

Abstract
Lipid droplets (LDs), the organelles central to alcoholic steatosis, are broken down by lipophagy, a specialized form of autophagy. Here, we hypothesize that ethanol administration retards lipophagy by down‐regulating dynamin 2 (Dyn2), a protein that facilitates lysosome re‐formation, contributing to hepatocellular steatosis. Primary hepatocytes were isolated from male Wistar rats fed Lieber–DeCarli control or ethanol (EtOH) liquid diets for 6‐8 weeks. Hepatocytes were incubated in complete medium (fed) or nutrient‐free medium (fasting) with or without the Dyn2 inhibitor dynasore or the Src inhibitor SU6656. Phosphorylated (active) forms of Src and Dyn2 and markers of autophagy were quantified using western blot analysis. Colocalization of LDs with autophagic machinery was determined using confocal microscopy. In hepatocytes from pair‐fed rats, LD breakdown was accelerated during fasting, as judged by smaller LDs and lower triglyceride (TG) content when compared with hepatocytes in complete media. Fasting‐induced TG loss in control hepatocytes was significantly blocked by either SU6656 or Dynasore. Compared with controls, hepatocytes from EtOH‐fed rats had 66% and 40% lower content of phosphorylated Src (pSrc) and phosphorylated Dyn2 (pDyn2), respectively, coupled with a lower rate of fasting‐induced TG loss. This slower rate of fasting‐induced TG loss was blocked in cells coincubated with Dynasore. Microscopic examination of EtOH‐fed rat hepatocytes revealed increased colocalization of the autophagosome marker LC3 on LDs with a concomitant decrease in lysosome marker LAMP1. Whole livers and LD fractions of EtOH‐fed rats exhibited simultaneous increase in LC3II and p62 over that of controls, indicating a block in lipophagy. Conclusion: Chronic ethanol administration slowed the rate of hepatocyte lipophagy, owing in part to lower levels of phosphorylated Src kinase available to activate its substrate, Dyn2, thereby causing depletion of lysosomes for LD breakdown. (Hepatology Communications 2017;1:501–512)
Abbreviations
- Dyn2
dynamin 2
- EtOH
ethanol
- LD
lipid droplet
- PBS
phosphate‐buffered saline
- pDyn2
phosphorylated Dyn2
- PNS
postnuclear supernatant
- pSrc
phosphorylated Src
- SEM
standard error of the mean
- TG
triglyceride
Liver damage from heavy drinking carries an enormous economic and health care burden worldwide.1 Alcohol‐induced liver damage is initially characterized by a buildup of fat (steatosis) in hepatocytes, which, if sustained by continued drinking, can progress to inflammation (steatohepatitis). If injury is prolonged by continued alcohol abuse, hepatic fibrogenesis (scar tissue formation) can occur, which may progress to alcoholic cirrhosis and ultimately liver failure.2 Steatosis is characterized by the abnormal accumulation of intracellular lipids in organelles called lipid droplets (LDs).3 Although steatosis is often benign and reversible if drinking ceases, it can progress to more advanced liver pathologies if drinking continues. The precise role of hepatic LD accumulation in the development of more severe pathologies associated with alcohol‐induced liver damage is not fully understood.
LDs are now recognized as highly dynamic organelles that play central roles in energy metabolism.4, 5 They are composed primarily of neutral lipids, including cholesteryl esters and triglycerides enclosed by a phospholipid monolayer, which has attached or embedded proteins. The hydrophobic cores of LDs are decorated with structural proteins, lipid‐synthesizing enzymes, lipases, and membrane‐trafficking proteins that regulate LD lipid storage and use. Proteins that are critical for vesicular transport, fusion, and fission have been implicated in LD interaction with other organelles, including the endoplasmic reticulum, endosomes, mitochondria, and peroxisomes for lipid exchange and metabolism.6 Under nutrient deprivation (fasting) conditions, LDs are an important source of energy, as they undergo a catabolic process called lipophagy, (a specific form of autophagy) to ultimately generate free fatty acids that undergo beta oxidation to generate energy for cellular demand.7, 8, 9
Macroautophagy (autophagy) is the general name of the catabolic process during which intracellular macromolecules and organelles are degraded in lysosomes to supply energy during stressful periods, such as nutrient deficiency. When an LD undergoes lipophagy, it is engulfed by an autophagosome (called a lipophagosome), which is then transported to and fuses with a lysosomes, forming an autolysosome in which the LD is degraded.8 After degradation of the autophagosome cargo in autolysosomes, new, smaller versions of lysosomes are generated from autolysosomes by recycling proto‐lysosomal membrane components through tubules that vesiculate (i.e., bud off) from autolysosomes to maintain a full complement of lysosomes. The aforementioned vesicles then mature into new lysosomes for further cycles of autophagy. This process, known as autophagic lysosome reformation, allows the cell to reuse the autolysosomal membrane and the contents within it for subsequent rounds of autophagy.9
Our recent studies report that lipophagy is regulated by membrane trafficking proteins including Rab710 and Dynamin2 (Dyn2).9 Dyn2 is a large GTPase that is activated via phosphorylation by Src kinase. Once phosphorylated, Dyn2 catalyzes constriction and scission of endocytic vesicles at the plasma membrane, thereby releasing early endosomes to the cell interior.11, 12, 13 The Src‐kinase is a nonreceptor protein tyrosine kinase that is activated during stress conditions and regulates cytoskeletal‐dependent membrane trafficking by phosphorylation of specific substrates, including Dyn2.14, 15, 16, 17 Inhibition of Dyn2 by small molecule inhibitors impairs lipophagy‐dependent LD breakdown in hepatic cells. Further studies indicate that Dyn2 activity is required for recycling lysosomes from autolysosomes during autophagic lysosome reformation,9 an important step for sustaining autophagy. Although there are data that indicate impaired autophagy after alcohol administration,18, 19, 20 there are no reports that have specifically examined lipophagy during or after chronic ethanol (EtOH) feeding. Our previous studies have focused on how EtOH exposure affects membrane protein trafficking, specifically that of the asialoglycoprotein receptor and its ligand interactions.21, 22 Additionally, we have examined several small Rab‐GTPases involved in vesicle trafficking and have demonstrated their decreased contents after alcohol exposure.23
Here, we report that, compared with fasted hepatocytes from control rats, the loss of triglycerides in fasted hepatocytes from alcohol‐fed animals was slower, and that this was associated with a partial loss of activated (phosphorylated) Src kinase, which was accompanied by a decline in Dyn2 activation in livers of EtOH‐fed rats.
