

Abstract
Hepatic macrophages (MΦs) are important in the development and progression of alcoholic liver disease (ALD). This study investigates the role of gp91phox (nicotinamide adenine dinucleotide phosphate oxidase 2) in the severity of ALD and specifically in regulating hepatic MΦ efferocytic capability and the subsequent reprogramming associated with resolution of inflammation. After 4 weeks of ethanol feeding, more severe ALD developed in gp91phox−/− mice than in wild‐type (WT) C57Bl/6J mice, evidenced by increased liver injury and inflammation. This phenomenon was not sex dependent, and thus the majority of experiments were performed with female mice. While total hepatic MΦ numbers did not differ between genotypes, hepatic infiltrating MΦs (IMs) were slightly more numerous in gp91phox−/− mice, and both IMs and resident Kupffer cells displayed enhanced proinflammatory and reduced tissue‐restorative programming compared with these cells from WT mice. The ratio of proinflammatory IMs with higher expression of Ly6C (Ly6Chi) to anti‐inflammatory IMs with lower expression of Ly6C (Ly6Clow) was significantly higher in gp91phox−/− mice compared to WT mice. Greater numbers of apoptotic cells accumulated in the liver of gp91phox−/− mice compared to WT mice, and receptors for binding and engulfing apoptotic cells were expressed at much lower levels on both Kupffer cells and IMs of gp91phox−/− mice. Interactions with apoptotic cells (binding and engulfment) in vitro were significantly fewer for gp91phox−/− MΦs than for WT MΦs, resulting in diminished expression of tissue restorative mediators by hepatic MΦs of gp91phox−/− mice. Conclusion: gp91phox plays a critical role in the differentiation of proinflammatory hepatic MΦs to a tissue‐restorative phenotype, likely through programming for efferocytosis, and thereby lessens the severity of ALD. These findings enhance our understanding of the tissue environmental cues that regulate MΦ phenotypes. This knowledge could help in designing MΦ‐targeting strategies to prevent and treat ALD. (Hepatology Communications 2017;1:765–779)
Abbreviations
- ALD
alcoholic liver disease
- ALT
alanine transaminase
- Arg1
arginase 1
- AT
apoptotic thymocyte
- CD
cluster of differentiation
- CFSE
carboxyfluorescein succinimidyl ester
- EtOH
ethanol
- FACS
fluorescence‐activated cell sorting
- HSC
hepatic stellate cells
- IFN‐γ
interferon gamma
- IL
interleukin
- IM
infiltrating macrophage
- iNOS
inducible nitric oxide synthase
- KC
Kupffer cell
- LSEC
liver sinusoidal endothelial cell
- MACS
magnetic‐activated cell sorting
- MCP‐1
monocyte chemoattractant protein‐1
- MerTK
MER proto‐oncogene, tyrosine kinase
- MMP
matrix metalloproteinase
- mRNA
messenger RNA
- MΦ
macrophage
- NADPH
nicotinamide adenine dinucleotide phosphate
- NK
natural killer
- NKT
natural killer T
- NOX
nicotinamide adenine dinucleotide phosphate oxidase
- NPC
nonparenchymal cell
- PCR
polymerase chain reaction
- PPAR
peroxisome proliferator activated receptor
- qPCR
real‐time polymerase chain reaction
- ROS
reactive oxygen species
- TIM
T‐cell immunoglobulin and mucin domain
- TNF‐α
tumor necrosis factor‐alpha
- VEGF
vascular endothelial growth factor
- WT
wild type
Introduction
Alcoholic liver disease (ALD) affects millions of people worldwide. The majority of heavy drinkers (>90%) develop steatosis, and approximately one third will develop cirrhosis.1 Alcohol‐related liver cirrhosis accounts for nearly half of liver cirrhosis‐associated deaths in the United States.1 Among patients with alcohol‐induced advanced liver cirrhosis, approximately 10% will develop hepatocellular carcinoma.2 Options for the prevention and treatment of ALD are limited due to the complexity and the incomplete understanding of the pathogenesis of the disease.
Macrophages (MΦs) have emerged as a critical player and therapeutic target in many chronic inflammatory diseases3 through their many proinflammatory and anti‐inflammatory activities. Evidence suggests that hepatic MΦs are important in the development and progression of ALD. Increased numbers of MΦs have been observed in all stages of ALD,4 and MΦs from patients with ALD appear to be activated and more sensitive to lipopolysaccharide stimulation.5 Using a murine model, we previously demonstrated that chronic ethanol feeding of mice causes not only activation of resident Kupffer cells (KCs) but also hepatic recruitment of infiltrating MΦs (IMs).6 The IMs consist of a proinflammatory Ly6Chi subset and an anti‐inflammatory tissue‐restorative Ly6Clow subset. Further, we reported that phagocytosis of dead cells (known as efferocytosis) promoted the Ly6Chi IMs to switch toward a phenotype similar to that of the tissue‐restorative Ly6Clow IMs, corresponding with decreased expression of proinflammatory factors and increased expression of anti‐inflammatory mediators, growth factors, and tissue‐remodeling genes.6 These data strongly suggest that efferocytosis may be critically important in reducing ethanol‐induced inflammation and initiating postinjury repair. Hence, hepatic MΦs may be a potential therapeutic target for ALD; however, heterogeneity and phenotype diversity of these cells require further investigation to guide the development of effective therapeutic strategies.
Ethanol (EtOH)‐induced reactive oxygen species (ROS) are critical in the causation of ALD, and both exogenous and endogenous antioxidants can ameliorate injury.7 Potential sources of ROS are many, not least of which are the nicotinamide adenine dinucleotide phosphate, reduced form, oxidases (NADPH oxidases or NOXs), which are multicomponent transmembrane enzymes that transport electrons across cellular membranes to reduce oxygen to superoxide. All NOX enzymes, including seven isoforms in humans (NOX1, NOX2, NOX3, NOX4, NOX5, DUOX1, and DUOX2) are able to reduce oxygen to superoxide, although they differ in subunit composition, tissue distribution, cellular localization, and activation. For example, NOX2 is composed of a transmembrane heterodimer, in which the catalytic subunit gp91phox is structurally stabilized by p22phox and four regulatory cytosolic subunits (p40phox, p47phox, p67phox, and the small guanosine triphosphatase Rac2). Notably, previous studies showed that p47phox−/− mice develop less severe ALD than wild‐type (WT) mice, consistent with the proposed mechanism that an NADPH oxidase contributes to ROS generation and cell damage during ALD.8, 9 However, while the p47phox subunit is expressed in many cells, including hepatocytes, and can couple with other NOXs, gp91phox is predominantly expressed by phagocytes, such as neutrophils and monocytes/MΦs. We therefore hypothesized that involvement of gp91phox in ALD might differ significantly from that of p47phox.
Our current investigation shows that gp91phox−/− mice develop significantly more severe injury than WT mice after chronic ethanol feeding. In the absence of gp91phox, ALD was associated with enhanced inflammation, exacerbated proinflammatory phenotypes of hepatic MΦs, and increased accumulation of apoptotic cells in the liver. Furthermore, we describe a critical role of gp91phox in enhancing MΦ efferocytic capabilities, altering expression levels of efferocytic receptors, and profoundly altering MΦ programming toward a tissue‐restorative phenotype.
