ instructions with modifications. Transfections of Dharmacon siGENOME SMARTpool siRNA against mouse Dicer (Dharmacon, Cat. No. MU-040892-01-0005), Dgcr8 (Dharmacon, Cat. No. MU-051365-00-0002) or Ago2 (Dharmacon, Cat. No. MU-058989-01-0005) and siGENOME non-targeting siRNA#2 (Dharmacon, Cat. No. D-001210-02-05) were performed with Lipofectamine 2000 according to the manufacturers’ instructions. The transfection was done in two rounds. The cells were plated at a density of 1 × 10ˆ5 ES cells per well of gelatinized 12-well plate. One day later the first transfection was done the following for each well of a 12 well plate: 50uM siRNA were added to 100 μl DMEM. 6 μl of Lipofectamin2000 were mixed with 100 μl DMEM. The mix was incubated for 5 min at room temperature. Afterward the two solutions were mixed and incubated at room temperature for 15 min. 200 μl of the siRNA and Lipofectamin2000 mix were added to each well of a 12 well plate. On the third day the medium was changed. On the fourth day the second transfection was done the following: 125uM siRNA were added to 250 μl DMEM. 7.5 μl of Lipofectamin2000 were added to 250 μl DMEM and incubated at room temperature for 5 min. The solutions were then mixed and again incubated for 15 min at room temperature. The cells were washed with PBS, trypsinized, inactivated and resuspended in ESC medium and plated on a gelatinized 6-well plate I a total volume of 1.8 mL each well. 500μl of siRNA and Lipofectamin2000 were added to each well. The ESCs were incubated at 37C for 6 hours and then the medium was changed.
Cells were harvested 48 h after the 2nd transfection and RNA was extracted and analyzed.
Quantitative Real-Time PCR
100 ng −1 μg of DNase treated RNA was reverse transcribed (Thermo RevertAid #K1622) using random hexamer primers. Endogenous controls (Atp5b, Hspcb, U1) were used to normalize expression. Primers are listed in Table S2.
CRISPR cKO and KO
guideRNAs (gRNAs) were constructed following https://chopchop.rc.fas.harvard.edu/ and http://crispr.mit.edu/ and cloned following the protocol by Ran et al. (2013) into pSpCas9(BB)-2A-GFP (Addgene plasmid ID: 48138) or pSpCas9(BB)-2A-hCD4, constructed by replacing the GFP in the pSpCas9(BB)-2A-GFP with human CD4. Cells were cultured on feeder plates and transfected with 1 μg gRNA and 100 ng donor DNA, where appropriate, using Lipofectamine 2000 transfection reagent. Cells were sorted for GFP in single cell colonies into 96 well plates using flow cytometry or CD4 expression plating on 10cm dishes as single cell colonies. Colonies were screened by PCR using MyTaq (Bioline, BIO-25044) and Sanger sequencing. See Figure S3 for knock out strategy and Table S3 for gRNAs, screening primers and donor DNA sequence.
Fluorescence-activated cell sorting (FACS)
Cells were trypsinized and resuspended in PBS + 1% FBS and analyzed on a LSR Fortessa Cell Analyzer (BD). Cells were gated for singlets and living cells were identified using the level of DAPI incorporation and the level of GFP signal was recorded for each cell.
CD4 pull down
Cells were trypsinized and resuspended in 70 μl 1 x PBS and stained with human CD4 Microbead antibody (Miltenyl Biotec, Cat. No. 130-045-101) according to manufacturers’ instructions. The CD4 positive cells were enriched using MACS columns. Negative cells were collected from flow through. The cells were eluted in 500 μl 1 x PBS.
In vivo PGC collection
All embryonic samples for library preparation were collected from timed mattings of C57BL/6J female mice PGCs that express the Oct4-GFP transgene in the developing gonad (Yoshimizu et al., 1999). E13.5 and E14.5 PGCs, male and female samples were collected separately and after collagenase digestion PGC samples were sorted for GFP positive cells using a FACSAria (BD) cell sorter with > 98% purity.
Cell lines and culture conditions
Mouse ESCs were cultured with or without feeders on gelatinized plates (0.1% gelatin) in serum-containing media (DMEM 4,500 mg/l glucose, 4 mM L-glutamine, 15% fetal bovine serum, 1 U/ml penicillin, 1 μg/ml streptomycin, 0.1 mM nonessential amino acids, 50 μM β-mercaptoethanol) supplemented with mouse LIF at 37°C and 5% CO2. Conditional deletion was induced by Cre mediated recombination, as described before (Sharif et al., 2016). Cre expression was induced in response to tamoxifen (4OHT, 800 nM).
