

Abstract
Protein glycosylation is essential for cell survival and regulates many cellular events. Reversible glycosylation is also dynamic in biological systems. The functions of glycoproteins are regulated by their dynamics to adapt the ever-changing inter- and intracellular environments. Glycans on proteins not only mediate a variety of protein activities, but also creates a steric hindrance for protecting the glycoproteins from degradation by proteases. In this work, a novel strategy integrating isotopic labelling, chemical enrichment and multiplexed proteomics was developed to simultaneously quantify the degradation and synthesis rates of many glycoproteins in human cells. We quantified the synthesis rates of 847 N-glycoproteins and the degradation rates of 704 glycoproteins in biological triplicate experiments, including many important glycoproteins such as CD molecules. Through comparing the synthesis and degradation rates, we found that most proteins have higher synthesis rates since cells are still growing throughout the time course, while a small group of proteins with lower synthesis rates mainly participate in adhesion, locomotion, localization, and signaling. This method can be widely applied in biochemical and biomedical research and provide insights into elucidating glycoprotein functions and the molecular mechanism of many biological events.
TOC image

INTRODUCTION
Protein glycosylation plays vital roles in a variety of cellular processes.1–4 The functions of glycoproteins are intrinsically related to their dynamics, and the presence of glycans on proteins create a steric hindrance that prevents proteases from approaching,5 thus impacting protein dynamics. Modern mass spectrometry (MS)-based proteomics has offered a unique opportunity for global analysis of glycoproteins,6–15 but it is still challenging due to the low abundance of glycoproteins and the heterogeneity of glycan structures.16–23 Studying protein glycosylation and its dynamics not only advances our knowledge of the underlying mechanisms of many cellular activities and diseases, but also enables us to identify glycoproteins as disease biomarkers and drug targets.24–28
There have been many reports about the global analysis of glycoproteins, and considerable progress has been made in recent years.6,29–34 However, few of them focused on glycoprotein dynamics on a large scale in spite of its importance.35 Stable isotope labelling with amino acid in cell culture (SILAC) has been widely used for protein turnover study.36,37 Using pulse-chase SILAC, the newly-synthesized proteins can be distinguished from the existing background through incorporation of the heavy (or light) isotopic amino acid residues. The labelling can allow us to generate valuable information about protein degradation and synthesis through mass spectrometric analysis.
In this work, we combined pulse-chase SILAC, chemical enrichment of glycopeptides, and multiplexed proteomics to globally quantify the degradation and synthesis rates of glycoproteins simultaneously. Pulse-chase labelling allowed us to track the protein abundance changes, and in combination with chemical enrichment of glycopeptides we were able to quantify glycoprotein dynamics. After enrichment, we labelled the enriched glycopeptides from multiple time points with the tandem mass tag (TMT) reagents38 for quantitation with MS-based proteomics. The abundance changes of glycoproteins as a function of time were used to calculate the degradation and synthesis rates.
EXPERIMENTAL SECTION
Cell Culture, Heavy Isotope Labeling, and Time Course-based Cell Collection
MCF-7 cells (ATCC) were grown in a humidified incubator at 37 °C and 5.0% CO2 in high glucose Dulbecco’s modified eagle’s medium (DMEM) (Sigma-Aldrich) with 10% fetal bovine serum (FBS) (Corning) for each of the triplicate experiments. Heavy isotope labeling of cells was performed with 13C6, 15N2 L-lysine (Lys8) and 13C6 L-arginine (Arg6) (Cambridge Isotopes) in SILAC DMEM with 10% dialyzed FBS for about six generations. Media was then switched to normal media with Lys0 and Arg0 to begin the time-course experiment. Cells were collected separately at five time points (0, 6, 12, 24 and 48 hours).
Cell Lysis and Protein Digestion
Cells were washed twice with phosphate buffered saline (PBS) and pelleted by centrifugation at 500 g for 3 minutes and washed twice with cold PBS. Cell pellets were lysed through end-to-end rotation at 4 °C for 45 minutes in the lysis buffer (50 mM N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid (HEPES) pH=7.4, 150 mM NaCl, 0.5% sodium deoxycholate (SDC), and 25 units/mL benzonase and 1 tablet/10 mL protease inhibitor). Lysates were centrifuged, and the resulting supernatant was transferred into new tubes. Proteins were subjected to disulfide reduction with 5 mM dithiothreitol (DTT) (56 °C, 25 minutes) and alkylation with 14 mM iodoacetamide (room temperature, 20 minutes in the dark). Detergent was removed by the methanol-chloroform protein precipitation method. The purified proteins were digested with 10 ng/μL Lys-C (Wako) in 50 mM HEPES pH 8.6, 1.6 M urea, 5% ACN at 31 °C for 16 hours, followed by further digestion with 8 ng/μL Trypsin (Promega) at 37 °C for 4 hours.