Materials and Methods
ANIMAL TREATMENT
Breeding pairs of C57Bl/6J and gp91phox−/− mice (on a C57Bl/6J background) were purchased from Jackson Laboratories (Bar Harbor, ME). The colonies were expanded and maintained under pathogen‐free conditions at the Center for Laboratory Animal Care, the University of Colorado Anschutz Medical Campus. Mice were fed with the Lieber–DeCarli liquid diet containing 5% EtOH (EtOH‐fed) or liquid control diet containing equal calories of maltose dextrin (pair‐fed) for 4 weeks, as described.6 Sex‐ and age‐matched (8‐12 weeks old) mice were randomly divided into pair‐fed and EtOH‐fed groups. In some experiments, female mice were fed with EtOH for 10 days followed by one EtOH binge (5 g/kg, Gao binge model).10
MEASUREMENT OF ALCOHOL‐INDUCED LIVER INJURY AND STEATOSIS
Serum levels of alanine transaminase (ALT) were measured using a Teco Diagnostics kit. To determine hepatic steatosis, 10‐μm‐thick frozen sections of liver tissue were prepared using a Leica Cryostat CM1850 (Leica Biosystems, Buffalo Grove, IL). Frozen sections were stained with Oil Red O and hematoxylin (Sigma‐Aldrich, St. Louis, MO). Quantitation of liver triglycerides was performed using a GPO assay kit (Teco Diagnostics). Hepatic triglyceride values were expressed as milligrams of triglyceride per gram of liver tissue.
LIVER NONPARENCHYMAL CELL ISOLATION, MΦ PURIFICATION, AND FLOW CYTOMETRY ANALYSIS
Liver nonparenchymal cells (NPCs) were isolated following an established method.11 Briefly, the liver was perfused with Hank's balanced salt solution followed by a digestion buffer (1× Hank's balanced salt solution supplemented with 0.04% collagenase [type IV; Sigma‐Aldrich], 1.25 mM CaCl2, 4 mM MgSO4, and 10 mM 4‐(2‐hydroxyethyl)‐1‐piperazine ethanesulfonic acid). After digestion, single cells were passed through a 70‐μm cell strainer, and the cells were fractionated using 30% (weight/volume) Nycodenz (Axis‐Shield PoC AS, Oslo, Norway) at 1.155 g/mL to yield liver NPCs and further purified using 35% Percoll (Sigma‐Aldrich) at 1.04 g/mL.
Liver MΦs (including both KCs and IMs) were purified from liver NPCs, which were pooled from three to four mice, by magnetic‐activated cell sorting (MACS) using PE anti‐F4/80 and EasySep PE positive selection kit (Stemcell Technologies, Vancouver, Canada).
To further purify KCs/IMs, liver NPCs pooled from four mice were stained with antibodies (anti‐clusters of differentiation 45 [anti‐CD45], anti‐Ly6C, and anti‐Ly6G [BD Biosciences]) and anti‐F4/80 and anti‐CD11b (eBioscience) and then sorted by fluorescence‐activated cell sorting (FACS) using a BD FACSAria II cell sorter (BD Biosciences). The sorting strategy was carried out as described6; KCs were CD45+Ly6GlowF4/80hiCD11blow and IMs were CD45+Ly6GlowF4/80lowCD11bhi. Ly6C expression levels on IMs were measured by flow cytometry (FACScan cytometer; Cytek Development, Fremont, CA) using FlowJo software (Tree Star, Ashland, OR).
REAL‐TIME POLYMERASE CHAIN REACTION AND ELISA ANALYSES
Total RNA was isolated from liver tissues or various types of cells using PureLink RNA Mini kit (Thermo Fisher Scientific, Waltham, MA) as described by the manufacturer. RNA (0.5 μg) was reverse transcribed to complementary DNA using Moloney murine leukemia virus reverse transcriptase and amplified using SYBR Green PCR Master Mix (Applied Biosystems, Grand Island, NY) and primers for specific genes. All polymerase chain reaction (PCR) products were measured using a 7500 Real‐time PCR (qPCR) System and SDS software (Applied Biosystems). The 18S ribosomal RNA was used as the control for qPCR analyses.
Protein expression levels of tumor necrosis factor‐alpha (TNF‐α), interferon gamma (IFN‐γ), and interleukin 6 (IL‐6) in the serum and liver were measured using enzyme‐linked immunosorbent assay kits (Biolegend, San Diego, CA) according to the manufacturer's instruction.
MEASUREMENT OF EFFEROCYTIC RECEPTORS ON HEPATIC MΦs
To measure efferocytic receptors on hepatic MΦs, liver NPCs were stained with the following antibodies: anti‐CD45 and anti‐Ly6G (BD Biosciences), anti‐F4/80 and anti‐CD11b (eBioscience), and anti‐MER proto‐oncogene, tyrosine kinase Mer (MerTK), anti‐T‐cell immunoglobulin and mucin domain 3 (TIM3), or anti‐TIM4 (Biolegend). Expressions were measured by flow cytometry (FACScan cytometer; Cytek Development) using FlowJo software (Tree Star).
MEASUREMENT OF IN VITRO EFFEROCYTOSIS AND EFFEROCYTIC INTERACTIONS BY LIVER MΦs
To generate apoptotic hepatocytes, primary mouse hepatocytes were isolated from EtOH‐fed WT mice, labeled with carboxyfluorescein succinimidyl ester (CFSE), and treated with anti‐CD95 overnight. This preparation yielded >95% trypan blue‐positive hepatocytes.6 For effercytosis imaging, hepatocyte debris was generated from dead hepatocytes by a forcing pipette.
To generate apoptotic thymocytes (ATs), primary mouse thymocytes were isolated from WT mice and labeled with CFSE; apoptosis was induced by incubating with 0.1 μM dexamethasone at 37°C overnight. This preparation yielded >95% annexin V‐positive ATs.
To measure efferocytosis and binding of apoptotic cells (efferocytic interaction [EI]) by fluorescence imaging, liver MΦs, purified by MACS, were cultured in a 12‐well removable silicone cultivation chamber (ibidi; Martinstied, Germany) for 2 hours. The MΦs were stained with CellMask Orange plasma membrane stain (Life Technology, Carlsbad, CA) and then cocultured with CFSE‐labeled hepatocyte debris or AT in a ratio of 1:5 (MΦ:AT). Following 30 minutes coculturing, loosely adherent dead cells were removed by washing with phosphate‐buffered saline. The cells were visualized by using a Nikon C1 confocal microscope with Nikon EZ‐C1 software (Nikon, Tokyo, Japan). EI was calculated according to the following formula: (number of engulfed plus bound AT/number of MΦs) × 100. A minimum of 200 MΦs were counted randomly.12
To measure efferocytosis by flow cytometry, liver NPCs were isolated from EtOH‐fed WT and gp91phox−/− mice. NPCs were cocultured with ATs in 1:5 ratios for 30 minutes, vigorously washed, and then stained with anti‐CD45, anti‐F4/80, anti‐Ly6G, and anti‐Ly6C to identify MΦs. Cells were then analyzed by flow cytometry.
To determine the effect of EI on MΦ phenotype, MACS‐purified liver MΦs (7 × 105) were cocultured with 7 × 105 dead hepatocytes for 16 hours at 37°C as described.6 Noningested hepatocytes were removed by washing 3 times with phosphate‐buffered saline, and residual adherent MΦs were used for qPCR analyses. Total RNA could not be extracted from dead hepatocytes as no RNA was detectable by nano‐drop. Therefore, qPCR was not performed in the samples containing hepatocyte alone.
STATISTICAL ANALYSIS
Data were expressed as mean ± SEM. Statistical analysis was performed using GraphPad Prism software (La Jolla, CA). The unpaired t test was used to compare differences among dual samples. Genotype and treatment effects were analyzed by two‐way analysis of variance with Tukey post‐hoc comparisons. Differences in values were considered significant at P < 0.05.
Results
DEFICIENCY OF gp91phox ENHANCES LIVER TISSUE INJURY AND INFLAMMATION AFTER CHRONIC ALCOHOL INGESTION
We found that the hepatic messenger RNA (mRNA) expression levels of gp91phox were dramatically increased in EtOH‐fed C57Bl/6J (WT) mice compared with naive or pair‐fed mice. This increase was observed in both male and female mice (Fig. 1A). To determine the role of gp91phox in ALD, WT and gp91phox−/− mice were subjected to 4 weeks of ethanol feeding. We genotyped the gp91phox−/− mice and determined the substrain specificity to be C57Bl/6J (data not shown), in agreement with the original report describing the generation of the mouse.13 Although we did not use littermates as WT controls, to minimize the possible variations caused by animal facility‐related environmental factors, we bred the WT and gp91phox−/− mice in the same room of our Center for Laboratory Animal Care at the University of Colorado Anschutz Medical Campus for the entire study.
