

Abstract
S282T in NS5B is the primary amino acid substitution associated with resistance to sofosbuvir (SOF) but has rarely been detected in patients treated with a SOF‐based regimen. Here, the emergence and fitness of the S282T substitution in virologic failure patients administered SOF‐based regimens across the SOF and ledipasvir (LDV)/SOF phase 2 and 3 programs was evaluated. Plasma samples collected at baseline and at virologic failure were amplified and deep sequenced (1% cutoff). To date, over 12,000 patients have been treated in SOF or LDV/SOF phase 2 and 3 studies. Of these, deep sequencing was available at baseline in 8598 patients (62.4% genotype [GT] 1, 10.7% GT2, 20.9% GT3, and 6.0% GT4‐6) and at virologic failure in 901 patients. In the 8598 patients, no S282T substitution was detected at baseline; at virologic failure, 10 of the 901 (1%) patients had S282T detected. The SOF‐based regimen associated with treatment‐emergent S282T was SOF monotherapy in two patients, retreatment with LDV/SOF in prior LDV/SOF failures in three patients, LDV/SOF for 8 weeks in 1 GT1 patient, LDV/SOF for 12 weeks in 1 patient each with GT3, GT4, and GT5, and LDV/SOF + ribavirin for 12 weeks in 1 GT6 patient. Nine of 10 patients with emergent S282T received an SOF‐based retreatment regimen, eight of whom achieved sustained virologic response 12 weeks after treatment and one of whom failed retreatment. Conclusion: The emergence of S282T substitution was rare in patients who fail SOF‐based regimens. Successful retreatment of prior SOF failure patients is possible in the presence of S282T substitution with SOF in combination with various direct‐acting antiviral agents. (Hepatology Communications 2017;1:538–549)
Abbreviations
- DAA
direct‐acting antiviral agent
- DMSO
dimethyl sulfoxide
- GT
genotype
- LDV
ledipasvir
- NI
nucleoside inhibitor
- PCR
polymerase chain reaction
- RAS
resistant associated substitution
- RBV
ribavirin
- SOF
sofosbuvir
- SVR
sustained virologic response
- SVR12
SVR 12 weeks after treatment
Chronic hepatitis C virus (HCV) infection is a major global health burden, with approximately 170 million individuals infected worldwide.1 In the last several years, there has been expansion in the development of direct‐acting antiviral agents (DAAs) for treatment of chronic HCV infection. By combining two or more DAAs, high rates of sustained virologic response (SVR) have been achieved.
The pangenotypic NS5B HCV inhibitor sofosbuvir (SOF) has demonstrated high efficacy in patients infected with HCV in combination with ledipasvir (LDV) for genotype (GT) 1, 2, 4, 5, and 62, 3, 4, 5, 6, 7 and in combination with ribavirin (RBV) with or without pegylated interferon for GT3 HCV.8, 9 SOF exhibits a high barrier to resistance in vivo. After in vitro selection, S282T in NS5B was the only substitution selected in all tested GTs, conferring 2.4‐ to 18.1‐fold reduced susceptibility to SOF, while also reducing the viral replication capacity by 89% to 99% when compared with wild type.10, 11, 12
Even though S282T was selected in vitro,10 this substitution is very rare in SOF clinical trials. Out of almost 2000 patients receiving SOF‐containing regimen in the SOF phase 2 and 3 registration studies, only a single patient developed S282T at virologic failure.13 Similarly, in LDV/SOF combination phase 2 and 3 registration studies, only one of almost 4000 patients developed S282T at virologic failure.14 This low rate of S282T development observed for patients treated with SOF is not consistent across the class of nucleoside inhibitors (NIs). S282T has been detected more frequently at virologic failure in patients treated with VX‐135 or mericitabine.15, 16
Recently, two other NS5B substitutions have been associated with SOF treatment. Treatment‐emergent L159F and V321A have been observed at time of virologic failure in a few patients in SOF phase 2 and 3 clinical trials.11 The 50% effective concentration (EC50) fold reduction of these substitutions to SOF was 1.2‐ to 1.3‐fold for L159F in GT1a, GT1b, and GT3a and 1.3‐fold for V321A in GT3a.11, 12 In an analysis of SOF and LDV/SOF studies, the presence of baseline L159F was shown to be associated with virologic failure in a subset of GT1b patients with advanced liver disease treated for a shorter duration with SOF plus RBV before liver transplantation but not for patients receiving LDV/SOF.17 Moreover, the substitution L320F, which is associated with low‐level resistance to mericitabine,18 has been observed in a few patients experiencing virologic failure in SOF studies, but in no patients in LDV/SOF studies. In vitro analyses of L320F and the combination of L320F with L159F showed a low EC50 fold reduction in SOF susceptibility (1.7‐ to 2.2‐fold for L320F and L320F+L159F, respectively).17
A concern of clinicians considering retreatment of patients who have not achieved SVR after earlier treatment with DAAs is the possible presence of resistant associated substitutions (RASs). Even the failure to detect RASs after treatment failure does not rule out the possibility that RASs may have emerged at the time of treatment failure before being replaced by wild type because of their relatively poor replication fitness. Two recent studies have demonstrated that previous virologic failure with SOF‐based treatment does not prevent successful retreatment with another SOF‐based regimen.19, 20 In both studies, all patients with HCV GT1 infection who had relapsed after SOF+RBV with or without pegylated interferon achieved SVR following 12 weeks LDV/SOF with or without RBV.
Here, a comprehensive analysis was performed to evaluate the prevalence of S282T across SOF and LDV/SOF phase 2 and phase 3 programs, including 12,012 patients treated with an SOF‐based regimen. We evaluated the emergence and fitness of the S282T substitution in patients who were administered SOF‐based regimens and experienced virologic failure. Moreover, in patients with emergent S282T, we investigated evolution at position 282 after treatment.
Materials and Methods
PATIENT SAMPLES
All studies were conducted in accordance with the Declaration of Helsinki, Good Clinical Practice guidelines, and local regulatory requirements. All patients provided written informed consent.