WGBS-seq libraries
For preparation of WGBS-seq libraries, genomic DNA was sonicated using a Covaris Sonicator, followed by end-repair, A-tailing and methylated adaptor (Illumina) ligation using NEBNext reagents (E6040S, NEB). Afterward the libraries were bisulfite treated using Imprint DNA modification kit (MOD50-1KT, Sigma), followed by library amplification with indexed primers using KAPA HiFi Uracil HotStart DNA Polymerase (KAPA HiFi Uracil+, KK2801/2). Subsequently, the amplified libraries were purified and assessed for quality and quantity using High-Sensitivity DNA chips on an Agilent Bioanalyzer. High-throughput sequencing of all libraries was carried out with a 75 bp or 50 bp paired-end (PE) sequencing on Illumina HiSeq 2500 instruments using TruSeq reagents (Illumina, San Diego, CA, USA), according to manufacturers’ instructions.
ChIP-seq libraries
ESCs were grown on 15 cm dishes coated with 0.1% gelatine until they were 80% confluent. Subsequently cells were cross-linked with 1% methanol-free formaldehyde in fresh medium for 10 minutes. To quench the cross-linking, 0.2 M final concentration of glycine was added. ESCs were washed twice with ice cold 1 x PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4 dissolved in 800 mL distilled H2O, pH was adjusted to 7.4 with HCl) and harvested using a cell scraper. Cells were then pelleted by centrifugation at 8,000 x g at 4 ◦C for 3 min. Pellets were resuspended in LB1 buffer (50 mM HEPES’ KOH, pH 7.5; 140 mM NaCl; 1 mM EDTA; 10% glycerol; 0.5% NP-40; 0.25% Triton X-100, protease inhibitors) for 10 minutes at 4°C, pelleted and resuspended in LB2 buffer (10 mM Tris/HCl, pH 8.0; 200 mM NaCl; 1 mM EDTA; 0.5 mM EGTA, protease inhibitors) for 10 minutes at 4 ◦C. Cells were pelleted and resuspended in LB3 buffer (10 mM Tris-HCl, pH 8; 100 mM NaCl; 1 mM EDTA; 0.5 mM EGTA; 0.1% Na/Deoxycholate; 0.5% N-Lauroylsarcosine, protease inhibitors). Next the cells were sonicated using Misonix Sonicator 3000. Triton X-100 was added to a final concentration of 1% and the lysate was centrifuged at 20,000 x g for 10 min to pellet the debris. The bead-antibody complexes were prepared before adding the sonicated DNA. Protein G-coupled Dynabeads (Thermo Fisher Scientific, Cat. No. 10003D) and the primary antibodies in PBS with 5 mg/ml BSA were incubated ON. Subsequently, the bead-antibody complexes were added to the sonicated chromatin and both were incubated at 4 ◦C ON. On the following day, beads were washed extensively with RIPA buffer (50 mM HEPES pH 7.6, 1 mM EDTA, 0.7% Na deoxycholate, 1% NP-40, 0.5M LiCl), once with 1x TE bu er (1 M Tris-HCl (pH approximately 8.0), 0.1 M EDTA) and eluted in 200 μL of buffer containing 1% SDS and 0.1 M NaHCO3. They were then incubated at 65°C ON for reverse cross-linking. RNase A treatment at 37°C was carried out for 1 h, then Proteinase K treatment at 55°C for 2 h. The DNA was then extracted with phenol/chloroform, followed by ethanol precipitation. ChIP-seq library preparation was performed using MicroPlex Library Preparation kit (Diagenode) following manufacturer’s instructions. Libraries were quantified using the High Sensitivity DNA Bioanalyzer kit and Kapa library quantification. High-throughput sequencing of all libraries was carried out with a 100 bp PE sequencing on Illumina HiSeq 2500 instruments.