Figure 1.
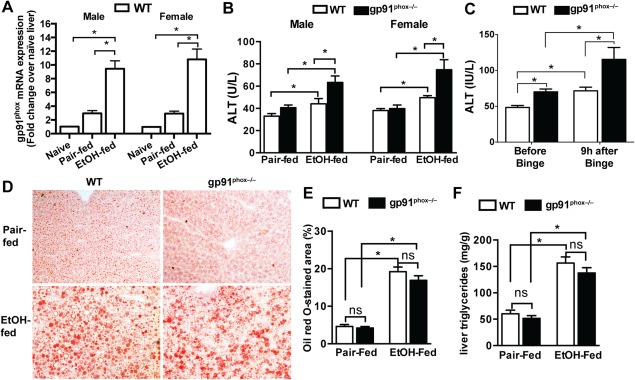
Chronic alcohol feeding causes more severe liver injury in gp91phox−/− mice. Female and male WT and gp91phox−/− mice were fed with the control liquid diet (pair‐fed) or a diet containing 5% EtOH (EtOH‐fed) for 4 weeks. (A) Hepatic gp91phox mRNA expression levels in naive, pair‐fed, and EtOH‐fed WT mice were measured. Data shown are from four mice per group. (B) Serum ALT activities were measured. *P < 0.05. Data shown are from 8 to 12 mice per group. (C) Female WT and gp91phox−/− mice were fed with EtOH for 10 days followed by one alcohol binge (5 g/kg). Serum ALT levels were measured before and 9 hours after the binge. *P < 0.05. Data shown are from six mice per group. (D,E) Liver cryosections were stained with Oil Red O. *P < 0.05. Data shown are representative of and quantified from six female mice per group (magnification ×100). (F) Liver triglyceride levels were measured from 12 female mice per group. *P < 0.05. Abbreviation: n.s., not significant. Error bars in the histograms stand for SEM.
Our data showed that serum ALT levels were significantly higher in both male and female gp91phox−/− mice compared with their WT counterparts (Fig. 1B). Because this phenomenon is not sex dependent, we performed the majority of experiments with female mice as female are more susceptible to ALD than male.14 To substantiate our finding of the differential susceptibilities to ALD between WT and gp91phox−/− mice, we used the Gao binge model, which produces more robust liver injury.10 After 10 days of EtOH feeding, mice were binged once with 5 g/kg of EtOH. Again, the ALT levels were much higher in gp91phox−/− mice than in WT mice (Fig. 1C). The degrees of alcohol‐induced hepatic steatosis were similar between WT and gp91phox−/− mice in either pair‐fed or EtOH‐fed groups, evidenced by comparable extents of Oil Red O staining and levels of liver triglycerides (Fig. 1D‐F). These results suggest that the gp91phox−/− mice are more susceptible than WT mice to developing liver injury, but not steatosis, after chronic ethanol ingestion.
The more severe liver injury in gp91phox−/− mice was accompanied by increased inflammatory responses. Protein expression levels of TNF‐α, IFN‐γ, and IL‐6 in the serum were significantly higher in EtOH‐fed gp91phox−/− mice than in WT mice (Fig. 2A). Moreover, the hepatic mRNA expression levels of IL‐1α, IL‐1β, TNF‐α, IFN‐γ, monocyte chemoattractant protein‐1 (MCP‐1), and IL‐6 were more than 3‐fold to 5‐fold higher in EtOH‐fed gp91phox−/− mice than in WT mice (Fig. 2B). Baseline levels of the majority of the mediators in pair‐fed mice were similar between the two genotypes. IFN‐γ and TNF‐α were expressed 2‐fold higher in pair‐fed gp91phox−/− mice than in WT mice. However, these levels in pair‐fed mice were 5‐fold to 15‐fold lower than those in EtOH‐fed mice, either WT or knockout mice (Supporting Fig. S1). Aside from the liver, circulating monocytes may also be activated during ALD, and the differential activation of these cells could contribute to the increased serum cytokine levels in gp91phox−/− mice. Therefore, we isolated circulating monocytes from EtOH‐fed WT and gp91phox−/− mice; however, we did not find significant differences in the mRNA expression levels of TNF‐α, IL‐1β, MCP‐1, IL‐6, and IFN‐γ, although there were trending increases of IL‐1β and MCP‐1 in monocytes from gp91phox−/− mice compared to WT mice (Supporting Fig. S2A).
Figure 2.
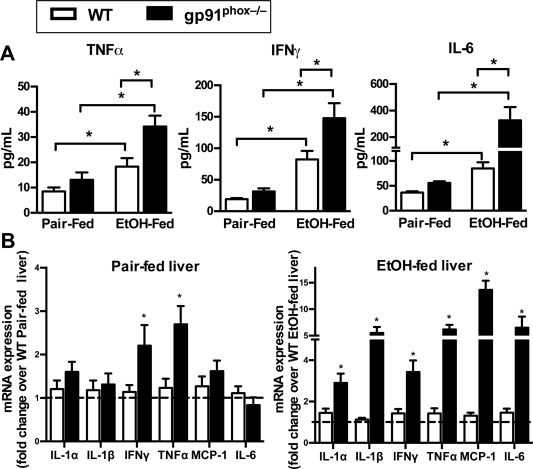
gp91phox−/− mice exhibit an increased inflammatory response to chronic alcohol treatment. Female WT and gp91phox−/− mice were treated as described in Fig. 1. (A) Serum levels of TNF‐α, IFN‐γ, and IL‐6 were determined by enzyme‐linked immunosorbent assay. *P < 0.05. Data shown are from 8 mice per group in pair‐fed groups and 10 mice per group in EtOH‐fed groups. (B) Hepatic mRNA expression levels of various proinflammatory mediators were measured. Fold changes were compared to WT mice of each respective group. *P < 0.05 compared with WT mice. Results shown are from 12 mice per group. Error bars in the histograms stand for SEM.
DELETION OF gp91phox CONFERS A MORE PROINFLAMMATORY PHENOTYPE OF HEPATIC MΦs
Given that NOX2 is important in the control of bacterial infection and that EtOH treatment increases intestinal permeability and affects gut microbiome overgrowth,15 we investigated the possibility that the enhanced hepatic inflammatory responses in gp91phox−/− mice were due to increased bacterial translocation. The serum levels of endotoxin were comparable between WT and gp91phox−/− mice (Supporting Fig. S2B). We also measured bacterial 16S RNA in the liver tissues, and the levels were similar between the two genotypes (Supporting Fig. S2C). Moreover, to determine whether EtOH feeding differentially affected Peyer's patches in the small intestine, we examined the numbers of various immune cells in the Peyer's patches of WT and gp91phox−/− mice. Our data showed no difference in the abundance of MΦs, neutrophils, dendritic cells, natural killer (NK) cells, natural killer T (NKT) cells, or T/B lymphocytes (Supporting Fig. S2D). Together, these data suggest that the enhanced hepatic inflammatory responses in the gp91phox−/− mice were not due to increased portal endotoxin but likely due to direct effects of gp91phox on cells that express it.