LABORATORY ASSESSMENTS
HCV RNA was determined at a central laboratory using the Roche High‐Pure‐System, COBAS TaqMan version 2 assay (Roche Molecular Diagnostics, Pleasanton, CA) with a lower limit of quantitation of 25 IU/mL. HCV genotype was determined using the VERSANT HCV Genotype 2.0 assay (LiPA) or by TRUGENE (both Siemens, Munich, Germany). The genotype results from LiPA and TRUGENE assay were confirmed or refined by direct sequencing results if available.
SEQUENCING ANALYSIS
For all patients, the HCV NS5B coding regions were amplified by DDL Diagnostic Laboratory (Rijswijk, Netherlands) using standard reverse transcription polymerase chain reaction (PCR) in available plasma/serum samples wherein the HCV RNA was >1000 IU/mL. For patients receiving NS5A inhibitor, the NS5A region was also amplified as described above. Deep sequencing using the MiSeq platform (Illumina, Inc., San Diego, CA) was performed with DDL or WuXi AppTec (Shanghai, China). Amino acid substitutions in the generated sequences from each time point were compared with the respective baseline sequence for each subject in the parent study. The baseline prevalence rate of NS5B S282T was evaluated in 8598 patients from 24 countries in SOF and LDV/SOF clinical trials. The prevalence rates of treatment‐emergent NS5B S282T, L159F, and V321A were assessed at the time of virologic failure. For patients receiving NS5A inhibitors, NS5A RASs at positions 24, 28, 30, 31, 32, 58, and 93 that conferred >2.5‐fold reduced susceptibility to LDV in vitro were included in the analysis; K24G/N/R, M28A/G/T, Q30E/G/H/L/K/R/T, L31I/F/M/V, P32L, S38F, H58D, A92K, and Y93C/F/H/N/S for patients with GT1a HCV infection and L31I/F/M/V, P32L, P58D, A92K, and Y93C/H/N/S for patients with GT1b infection.21, 22 A genotype‐specific reference was used for each HCV genotype (HCV1a_H77_ NC_004102, HCV1b_Con1_AJ238799, HCV2a_JFH1_AB047639, HCV2b_MD2b10_AY232748, HCV3a_S52_GU814263, HCV4a_ED43_GU814265, HCV5a_SA13_AF064490, HCV6a_EUHK2_Y12083).
Standard population sequencing was performed on patient samples if deep sequencing was not successful. Population sequencing of the full‐length HCV NS5A coding region was performed by DDL Diagnostic Laboratory or Monogram Biosciences (San Francisco, CA) using reverse transcription PCR and standard Sanger sequencing of the bulk PCR product. The sensitivity for detection of resistant variants is approximately 10%‐20%.
SOF SUSCEPTIBILITY ANALYSES
RASs were introduced into the GT1a or GT1b replicon by site‐directed mutagenesis and tested in transient transfections as described previously.23 Briefly, NS5B mutations were introduced into a plasmid encoding the PI‐hRluc replicon using a QuikChange II XL mutagenesis kit, following the manufacturer's instructions (Stratagene, La Jolla, CA). Mutations were confirmed by DNA sequencing. Replicon RNAs were transcribed in vitro from replicon‐encoding plasmids using a MEGAscript kit (Ambion, Austin, TX). RNA was transfected into Huh‐lunet cells using the method of Lohmann et al.24 Briefly, cells were trypsinized and washed twice with phosphate‐buffered saline. A suspension of 4 × 106 cells in 400 μL of phosphate‐buffered saline was mixed with 5 μg of RNA and subjected to electroporation using settings of 960 μF and 270 V. Cells were transferred into 40 mL of prewarmed culture medium and then seeded into 96‐well plates (100 μL/well). Compounds were 3‐fold serially diluted in 100% dimethyl sulfoxide (DMSO) and added to cells at a 1:200 dilution, achieving a final DMSO concentration of 0.5% in a total volume of 200 μL/well. Cells were treated for 3 days, after which culture media were removed, cells were lysed, and Renilla luciferase activity was quantified using a commercially available assay (Promega, Madison, WI) and a Top Count instrument (Perkin Elmer, Waltham, MA). EC50 values were calculated as the compound concentration at which a 50% reduction in the level of Renilla reporter activity was observed when compared with control samples with DMSO. Dose‐response curves and EC50 values were generated using GraphPad Prism software package (GraphPad Software, La Jolla, CA) by nonlinear regression analysis. The replication level of either reference strains (1b‐Con1 or 1a‐H77) or chimeric replicons derived transiently from clinical isolates was determined as the ratio of the Renilla luciferase signal at day 4 to that at 4 hours after electroporation to normalize for transfection efficiency. The replication capacity of each replicon was expressed as their normalized replication efficiency compared with that of the reference strain (1b‐Con1 or 1a‐H77) within the same experiment. SOF susceptibility and replication capacity were tested by subcloning of patient isolates.
Results
PREVALENCE OF BASELINE AND TREATMENT‐EMERGENT S282T IN PATIENT TREATED WITH SOF
Baseline NS5B sequences were available by deep sequencing (1% cutoff) in all 8598 patients before SOF therapy, including 4766 patients from North America, 1767 patients from Europe, 1094 patients from Oceania, 954 patients from Asia, and 17 patients from Africa. Of these patients, 62.4% had GT1 infection, 10.7% had GT2 infection, 20.9% had GT3 infection, 4.0% had GT4 infection, 0.9% had GT5 infection, and 1.2% had GT6 infection.
In the SOF or LDV/SOF in clinical studies, 1025 patients experienced virologic failure, of whom 901 patients were successfully sequenced by deep sequencing. Of the remaining 124 patients, 77 were able to be sequenced using standard population sequencing, whereas no sequencing was possible in 47 (<5% of virologic failures). Thus, sequencing results were obtained from 978 patients. A total of 10 patients had S282T at the virologic failure time point, which is 1% (10/978) of virologic failures.