Small RNA-seq libraries
Small RNA-seq libraries were produced according to the Illumina protocol (RS-200-0012), with the following changes: 10 ng or 1 μg RNA (RIN of 8-10) were used as input material. The instructions were followed until the cDNA purification. In order to purify the cDNA, the samples were run on 10% Novex PAGE gel. The entire area between the 145 and 160 bp markers was excised, gel purified by addition of 0.3 M NaCl and the DNA was eluted from the gel by rotation over night at 4°C. The DNA was precipitated in EtOH overnight and the library was quantified using the HighSensitivity Bioanalyzer kit. The small RNA-seq libraries were additionally quantified by Kapa Library Quantification. The libraries were pooled according to their molecular weight. High-throughput sequencing of all libraries was carried out with a 50 bp SE on Miseq or SE and PE on Illumina HiSeq 2500 instruments.
Total RNA-seq libraries
Stranded Total RNaseq libraries were prepared according to manufacturers’ protocols using the Illumina stranded Total RNaseq library preparation after Ribo-zero depletion. High-throughput sequencing of all libraries was carried out with a 100 bp PE on Illumina HiSeq 2500 instruments.
Quantification and Statistical Analysis
WGBS-seq mapping and analysis
Raw sequence reads from WBGS libraries were trimmed to remove poor quality reads and adaptor contamination, using Trim Galore (v0.4.1, http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) with default parameters. The remaining sequences were mapped using Bismark (v0.14.4) (Krueger and Andrews, 2011) with default parameters to the mouse reference genome Ensembl v67 NCBIM37 in paired-end mode. Reads were then deduplicated and CpG methylation calls were extracted from the deduplicated mapping output using the Bismark methylation extractor (v0.14.4) in paired end mode. CpG methylation calls were analyzed using R and SeqMonk software (http://www.bioinformatics.babraham.ac.uk/projects/seqmonk/). The custom R scripts can be found in Data S1. Global CpG methylation levels of pooled replicates were calculated in windows of 50 CpGs with a coverage of at least 3, illustrated using bean plots. Methylation over a given genomic feature was calculated by averaging the individual methylation levels of CpGs covered by at least 3 reads and only features with at least 50 CpGs were used. Promoters were defined as the region −1 kb to the transcription start site as annotated in Ensembl NCBIM37 v67. For analysis of specific genome features these were defined as follows: Gene bodies (probes overlapping genes), CGI promoters (promoters containing a CGI) (Illingworth and Bird, 2009), non-CGI promoters (all other promoters).
RNA-seq mapping and analysis
RNA-seq sequences were trimmed using Trim Galore using default settings. Trimmed sequencing reads were aligned to mouse genome assembly NCBIM37 using TopHat (Trapnell et al., 2009) and reads with MAPQ scores < 20 were discarded. Mapped RNA-seq data were quantitated using the RNA-seq quantitation pipeline in SeqMonk software to generate log2 RPM (reads per million reads of library) expression values. Genes were considered to be differentially expressed if they were significantly different (p < 0.05 after Benjamini and Hochberg multiple testing correction) when analyzed with both DESeq2 and Intensity difference (SeqMonk) statistical tests.
Global pervasive transcription, was calculated as following: Genes with significant antisense expression were identified by initially counting both sense and antisense reads over all genes in the genome. A global expected antisense level was defined by the total proportion of antisense reads across all genes. Individual genes were considered to show significant antisense expression if they had a binomial p value < 0.05 following multiple testing correction (FDR) using the global antisense proportion as the expected success rate, the total reads for that gene as the trials and the total antisense reads for that gene as successes. Additionally, the raw antisense transcription counts for all samples was calculated and significant differential antisense expression was calculated using DESeq2 with an FDR < 0.05. The overlap of the two quantifications was used to define pervasive transcription, and the difference in antisense transcription between WT and KO samples at each time point was plotted using R.
ChIP-seq mapping and analysis
ChIP-seq sequencing data was trimmed to remove poor quality reads, adaptor and barcodes sequences using Trim Galore. Trimmed data were mapped using Bowtie2 (Langmead and Salzberg, 2012) against the mouse reference genome Ensembl v67 NCBIM37 and reads with a MAPQ value < 20 were discarded. Mapped ChIP-seq data were quantitated creating 1kb tiles of the whole genome and calculating the log2 observed/expected value comparing the observed read count with the expected count had all reads been uniformly distributed over the genome.