Although gp91phox is known to be predominantly expressed by phagocytes, such as neutrophils and MΦs, the presence of gp91phox on multiple cell types of the liver has been reported.16 In the naive liver, gp91phox mRNA was detected in MΦs, hepatocytes, hepatic stellate cells (HSCs), and liver sinusoidal endothelial cells (LSECs); however, the expression level was higher in MΦs than in other cell types (Supporting Fig. S3). EtOH feeding up‐regulated gp91phox expression to 12‐fold and 4‐fold in hepatic MΦs and neutrophils, respectively (Supporting Fig. S3), but strikingly, the gp91phox expression levels in hepatocytes, HSCs, and LSCEs remained unchanged (Supporting Fig. S3). This finding is consistent with the notion that gp91phox is a phagocytic‐specific NADPH oxidase.
Because hepatic MΦs play a critical role in regulating liver inflammation and injury during ALD, we examined the impact of gp91phox deletion on the phenotype and subset composition of liver MΦs. Regardless of the treatment (pair‐fed or EtOH‐fed), the WT and gp91phox−/− mice did not show significant differences in the numbers of total hepatic MΦs, resident KCs (CD45+Ly6GlowF4/80hiCD11blow), or recruited IMs (CD45+Ly6GlowF4/80lowCD11bhi) (Fig. 3A,B). As reported in other studies,6, 17 EtOH feeding resulted in a decrease in the number of KCs and an increase in IMs in both genotypes (Fig. 3B). Interestingly, the proportion of the proinflammatory Ly6Chi IMs, which express higher levels of proinflammatory cytokines and M1 markers,6 is much higher in gp91phox−/− mice (61.15% ± 3.677%) than in WT mice (32.30% ± 3.163%). Further, the ratio of Ly6Chi/Ly6Clow increased from approximately 1:2 in WT mice to almost 2:1 in gp91phox−/− mice (Fig. 3C). We have demonstrated that the higher ratio of Ly6Chi/Ly6Clow correlates with more severe liver injury.6 The data suggest that the impaired differentiation of Ly6Chi IMs into Ly6Clow IMs in EtOH‐fed gp91phox−/− mice may contribute to the more severe inflammation and liver injury in these mice.
Figure 3.
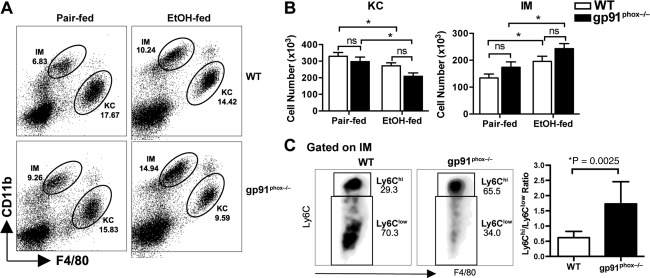
Comparison of the numbers of total hepatic MΦs and each subpopulation in WT and gp91phox −/− mice. Female WT and gp91phox−/− mice were treated as described in Fig. 1. (A) Liver NPCs from pair‐fed or EtOH‐fed WT and gp91phox−/− mice were analyzed by flow cytometry. KCs and IMs are distinguished by differential CD11b and F4/80 expression. Dot plots shown are representative of eight mice per group from two independent experiments. (B) The absolute numbers of KCs and IMs were calculated from eight mice per group. (C) The proportions of Ly6Chi IMs and Ly6Clow IMs among total IMs are compared between EtOH‐fed WT and gp91phox−/− mice (eight mice per group). *P < 0.05 compared to WT mice. Abbreviation: n.s., not significant. Error bars in the histograms stand for SEM.
To compare the phenotypes of MΦs, we purified KCs and IMs from pair‐fed as well as EtOH‐fed WT and gp91phox−/− mice. In pair‐fed mice, the gp91phox−/− MΦs did not show an enhanced inflammatory phenotype; instead, the expression levels of both the proinflammatory and tissue‐reparative genes were 2‐fold lower in gp91phox−/− MΦs than in WT MΦs (Fig. 4A). Similarly, when compared to naive mice, hepatic MΦs from gp91phox−/− mice expressed slightly lower levels of proinflammatory mediators (data not shown). These data suggest that the gp91phox−/− MΦs from the liver do not have an inflammatory phenotype at baseline. However, chronic EtOH feeding triggered much more severe inflammatory responses in KCs and IMs from gp91phox−/− mice than the respective MΦs from WT mice. The proinflammatory genes (IL‐1α, IL‐1β, IFN‐γ, TNF‐α, IL‐6, IL‐12p40, inducible nitric oxide synthase [iNOS]) were induced to greater levels in gp91phox‐deficient MΦs than WT MΦs. In contrast, genes related to tissue repair and anti‐inflammation (mannose receptor C‐type 1, IL‐1R2, arginase 1 [Arg1], vascular endothelial growth factor [VEGF], IL‐4, and IL‐13) were up‐regulated to a greater extent in WT MΦs than in gp91phox‐deficient MΦs (Supporting Fig. S4).
Figure 4.
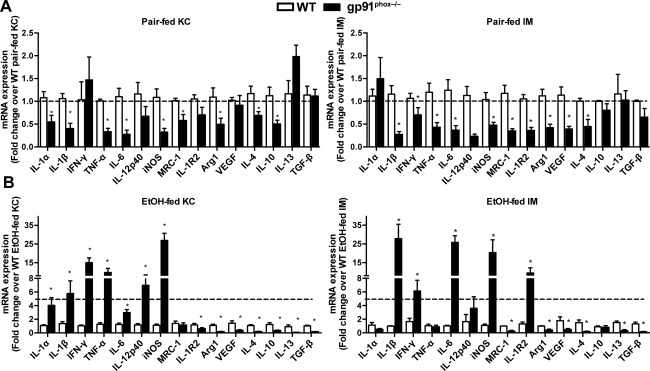
Both KCs and IMs in gp91phox−/− mice show a proinflammatory phenotype after chronic alcohol ingestion. Female WT and gp91phox−/− mice were treated as described in Fig. 1. After 4 weeks, KCs and IMs were purified by FACS from (A) pair‐fed and (B) EtOH‐fed mice. Expression levels of various proinflammatory, anti‐inflammatory, and tissue reparative genes were compared. *P < 0.05 compared with WT mice. Results shown are from 12 mice per group of three independent experiments. Abbreviation: MRC‐1, mannose receptor C‐type 1. Error bars in the histograms stand for SEM.
We further compared the gene expression levels of each subset of MΦs between the two genotypes. Compared with WT KCs, the gp91phox−/− KCs expressed more than 5‐fold higher levels of IL‐1β, IFN‐γ, TNF‐α, IL‐12p40, and iNOS (Fig. 4B). In contrast, gp91phox−/− KCs expressed significantly lower levels of anti‐inflammatory and tissue reparative genes, such as IL‐1R2, Arg1, VEGF, IL‐4, IL‐10, IL‐13, and transforming growth factor‐β. Similar to KCs, the gp91phox−/− IMs expressed much higher levels of IL‐1β, IFN‐γ, IL‐6, and iNOS but lower levels of mannose receptor C‐type 1, Arg‐1, VEGF, IL‐4, IL‐10, IL‐13, and transforming growth factor‐β than WT IMs. Interestingly, the gp91phox−/− IMs expressed approximately 10‐fold higher levels of IL‐1R2 than WT IMs. IL‐1R2 is a decoyed receptor that blocks IL‐1β signaling, and its expression level tracks that of IL‐1β.18 These results suggest that compared with their WT counterparts, both KCs and IMs of gp91phox−/− mice displayed a phenotype of exacerbated inflammation and compromised tissue restorative ability.
EtOH‐FED gp91phox−/− MICE EXHIBIT INCREASED ACCUMULATION OF APOPTOTIC CELLS IN THE LIVER
Compared with EtOH‐fed WT mice, we found a significantly higher number of apoptotic cells in EtOH‐fed gp91phox−/− mice (Fig. 5A,B). We hypothesize that gp91phox deletion in MΦs results in impaired removal of apoptotic cells (efferocytosis), thereby enhancing inflammation and exacerbating tissue injury. This is consistent with impaired efferocytosis described in other inflammatory settings in these mice.12, 19 We also observed some apoptotic cells in pair‐fed mice, although there was no significant difference between the genotypes. The pair‐feeding‐induced apoptosis may be due to the high caloric content of the Lieber–DeCarli liquid diet (35% fat) as a high‐fat diet has been reported to cause apoptosis in the liver.20
Figure 5.