Glycopeptide Enrichment, TMT Labeling, and Deglycosylation
Protein digestions were quenched by addition of trifluoroacetic acid (TFA) to a final concentration of 0.1%, followed by centrifugation to remove the precipitate. The peptides were desalted using a tC18 Sep-Pak cartridge (Waters) and lyophilized, subjected to boronic acid-conjugated beads-based enrichment as described previously.39 The peptides were then eluted twice by incubating the beads in a solution containing acetonitrile, water, and trifluoroacetic acid at a respective ratio of 50:49:1 for 30 minutes at 37 °C. Eluates were desalted using tC18 Sep-Pak cartridges and lyophilized. Purified glycopeptides from each time point were labeled with each channel (126, 128, 129, 130, or 131) of the multiplexed TMT reagents (Thermo) following the manufacturer’s protocol. Briefly, purified and lyophilized peptides were dissolved in 100 μL of 100 mM triethylammonium bicarbonate (TEAB) buffer, pH= 8.5. Each tube of TMT reagents was dissolved in 41 μL of anhydrous ACN, and 7 μL was transferred into the peptide tube with another 34 μL of ACN. The reaction was performed for 1 hour at room temperature, quenched by adding 8 μL of 5% hydroxylamine and shaking for 15 min. Peptides from all tubes were then mixed, desalted using a tC18 Sep-Pak cartridge, and lyophilized overnight. The dried peptides were deglycosylated with three units of peptide-N-glycosidase F (PNGase F, Sigma-Aldrich)40 in 60 μL buffer containing 40 mM NH4HCO3 (pH=9) in heavy-oxygen water (H218O) for 3 hours at 37 °C. The reaction was quenched by adding formic acid (FA) to a final concentration of 1%, and peptides were desalted again using tC18 Sep-Pak cartridges and dried.
Glycopeptide Fractionation and LC-MS/MS Analysis
Purified and dried peptides were separated by high pH reversed-phase high-performance liquid chromatography (HPLC) into 10 fractions with a 40-min gradient of 5–55% ACN in 10 mM ammonium acetate (pH=10), dried and purified using the stage-tip method, and dissolved in a 10 μL solution with 5% ACN and 4% FA. 4 μL were loaded onto a microcapillary column packed with C18 beads (Magic C18AQ, 3 μm, 200 Å, 100 μm × 16 cm, Michrom Bioresources) by a Dionex WPS-3000TPLRS autosampler (UltiMate 3000 thermostatted Rapid Separation Pulled Loop Wellplate Sampler). Peptides were separated by reversed-phase chromatography using an UltiMate 3000 binary pump with a 128 min gradient. Peptides were detected with a data-dependent Top15 method41 in a hybrid dual-cell quadrupole linear ion trap - Orbitrap mass spectrometer (LTQ Orbitrap Elite, Thermo Scientific, with Xcalibur 3.0.63 software). For each cycle, one full MS scan (resolution: 60,000) in the Orbitrap at 106 automatic gain control (AGC) target was followed by up to 15 MS/MS in the Orbitrap again for the most intense ions. The selected ions were excluded from further analysis for 90 seconds. Ions with singly or unassigned charge were not sequenced.