DEMOGRAPHICS OF PATIENTS WITH EMERGENT S282T
The mean age of the 10 patients with emergent S282T was 56 years. Nine patients were men, eight were white, and two were black. Of the nine patients who had IL28B genotyping available, eight had non‐CC genotypes. Five had cirrhosis and five were naïve to prior DAA treatment (Table 1). Eight patients were treated with LDV/SOF±RBV, and the remaining two patients were treated with SOF monotherapy. The mean HCV RNA at time of treatment was 6.6 log10 IU/mL. Four patients had HCV GT1a infection and one patient each had GT1b, GT2, GT3, GT4, GT5, and GT6 infection. In comparison, 64% of all patients treated in the SOF and LDV/SOF clinical studies had HCV GT1a infection.
Table 1.
Characteristics of Patients With S282T
| Characteristic | Overall (n = 10) |
|---|---|
| Age, years, mean (range) | 55 (34‐72) |
| Sex, n (%) | |
| Men | 8 (80) |
| Women | 2 (20) |
| Race, n (%) | |
| White | 8 (80) |
| Black | 2 (20) |
| Body mass index, kg/m2, mean (range) | 31 (22‐37) |
| Cirrhosis, n (%) | 5 (50) |
| IL28B non‐CC, n (%) | 8 (89)a |
| Mean baseline HCV RNA, log10 IU/mL (range) | 6.7 (6.0‐7.8)b |
| Genotype, n (%) | |
| GT1a | 4 (40) |
| GT1b | 1 (10) |
| GT2b | 1 (10) |
| GT3a | 1 (10) |
| GT4r | 1 (10) |
| GT5a | 1 (10) |
| GT6l | 1 (10) |
| Prior treatment status, n (%): | |
| Treatment‐naïve | 4 (40) |
| Treatment‐experienced, DAA‐naive | 1 (10) |
| DAA‐experienced | 5 (50) |
| Treatment, n (%) | |
| LDV/SOF | 8 (80) |
| SOF | 2 (20) |
Data were available for nine patients.
Based on data from eight patients.
HCV RNA DYNAMICS AND S282T FREQUENCY IN PATIENTS WITH EMERGENT S282T
The initial reported case of S282T was a 52‐year‐old white woman with HCV GT2b infection (patient 1). She received SOF monotherapy for 12 weeks in the phase 2 ELECTRON study and relapsed at week 4 posttreatment with S282T detected at a frequency of >99%. The frequency of S282T rapidly decreased to 27.6% at week 8 and <1% at week 12 posttreatment. This patient was retreated with SOF+RBV for 12 weeks and subsequently achieved SVR 12 weeks after treatment (SVR12) (Fig. 1A).
Figure 1.
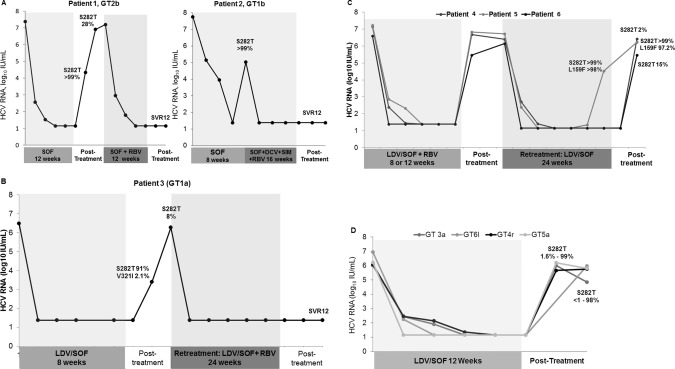
RNA dynamics and S282T emergence in SOF‐ or LDV/SOF‐treated patients. (A) Emergent S282T substitution in patient 1 (GT2b infection) and patient 2 (GT1b infection), who were treated with SOF monotherapy. (B) Emergent S282T substitution in patient 3 (GT1a infection) treated with LDV/SOF. (C) Emergent S282T substitution in three patients with GT1a infection (patients 4, 5, and 6) retreated with LDV/SOF. (D) Emergent S282T substitution in patients infected with non‐GT1 HCV (patients 7, 8, 9, and 10) who were treated with LDV/SOF.
The second case was a 56‐year‐old white man who was a post‐liver transplantation recipient with decompensated cirrhosis with recurrent HCV GT1b infection (patient 2). In 2013, he received compassionate supply of SOF monotherapy for 8 weeks, but experienced virologic breakthrough during treatment with S282T detected at a frequency of >99%. After virologic failure, he was immediately retreated with SOF+ DCV+SIM+RBV for 16 weeks and subsequently achieved SVR12 (Fig. 1A).
Four patients with GT1a infection developed treatment‐emergent S282T after LDV/SOF. The first patient (patient 3) was a 60‐year‐old white man treated with LDV/SOF for 8 weeks and relapsed posttreatment with S282T. At baseline, S282T was not detected but emerged as the dominant population following virologic failure at 8 weeks posttreatment, with a frequency of 91% of the quasispecies. Two weeks later, S282T population had decreased to only 8% (Fig. 1B). At baseline, the NS5A RAS L31M was detected at 25.5% of the viral quasispecies. This mutation confers high level resistance (EC50 >500 fold shift) to LDV. At week 8 posttreatment, the frequency of L31M had increased to >99%. Additional treatment emergent RASs included Q30L (4.5%) and Y93H (96.7%). The frequencies of the 3 NS5A RASs remained stable at week 10 posttreatment (Table 2). This patient was retreated with LDV/SOF+RBV for 24 weeks and achieved SVR12 (Fig. 1B).
Table 2.