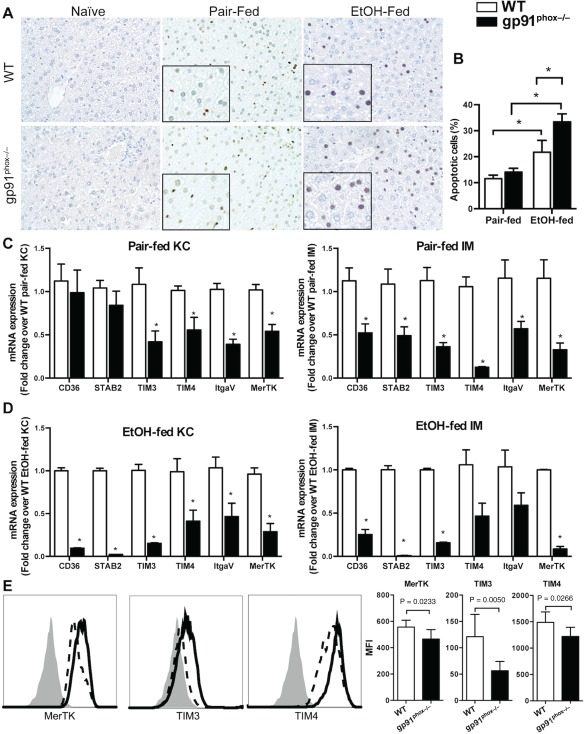
Increased apoptotic cell accumulation in the liver of EtOH‐fed gp91phox−/− mice and reduced efferocytic receptor expression in gp91phox−/− MΦs. (A,B) Naive, pair‐fed, and EtOH‐fed WT and gp91phox−/− mice were killed after 4 weeks of treatments. Apoptotic cells were visualized by TUNEL staining and quantified with ImageJ (magnification ×200, magnification of the inset ×400). *P < 0.05. Data shown are from six mice per group. (C,D) Liver KCs and IMs were purified from pair‐fed and EtOH‐fed WT and gp91phox−/− mice. The mRNA expression levels of various efferocytic receptors were measured. *P < 0.05 compared with WT mice. Data shown are from 12 mice per group in three independent experiments. (E) Comparison of protein levels of the receptors expressed on MΦs. In the graphs, solid line indicates EtOH‐fed WT MΦs, dash line indicates gp91phox−/− MΦs, and isotype controls are shown as solid gray histograms. MFI values are shown. *P < 0.05 compared with WT mice. Data shown are from seven mice per group. Abbreviations: ItgaV, integrin alpha V; MFI, mean fluorescence intensity; STAB2, stabilin 2; TUNEL, terminal deoxynucleotidyl transferase‐mediated deoxyuridine triphosphate nick‐end labeling. Error bars in the histograms stand for SEM.
Apoptotic cells express “eat‐me” signals, such as phosphatidylserine, that can be recognized by MΦs through efferocytic receptors. These receptors include stabilin 2 and members of the TIM protein family, such as TIM3 and TIM4. In addition to the direct recognition of eat‐me signals by receptors, extracellular bridge molecules can facilitate efferocytosis through receptors on phagocytes, such as integrin αVβ3, scavenger receptors (e.g., CD36), and the receptor MerTK.21, 22 We have demonstrated that chronic ethanol feeding can up‐regulate the expression of efferocytic receptors by both KCs and IMs.6 In the present study, we investigated whether gp91phox deficiency affects the expression of these receptors by MΦs. The data showed that ethanol feeding significantly up‐regulated all the receptors in WT‐KCs and WT‐IMs (Supporting Fig. S5). However, ethanol feeding increased the expression levels of some, but not all, receptors in gp91phox−/− KCs and IMs (Supporting Fig. S5). More importantly, in both pair‐fed and EtOH‐fed mice, the receptors were expressed at much lower levels in gp91phox−/− KCs and gp91phox−/− IMs than in the respective cell types of WT mice (Fig. 5C,D). Moreover, the surface protein expression levels of MerTK, TIM3, and TIM4 were lower on gp91phox−/− MΦs than on WT MΦs (Fig. 5E).
DEFICIENCY OF gp91phox IMPAIRS THE EFFEROCYTIC CAPABILITY OF HEPATIC MΦs
Bone marrow‐derived MΦs, peritoneal MΦs, and alveolar MΦs from gp91phox−/− mice have been demonstrated to exhibit impaired efferocytosis compared with their counterparts from WT mice.12 We investigated the efferocytic capability of the hepatic MΦs purified from EtOH‐fed gp91phox−/− and WT mice. The isolated MΦs were incubated with hepatocyte debris or ATs. We found that the liver MΦs from gp91phox−/− mice engulfed far fewer dead cells than WT MΦs (Fig. 6A; Supporting Fig. S6).
Figure 6.
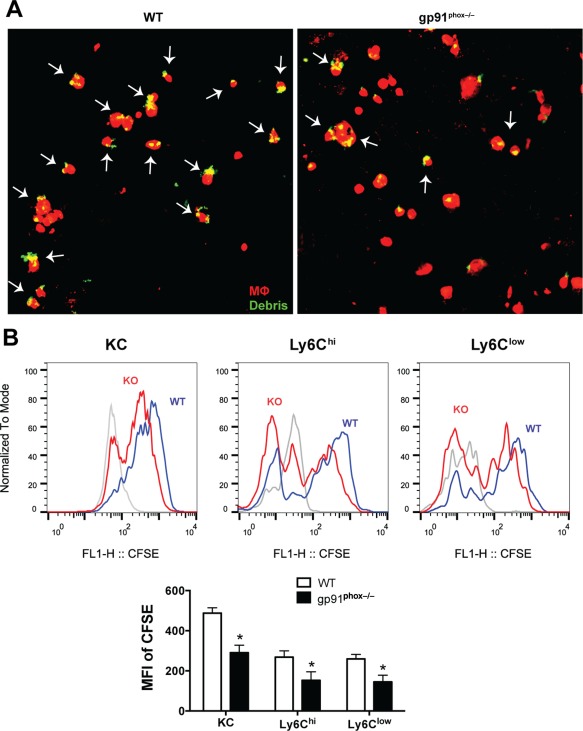
Hepatic MΦs from gp91phox−/− mice exhibit impaired efferocytic capability. (A) Hepatic MΦs (mixture of KCs and IMs) were isolated from female EtOH‐fed WT and gp91phox−/− mice and cocultured with CFSE‐labeled hepatocyte debris. After 30 minutes, the cells were visualized by confocal microscopy. Arrows indicate hepatocyte debris (green) captured/engulfed by MΦs (red). Data shown are from three independent experiments of a total of six mice per group. (B) Efferocytic interaction measured by flow cytometry as described in the Materials and Methods. Representative histogram shows CFSE intensities in KCs (gated on CD45+Ly6GlowF4/80hi), Ly6Chi IMs (gated on CD45+Ly6GlowF4/80low Ly6Chi), and Ly6Clow IMs (gated on CD45+Ly6GlowF4/80lowLy6Clow) from EtOH‐fed WT mice (WT, blue line) and gp91phox−/− mice (KO, red line) after 30 minutes coculture with CFSE‐labeled ATs. The histogram in solid gray represents control cells without incubation with ATs. The MFIs of CFSE are shown in the bar graph. *P < 0.05 compared with WT mice. Results shown are from four mice per group. Abbreviations: KO, knockout; MFI, mean fluorescence intensity. Error bars in the histograms stand for SEM.