Database Search and Data Filtering
All MS2 spectra were converted into a mzXML format and searched using the SEQUEST algorithm (version 28).42 Spectra were matched against a database containing sequences of all proteins in the Human (Homo sapiens) database downloaded from the UniProt. The following parameters were used during the search: 10 ppm precursor mass tolerance; 0.1 Da product ion mass tolerance; fully digested with trypsin; up to three missed cleavages; fixed modification: carbamidomethylation of cysteine (+57.0214); variable modifications: oxidation of methionine (+15.9949), O18 tag of asparagine (+2.9883). For heavy TMT-labeled proteins, these following fixed modifications were also added to the search: TMT plus heavy isotope for lysine (+237.1771), heavy arginine (+6.0201), N-terminal TMT (229.1629). For light TMT-labeled proteins, TMT (+229.1629) was added to both lysine and N-terminal as fixed modification. False discovery rates (FDR) of glycopeptide and glycoprotein identifications were evaluated and controlled to less than 1% by the target-decoy method43 through linear discriminant analysis (LDA),44 using parameters such as XCorr, precursor mass error, and charge state, to control the glycopeptide identification quality.45 The consensus motif N#X[S/T/C] (# stands for the glycosylation site and X represents any amino acid residues other than proline) was also required to guarantee the reliability of the N-glycosylation analysis. Peptides fewer than seven amino acid residues in length were deleted. The dataset was restricted to glycopeptides when determining FDRs for glycopeptide identification.46
Glycosylation Site Localization
We assigned and measured the confidence of glycosylation site localizations by calculating their ModScores, which applies a probabilistic algorithm46 that considers all possible glycosylation sites in a peptide and uses the presence of experimental fragment ions unique to each site to assess the localization confidence. Sites with ModScore > 13 (P < 0.05) were considered as confidently localized. If the same glycopeptide was quantified several times, the median value was used as the glycopeptide abundance change.
RESULTS AND DISCUSSION
Experimental Procedure for Simultaneous Measurement of Glycoprotein Degradation and Synthesis Rates
The experimental procedure is shown in Fig. 1, and the detailed description is included in the Experimental Section. Briefly, MCF-7 cells were cultured with SILAC Dulbecco’s Modified Eagle’s Medium (DMEM) containing Lys8 and Arg6 for six generations for full heavy isotope incorporation, and then were equally passaged for the time-course experiments. When the cells were approaching nearly full confluency (to minimize the dilution effect from cell growth), we switched the media to normal DMEM with Lys0 and Arg0 and began the time course. Upon the media switch (0 h), the numbers of cells across different groups were kept as similar as possible. For each sample, there were also very similar amount of heavy isotope-labelled proteins (heavy proteins) and nearly no light isotope-labelled proteins (light proteins). We then harvested cells at each time point until the completion of the 48 h time course. As time went by, heavy proteins were degraded and newly-synthesized proteins were theoretically all light proteins. Therefore, the abundance changes of heavy glycoproteins can be used to calculate the degradation rates while the abundance changes of light glycoproteins as a function of time are glycoprotein synthesis rates. We performed biological triplicate experiments to evaluate the reproducibility and ensure the technical rigor.
Figure 1.
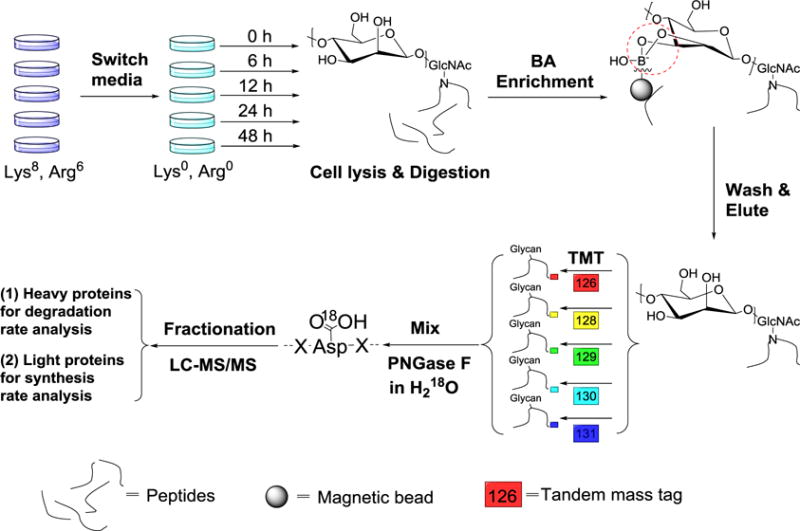
The experimental procedure for the simultaneous quantification of the glycoprotein degradation/synthesis rates.
Proteins were reduced, alkylated, and digested by Lys-C and trypsin. Purified peptides were subjected to the chemical enrichment of glycopeptides through incubation with boronic acid-conjugated magnetic beads, as reported previously.39,47 The beads were then washed to remove nonglycopeptides, and elution was performed twice using a buffer containing water: acetonitrile: trifluoroacetic acid= 49:50:1. Glycopeptides from each time point were purified using C18 cartridges, labelled with the TMT reagents and mixed. Glycopeptides were treated with PNGase F in heavy oxygen water (H218O) to create a common tag on the N-glycosylation sites for MS analysis.48,49 After purification, the deglycosylated peptides from each experiment were separated into 10 fractions using high-pH reversed-phase high performance liquid chromatography. Each fraction was further purified by the stage-tip method, followed by LC-MS analysis.