Materials and Methods
MATERIALS
EtOH was purchased from Pharmaco‐AAPER (Brookfield, CT). IRDye infrared secondary antibodies and blocking buffer were from Li‐COR Biosciences (Lincoln, NE). BODIPY 493/503 was obtained from Invitrogen (Carlsbad, CA). Protease inhibitor cocktail, phosphatase inhibitor cocktail‐3, SU6656 (selective inhibitor of Src kinase), and dynasore hydrate (a noncompetitive inhibitor of the Dyn2 GTPase) were obtained from Sigma‐Aldrich (St. Louis, MO). Rabbit polyclonal anti‐Src antibody and mouse monoclonal anti‐LAMP1 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit polyclonal anti‐phospho Src (Tyr418) antibody and mouse anti‐actin antibody were from Millipore (Billerica, MA). Rabbit polyclonal anti‐LC3B antibody was obtained from Cell Signaling (Danvers, MA). Anti‐p62/SQSTM1 was purchased from Medical and Biological Laboratories Ltd (Nayoga, Japan). The rabbit polyclonal antibodies rose against Dyn2 and pDyn2 were kindly provided by Mark A. McNiven (Mayo Clinic, Rochester, MN). All other chemicals were obtained from Sigma Chemical Co. (St. Louis, MO) unless stated otherwise.
ANIMALS, DIET ADMINISTRATION, AND SAMPLE COLLECTION
All animals received humane care in accordance with guidelines established by the American Association for the Accreditation of Laboratory Animal Care. All protocols were approved by the Institutional Animal Care and Use Committee at the VA NWIHCS Research Service. Male Wistar rats weighing 175‐200 g, purchased from Charles River Laboratories (Portage, MI) were paired according to weight and fed control or EtOH‐containing Lieber–DeCarli diets24 for 6‐8 weeks as described previously.22 At the termination of feeding, rats were anesthetized with isofluorane, blood samples were collected from the vena cava, and euthanasia was complete after exsanguination. In some animals, hepatocytes were isolated and used in cell culture experiments, while in others, livers were excised, rinsed in TE buffer (10 mM Tris‐HCl, 1 mM EDTA, pH 7.4), weighed, and divided for various analyses. Portions of liver tissue were frozen immediately in liquid nitrogen and stored at −80°C until needed, or were freshly processed immediately to obtain liver postnuclear supernatants (PNSs) and LDs.
HEPATOCYTE CULTURE AND TREATMENT
Primary hepatocytes, isolated from control and EtOH‐fed rats were cultured as described previously.22 Briefly, hepatocytes suspended in William's media were seeded onto collagen‐coated 6‐well plates with or without coverslips. After 2 hours at 37°C in a 5% CO2 atmosphere, cells were washed with phosphate‐buffered saline (PBS) and further incubated at 37°C in either Williams media with 5% fetal bovine serum (fed cells) or in nutrient‐free Krebs‐Ringer‐HEPES (KRH‐fasted cells) for 4 hours as described previously.25, 26, 27 The latter incubations were conducted in the presence or absence of 15 μM SU6656 (Src kinase inhibitor) or 40 μM dynarose (Dyn2 inhibitor). The optimal concentrations of the aforementioned inhibitors were determined previously.9, 28, 29
ISOLATION OF LDs
LDs were isolated from freshly homogenized livers as described previously.23, 30 Briefly, PNS fractions were obtained by way of centrifugation (1000g for 10 minutes) of 20% homogenates. LDs were isolated by subjecting PNS fractions to discontinuous sucrose gradient ultracentrifugation as described previously.31 The white band (LD fraction) at the top of the gradient was collected and further purified by way of centrifugation (20,800g) for 10 minutes and then, the clear buffer underlying the LDs white band was removed and the LD fraction was brought up to 200 μL with TE buffer and frozen at −70°C for western blot analysis.
WESTERN BLOT ANALYSIS
Briefly, PNS and LD samples (diluted 1:20 and 1:4, respectively), were resolved under reducing conditions on 12% gels by SDS‐PAGE and transferred onto nitrocellulose membranes. Relevant protein bands on the blots were detected by probing with primary antibodies and fluorescent‐tagged secondary antibodies and blots were scanned by using the Odyssey infrared imaging system (Li‐Cor, Inc.). The band intensities were normalized by dividing their intensities by that of β‐actin in samples from PNS fractions. For LDs, we normalized the band intensities to perilipin‐2 as described previously.23
TRIGLYCERIDE MEASUREMENT
Measurement of triglyceride (TG) loss was performed as described previously, with minor modifications.32 Briefly, hepatocytes were collected by scraping 4 hours after treatment. Cells were centrifuged to obtain the cell pellets, which were resuspended in PBS and lysed by sonication. An aliquot of the cell lysate was saved for protein/DNA determination and the remaining lysate was used for total lipid extraction.33 Aliquots of the lipid extracts were saponified to quantify triglycerides using a diagnostic kit #TR22421 from Thermo Fisher Scientific (Middletown, VA). Triglyceride levels were normalized to total DNA content. The loss of triglyceride (TG) from the cells was determined by comparing the amount of TG in cells before (zero time) and 4 hours after treatment and calculated as the percent loss from their initial TG values at zero time.