As an additional approach to evaluate MΦ efferocytosis, we performed flow cytometric analyses. Efferocytic MΦs were identified as CFSE‐positive MΦs after a vigorous washing to remove unbound cells. When gated on KCs, Ly6Chi IMs, and Ly6Clow IMs, the fluorescent intensity of CFSE, reflecting the number of ingested ATs, shifted further to the right in the case of WT MΦs compared to gp91phox−/− MΦs (Fig. 6B). The mean fluorescence intensity of CFSE in gp91phox−/− MΦs was 40% lower than that in WT MΦs (Fig. 6B). Together, these data indicate an impaired efferocytic ability of the gp91phox−/− hepatic MΦs compared with the WT hepatic MΦs.
EFFEROCYTOSIS‐INDUCED MΦ REPROGRAMMING IS ABROGATED IN gp91phox−/− MICE
We previously demonstrated that co‐incubation of hepatic MΦs from EtOH‐fed mice with apoptotic hepatocytes altered MΦ programming with significant increases in the levels of tissue restorative factors produced.6 To examine the impact of gp91phox deficiency on this reprogramming of MΦs, we cocultured liver MΦs isolated from EtOH‐fed WT and gp91phox−/− mice with apoptotic hepatocytes. In cultures of MΦs alone, the levels of proinflammatory mediators IL‐1β and TNF‐α were markedly higher in gp91phox−/− MΦs than in WT MΦs, demonstrating stability of their proinflammatory phenotype (as shown in Fig. 3) during culture. Tissue restorative/anti‐inflammatory genes were minimally expressed at baseline by WT or gp91phox−/− MΦs except that the levels of matrix metalloproteinase 9 (MMP9) and IL‐1R2 were higher in gp91phox−/− MΦs (Fig. 7). Interactions with apoptotic hepatocytes dramatically increased tissue restorative and anti‐inflammatory genes in WT MΦs. For example, MMP9 and MMP12 were increased more than 30‐fold; levels of Arg1 and VEGF were increased more than 10‐fold; levels of hepatocyte growth factor and IL‐1R2 were increased more than 5‐fold. In contrast, following incubation with apoptotic hepatocytes, gp91phox−/− MΦs showed less than 2‐fold or 3‐fold increases of MMP9, MMP12, Arg1, and VEGF, while the levels of hepatocyte growth factor and IL‐1R2 were not changed or even decreased (Fig. 7). These data demonstrate that gp91phox deficiency impairs efferocytosis‐induced MΦ reprogramming.
Figure 7.
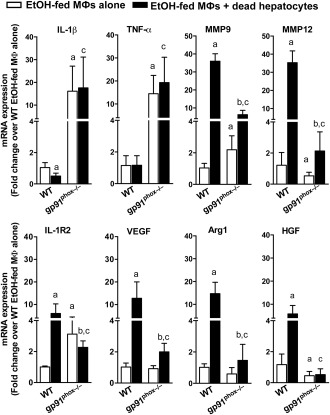
Comparison of the effects of efferocytic interaction on WT and gp91phox−/− hepatic MΦs. Liver MΦs were isolated from EtOH‐fed WT and gp91phox−/− mice. MΦs were cultured in the presence or absence of apoptotic hepatocytes for 16 hours. Expression levels of various genes associated with tissue repair and inflammation were compared. a P < 0.05 compared with WT MΦs alone. b P < 0.05 compared with gp91phox−/− MΦs alone. c P < 0.05 compared with WT MΦs plus hepatocytes. Results shown are from three independent experiments of 12 mice per group. Abbreviation: HGF, hepatocyte growth factor. Error bars in the histograms stand for SEM.
Discussion
This study highlighted a protective role of gp91phox in ALD. Increased accumulation of apoptotic cells and exacerbated inflammatory responses and tissue injury were observed in the liver of gp91phox−/− compared to that of WT mice. EtOH causes oxidative stress in hepatocytes, which could lead to cell death.23 However, EtOH‐induced hepatic inflammation is a critical cause of tissue injury during ALD.2, 24 The data presented demonstrate that the hepatic inflammatory environment is exacerbated in gp91phox−/− mice compared to WT mice, resulting in enhanced liver injury. Our data also suggest that this exacerbation of inflammation is due to impaired clearance of dead cells by hepatic MΦs.
Our finding of an anti‐inflammatory and protective role for gp91phox in ALD may appear to be paradoxical and certainly is at odds with the simplistic hypothesized role for ROS in this disorder; however, a large number of publications in recent years provide evidence to support anti‐inflammatory activities of gp91phox/NOX2. These studies show that under sterile conditions, gp91phox−/− mice exhibit prolonged hyperinflammation.19 Multiple mechanisms may contribute to these hyperinflammatory responses in gp91phox−/− mice, but substantial data support that various MΦs from gp91phox−/− mice exhibit impaired efferocytosis of apoptotic cells and poor suppression of proinflammatory mediator production associated with efferocytosis.12, 25 Consistent with these observations, we found greater accumulation of apoptotic cells in the livers of the ethanol‐treated gp91phox−/− mice and impaired interaction of gp91phox−/−‐deficient MΦs with apoptotic cells in vitro.
Here, we show deficient expression in gp91phox−/− MΦs of a number of efferocytic receptors known to mediate binding and/or engulfment of apoptotic cells. This is true for both KCs and IMs, suggesting that a functioning NOX2 is required for the receptor expression in heterogeneous and ontogenically diverse MΦ populations. As result of the diminished efferocytic receptor expression in the gp91phox−/− hepatic MΦs, we saw decreased efferocytosis and, in addition, marked impairment of efferocytosis‐induced reprogramming and anti‐inflammatory mediator production in the EtOH‐fed gp91phox−/− mice. Expression of many of these efferocytic receptors is governed by the nuclear receptor family that sense oxidized lipids, e.g., peroxisome proliferator activated receptors (PPARs) and liver X receptors.26, 27 Further, it is known that efferocytosis itself can also enhance expression of these receptors on MΦs.26, 28 These observations raise the hypothesis that NOX2 provides oxidized lipid ligands, potentially derived from ingested apoptotic cells, that activate and enhance the expression of the nuclear receptors,19, 27 and they, in turn, up‐regulate the efferocytic receptors in an autocrine feed‐forward manner to promote efferocytosis and MΦ reprogramming (Fig. 8). In support of this hypothesis are several additional observations: i) deficient activity of PPARγ has been described in inflammatory MΦs of gp91phox−/− mice,29 ii) absent ROS production in gp91phox‐deficient phagocytes is associated with decreased capacity of degrading phagocytosed material,30 iii) gp91phox deficiency is associated with more proinflammatory Ly6Chi MΦs and fewer restorative Ly6Clow MΦs in vivo, a programming switch that can be driven in WT cells by productive interaction with apoptotic cells.6
Figure 8.
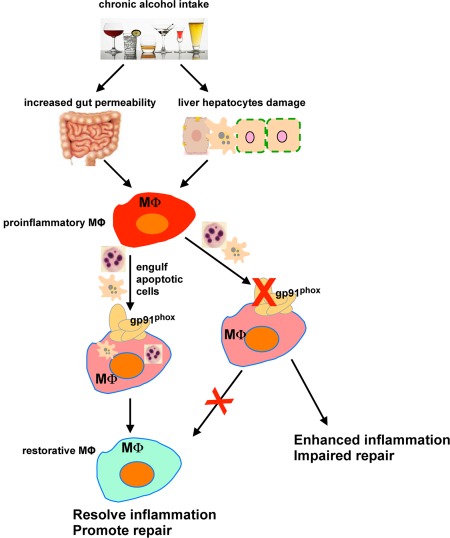
Schematic for the role of gp91phox in regulating hepatic MΦs and attenuating ALD. Chronic alcohol ingestion triggers the proinflammatory activation of hepatic MΦs. Phagocytosis of dead cells leads to the differentiation of proinflammatory MΦs to tissue‐restorative MΦs, which promotes the resolution of inflammation and tissue repair. gp91phox plays a critical role in regulating the efferocytic ability and phenotypic programing of hepatic MΦs. Deletion of gp91phox impairs the efferocytic capability of hepatic MΦs and prevents their switching from a proinflammatory state to a tissue‐restorative phenotype, thereby resulting in enhanced liver inflammation and tissue repair.