Deep Sequencing Results of NS5B and NS5A of Patients with S282T and/or footprint codon
| Patient No. | Genotype | Treatment | Visit Name | Baseline HCV RNA, IU/mL | 282 codon (Amino Acid) Frequency | NS5B RASs Other Than S282T (Frequency) | NS5A RASs (Frequency) |
|---|---|---|---|---|---|---|---|
| 1 | 2b | SOF monotherapy, 12 weeks | Baseline | 23,600,000 | AGU (S) 54.9%; AGC (S) 44.8% | None | ND |
| 4 weeks posttreatment | 21,700 | ACU (T) >99% | None | ND | |||
| 8 weeks posttreatment | 647,000 | AGU (S) 68%; UCU (S) 4%; ACU (T) 27% | None | ND | |||
| 12 weeks posttreatment | 8,140,000 | AGU (S) 90%; UCU (S) 8% | None | ND | |||
| 24 weeks posttreatment | 11,400,000 | AGT (S) 56.7%; UCU (S) 40.6% | None | ND | |||
| 48 weeks posttreatment | 24,300,000 | AGU (S) 77%; UCU (S) 23% | None | ND | |||
| 2 | 1b | SOF monotherapy, 8 weeks | Baseline | 57,700,000 | AGC (S) 99.0% | None | ND |
| 8 weeks posttreatment | 107,000 | ACC (T) >99% | None | ND | |||
| 3 | 1a | LDV/SOF, 8 weeks | Baseline | 2,940,000 | AGC (S) >99% | None | L31M (25.5%) |
| 8 weeks posttreatment | UCC (S) 8.4%; ACC (T) 91.2% | None | Q30L (4.5%); L31M (>99%); Y93H (96.7%) | ||||
| 10 weeks posttreatment | 1,879,337 | UCC (S) 88.5%; ACC (T) 8.0%; AGC (S) 3.2% | None | Q30L (3.5%); L31M (94.4%); L31V (4.7%); Y93H (98.2%) | |||
| 4 | 1a | LDV/SOF, 24 weeks | Baseline | 2,520,000 | AGC (S) >99% | None | M28T (>99%); Q30R (>99%) |
| 4 weeks posttreatment | 2,610,000 | ACC (T) 1.7%; AGC (S) 97.9% | None | M28T (>99%); Q30R (>99%) | |||
| 6 weeks posttreatment | 4,330,000 | AGC (S) 99.0% | None | M28T (>99%); Q30R (>99%) | |||
| 12 weeks posttreatment | 3,860,000 | AGC (S) >99% | None | M28T (>99%); Q30R (>99%) | |||
| 5 | 1a | LDV/SOF, 24 weeks | Baseline | 5,170,000 | AGC (S) >99% | None | M28V (8.1%); Q30K (10.1%); Q30R (3.8%); Q30T (73.9%) |
| Week 18 | 33,000 | ACC (T) >99% | L159F (97.7%) | K24N (>99%); Q30K (>99%) | |||
| Week 20a | 625,000 | ACC (T) 98.8% | L159F (99.5%) | K24N (98.0%); Q30K (99.6%) | |||
| 4 weeks posttreatmenta | 1,690,000 | ACC (T) >99% | L159F (97.2%) | K24N (98.7%); Q30K (98.0%) | |||
| 6 | 1a | LDV/SOF, 24 weeks | Baseline | 1,370,000 | AGC (S) >99% | None | Y93N (>99%) |
| 5 weeks posttreatment | 288,000 | UCC (S) 84.6%; ACC (T) 14.5% | None | Y93N (>99%) | |||
| SOF/VEL+VOX, 12 weeks | Baseline of retreatmentb | 4,900,000 | UCC (S) 98.1% | None | Y93N (>99%) | ||
| 7 | 3a | LDV/SOF, 12 weeks | Baseline | 1,037,993 | AGU (S) >99% | None | None |
| 2 weeks posttreatment | 437,353 | AGU (S) 10.1%; ACU (T) 89.7% | None | ND | |||
| 4 weeks posttreatment | 990,822 | AGU (S) 62.0% ACU (T) 37.6% | None | None | |||
| 8 weeks posttreatment | 309,023 | AGU (S) 99.4% | None | None | |||
| 12 weeks posttreatment | 70,894 | AGU (S) 89.9% UCU (S) 9.6% | None | ND | |||
| 8 | 4r | LDV/SOF, 12 weeks | Baseline | AGC (S) >99% | None | L30R (>99%); M31M (>99%) | |
| 4 weeks posttreatment | ACC (T) >99% | None | L30R (>99%); M31M (>99%); Y93C (7.8%) | ||||
| 5 weeks posttreatment | ACC (T) 69.1%; AGC (S) 14.9%; UCC (S) 15.4% | None | ND | ||||
| 9 | 5a | LDV/SOF, 12 weeks | Baseline | AGC (S) >99% | None | L31M (>99%) | |
| 4 weeks posttreatment | ACC (T) 1.6%; AGC (S) 98.1% | None | L31M (>99%) | ||||
| 10 | 6l | LDV/SOF+RBV, 12 weeks | Baseline | 6,093,007 | AGU (S) >99% | None | Q24K (>99%); F28V (>99%); R30A (>99%); T58P (>99%) |
| 12 weeks posttreatment | 933,009 | ACU (T) 98.0%; UCU (S) 1.5% | None | Q24K (>99%); F28V (>99%); R30A (>99%); T58P (>99%) | |||
| 11‡ | 1a | LDV/SOF, 12 weeks | Baseline | AGC (S) | None | Y93F (1.22%); Y93N (9.93%) | |
| 12 weeks posttreatment | UCC (S) | None | Y93N (>99%) |
Footprint codons are shown in bold.
Full genome sequencing results.
Baseline sample of retreatment was collected 16 months after failure of LDV/SOF treatment. This patient has achieved SVR12 after treatment with SOF/VEL+VOX.
Patient 11, did not have S282T observed at posttreatment but the footprint codon UCC at 12 weeks posttreatment suggests that this patient may have had emergent S282T after an SOF‐based regimen that already had reverted to wild type.
Abbreviations: ND, not done; S, serine; T, threonine.
The three additional patients with GT1a infection (patients 4, 5, and 6) were treated with LDV/SOF+RBV for 8 or 12 weeks and relapsed posttreatment without S282T. They were retreated with LDV/SOF for a longer duration of 24 weeks (Fig. 1C). Patients 4 and 6, a 64‐year‐old white man and a 65‐year‐old white man, respectively, relapsed at 4‐5 weeks after retreatment with S282T detected at a frequency of 1.7% and 14.5%, respectively. Patient 5, a 54‐year‐old black woman, experienced virologic breakthrough after 16 weeks of retreatment with dual NS5B RASs S282T and L159F at a frequency of >99% and 97.7%, respectively. S282T and L159F remained at high frequencies at week 4 posttreatment (>99% and 97.2%, respectively; Table 2, Fig. 1C). After the initial course of LDV/SOF, all three patients developed NS5A RASs conferring high‐level (EC50 >1000‐fold shift) LDV resistance; M28T (>99%) and Q30R (>99%) in patient 4, M28V (8.1%) and Q30K/R/T (87.8%) in patient 5, and Y93N (>99%) in patient 6. All three patients were retreated with a third course of an SOF‐based regimen. Patients 5 and 6 received SOF/VEL+VOX for 12 weeks, and both achieved SVR12. Patient 4 received LDV/SOF for 24 weeks and failed to achieve SVR12.