HISTOLOGICAL ANALYSIS OF LDs USING FLUORESCENCE MICROSCOPY
Hepatocytes plated onto glass coverslips were subjected to fasted and fed conditions as indicated above and fixed for 20 minutes with 4% formaldehyde. The cells were made permeable with 0.1% Triton X‐100 for 2 minutes, washed with PBS, and then stained with BODIPY (1 μg/mL) in PBS for 20 minutes at room temperature, followed by three washes with PBS. Coverslips were mounted onto glass slides using a DAPI mounting media for nuclear stain (Vector Laboratories, Burlingame, CA). Images were acquired using a Zeiss 510 Meta Confocal Laser Scanning Microscope (Carl Zeiss, Thornwood, NY). Quantifications of LD number and size were performed using ImageJ software (National Institutes of Health, Bethesda, MA). For quantification, five different fields were randomly selected from each coverslip and data derived were pooled from samples obtained from four sets of control and EtOH‐fed animal pairs.
STATISTICAL ANALYSIS
The results are expressed as mean ± standard error of the mean (SEM). Comparisons between two groups were analyzed using the Student t test. P ≤ 0.05 was considered statistically significant.
Results
CHRONIC EtOH FEEDING RESULTED IN LIVER ENLARGEMENT AND STEATOSIS
After 6‐8 weeks of feeding rats with isocaloric control or EtOH‐containing liquid diets, we observed no difference in mean body weight between EtOH‐fed rats and their pair‐fed controls (data not shown). However, the liver/body weight ratio of EtOH‐fed rats was significantly higher than that of controls (control, 3.4 ± 0.07 g liver/100 g body weight; EtOH‐fed rats, 3.9 ± 0.13 g liver/100 g body weight; P = 0.003), indicating EtOH‐induced hepatomegaly, which was contributed in part by nearly 3‐fold higher levels of hepatic triglycerides compared with those in controls (control, 104 ± 11 mg TG/100 g body weight; EtOH‐fed rats, 305 ± 36 mg TG/100 g body weight; P < 0.02).
EtOH ADMINISTRATION IMPAIRED Dyn2 ACTIVITY AND REDUCED LD BREAKDOWN
We sought to determine whether chronic EtOH administration affected the activity and/or hepatic content of Dyn2, which can regulate autophagy and may control lipid homeostasis by regulating lipophagy of lipid droplets. Because Dyn2 is phosphorylated by an active Src kinase (pSrc) to achieve its biologically active form (pDyn2),16, 34 we quantified the total and phosphorylated forms of Src and Dyn2 in liver homogenates. As shown in Figure 1A, chronic EtOH administration did not affect the total content of either protein (bottom band in each panel). However, the active (phosphorylated) forms of Src and Dyn2 in EtOH‐fed rats were 66% and 40% lower, respectively, than those of pair‐fed control rats, when normalized to β‐actin (Fig. 1C,F). Normalization of pSrc and pDyn2 to their total (unphosphorylated) forms showed that their respective levels of phosphorylation of were 60% and 28% lower than controls (Fig. 1D,G).
Figure 1.
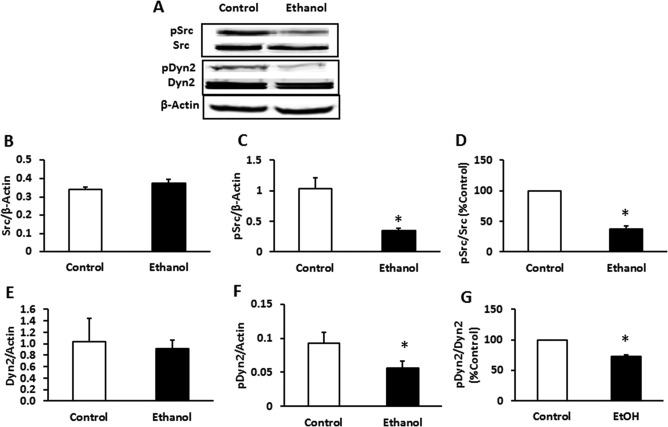
pSrc and pDyn2 content were reduced in the livers of EtOH‐fed rats. Male Wistar rats were pair‐fed a nutritionally balanced isocaloric control or EtOH Lieber–DeCarli diet for 6‐8 weeks. Liver PNS fractions were subjected to western blot analysis. (A) Representative western blot and mean densitometric ratios of (B) Src to actin, (C) pSrc to actin, (D) pSrc/Src, (E) Dyn to actin, (F) pDyn to actin, and (G) pDyn to Dyn. EtOH administration did not affect total Src and Dyn2 protein content, whereas pSrc and pDyn2 content were significantly decreased after EtOH feeding. Data are expressed as the mean ± SEM for eight pairs of animals. *P < 0.05.
We then tested whether the loss of Src kinase and Dyn2 activities influenced the rate of TG loss (LD breakdown) in fed and fasted hepatocytes from control and EtOH‐fed rats. As shown in Figure 2, neither the fed nor the fasting conditions affected the total Src or Dyn2 protein levels in hepatocytes of both groups of animals. However, the phosphorylated levels of these proteins were increased in hepatocytes of pair‐fed rats subjected to fasting but not of EtOH‐fed rats subjected to the same condition.
Figure 2.
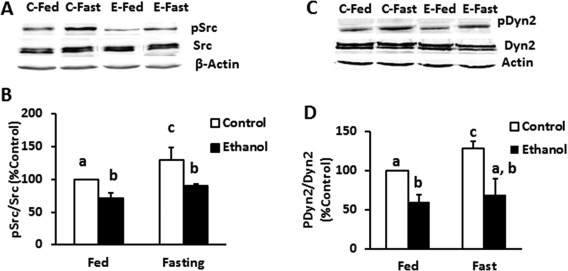
Ethanol administration impaired fasting‐induced Src kinase and Dyn2 activity. Representative western blots (A, C) and densitometric ratios (B, D) of total and active (phosphorylated) Src and Dyn in hepatocytes of control and EtOH‐fed rats exposed to either fed or fasting media as described in Materials and Methods. Data are expressed as the mean ± SEM (n = 3; P < 0.05).