Small RNA-seq mapping and analysis
For small RNA-seq data analysis trimmed sequencing reads were filtered to 20-24nt length and mapped to the mouse NCBIM37 genome assembly using Bowtie2. Raw overlap counts for each small RNA molecule were quantitated using SeqMonk. Graphing and statistics was performed using Excel or R. For consensus sequence mapping the piPipes small RNA pipeline was used (https://github.com/bowhan/piPipes) (Han et al., 2015). IAPEZ consensus sequences were used from repeatmasker libraries (repeatmasker v4.0.3, library version 20130422). Additionally, the small RNA-seq data processing was performed using the freely available piRNA pipeline piPipes. For repeat mapping, trimmed data were mapped using Bowtie2 against repeats as defined in the analysis by using the mouse repeatmasker annotation. The plots shown were generated as described below: The distribution of small RNAs was computed by mapping all small RNA-seq reads to the individual genomic features. The length distribution was calculated taking all uniquely mapped small RNAs into account, excluding small RNA-seq mapping to ribosomal RNAs (rRNAs). For all subsequent analysis, small RNA reads were pre-filtered as follows: reads mapping to rRNAs and miRNAs were excluded, then reads aligning to the repeat masked mm9 genome (all annotated repeats were masked/replaced by Ns) were removed, too. The remaining small RNAs reads were mapped to the mouse repeatmasker annotation. The 5′ end nucleotide composition was computed from the uniquely mapped small RNAs. Similarly, analysis of the position of 5′ to 5′ overlap was performed on the mapped small RNAs reads and the length distribution and strand orientation of small RNAs shown was generated using uniquely mapped small RNA reads.
Transposon analysis
Repeat locations for a pre-defined set of repeat classes of interest were extracted from the pre-masked repeatmasker 4.0.3-20130422 library in the mm9 genome. Repeat instances within 2 kb of an annotated gene in the Ensembl v67 NCBIM37 gene set were removed to avoid mixing signals from genic expression with specific expression of repetitive sequences. RNA-seq data were processed and mapped as described above (RNA-seq mapping and analysis). We set a standard outlier filtering approach with a cutoff of counts > 3. Overlaps were quantitated between the mapped RNA-seq reads and the repeat instances. This allowed an unbiased identification of TEs depending on Dnmt1 KO as well as Dicer KO, which we followed throughout this manuscript. Summed counts for all instances of each class of repeat were calculated and these were corrected for both the total length of all TEs and the size of the individual libraries to generate log2 RPM expression values. The matrix of expression values and samples were plotted using the R pheatmap library allowing the repeat classes to cluster using default parameters. WGBS-seq libraries were processed and mapped as described above (WGBS-seq mapping and analysis). Methylation levels at the repeat instances were quantitated by summing up all methylation calls and non-methylation calls for all instances of each class of repeat and calculating the percentage of methylated Cs over all Cs. Only TEs with at least 1000 observations in all samples were used for the analysis and calculation of percentage methylation. For major satellite methylation analysis Bismark (Krueger and Andrews, 2011) was used to map all reads against the mouse major satellite consensus sequence (GSAT from repeatmasker) and the methylation calls from these results were analyzed directly. The custom R scripts can be found in Data S1.
Statistics
Statistical values including the exact number of replicates (n), the definition of standard deviation and statistical significance are reported in the Figure Legends.
WGBS-seq
For statistical analysis WGBS-seq of Figures 1B and S1 of WT versus Dnmt1 KO data we used the Wilcoxon rank sum test with Bonferroni correction testing with a p value threshold of < 0.05. The code of the analysis of the retained methylation over TEs can be found in Data S1.
Total RNA-seq
To call differentially expressed mRNAs, we applied the SeqMonk intensity difference filter with Benjamini and Hochberg correction for multiple testing with a p value threshold of < 0.05 and overlapped them with the genes called differentially expressed by DESeq2 with a p value threshold of < 0.05 and multiple testing correction.
For TE analysis we only considered significantly differentially expressed TEs p < 0.05 of Dnmt1 KO over WT samples into account. The code of the analysis can be found in Data S1.
small RNA-seq
To call differentially expressed miRNAs we overlapped the differentially expressed miRNAs using DESeq2 with multiple testing correction and SeqMonk intensity difference filter with Benjamini and Hochberg correction with a p value of < 0.05.
To call differential amount of mapped small RNAs to TEs we used Students t test to compare day 8 to day 0 enrichment of small RNAs with a p value of < 0.05.