Four patients (7, 8, 9, and 10) with non‐GT1 infection (GT3a, GT4r, GT5a, or GT6l, respectively) relapsed after receiving LDV/SOF for 12 weeks with S282T (Fig. 1D). The frequency of NS5A RASs is high in untreated patients with non‐GT1 genotypes, which is consistent with the observed reduced antiviral activity of LDV against genotypes 2, 3, and 6.22 Patient 7, a 45‐year‐old white man with GT3a infection, had no NS5A RASs detected at baseline or posttreatment. S282T was detected at a frequency of 89.7% at week 2 posttreatment, which declined to 37.6% at week 4 posttreatment and to undetectable levels at week 8 posttreatment. This patient was retreated with LDV/SOF+RBV for 24 weeks and achieved SVR12. Patient 8 was a 50‐year‐old black man with GT4r infection and both NS5A L30R and L31M detected at baseline with frequencies >99%, which remained stable through week 4 posttreatment. S282T was observed at a frequency of >99% at week 4 posttreatment, which declined to 69.1% at week 5 posttreatment. In addition, this patient developed treatment‐emergent NS5A Y93C (frequency of 7.8%) at virologic failure. This patient was retreated with SOF+SIM+RBV for 24 weeks and subsequently achieved SVR12. Patient 9, a 72‐year‐old white man with GT5a infection, had NS5A L31M detected at baseline at frequency >99%, which was maintained at week 4 posttreatment, without any treatment‐emergent NS5A RASs. S282T was observed at a frequency of 1.6% at week 4 posttreatment. This patient was retreated with LDV/SOF+RBV for 24 weeks and achieved SVR12. Patient 10, a 34‐year‐old white man with GT6l infection, had NS5A Q24K, F28V, R30A, and T58P detectable at baseline at frequency >99%, which was maintained at week 12 posttreatment. S282T was observed at a frequency of 98% at week 12 posttreatment. This patient has not yet been retreated.
SOF SUSCEPTIBILITY ANALYSIS
The impact on SOF susceptibility of S282T alone or in combination with L159F was assessed by constructing site‐directed mutants in a replicon vector. In GT1a replicon, S282T and L159F alone conferred 17 and 1.6 reduced susceptibility to SOF. The reduction in susceptibility was slightly increased in the S282T/L159F double mutant, which conferred 24‐fold reduced susceptibility to SOF. Replication capacity of site‐directed mutants ranged from 3% to 11% compared with the wild‐type replicon (Fig. 2). SOF susceptibility and replication capacity were tested by subcloning of patient isolates. Replication capacity testing in vitro did not identify any restoration of the replicative defect of S282T.
Figure 2.
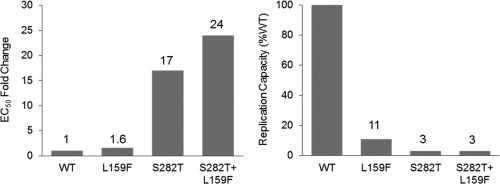
SOF susceptibility and replication capacity of S282T alone and in combination with L159F. EC50 fold change and replication capacity were calculated using GT1a replicon values. WT, wild type.
TREATMENT‐EMERGENT S282T AND ASSOCIATED REVERSION OF S282T TO WILD‐TYPE FOOTPRINT CODONS
Sequence analysis was conducted on posttreatment samples of patients with emergent S282T to investigate evolution at position 282 in absence of drug selection pressure. In the 10 patients with emergent S282T during SOF‐based treatment in this study, the baseline codon at position 282 was AGU (n = 2), AGC (n = 7), or a combination of AGU and AGC (n = 1), which all codes for serine. In six of eight patients with an observed decline of S282T in samples at posttreatment visits, new serine codons not observed at baseline emerged posttreatment; UCU (n = 3) and UCC (n = 3) (Table 2, Fig. 3). These serine codons are a result of reversion of S282T to wild type, and from here on we refer to them as “footprint codons.” Patients 4, 5, and 6 did not have footprint codon observed after their initial failure with LDV/SOF treatment. After the second course of LDV/SOF treatment, the footprint codon observed in patient 6 persisted for up to 16 months (Table 2). Despite the presence of footprint codon in this patient, SVR12 was achieved after treatment with SOF/VEL+VOX for 12 weeks.
Figure 3.
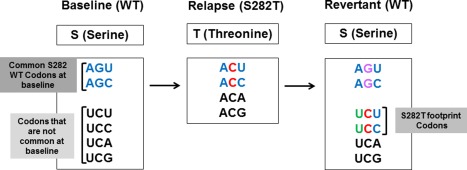
Evolution of NS5B 282 codons usage in patient who developed S282T. WT, wild type.
Interestingly, investigation of codons at position 282 in 10,000 SOF‐naïve patients showed that only two patients had these serine footprint codons at baseline; UCU (n = 1) and UCC (n = 1). Both of these patients were treated previously with the nucleotide inhibitor VX‐135. All other SOF‐naïve patients had the serine codons AGU (n = 7578) and AGC (n = 2221).
Based on these results, we investigated all patients experiencing virologic failure in the SOF clinical trials (n = 901) for the presence of footprint codons. In addition to the six patients described above, one more patient (patient 11) had a footprint codon at virologic failure. This patient was infected with GT1a and had the serine codon AGC at baseline and the footprint codon UCC at 12 weeks posttreatment (Table 2). This patient was treated with LDV/SOF for 12 weeks. The footprint codon observed in this patient at virologic failure suggests that this patient may have had emergent S282T after an SOF‐based regimen that already had reverted to wild type, leaving a footprint at time of sampling. In addition to the serine footprint codon, the NS5A Y93N was detected at virologic failure in this patient.