Triglyceride (TG) loss from the cells was measured by their TG content that remained after 4 hours of incubation and was calculated as the percent difference from the TG content in unincubated hepatocytes. Cells from both groups of animals incubated under fed conditions exhibited little to no net loss of TGs after 4 hours of incubation. However, when the cells were exposed to fasting conditions, there was a 30% loss of TGs in hepatocytes from control rats, whereas fasted hepatocytes from EtOH‐fed rats lost only 10% of their triglyceride mass (Fig. 3A). When hepatocytes from both animal groups were incubated with the Src inhibitor SU6656, this fasting‐induced TG loss was significantly impaired in the control cells, but only minimally altered in cells from EtOH‐fed animals, likely due to the already‐impaired lipophagy in the EtOH‐fed animals. When hepatocytes were incubated with the Dyn2 inhibitor dynasore in the nutrient‐free media, dynasore almost completely blocked fasting‐enhanced lipid loss in cells from both groups of animals.
Figure 3.
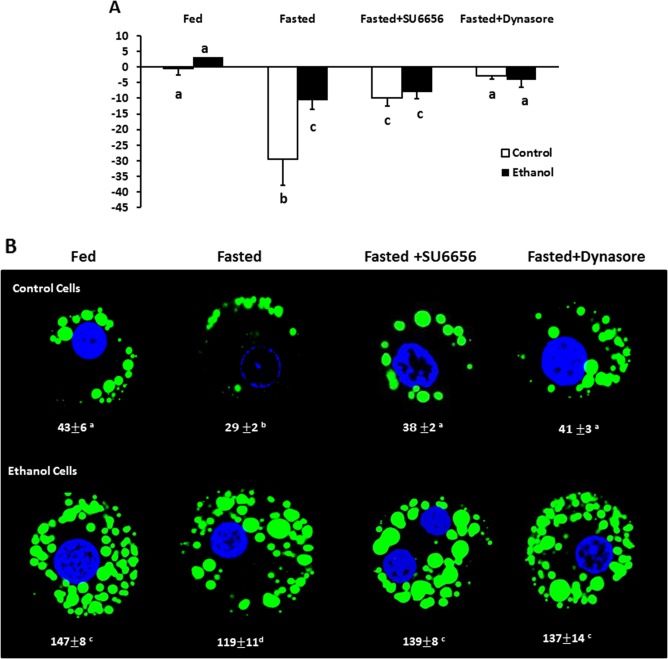
Chemical inhibition of Src kinase or Dyn2 equalized differences in TG clearance between control and EtOH‐fed rats. (A) Percent TG loss after 4 hours of fasting. Hepatocytes from control and EtOH‐fed rats were cultured for 4 hours in either fed or fasting media with and without 15 μM SU6656 (Src kinase inhibitor) or 40 μM Dynasore (a Dyn2 inhibitor). To quantify fat loss, TG content to μg DNA in cell pellets was measured before and after treatment and fat loss was calculated by comparing the amount of TG in cells before and after starvation and calculated percent loss from initial TG (0 hours) values. Loss of fat was more dramatic in fasted control cells and was inhibited by SU6656 and dynasore. Cells from EtOH‐fed animals demonstrated an impaired loss of fat with Dynasore treatment completely blocking the loss of fat. Data are expressed as the mean ± SEM for five pairs of animals (P < 0.05). (B) Representative micrographs of BODIPY staining of cells treated as indicated in the images. LDs in hepatocytes were stained with 1 μg/mL of BODIPY and visualized using confocal microscopy. Images were analyzed using ImageJ software to determine LD area. Results were obtained from five randomly selected cells from each slide, and data pooled from three independent experiments are expressed as the mean ± SEM (P < 0.05). Values not sharing a common letter are statistically different.
The TG loss in hepatocytes incubated in nutrient‐free media correlated with a parallel loss in BODIPY‐stained LDs, as judged by the cellular area occupied by LDs in hepatocytes from control and EtOH‐fed rats (Fig. 3B). Similar to the results shown in Fig. 3A, exposure of fasted hepatocytes to SU6656 or dynasore caused retention of LDs in cells from both groups of rats, indicating that the Src kinase and Dyn2 each strongly influences LD clearance, regardless of prior treatment in vivo. Our quantitative analyses of LD areas confirmed this, showing a 38% loss of LD area in fasted hepatocytes from pair‐fed control rats, compared with a 19% loss in identically treated cells from EtOH‐fed rats when calculated relative to their respective fed hepatocytes. SU6656 and dynasore exposure attenuated the loss due to nutrient deprivation, resulting in 11% and 5% loss, respectively, of LDs in cells from pair‐fed controls and a 5% and 7% loss, respectively, of LDs in hepatocytes of EtOH‐fed rats (Fig. 3B).
AUTOPHAGOSOME MARKER CONTENT IS HIGHER IN LDs FROM EtOH‐FED RATS
Based on the findings shown in Figure 3, previous alcohol administration caused delayed LD breakdown even in fasted (i.e., nutrient‐deprived) hepatocytes. We further tested whether this delay represents an alcohol‐induced slowdown of lipophagy by quantifying the autophagosome marker LC3 and p62/SQSTM1, an adapter protein that is ultimately degraded in the lysosome and is a sensitive indicator of autophagic activity, in liver PNS fractions and in partially purified LDs. EtOH‐fed rats had 1.7‐fold higher LC3II content in liver PNS (Fig. 4B) and 2.6‐fold higher LC3II in LD fractions than pair‐fed controls (Fig. 4D). Interestingly, we also observed a 1.32‐ and a 2.5‐fold elevation in LC3I (the unlipidated form of LC3II) in both hepatic PNS and LD fractions, respectively, of EtOH‐fed rats. In addition to exhibiting increased LC3II levels, both the PNS and LD fractions of EtOH‐fed rats simultaneously exhibited a 1.4‐ and 2.4‐fold increase in p62 protein level, respectively, over that of pair‐fed controls (Fig. 4F,H). P62 is a signaling adaptor protein that identifies the cargo to be sequestered by the autophagosomes, eventually becomes part of the cargo, and is degraded in the lysosomes when autophagy is enhanced. Here, simultaneous increase in the contents of both LC3II and p62 in PNS of EtOH‐fed rats suggested that chronic EtOH feeding impaired lysosomal degradation of autophagosomes, thus contributing to accumulation of autophagosomes (LC3II) in the liver. Similar increase of LC3II and p62 in LD fractions of EtOH‐fed rats suggested accumulation of undegraded autophagosomes containing LDs, a sign of defective lipophagy.