ChIP-seq
As we only have data from one measurement we could not call significant differences of histone modification enrichment but show TEs which have at least 2 times higher enrichment in Dnmt1 KO versus WT samples. The code of the analysis can be found in Data S1.
Quantitative Real-Time PCR
Each quantitative real-time PCR was done with 3 technical replicates. Differences between conditions that are statistically significant are denoted by ∗p value < 0.05, ∗∗p value < 0.005 using the standard distributed two tailed t test.
siRNA knock-down
Every siRNA knock-down was done in 3 technical replicates. Differences between conditions that are statistically significant are denoted by ∗p value < 0.05, ∗∗ p value < 0.005 using the standard distributed two tailed t test.
Data and Software Availability
The accession number for the next-generation-sequencing data reported in this study is GEO: GSE89698. The software of this study can be found in Data S1.
Author Contributions
R.V.B. conceived and designed the study, performed experiments, analyzed data, and wrote the paper; S.A. analyzed data; D.S., W.D., P.G., J.S., and F.S. performed experiments; N.O. and T.C. helped to design the project; J.S. and H.K. generated original conditional Dnmt1 knockout ESCs; F.v.M. designed and supervised the study and wrote the paper; and W.R. conceived, designed, and supervised the study and wrote the paper.
Acknowledgments
We thank all members of the Reik lab for helpful discussions, Felix Krueger for bioinformatics support, the sequencing facilities at Babraham Institute (BI) and Sanger Institute, and the flow cytometry facility at BI for support. We thank Jon Houseley, Andrea Schorn, and Rob Martienssen for helpful discussions, and Dónal O’Carroll for providing the AGO2 antibody and sharing the AGO2 IP protocol. F.v.M. was supported by the Swiss National Science Foundation. R.V.B. is funded by the Gates Cambridge Trust. W.R. is supported by the BBSRC (BB/K010867/1), Wellcome Trust (095645/Z/11/Z), EU BLUEPRINT, and EpiGeneSys. W.R. is a consultant and shareholder of CEGX.
Published: November 2, 2017
Footnotes
Supplemental Information includes four figures, three tables, and one data file and can be found with this article online at https://doi.org/10.1016/j.stem.2017.10.004.
Contributor Information
Rebecca V. Berrens, Email: rebecca.berrens@gmail.com.
Ferdinand von Meyenn, Email: vonmeyenn@babraham.ac.uk.
Wolf Reik, Email: wolf.reik@babraham.ac.uk.
Supplemental Information
Differentially expressed genes were called using the overlap between the SeqMonk Intensity difference as well as DESeq2.
References
- Aravin A.A., Sachidanandam R., Bourc’his D., Schaefer C., Pezic D., Toth K.F., Bestor T., Hannon G.J. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol. Cell. 2008;31:785–799. doi: 10.1016/j.molcel.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiarz J.E., Ruby J.G., Wang Y., Bartel D.P., Blelloch R. Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes Dev. 2008;22:2773–2785. doi: 10.1101/gad.1705308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodak M., Cirera-Salinas D., Yu J., Ngondo R.P., Ciaudo C. Dicer, a new regulator of pluripotency exit and LINE-1 elements in mouse embryonic stem cells. FEBS Open Bio. 2017;7:204–220. doi: 10.1002/2211-5463.12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.Y., Morris Q., Mitchell J.A. Enhancer identification in mouse embryonic stem cells using integrative modeling of chromatin and genomic features. BMC Genomics. 2012;13:152. doi: 10.1186/1471-2164-13-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz C., Houseley J. Endogenous RNA interference is driven by copy number. eLife. 2014;3:e01581. doi: 10.7554/eLife.01581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi N., Zenno S., Ueda R., Ohki-Hamazaki H., Ui-Tei K., Saigo K. Short-interfering-RNA-mediated gene silencing in mammalian cells requires Dicer and eIF2C translation initiation factors. Curr. Biol. 2003;13:41–46. doi: 10.1016/s0960-9822(02)01394-5. [DOI] [PubMed] [Google Scholar]
- Ferguson-Smith A.C. Genomic imprinting: the emergence of an epigenetic paradigm. Nat. Rev. Genet. 2011;12:565–575. doi: 10.1038/nrg3032. [DOI] [PubMed] [Google Scholar]