Glycoprotein Identification
After glycopeptides were treated with PNGase F in heavy oxygen water (H218O), a common and unique tag (+2.9883 D) on the N-glycosylation sites was created for MS analysis. The deamidation of asparagine happens in vivo and in vitro. The tag containing the heavy oxygen can allow us to distinguish the real N-glycosylation sites from spontaneous deamidation of asparagine. However, the spontaneous deamidation of asparagine could occur during the PNGase F treatment in heavy water, resulting in the false positive identification. In order to minimize this, we carried out the reaction for only three hours. As tested previously, the spontaneous deamidation of asparagine is negligible for three hours under the mild enzymatic reaction conditions.48
The glycopeptides were filtered to <1% false discovery rate. Additionally they are required to have the consensus motif, i.e. N#X[S/T/C] (# stands for the glycosylation site and X represents any amino acid residues other than proline). Among biological triplicate experiments, we identified 1,373, 1,342, and 1,280 unique light glycopeptides (Table S1), respectively. They overlapped very well, and 790 glycopeptides were found in all three experiments (Fig. 2a). Totally, 1,875 unique light glycopeptides were identified (Table S1). Slightly fewer number of heavy glycopeptides were identified, i.e. 866, 1,048 and 1,097 in each of the three experiments (Table S2). Finally, 1,515 unique heavy glycopeptides (Table S2) were identified with site-specific information, and the comparison is displayed in Fig. 2b.
Figure 2.
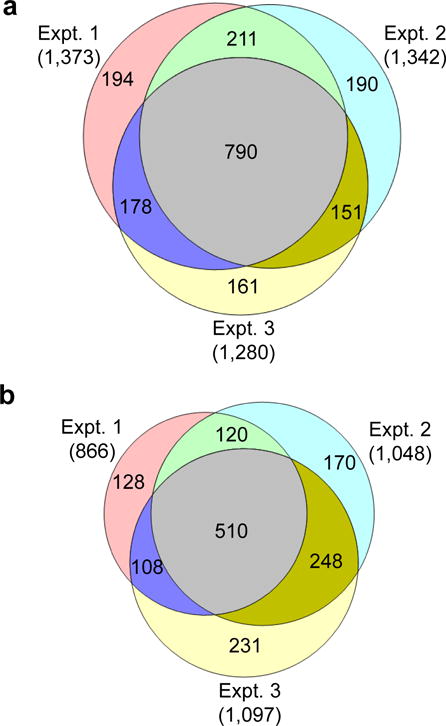
The overlap of the unique glycopeptides identified in the biological triplicate experiments: (a) light glycopeptides; (b) heavy glycopeptides.
Calculation of the Glycoprotein Degradation and Synthesis Rates
We calculated the degradation/synthesis rates based on the abundance changes of glycopeptides as a function of time simulated by the following exponential decay/growth equation (1) or (2), as performed previously:50,51
Based on the abundance changes of heavy glycopeptides, the degradation rates were calculated:
| (1) |
According to the abundance changes of light glycopeptides, the synthesis rates were obtained using the following equation:
| (2) |
where Ph0 or Pl0 is the abundance of the heavy or light glycopeptide at the first time point (represented by the intensity of the reporter ion), Ph(t) or Pl(t) is the abundance of the heavy or light glycopeptide at each subsequent time point, t stands for time. kd is the degradation rate constant while ks is the synthesis rate constant. The different time points at 0, 6, 12, 24, and 48 h were also designed to provide convenience for the exponential simulation and to be compatible with the measurement of relatively long half-lives of glycoproteins.48,49
An example of glycopeptide quantification is shown in Fig. S1. Glycopeptide KWGHN#ITEFQQR is from protein ERO1A, an oxidoreductase involved in disulfide bond formation and preventing the accumulation of reactive oxygen species in the endoplasmic reticulum (ER). This glycopeptide was confidently identified with an XCorr of 3.8, and the glycosylation site was localized on N280, which was also reported on the UniProt (www.uniprot.org). We quantified its synthesis rate based on the reporter ion intensities. Through using this method, we quantified the synthesis rates of 847 glycoproteins (Table S3) and the degradation rates of 704 glycoproteins (Table S4) from the three experiments.