Figure 4.
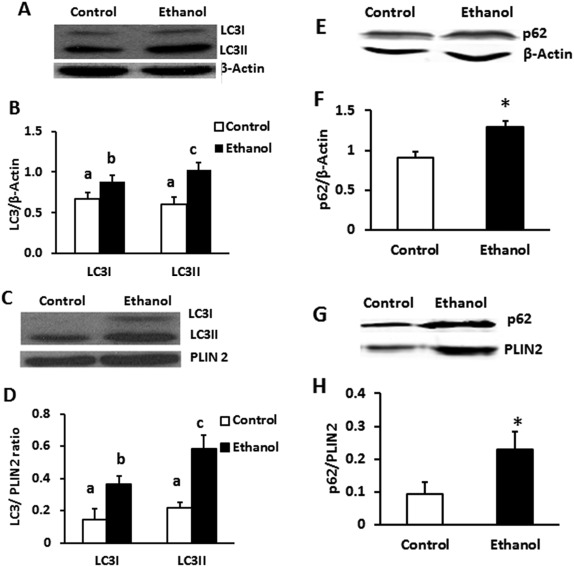
Hepatic PNS and LD fractions from EtOH‐fed rats showed higher LC3 and p62 content than controls. LDs were purified as outlined in Materials and Methods. (A,E) Liver PNS and (C,G) purified LD fraction from control and EtOH‐fed animals were subjected to western blot analysis. EtOH administration significantly increased both LC3 and p62 contents in both total liver (B,F) as well as LDs (D,H), respectively, compared with controls. Data are expressed as the mean ± SEM from 4‐8 pairs of animals (P < 0.05). Values not sharing a common letter are statistically different.
To further examine these possibilities, we performed quantitative immunochemistry using freshly seeded hepatocytes subjected to 4 hours of either the fed or fasted conditions described earlier. Cells were then immunostained for LC3 and LAMP1 (lysosomal marker) and stained with BODIPY to detect LDs.
Compared with fed cells, fasted hepatocytes from control rats had a significantly greater area covered by LC3‐positive puncta, indicating that fasting enhanced autophagosome biogenesis (Fig. 5A). In fed and fasted cells from EtOH‐fed rats, LC3‐positive puncta were 2‐ and 1.4‐fold higher, respectively, than those from corresponding controls (Fig. 5B). However, the cell area occupied by the lysosomal marker LAMP1 was 1.6‐ and 2‐fold lower in fed and fasted hepatocytes, respectively, from EtOH‐fed rats compared with controls (Fig. 5C). Furthermore, the incidence of LC3II‐LD colocalization was 2.8‐ and 1.8‐fold higher, respectively, in both fed and fasted hepatocytes from EtOH‐fed rats (Fig. 6B). LAMP1‐LD colocalization was significantly elevated in fasted control hepatocytes (Fig. 5C), whereas fasted cells from EtOH‐fed rats exhibited no such increase over fed hepatocytes and showed a 2.5‐fold lower incidence of LD–lysosome colocalization (Fig. 6C). Thomes et al.35 reported higher levels of LC3 after 24 hours of EtOH exposure to EtOH‐metabolizing HepG2 (VL‐17A) cells, indicating impaired degradation of LC3 by lysosomes, the number of which were lower, consistent with the lower lysosome numbers reported here.
Figure 5.
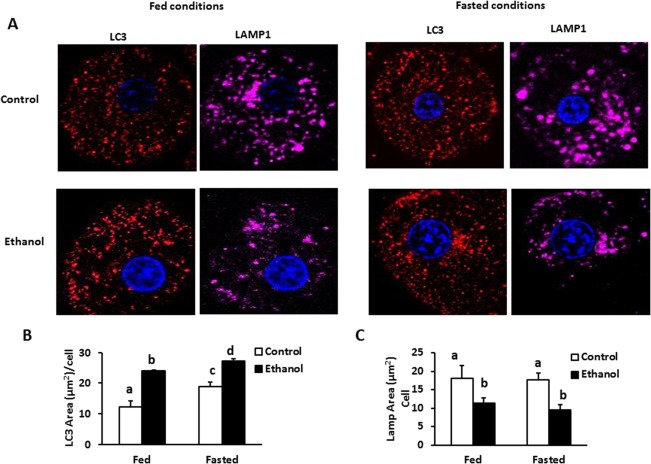
Hepatocytes from control and EtOH‐fed rats were treated with either fed (Williams media) or fasting media (KRH; Krebs‐Ringers HEPES buffer, a starvation media) for 4 hours and stained for LC3 (red) and LAMP1 (pink) proteins. (A) Control and EtOH hepatocytes treated with fed and fasting media. (B,C) Staining intensities of LC3 and Lamp1 were performed using ImageJ. Data pooled from three independent experiments are expressed as the mean ± SEM (P < 0.05). Values not sharing a common letter are statistically different.
Figure 6.