Discussion
SOF‐based regimens have achieved high rates of SVR in patients with chronic HCV infection. Although S282T is the signature substitution associated with in vitro resistance to SOF, this has rarely been detected in patients treated with an SOF‐based regimen.13, 14 Here, we evaluated the emergence and fitness of the S282T in SOF and LDV/SOF‐treated patients and the posttreatment evolution at position 282 in patients with emergent S282T.
Across the SOF development program, S282T was not detected by deep sequencing (1% cutoff) in any of the 8598 baseline samples, which is consistent with the findings of previous studies.11, 25, 26 This suggests that S282T has a poor fitness in the absence of drug pressure, and S282T is therefore unlikely to be detected at measurable frequency in untreated patients. In comparison, baseline NS3, NS5A, and NS5B non‐nucleoside inhibitor (NNI) RASs has been detected in 10%‐90% of DAA‐naïve patients depending on genotype and subtype27, 28, 29, 30, 31; for example, NS5A L31M has been detected in >50% of GT2a patients.32 Due to the high error rate of HCV polymerase, substitutions at all sites in the HCV genome can exist within the viral quasispecies33, 34; however, the lack of S282T compared with other RASs suggests that not all positions in the HCV genome has the same allowance for genetic variability. Across HCV genotypes, the NS5B position 282 is highly conserved, both at nucleotide and amino acid level, resulting in the presence of only two of six serine codons at baseline (AGU and AGC). The preservation of these specific serine codons could be a result of RNA secondary structures involving the 282 position, reducing genetic variability at this position due to fitness cost. This could also explain the rare occurrence of S282T development and high barrier to resistance for NI drugs.
More than 12,000 patients have received SOF in clinical trials, of whom, just over 1000 experienced virologic failure. S282T has been detected at the time of virologic failure in only 10 patients (<1% virologic failures). This suggests that SOF‐based regimens have an exceptionally high in vivo resistance barrier. The reason for virologic failure in the patients without S282T development is not completely clear, but the most likely explanation is persistence of a wild‐type virus that was not completely eradiated by the treatment. Moreover, several studies have demonstrated that virologic failures of an SOF‐based regimen can be successfully retreated with another SOF‐based regimen.19, 20 In the current study, eight of nine patients who failed SOF or LDV/SOF with treatment‐emergent S282T achieved SVR following retreatment with a second course of SOF‐based regimen, with either a second or third DAA or longer treatment duration. The successful retreatment of patients despite previous development of S282T is in part due to the rapid decline of S282T in absence of drug selection pressure and the high intracellular concentration of SOF in human hepatocytes in treated patients (73 μM),35 which greatly exceeds the inhibition constant of HCV NS5B RNA‐dependent RNA polymerase (Ki =0.42 μM).36 The low fold change in susceptibility of S282T to SOF (<20‐fold) suggests that the intracellular concentration of SOF may be sufficient for suppression of S282T in most patients. Longitudinal posttreatment samples were available in eight of 10 patients; S282T was shown to decline over time. Interestingly, by studying the evolution at position 282 in the patients with emergent S282T at virologic failure of SOF‐based regimen, we observed reversion of S282T to wild type resulting in generation of new serine codons not present at baseline, suggesting a footprint of S282T reversion, as described previously for one patient who developed S282T after SOF monotherapy.37
Here, we show that these serine footprint codons emerged in six of eight patients with decline of S282T at posttreatment. In one patient, the footprint codon was shown to persist for up to 16 months posttreatment. Encouragingly, despite the presence of footprint codon, this patient achieved SVR12 after treatment with SOF/VEL+VOX for 12 weeks. Moreover, in over 10,000 SOF‐naïve patients, the serine footprint codons at position 282 were only found in two patients at baseline (UCU and UCC). These two patients were treated previously with the NI VX‐135, and we cannot exclude possible S282T emergence during that treatment resulting in reversion of S282T posttreatment and generation of footprint codons in these patients. Because S282T declines rapidly in the absence of drug pressure, it is possible that presence of footprint codons can be used as a tool to identify patients who developed S282T during SOF or other NI‐based regimens but in which S282T already reverted to wild type at the time of sampling. By analyzing all patients experiencing virologic failure in the SOF clinical trials (n = 901) for the presence of serine footprint codons, we found one additional patient with serine footprint codon at virologic failure after treatment with SOF‐based regimen. This patient had the serine codon AGC at position 282 at baseline and the footprint codon UCC at 12 weeks posttreatment, suggesting that this patient had emergent S282T after an SOF‐based regimen that had reverted to wild type at time of sampling. It is important to continue to use deep sequencing to enable analysis of posttreatment HCV evolution, because standard population sequencing may be unable to resolve mixtures of codons, as described previously.37
In conclusion, SOF‐based regimens have an exceptionally high in vivo resistance barrier. The S282T substitution is not seen in untreated patients and is rarely detected (1%) in patients with virologic failure after treatment with an SOF‐based regimen. In addition, the S282T substitution is unfit in vivo and will disappear in the majority of patients in the absence of drug selection pressure. Therefore, successful retreatment of previous SOF‐failure patients is possible in the presence of S282T substitution with SOF‐based regimens.
Acknowledgment
We thank the patients, their families, and all participating investigators in the below studies: GS‐US‐334‐0107, GS‐US‐334‐0125, GS‐US‐337‐0109, GS‐US‐337‐1118, GS‐US‐334‐0108, GS‐US‐334‐0126, GS‐US‐337‐0113, GS‐US‐337‐1119, GS‐US‐334‐0109, GS‐US‐334‐0133, GS‐US‐337‐0115, GS‐US‐337‐1406, GS‐US‐334‐0110, GS‐US‐334‐0138, GS‐US‐337‐0118, GS‐US‐337‐1468, GS‐US‐334‐0114, GS‐US‐334‐0139, GS‐US‐337‐0121, GS‐US‐337‐1512, GS‐US‐334‐0115, GS‐US‐334‐0151, GS‐US‐337‐0122, P7977‐0523, GS‐US‐334‐0116, GS‐US‐334‐0153, GS‐US‐337‐0123, P7977‐0724, GS‐US‐334‐0118, GS‐US‐334‐0154, GS‐US‐337‐0124, P7977‐1231, GS‐US‐334‐0119, GS‐US‐334‐1274, GS‐US‐337‐0125, P7977‐2025, GS‐US‐334‐0123, GS‐US‐337‐0102, GS‐US‐337‐0131, GS‐US‐334‐0124, GS‐US‐337‐0108, and GS‐US‐337‐0133.