Aside from diminished efferocytosis, the absence of ROS from a functional NOX2 may alter MΦ inflammatory programming through changes in signal transduction31 and gene expression.32 For instance, it is widely reported that PPARγ is a key regulator of MΦ anti‐inflammatory phenotypes.33 Additionally, nuclear factor erythroid 2‐related factor 2 expression is also impaired in the absence of a functioning NOX2,34 and nuclear factor erythroid 2‐related factor 2 has been shown to ameliorate ALD.35 Defects in endogenous braking systems that attenuate inflammation and promote the resolution of inflammation may also explain exaggerated inflammation in the gp91phox−/− mice. Lower levels of cyclic adenosine monophosphate, adenosine, and hypoxia‐inducible factor have been described.36, 37 NOX2‐generated ROS is necessary for autophagy during phagocytosis.38, 39 Interestingly, defective autophagy in NOX2‐deficient MΦs has been mechanistically linked to enhanced inflammasome activity and increased IL‐1β release.39, 40 Moreover, the impairment of ROS‐dependent attenuation of Ca++ signaling and ROS‐dependent inactivation of proinflammatory mediators may also contribute to enhanced inflammation.41, 42
The role of NOX2 in nonalcoholic fatty liver disease and liver fibrosis has also been investigated in murine models. One report described a significantly worsened nonalcoholic fatty liver disease in gp91phox−/− mice compared to WT mice after high‐fat diet feeding43; however, another showed no effect on liver steatosis.44 Three studies have investigated the role of NOX2 in liver fibrosis, and the results suggest a profibrogenic role of NOX2 expressed by hepatic stellate cells but not hepatic MΦs.16, 45 Although the underlying molecular mechanisms are not completely understood, there is mounting evidence supporting an important anti‐inflammatory function of NOX2 in liver injury and repair. These findings in animal studies correlate with clinical observations in patients with chronic granulomatous disease. These patients have deficient NOX2 function and, in addition to being immunodeficient, suffer from a variety of inflammatory conditions,46 including various hepatic abnormalities.47
Using whole‐body rather than cell‐specific gp91phox knockout mice poses potential limitations, as the expression of NOX in various types of hepatic cells has been reported.16 However, our data revealed that the expression of gp91phox was 2‐fold lower in hepatocytes, HSCs, and LSECs than in MΦs (Supporting Fig. S3). More importantly, EtOH feeding did not increase gp91phox in these cells. In contrast, gp91phox was dramatically up‐regulated in MΦs and neutrophils of the liver after EtOH feeding. Although the current study focused on hepatic MΦs, the role of gp91phox deletion in neutrophils in modulating the susceptibility to ALD warrants investigation in future studies. Deficiency in gp91phox reportedly impairs the externalization of phosphatidylserine by neutrophils and thus inhibits the clearance of neutrophils by MΦs.48 Increased accumulation of neutrophils in the liver is a hallmark for alcoholic steatohepatitis.49 In murine models of ALD, clear evidence suggests that neutrophils contribute to ethanol‐induced inflammation and injury in the liver.50
In summary, hepatic MΦs have emerged as an important player in multiple pathologic events during ALD, including bacterial overload, liver inflammation, and fibrosis, and poor regenerative responses to acute‐on‐chronic liver injury.4, 5 However, whether MΦs play a pathologic or protective role in the pathogenesis of ALD is likely dependent on the predominant phenotype/programming of MΦs. The present study provides evidence to support that gp91phox plays a critical role in promoting efferocytosis by hepatic MΦs and diverting programming of these cells from a proinflammatory to a tissue‐restorative phenotype, thereby protecting the liver from alcohol‐induced injury (Fig. 8). To better understand the mechanistic involvement of hepatic MΦs in ALD and to better target these cells for therapy, it is critical to gain insights into the tissue environmental cues that dictate the MΦ phenotypes and molecular signaling pathways that regulate MΦ functions.
Author names in bold designate shared co‐first authorship.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1078/full.
Supporting Information
Acknowledgment
We thank Dr. Brent Palmer for assistance with FACS.
Potential conflict of interest: Nothing to report.
Supported by the National Institute on Alcohol Abuse and Alcoholism (NIAAA) (grants U01 AA021723: 09/10/2013‐08/31/2014, R21 AA022387: 09/20/2013‐08/31/2015, R21AA024636: 08/01/2016‐07/31/2018 to C.J.), National Institute of Allergy and Infectious Diseases (NIAID) (grant R01AI110408 to D.L.B.), National Natural Science Foundation of China (NSFC) (grant NSFC81428006 to C.J.), and National Heart, Lung, and Blood Institute (NHLBI) (grant P01HL034303 to D.L.B.).
REFERENCES
- 1. O'Shea RS, Dasarathy S, McCullough AJ; Practice Guideline Committee of the American Association for the Study of Liver Diseases; Practice Parameters Committee of the American College of Gastroenterology . Alcoholic liver disease. Hepatology 2010;51:307‐328. [DOI] [PubMed] [Google Scholar]
- 2. Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 2011;141:1572‐1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 2012;122:787‐795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Karakucuk I, Dilly SA, Maxwell JD. Portal tract macrophages are increased in alcoholic liver disease. Histopathology 1989;14:245‐253. [DOI] [PubMed] [Google Scholar]
- 5. Ju C, Mandrekar P. Macrophages and alcohol‐related liver inflammation. Alcohol Res 2015;37:251‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang M, You Q, Lor K, Chen F, Gao B, Ju C. Chronic alcohol ingestion modulates hepatic macrophage populations and functions in mice. J Leukoc Biol 2014;96:657‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Han KH, Hashimoto N, Fukushima M. Relationships among alcoholic liver disease, antioxidants, and antioxidant enzymes. World J Gastroenterol 2016;22:37‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kono H, Rusyn I, Yin M, Gabele E, Yamashina S, Dikalova A, et al. NADPH oxidase‐derived free radicals are key oxidants in alcohol‐induced liver disease. J Clin Invest 2000;106:867‐872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Levin I, Petrasek J, Szabo G. The presence of p47phox in liver parenchymal cells is a key mediator in the pathogenesis of alcoholic liver steatosis. Alcohol Clin Exp Res 2012;36:1397‐1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc 2013;8:627‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Holt MP, Cheng L, Ju C. Identification and characterization of infiltrating macrophages in acetaminophen‐induced liver injury. J Leukoc Biol 2008;84:1410‐1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fernandez‐Boyanapalli RF, Frasch SC, McPhillips K, Vandivier RW, Harry BL, Riches DW, et al. Impaired apoptotic cell clearance in CGD due to altered macrophage programming is reversed by phosphatidylserine‐dependent production of IL‐4. Blood 2009;113:2047‐2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, et al. Mouse model of X‐linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet 1995;9:202‐209. [DOI] [PubMed] [Google Scholar]
- 14. Thurman RG. Sex‐related liver injury due to alcohol involves activation of Kupffer cells by endotoxin. Can J Gastroenterol 2000;14(Suppl. D):129D‐135D. [DOI] [PubMed] [Google Scholar]