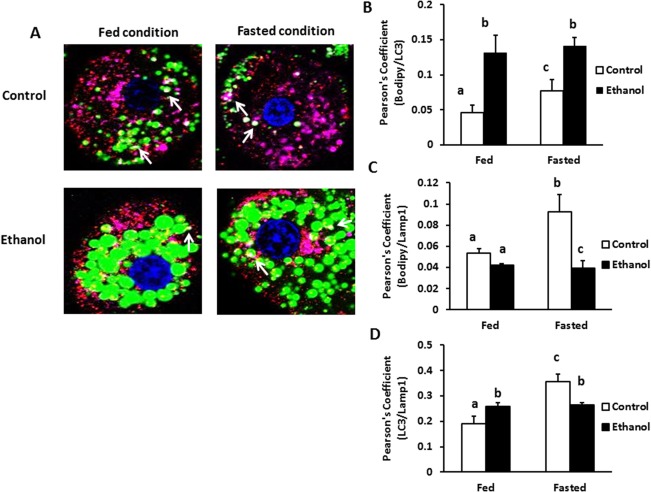
EtOH administration decreased colocalization of lysosomes with autophagosomes and LDs. Hepatocytes from control and EtOH‐fed animals were treated with either fed or fasting media for 4 hours and stained for colocalization of LDs (BODIPY stain, green) with LC3 (red) and LAMP1 (pink) proteins. (A) Images show colocalization of LC3 and Lamp1 on the LDs surface, as indicated by the white regions in the images. Colocalization was performed by ImageJ and calculating the Pearson's overlap coefficient, corresponding to the fraction of green pixels that overlap with red or pink pixels. (B) Overlap coefficient for LC3 and BODIPY in fed and fasted conditions. (C) Overlap coefficient for LAMP1 and BODIPY in fed and fasted conditions. (D) Overlap coefficient for LC3 and LAMP1 in fed and fasted conditions. Values not sharing a common letter are statistically different. Data pooled from three independent experiments are expressed as the mean ± SEM (P < 0.05).
Discussion
Alcohol exposure typically promotes TG synthesis and slows the breakdown of LDs, leading to accumulation of fat in liver cells.32 Here we show that chronic EtOH exposure inhibited (i.e., decreased the active form of) Dyn2, a membrane pinchase, which plays a critical role in repopulating lysosomes.9 Furthermore, such inhibition of Dyn2 after alcohol consumption likely compromised autophagic degradation of LDs in lysosomes, contributing to EtOH‐induced hepatic lipid accumulation.
Lower Dyn2 activity in livers of EtOH‐fed rats was associated with decreased phosphorylation of Src kinase. The latter enzyme phosphorylates Dyn2 to suggest that EtOH consumption likely blocked the phosphorylation activity of Src kinase, thereby exerting a downstream inhibitory effect on Dyn2. Indeed, treatment with the Dyn2 pharmacological inhibitors dyansore and MiTMAB (data not shown) completely blocked starvation‐induced triglyceride loss from hepatocytes of control and EtOH‐fed rats (Fig. 2A,B), supporting the concept that Dyn2 plays a crucial role in LD catabolism. Moreover, it signifies that Dyn2 inhibition by chronic EtOH administration plausibly contributed to hepatic fat accumulation in these animals.
Consistent with our earlier reports on hepatic autophagy in EtOH‐fed mice,36 EtOH‐fed rats similarly exhibited higher levels of autophagosomes in whole livers relative to pair‐fed controls. Interestingly, autophagosomes were tightly associated with partially purified LDs isolated from the livers of both groups of rats, and such association was still significantly higher in rats fed EtOH. Immunostaining studies further confirmed that hepatocytes from EtOH‐fed rats exhibited greater LD–autophagosome colocalization. Although the higher autophagosome content in EtOH‐fed rats may have occurred due to either increased autophagic flux or a block in lysosomal degradation,35, 36 the finding that autophagosomes were physically associated with LDs of EtOH‐fed rats suggests that autophagosomes were trafficked/directed to LDs and they attempted or perhaps sequestered the LDs for degradation. Using electron microscope imaging of liver tissue, others have shown that LDs were the predominant cargo of autophagosomes in livers of EtOH‐fed rats,37 suggesting that at least a subset of autophagosomes in EtOH‐fed rats sequestered LDs for subsequent degradation in lysosomes.
Despite attempts by hepatocytes to initiate lipophagy by recruiting autophagosomes to the LDs, greater numbers of LDs associating with p62 accumulated in cells of EtOH‐fed rats, indicating a block in lipophagy of LDs marked by p62 for degradation. Subsequently, when we analyzed the fusion events between autophagosomes–lysosomes and LD–lysosomes, we found striking reductions in both the aforementioned events in hepatocytes of EtOH‐fed rats under fasted conditions. The latter findings suggest that chronic EtOH exposure disturbed the fusion of LD‐ containing autophagosomes with lysosomes as well as the direct interaction between LDs and lysosomes. Findings that we reported recently suggest that disruption in fusion of these organelles may have occurred due to EtOH‐induced defects in the activities of LD‐associated Rab GTPases,23 which assist in the transport, docking, and fusion of vesicles with target membranes. Interestingly, EtOH exposure not only reduced the frequency of fusion of these organelles but also decreased the total area of lysosomes (Fig. 4) suggesting that EtOH exposure overwhelmed the hepatocytes with autophagosomal cargo by simultaneously decreasing the lysosome numbers to levels that were inadequate to perform LD breakdown. These findings clearly indicate that accumulation of autophagosomes in livers of EtOH‐fed rats reflects a slowdown in their degradation due to reduced numbers of lysosomes that are also functionally deficient.38, 39 The decrease in lysosome numbers reported here is consistent with previous reports that lysosomes from EtOH‐fed rats are functionally deficient38 because of impaired transit of lysosomal hydrolases (e.g., cathepsin L) to the lysosome compartment, indicating retarded lysosome biogenesis.39 There is also more recent evidence that lower lysosome content resulting from chronic EtOH administration is caused by defective lysosome biogenesis, which is regulated by the transcription factor TFEB.36
Our current findings, corroborating with foregoing reports on EtOH‐induced impairments of lysosome biogenesis, were simultaneously associated with down‐regulation of Dyn2 involved in autophagic lysosome reformation, revealing another novel target responsible for maintaining lysosomal homeostasis. Dyn2, which executes the scission of tubular structures from the autolysosomal compartment,9 thereby repopulating the cellular lysosomal pool, is a membrane trafficking protein that is indispensable for the autophagic pathway and recycling of intracellular macromolecules.9 Our findings suggest that the decrease in lysosome numbers induced by EtOH exposure, reported here, is partially due to the down‐regulation of Dyn2, which has a significant if not crucial function in maintaining adequate lysosome numbers in hepatocytes. This was illustrated not only by lower levels of active (phosphorylated) Dyn2 in EtOH‐fed rats but also by a nearly complete abolition of lipophagy after exposure to either the Src or Dyn2 inhibitor and as judged by LD retention and lower TG turnover. Also significant to this study are reports of EtOH‐induced phosphodiesterase activity, leading to decreased hepatic cyclic AMP, which is a known regulator for Src kinase activation.40, 41, 42, 43, 44, 45 Decreased cyclic adenosine monophosphate content after chronic EtOH consumption is likely responsible for the findings reported here, that of impaired activity of Src kinase and its substrate Dyn2, which cause downstream impairments in Dyn2 mediated lipophagy.