Potential conflict of interest: Nothing to report.
This study was funded by Gilead Sciences, Inc.This study was presented in part at the 65th Annual Meeting of the American Association for the Study of Liver Diseases, San Francisco, CA, November 13‐17, 2015.
REFERENCES
- 1. Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis 2005;5:558‐567. [DOI] [PubMed] [Google Scholar]
- 2. Afdhal N, Zeuzem S, Kwo P, Chojkier M, Gitlin N, Puoti M, et al. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 2014;370:1889‐1898. [DOI] [PubMed] [Google Scholar]
- 3. Afdhal N, Reddy KR, Nelson DR, Lawitz E, Gordon SC, Schiff E, et al. Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N Engl J Med 2014;370:1483‐1493. [DOI] [PubMed] [Google Scholar]
- 4. Kohli A, Kapoor R, Sims Z, Nelson A, Sidharthan S, Lam B, et al. Ledipasvir and sofosbuvir for hepatitis C genotype 4: a proof‐of‐concept, single‐centre, open‐label phase 2a cohort study. Lancet Infect Dis 2015;15:1049‐1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gane EJ HR, Yang Y, Svarovskaia E, Pang PS, McHutchison JG, Stedman CA. ISVHLD: ledipasvir/sofosbuvir single tablet regimen is effective in patients with HCV genotype 2 infection [Abstract]. Presented at the 15th International Symposium on Viral Hepatitis and Liver Diseases (ISVHLD); Berlin, Germany; June 26‐28, 2015. Abstract O-25.
- 6. Abergel A, Metivier S, Samuel D, Jiang D, Kersey K, Pang PS, Svarovskaia E, et al. Ledipasvir plus sofosbuvir for 12 weeks in patients with hepatitis C genotype 4 infection. Hepatology 2016;64:1049‐1056. [DOI] [PubMed] [Google Scholar]
- 7. Abergel A, Asselah T, Metivier S, Kersey K, Jiang D, Mo H, et al. Ledipasvir‐sofosbuvir in patients with hepatitis C virus genotype 5 infection: an open‐label, multicentre, single‐arm, phase 2 study. Lancet Infect Dis 2016;16:459‐464. [DOI] [PubMed] [Google Scholar]
- 8. Lawitz E, Lalezari JP, Hassanein T, Kowdley KV, Poordad FF, Sheikh AM, et al. Sofosbuvir in combination with peginterferon alfa‐2a and ribavirin for non‐cirrhotic, treatment‐naive patients with genotypes 1, 2, and 3 hepatitis C infection: a randomised, double‐blind, phase 2 trial. Lancet Infect Dis 2013;13:401‐408. [DOI] [PubMed] [Google Scholar]
- 9. Zeuzem S, Dusheiko GM, Salupere R, Mangia A, Flisiak R, Hyland RH, et al. Sofosbuvir and ribavirin in HCV genotypes 2 and 3. N Engl J Med 2014;370:1993‐2001. [DOI] [PubMed] [Google Scholar]
- 10. Lam AM, Espiritu C, Bansal S, Micolochick Steuer HM, Niu C, Zennou V, et al. Genotype and subtype profiling of PSI‐7977 as a nucleotide inhibitor of hepatitis C virus. Antimicrob Agents Chemother 2012;56:3359‐3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Svarovskaia ES, Dvory‐Sobol H, Parkin N, Hebner C, Gontcharova V, Martin R, et al. Infrequent development of resistance in genotype 1‐6 hepatitis C virus‐infected subjects treated with sofosbuvir in phase 2 and 3 clinical trials. Clin Infect Dis 2014;59:1666‐1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu S, Rajyaguru S, Chiu S, Hebner C, Svaroskaia E, Gontcharova V, Doehle B, et al. Sofosbuvir selects the NS5B S282T mutation in vitro in genotype 1‐6 replicons and is not cross‐resistant to resistance‐associated variants selected by other classes of antiviral inhibitors In: 64th Annual Meeting of the American Association for the Study of Liver Diseases: The Liver Meeting (AASLD); Washington, DC; November 1‐5, 2013. Poster 1094. [Google Scholar]
- 13. Gane EJ, Stedman CA, Hyland RH, Ding X, Svarovskaia E, Symonds WT, et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N Engl J Med 2013;368:34‐44. [DOI] [PubMed] [Google Scholar]
- 14. Lawitz E, Poordad FF, Pang PS, Hyland RH, Ding X, Mo H, et al. Sofosbuvir and ledipasvir fixed‐dose combination with and without ribavirin in treatment‐naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open‐label, randomised, phase 2 trial. Lancet 2014;383:515‐523. [DOI] [PubMed] [Google Scholar]
- 15. Gane E, Stedman C, Garg V, George S, Kieffer T, Krop J, et al. An interferon‐ and ribavirin‐free 12‐week regimen of once‐daily VX‐135 and daclatasvir in treatment‐naïve patients with genotype 1 HCV infection In: The International Liver Congress 2014, 49th Annual Meeting of the European Association for the Study of the Liver; London, UK; April 9‐13, 2014. [Google Scholar]
- 16. Gane EJ, Pockros PJ, Zeuzem S, Marcellin P, Shikhman A, Bernaards C, et al. Mericitabine and ritonavir‐boosted danoprevir with or without ribavirin in treatment‐naive HCV genotype 1 patients: INFORM‐SVR study. Liver Int 2015;35:79‐89. [DOI] [PubMed] [Google Scholar]
- 17. Svarovskaia ES, Gane E, Dvory‐Sobol H, Martin R, Doehle B, Hedskog C, et al. L159F and V321A sofosbuvir‐associated hepatitis C virus NS5B substitutions. J Infect Dis 2016;213:1240‐1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tong X, Le Pogam S, Li L, Haines K, Piso K, Baronas V, et al. In vivo emergence of a novel mutant L159F/L320F in the NS5B polymerase confers low‐level resistance to the HCV polymerase inhibitors mericitabine and sofosbuvir. J Infect Dis 2014;209:668‐675. [DOI] [PubMed] [Google Scholar]