- Fire A., Xu S., Montgomery M.K., Kostas S.A., Driver S.E., Mello C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Flemr M., Malik R., Franke V., Nejepinska J., Sedlacek R., Vlahovicek K., Svoboda P. A retrotransposon-driven dicer isoform directs endogenous small interfering RNA production in mouse oocytes. Cell. 2013;155:807–816. doi: 10.1016/j.cell.2013.10.001. [DOI] [PubMed] [Google Scholar]
- Ghildiyal M., Zamore P.D. Small silencing RNAs: an expanding universe. Nat. Rev. Genet. 2009;10:94–108. doi: 10.1038/nrg2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier J.L., Kazazian H.H., Jr. Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell. 2008;135:23–35. doi: 10.1016/j.cell.2008.09.022. [DOI] [PubMed] [Google Scholar]
- Haig D. Transposable elements: Self-seekers of the germline, team-players of the soma. BioEssays. 2016;38:1158–1166. doi: 10.1002/bies.201600125. [DOI] [PubMed] [Google Scholar]
- Hajkova P., Ancelin K., Waldmann T., Lacoste N., Lange U.C., Cesari F., Lee C., Almouzni G., Schneider R., Surani M.A. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature. 2008;452:877–881. doi: 10.1038/nature06714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B.W., Wang W., Zamore P.D., Weng Z. piPipes: a set of pipelines for piRNA and transposon analysis via small RNA-seq, RNA-seq, degradome- and CAGE-seq, ChIP-seq and genomic DNA sequencing. Bioinformatics. 2015;31:593–595. doi: 10.1093/bioinformatics/btu647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutnick L.K., Huang X., Loo T.-C., Ma Z., Fan G. Repression of retrotransposal elements in mouse embryonic stem cells is primarily mediated by a DNA methylation-independent mechanism. J. Biol. Chem. 2010;285:21082–21091. doi: 10.1074/jbc.M110.125674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illingworth R.S., Bird A.P. CpG islands--‘a rough guide’. FEBS Lett. 2009;583:1713–1720. doi: 10.1016/j.febslet.2009.04.012. [DOI] [PubMed] [Google Scholar]
- Iurlaro M., von Meyenn F., Reik W. DNA methylation homeostasis in human and mouse development. Curr. Opin. Genet. Dev. 2017;43:101–109. doi: 10.1016/j.gde.2017.02.003. [DOI] [PubMed] [Google Scholar]
- Karimi M.M., Goyal P., Maksakova I.A., Bilenky M., Leung D., Tang J.X., Shinkai Y., Mager D.L., Jones S., Hirst M., Lorincz M.C. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell. 2011;8:676–687. doi: 10.1016/j.stem.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley D., Rinn J. Transposable elements reveal a stem cell-specific class of long noncoding RNAs. Genome Biol. 2012;13:R107. doi: 10.1186/gb-2012-13-11-r107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H., Sakurai T., Miura F., Imai M., Mochiduki K., Yanagisawa E., Sakashita A., Wakai T., Suzuki Y., Ito T. High-resolution DNA methylome analysis of primordial germ cells identifies gender-specific reprogramming in mice. Genome Res. 2013;23:616–627. doi: 10.1101/gr.148023.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F., Andrews S.R. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei H., Oh S.P., Okano M., Jüttermann R., Goss K.A., Jaenisch R., Li E. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development. 1996;122:3195–3205. doi: 10.1242/dev.122.10.3195. [DOI] [PubMed] [Google Scholar]
- Lerat E., Rizzon C., Biémont C. Sequence divergence within transposable element families in the Drosophila melanogaster genome. Genome Res. 2003;13:1889–1896. doi: 10.1101/gr.827603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksakova I.A., Romanish M.T., Gagnier L., Dunn C.A., van de Lagemaat L.N., Mager D.L. Retroviral elements and their hosts: insertional mutagenesis in the mouse germ line. PLoS Genet. 2006;2:e2. doi: 10.1371/journal.pgen.0020002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murchison E.P., Partridge J.F., Tam O.H., Cheloufi S., Hannon G.J. Characterization of Dicer-deficient murine embryonic stem cells. Proc. Natl. Acad. Sci. USA. 2005;102:12135–12140. doi: 10.1073/pnas.0505479102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reik W., Surani M.A. Germline and pluripotent stem cells. Cold Spring Harb. Perspect. Biol. 2015;7:a019422. doi: 10.1101/cshperspect.a019422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert V.J.P., Sijen T., van Wolfswinkel J., Plasterk R.H.A. Chromatin and RNAi factors protect the C. elegans germline against repetitive sequences. Genes Dev. 2005;19:782–787. doi: 10.1101/gad.332305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe H.M., Trono D. Dynamic control of endogenous retroviruses during development. Virology. 2011;411:273–287. doi: 10.1016/j.virol.2010.12.007. [DOI] [PubMed] [Google Scholar]