In conclusion, we have identified a novel mechanism by which EtOH exposure decreases hepatocyte lysosome population by down‐regulating Dyn2, contributing to a slowdown of lipophagy thereby resulting in accumulation of LDs. We show that chronic EtOH feeding decreased the content of active pSrc and pDyn2, and that such decreases were likely responsible for lowering lysosome numbers, as lysosome regeneration partly relies on Dyn2 activity. Further work is warranted to examine whether EtOH feeding causes defects in Dyn2 recruitment to autolysosomal membranes, and whether EtOH consumption disrupts cytosolic lipases that affect LD breakdown. Such investigations will further broaden our understanding of mechanisms governing LD turnover and alcoholic fatty liver disease.
Potential conflict of interest: Nothing to report.
This study was supported by the National Institute on Alcohol Abuse and Alcoholism (grants 5RC1AA019032 and 1R01 AA020735‐01) and the Department of Veterans Affairs.
REFERENCES
- 1. Singal AK, Anand BS. Recent trends in the epidemiology of alcoholic liver disease. Clin Liver Dis 2013;2:53‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ishak KG, Zimmerman HJ, Ray MB. Alcoholic liver disease: pathologic, pathogenetic and clinical aspects. Alcohol Clin Exp Res 1991;15:45‐66. [DOI] [PubMed] [Google Scholar]
- 3. Lieber CS, DeCarli LM. Animal models of chronic ethanol toxicity. Methods Enzymol 1994;233:585‐594. [DOI] [PubMed] [Google Scholar]
- 4. Guo Y, Cordes KR, Farese RV Jr, Walther TC. Lipid droplets at a glance. J Cell Sci 2009;122:749‐752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Krahmer N, Guo Y, Farese RV Jr, Walther TC. SnapShot: lipid droplets. Cell 2009;139:1024‐1024. [DOI] [PubMed] [Google Scholar]
- 6. Fujimoto Y, Itabe H, Sakai J, Makita M, Noda J, Mori M, et al. Identification of major proteins in the lipid droplet‐enriched fraction isolated from the human hepatocyte cell line HuH7. Biochim Biophys Acta 2004;1644:47‐59. [DOI] [PubMed] [Google Scholar]
- 7. Walther TC, Farese RV Jr. Lipid droplets and cellular lipid metabolism. Annu Rev Biochem 2012;81:687‐714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature 2009;458:1131‐1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schulze RJ, Weller SG, Schroeder B, Krueger EW, Chi S, Casey CA, et al. Lipid droplet breakdown requires dynamin 2 for vesiculation of autolysosomal tubules in hepatocytes. J Cell Biol 2013;203:315‐326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schroeder B, Schulze RJ, Weller SG, Sletten AC, Casey CA, McNiven MA. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology 2015;61:1896‐1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Koenig JH, Ikeda K. Disappearance and reformation of synaptic vesicle membrane upon transmitter release observed under reversible blockage of membrane retrieval. J Neurosci 1989;9:3844‐3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van der Bliek AM, Meyerowitz EM. Dynamin‐like protein encoded by the Drosophila shibire gene associated with vesicular traffic. Nature 1991;351:411‐414. [DOI] [PubMed] [Google Scholar]
- 13. Hinshaw JE. Dynamin and its role in membrane fission. Annu Rev Cell Dev Biol 2000;16:483‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ahn S, Maudsley S, Luttrell LM, Lefkowitz RJ, Daaka Y. Src‐mediated tyrosine phosphorylation of dynamin is required for beta2‐adrenergic receptor internalization and mitogen‐activated protein kinase signaling. J Biol Chem 1999;274:1185‐1188. [DOI] [PubMed] [Google Scholar]
- 15. Ahn S, Kim J, Lucaveche CL, Reedy MC, Luttrell LM, Lefkowitz RJ, Daaka Y. Src‐dependent tyrosine phosphorylation regulates dynamin self‐assembly and ligand‐induced endocytosis of the epidermal growth factor receptor. J Biol Chem 2002;277:26642‐26651. [DOI] [PubMed] [Google Scholar]
- 16. Shajahan AN, Timblin BK, Sandoval R, Tiruppathi C, Malik AB, Minshall RD. Role of Src‐induced dynamin‐2 phosphorylation in caveolae‐mediated endocytosis in endothelial cells. J Biol Chem 2004;279:20392‐20400. [DOI] [PubMed] [Google Scholar]
- 17. Cao H, Chen J, Krueger EW, McNiven MA. SRC‐mediated phosphorylation of dynamin and cortactin regulates the “constitutive” endocytosis of transferrin. Mol Cell Biol 2010;30:781‐792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Donohue TM Jr, Zetterman RK, Tuma DJ. Effect of chronic ethanol administration on protein catabolism in rat liver. Alcohol Clin Exp Res 1989;13:49‐57. [DOI] [PubMed] [Google Scholar]
- 19. Kharbanda KK, McVicker DL, Zetterman RK, MacDonald RG, Donohue TM Jr. Flow cytometric analysis of vesicular pH in rat hepatocytes after ethanol administration. Hepatology 1997;26:929‐934. [DOI] [PubMed] [Google Scholar]
- 20. von Haefen C, Sifringer M, Menk M, Spies CD. Ethanol enhances susceptibility to apoptotic cell death via down‐regulation of autophagy‐related proteins. Alcohol Clin Exp Res 2011;35:1381‐1391. [DOI] [PubMed] [Google Scholar]
- 21. McVicker BL, Tuma DJ, Kubik JA, Hindemith AM, Baldwin CR, Casey CA. The effect of ethanol on asialoglycoprotein receptor‐mediated phagocytosis of apoptotic cells by rat hepatocytes. Hepatology 2002;36:1478‐1487. [DOI] [PubMed] [Google Scholar]