- 15. Yan AW, Fouts DE, Brandl J, Starkel P, Torralba M, Schott E, et al. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 2011;53:96‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Paik YH, Iwaisako K, Seki E, Inokuchi S, Schnabl B, Osterreicher CH, et al. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) homologues NOX1 and NOX2/gp91(phox) mediate hepatic fibrosis in mice. Hepatology 2011;53:1730‐1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zigmond E, Samia‐Grinberg S, Pasmanik‐Chor M, Brazowski E, Shibolet O, Halpern Z, et al. Infiltrating monocyte‐derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J Immunol 2014;193:344‐353. [DOI] [PubMed] [Google Scholar]
- 18. Peters VA, Joesting JJ, Freund GG. IL‐1 receptor 2 (IL‐1R2) and its role in immune regulation. Brain Behav Immun 2013;32:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fernandez‐Boyanapalli R, Frasch SC, Riches DW, Vandivier RW, Henson PM, Bratton DL. PPARgamma activation normalizes resolution of acute sterile inflammation in murine chronic granulomatous disease. Blood 2010;116:4512‐4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang Y, Ausman LM, Russell RM, Greenberg AS, Wang XD. Increased apoptosis in high‐fat diet‐induced nonalcoholic steatohepatitis in rats is associated with c‐Jun NH2‐terminal kinase activation and elevated proapoptotic Bax. J Nutr 2008;138:1866‐1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arandjelovic S, Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol 2015;16:907‐917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fadok VA, Warner ML, Bratton DL, Henson PM. CD36 is required for phagocytosis of apoptotic cells by human macrophages that use either a phosphatidylserine receptor or the vitronectin receptor (alpha v beta 3). J Immunol 1998;161:6250‐6257. [PubMed] [Google Scholar]
- 23. Kurose I, Higuchi H, Miura S, Saito H, Watanabe N, Hokari R, et al. Oxidative stress‐mediated apoptosis of hepatocytes exposed to acute ethanol intoxication. Hepatology 1997;25:368‐378. [DOI] [PubMed] [Google Scholar]
- 24. Seki E, Brenner DA. Toll‐like receptors and adaptor molecules in liver disease: update. Hepatology 2008;48:322‐335. [DOI] [PubMed] [Google Scholar]
- 25. Zeng MY, Pham D, Bagaitkar J, Liu J, Otero K, Shan M, et al. An efferocytosis‐induced, IL‐4‐dependent macrophage‐iNKT cell circuit suppresses sterile inflammation and is defective in murine CGD. Blood 2013;121:3473‐3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mukundan L, Odegaard JI, Morel CR, Heredia JE, Mwangi JW, Ricardo‐Gonzalez RR, et al. PPAR‐delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med 2009;15:1266‐1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hong C, Kidani Y, A‐Gonzalez N, Phung T, Ito A, Rong X, et al. Coordinate regulation of neutrophil homeostasis by liver X receptors in mice. J Clin Invest 2012;122:337‐347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. A‐Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 2009;31:245‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fernandez‐Boyanapalli RF, Frasch SC, Thomas SM, Malcolm KC, Nicks M, Harbeck RJ, et al. Pioglitazone restores phagocyte mitochondrial oxidants and bactericidal capacity in chronic granulomatous disease. J Allergy Clin Immunol 2015;135:517‐527.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brown JR, Goldblatt D, Buddle J, Morton L, Thrasher AJ. Diminished production of anti‐inflammatory mediators during neutrophil apoptosis and macrophage phagocytosis in chronic granulomatous disease (CGD). J Leukoc Biol 2003;73:591‐599. [DOI] [PubMed] [Google Scholar]
- 31. Forman HJ, Torres M. Reactive oxygen species and cell signaling: respiratory burst in macrophage signaling. Am J Resp Crit Care Med 2002;166:S4‐S8. [DOI] [PubMed] [Google Scholar]
- 32. Kobayashi SD, Voyich JM, Braughton KR, Whitney AR, Nauseef WM, Malech HL, et al. Gene expression profiling provides insight into the pathophysiology of chronic granulomatous disease. J Immunol 2004;172:636‐643. [DOI] [PubMed] [Google Scholar]
- 33. Chawla A. Control of macrophage activation and function by PPARs. Circ Res 2010;106:1559‐1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Segal BH, Han W, Bushey JJ, Joo M, Bhatti Z, Feminella J, et al. NADPH oxidase limits innate immune responses in the lungs in mice. PLoS One 2010;5:e9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gao Y, Chu SF, Xia CY, Zhang Z, Zhang S, Chen NH. Rg1 attenuates alcoholic hepatic damage through regulating AMP‐activated protein kinase and nuclear factor erythroid 2‐related factor 2 signal pathways. J Asian Nat Prod Res 2016;18:765‐778. [DOI] [PubMed] [Google Scholar]
- 36. Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med 2011;364:656‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med 2012;367:2322‐2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MA, et al. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci U S A 2009;106:6226‐6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. de Luca A, Smeekens SP, Casagrande A, Iannitti R, Conway KL, Gresnigt MS, et al. IL‐1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci U S A 2014;111:3526‐3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. van de Veerdonk FL, Smeekens SP, Joosten LA, Kullberg BJ, Dinarello CA, van der Meer JW, et al. Reactive oxygen species‐independent activation of the IL‐1 beta inflammasome in cells from patients with chronic granulomatous disease. Proc Natl Acad Sci U S A 2010;107:3030‐3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Geiszt M, Kapus A, Nemet K, Farkas L, Ligeti E. Regulation of capacitative Ca2+ influx in human neutrophil granulocytes. Alterations in chronic granulomatous disease. J Biol Chem 1997;272:26471‐26478. [DOI] [PubMed] [Google Scholar]
- 42. Harrison CA, Raftery MJ, Walsh J, Alewood P, Iismaa SE, Thliveris S, et al. Oxidation regulates the inflammatory properties of the murine S100 protein S100A8. J Biol Chem 1999;274:8561‐8569. [DOI] [PubMed] [Google Scholar]
- 43. Costford SR, Castro‐Alves J, Chan KL, Bailey LJ, Woo M, Belsham DD, et al. Mice lacking NOX2 are hyperphagic and store fat preferentially in the liver. Am J Physiol Endocrinol Metab 2014;306:E1341‐E1353. [DOI] [PubMed] [Google Scholar]
- 44. dela Pena A, Leclercq IA, Williams J, Farrell GC. NADPH oxidase is not an essential mediator of oxidative stress or liver injury in murine MCD diet‐induced steatohepatitis. J Hepatol 2007;46:304‐313. [DOI] [PubMed] [Google Scholar]
- 45. Jiang JX, Venugopal S, Serizawa N, Chen X, Scott F, Li Y, et al. Reduced nicotinamide adenine dinucleotide phosphate oxidase 2 plays a key role in stellate cell activation and liver fibrogenesis in vivo. Gastroenterology 2010;139:1375‐1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Winkelstein JA, Marino MC, Johnston RB Jr, Boyle J, Curnutte J, Gallin JI, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore) 2000;79:155‐169. [DOI] [PubMed] [Google Scholar]
- 47. Hussain N, Feld JJ, Kleiner DE, Hoofnagle JH, Garcia‐Eulate R, Ahlawat S, et al. Hepatic abnormalities in patients with chronic granulomatous disease. Hepatology 2007;45:675‐683. [DOI] [PubMed] [Google Scholar]
- 48. Sanmun D, Witasp E, Jitkaew S, Tyurina YY, Kagan VE, Ahlin A, et al. Involvement of a functional NADPH oxidase in neutrophils and macrophages during programmed cell clearance: implications for chronic granulomatous disease. Am J Physiol Cell Physiol 2009;297:C621‐C631. [DOI] [PubMed] [Google Scholar]
- 49. Bautista AP. Neutrophilic infiltration in alcoholic hepatitis. Alcohol 2002;27:17‐21. [DOI] [PubMed] [Google Scholar]
- 50. Jaeschke H. Neutrophil‐mediated tissue injury in alcoholic hepatitis. Alcohol 2002;27:23‐27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep4.1078/full.
Supporting Information