- 19. Wyles D, Pockros P, Morelli G, Younes Z, Svarovskaia E, Yang JC, et al. Ledipasvir‐sofosbuvir plus ribavirin for patients with genotype 1 hepatitis C virus previously treated in clinical trials of sofosbuvir regimens. Hepatology 2015;61:1793‐1797. [DOI] [PubMed] [Google Scholar]
- 20. Osinusi A, Kohli A, Marti MM, Nelson A, Zhang X, Meissner EG, et al. Re‐treatment of chronic hepatitis C virus genotype 1 infection after relapse: an open‐label pilot study. Ann Intern Med 2014;161:634‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sarrazin C, Dvory‐Sobol H, Svarovskaia ES, Doehle B, Pang PS, Chuang SM, et al. Prevalence of resistance‐associated substitutions in HCV NS5A, NS5B, or NS3 and outcomes of treatment with ledipasvir and sofosbuvir. Gastroenterology 2016;151:501‐512. [DOI] [PubMed] [Google Scholar]
- 22. Cheng G, Tian Y, Doehle B, Peng B, Corsa A, Lee YJ, Gong R, et al. In vitro antiviral activity and resistance profile characterization of the hepatitis C virus NS5A inhibitor ledipasvir. Antimicrob Agents Chemother 2016;60:1847‐1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shih IH, Vliegen I, Peng B, Yang H, Hebner C, Paeshuyse J, et al. Mechanistic characterization of GS‐9190 (tegobuvir), a novel non‐nucleoside inhibitor of hepatitis C virus NS5B polymerase. Antimicrobial Agents and Chemotherapy 2011;55:4196‐4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999;285:110‐113. [DOI] [PubMed] [Google Scholar]
- 25. Bartels DJ, Sullivan JC, Zhang EZ, Tigges AM, Dorrian JL, De Meyer S, et al. Hepatitis C virus variants with decreased sensitivity to direct‐acting antivirals (DAAs) were rarely observed in DAA‐naive patients prior to treatment. J Virol 2013;87:1544‐1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nguyen LT, Hall N, Sheerin D, Carr M, De Gascun CF; Irish Hepatitis C Outcomes Research Network . Naturally occurring HCV NS5A/B inhibitor resistance‐associated mutations to direct‐acting antivirals. Antivir Ther 2016;21:447‐453. [DOI] [PubMed] [Google Scholar]
- 27. Svarovskaia ES, Martin R, McHutchison JG, Miller MD, Mo H. Abundant drug‐resistant NS3 mutants detected by deep sequencing in hepatitis C virus‐infected patients undergoing NS3 protease inhibitor monotherapy. J Clin Microbiol 2012;50:3267‐3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pawlotsky JM. Treatment failure and resistance with direct‐acting antiviral drugs against hepatitis C virus. Hepatology 2011;53:1742‐1751. [DOI] [PubMed] [Google Scholar]
- 29. Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 2010;465:96‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lawitz EJ, Gruener D, Hill JM, Marbury T, Moorehead L, Mathias A, et al. A phase 1, randomized, placebo‐controlled, 3‐day, dose‐ranging study of GS‐5885, an NS5A inhibitor, in patients with genotype 1 hepatitis C. J Hepatol 2012;57:24‐31. [DOI] [PubMed] [Google Scholar]
- 31. Chen ZW, Li H, Ren H, Hu P. Global prevalence of pre‐existing HCV variants resistant to direct‐acting antiviral agents (DAAs): mining the GenBank HCV genome data. Sci Rep 2016;6:20310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vince B, Hill JM, Lawitz EJ, O'Riordan W, Webster LR, Gruener DM, et al. A randomized, double‐blind, multiple‐dose study of the pan‐genotypic NS5A inhibitor samatasvir in patients infected with hepatitis C virus genotype 1, 2, 3 or 4. J Hepatol 2014;60:920‐927. [DOI] [PubMed] [Google Scholar]
- 33. Rong L, Dahari H, Ribeiro RM, Perelson AS. Rapid emergence of protease inhibitor resistance in hepatitis C virus. Sci Transl Med 2010;2:30ra32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sarrazin C, Zeuzem S. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 2010;138:447‐462. [DOI] [PubMed] [Google Scholar]
- 35. Babusis D, Curry MP, Denning J, Park Y, Murakami E, Afdhal N, et al. Nucleotide analog levels in liver explants from HCV infected subjects undergoing liver transplantation after up to 24 weeks sofosbuvir (GS‐7977) with ribavirin treatment. Presented at the 64th Annual Meeting of the American Association for the Study of Liver Diseases (AASLD); Washington, DC; November 1‐5, 2013.
- 36. Murakami E, Niu C, Bao H, Micolochick Steuer HM, Whitaker T, Nachman T, et al. The mechanism of action of beta‐D‐2′‐deoxy‐2′‐fluoro‐2′‐C‐methylcytidine involves a second metabolic pathway leading to beta‐D‐2′‐deoxy‐2′‐fluoro‐2′‐C‐methyluridine 5′‐triphosphate, a potent inhibitor of the hepatitis C virus RNA‐dependent RNA polymerase. Antimicrob Agents Chemother 2008;52:458‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hedskog C, Dvory‐Sobol H, Gontcharova V, Martin R, Ouyang W, Han B, et al. Evolution of the HCV viral population from a patient with S282T detected at relapse after sofosbuvir monotherapy. J Viral Hepat 2015;22:871‐881. [DOI] [PubMed] [Google Scholar]