- 22. Casey CA, McVicker BL, Donohue TM Jr, McFarland MA, Wiegert RL, Nanji AA. Liver asialoglycoprotein receptor levels correlate with severity of alcoholic liver damage in rats. J Appl Physiol 2004;96:76‐80. [DOI] [PubMed] [Google Scholar]
- 23. Rasineni K, McVicker BL, Tuma DJ, McNiven MA, Casey CA. Rab GTPases associate with isolated lipid droplets (LDs) and show altered content after ethanol administration: potential role in alcohol‐impaired LD metabolism. Alcohol Clin Exp Res 2014;38:327‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lieber CS, DeCarli LM. Liquid diet technique of ethanol administration: 1989 update. Alcohol Alcohol 1989;24:197‐211. [PubMed] [Google Scholar]
- 25. Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004;23:2891‐2906. [DOI] [PubMed] [Google Scholar]
- 26. Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA, et al. Nutrient‐sensing nuclear receptors coordinate autophagy. Nature 2014;516:112‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Prokesch A, Graef FA, Madl T, Kahlhofer J, Heidenreich S, Schumann A, et al. Liver p53 is stabilized upon starvation and required for amino acid catabolism and gluconeogenesis. FASEB J 2017;31:732‐742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blake RA, Broome MA, Liu X, Wu J, Gishizky M, Sun L, et al. SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Mol Cell Biol 2000;20:9018‐9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kockx M, Karunakaran D, Traini M, Xue J, Huang KY, Nawara D, et al. Pharmacological inhibition of dynamin II reduces constitutive protein secretion from primary human macrophages. PLoS One 2014;9:e111186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu W, Cassara J, Weller PF. Phosphatidylinositide 3‐kinase localizes to cytoplasmic lipid bodies in human polymorphonuclear leukocytes and other myeloid‐derived cells. Blood 2000;95:1078‐1085. [PubMed] [Google Scholar]
- 31. Ontko JA, Perrin LW, Horne LS. Isolation of hepatocellular lipid droplets: the separation of distinct subpopulations. J Lipid Res 1986;27:1097‐1103. [PubMed] [Google Scholar]
- 32. McVicker BL, Rasineni K, Tuma DJ, McNiven MA, Casey CA. Lipid droplet accumulation and impaired fat efflux in polarized hepatic cells: consequences of ethanol metabolism. Int J Hepatol 2012;2012:978136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 1957;226:497‐509. [PubMed] [Google Scholar]
- 34. Warnock DE, Baba T, Schmid SL. Ubiquitously expressed dynamin‐II has a higher intrinsic GTPase activity and a greater propensity for self‐assembly than neuronal dynamin‐I. Mol Biol Cell 1997;8:2553‐2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thomes PG, Ehlers RA, Trambly CS, Clemens DL, Fox HS, Tuma DJ, et al. Multilevel regulation of autophagosome content by ethanol oxidation in HepG2 cells. Autophagy 2013;9:63‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thomes PG, Trambly CS, Fox HS, Tuma DJ, Donohue TM Jr. Acute and chronic ethanol administration differentially modulate hepatic autophagy and transcription factor EB. Alcohol Clin Exp Res 2015;39:2354‐2363. [DOI] [PubMed] [Google Scholar]
- 37. Eid N, Ito Y, Maemura K, Otsuki Y. Elevated autophagic sequestration of mitochondria and lipid droplets in steatotic hepatocytes of chronic ethanol‐treated rats: an immunohistochemical and electron microscopic study. J Mol Histol 2013;44:311‐326. [DOI] [PubMed] [Google Scholar]
- 38. Kharbanda KK, McVicker DL, Zetterman RK, Donohue TM Jr. Ethanol consumption reduces the proteolytic capacity and protease activities of hepatic lysosomes. Biochim Biophys Acta 1995;1245:421‐429. [DOI] [PubMed] [Google Scholar]
- 39. Kharbanda KK, McVicker DL, Zetterman RK, Donohue TM Jr. Ethanol consumption alters trafficking of lysosomal enzymes and affects the processing of procathepsin L in rat liver. Biochim Biophys Acta 1996;1291:45‐52. [DOI] [PubMed] [Google Scholar]
- 40. Zederman R, Low H, Hall K. Effect of ethanol and lactate on the basal and glucagon‐activated cyclic AMP formation in isolated hepatocytes. FEBS Lett 1977;75:291‐294. [DOI] [PubMed] [Google Scholar]
- 41. Pennington S. Ethanol‐induced growth inhibition: the role of cyclic AMP‐dependent protein kinase. Alcohol Clin Exp Res 1988;12:125‐129. [DOI] [PubMed] [Google Scholar]
- 42. Diehl AM, Yang SQ, Cote P, Wand GS. Chronic ethanol consumption disturbs G‐protein expression and inhibits cyclic AMP‐dependent signaling in regenerating rat liver. Hepatology 1992;16:1212‐1219. [PubMed] [Google Scholar]
- 43. Cao W, Luttrell LM, Medvedev AV, Pierce KL, Daniel KW, Dixon TM, et al. Direct binding of activated c‐Src to the beta 3‐adrenergic receptor is required for MAP kinase activation. J Biol Chem 2000;275:38131‐38134. [DOI] [PubMed] [Google Scholar]
- 44. Ram PT, Iyengar R. G protein coupled receptor signaling through the Src and Stat3 pathway: role in proliferation and transformation. Oncogene 2001;20:1601‐1606. [DOI] [PubMed] [Google Scholar]
- 45. Forget MA, Sisson JH, Spurzem JR, Wyatt TA. Ethanol increases phosphodiesterase 4 activity in bovine bronchial epithelial cells. Alcohol 2003;31:31‐38. [DOI] [PubMed] [Google Scholar]