- Schorn A.J., Gutbrod M.J., LeBlanc C., Martienssen R. LTR-retrotransposon control by tRNA-derived small RNAs. Cell. 2017;170:61–71.e11. doi: 10.1016/j.cell.2017.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seisenberger S., Andrews S., Krueger F., Arand J., Walter J., Santos F., Popp C., Thienpont B., Dean W., Reik W. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol. Cell. 2012;48:849–862. doi: 10.1016/j.molcel.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif J., Endo T.A., Nakayama M., Karimi M.M., Shimada M., Katsuyama K., Goyal P., Brind’Amour J., Sun M.A., Sun Z. Activation of endogenous retroviruses in Dnmt1(-/-) ESCs involves disruption of SETDB1-mediated repression by NP95 binding to hemimethylated DNA. Cell Stem Cell. 2016;19:81–94. doi: 10.1016/j.stem.2016.03.013. [DOI] [PubMed] [Google Scholar]
- Stadler M.B., Murr R., Burger L., Ivanek R., Lienert F., Schöler A., van Nimwegen E., Wirbelauer C., Oakeley E.J., Gaidatzis D. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–495. doi: 10.1038/nature10716. [DOI] [PubMed] [Google Scholar]
- Svoboda P., Stein P., Anger M., Bernstein E., Hannon G.J., Schultz R.M. RNAi and expression of retrotransposons MuERV-L and IAP in preimplantation mouse embryos. Dev. Biol. 2004;269:276–285. doi: 10.1016/j.ydbio.2004.01.028. [DOI] [PubMed] [Google Scholar]
- Tam O.H., Aravin A.A., Stein P., Girard A., Murchison E.P., Cheloufi S., Hodges E., Anger M., Sachidanandam R., Schultz R.M., Hannon G.J. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature. 2008;453:534–538. doi: 10.1038/nature06904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W.W.C., Kobayashi T., Irie N., Dietmann S., Surani M.A. Specification and epigenetic programming of the human germ line. Nat. Rev. Genet. 2016;17:585–600. doi: 10.1038/nrg.2016.88. [DOI] [PubMed] [Google Scholar]
- Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Lagemaat L.N., Medstrand P., Mager D.L. Multiple effects govern endogenous retrovirus survival patterns in human gene introns. Genome Biol. 2006;7:R86. doi: 10.1186/gb-2006-7-9-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Meyenn F., Iurlaro M., Habibi E., Liu N.Q., Salehzadeh-Yazdi A., Santos F., Petrini E., Milagre I., Yu M., Xie Z. Impairment of DNA methylation maintenance is the main cause of global demethylation in naive embryonic stem cells. Mol. Cell. 2016;62:848–861. doi: 10.1016/j.molcel.2016.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C.P., Chaillet J.R., Bestor T.H. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 1998;20:116–117. doi: 10.1038/2413. [DOI] [PubMed] [Google Scholar]
- Walter M., Teissandier A., Pérez-Palacios R., Bourc’his D. An epigenetic switch ensures transposon repression upon dynamic loss of DNA methylation in embryonic stem cells. eLife. 2016;5:e11418. doi: 10.7554/eLife.11418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T., Takeda A., Tsukiyama T., Mise K., Okuno T., Sasaki H., Minami N., Imai H. Identification and characterization of two novel classes of small RNAs in the mouse germline: retrotransposon-derived siRNAs in oocytes and germline small RNAs in testes. Genes Dev. 2006;20:1732–1743. doi: 10.1101/gad.1425706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimizu T., Sugiyama N., De Felice M., Yeom Y.I., Ohbo K., Masuko K., Obinata M., Abe K., Schöler H.R., Matsui Y. Germline-specific expression of the Oct-4/green fluorescent protein (GFP) transgene in mice. Dev. Growth Differ. 1999;41:675–684. doi: 10.1046/j.1440-169x.1999.00474.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Differentially expressed genes were called using the overlap between the SeqMonk Intensity difference as well as DESeq2.