Evaluation of the Experimental Reproducibility
The reproducibility was evaluated based on the biological triplicate experiments (Fig. 3 and Fig. S2). The comparison of the synthesis rates from triplicate experiments is shown in Fig. 3. A total of 1,330 (71%) unique light glycopeptides were identified in at least two experiments (Fig. 2a) displaying high reproducibility given that all experiments were performed independently from cell culture to LC-MS/MS analysis. In addition, the calculated synthesis rates of the light glycoproteins from the three experiments were in reasonably good agreement (Fig 3a, b). The heavy glycopeptide identification and quantification were also proved to be reproducible (Fig. 2b, S2a, S2b). Compared to the protein synthesis rates, their degradation rates are generally lower, resulting in a more condensed distribution pattern for the heavy glycoproteins, which is further discussed below.
Figure 3.
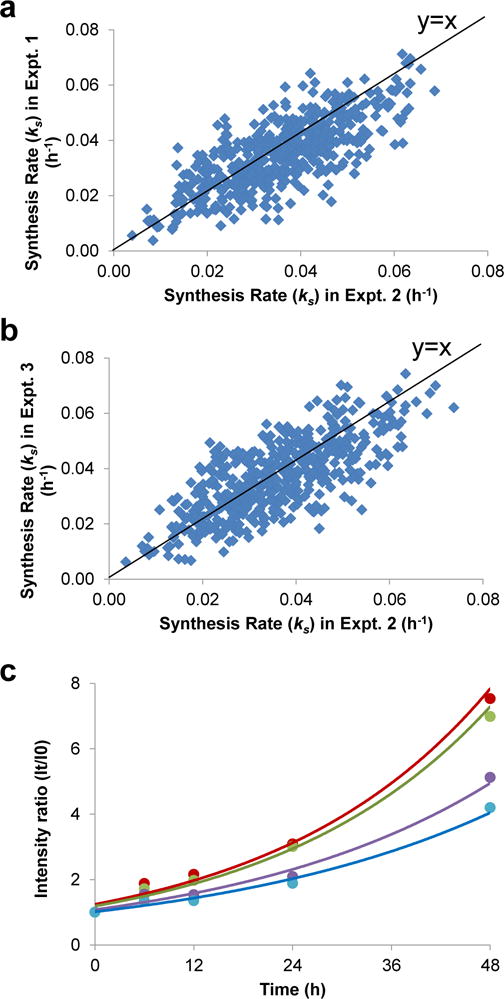
(a) Comparison of the synthesis rates of the glycoproteins quantified in experiments 1 & 2. (b) Comparison of the synthesis rates of the glycoproteins quantified in experiment 2 & 3. (c) Examples of glycopeptide quantification: red- KPN#ATAEPTPPDR from protein MRC2, green- RELYN#GTADITLR from protein RPIEZO1, purple- TCDWLPKPN#MSASCK from protein PSAP, and blue- QPMAPNPCEANGGQGPCSHLCLINYN#R from protein LRP1.
We quantified both the synthesis (Table S3) and degradation rates (Table S4) for 639 glycoproteins (Fig. S3). We illustrated the dynamic abundance changes over the time course for several light glycopeptides as examples in Figure 3c. Due to the fact that the raw intensities for each peptide can vary within a range of several orders of magnitudes, we used the normalized ratio of intensity at each time point, i.e. the intensity (It) divided by the initial intensity (I0). Therefore, the values of all glycopeptides are 1 at the first time point. The glycopeptides are from proteins MRC2, PIEZO1, PSAP, and LRP1, representing glycoproteins from a variety of subcellular locations and with various molecular functions. Since the time course lasted for 48 h, the accumulation of light glycoproteins made their abundance higher than heavy glycoproteins, rendering slightly fewer heavy glycoproteins being identified. Although the current time course was relatively long, a small group of proteins with a very slow degradation or synthesis rate (k<0.0034 h−1, t1/2>200 h) were still not able to be accurately quantified, thus we annotated the rates for these proteins as “very slow” in the Supplementary Tables. For glycoproteins quantified in multiple experiments, we used their median synthesis and degradation rates for further data analysis in this work.
Clustering of Glycoproteins
We clustered the identified glycoproteins according to cellular compartment using the Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.8 (Fig. 4a).52 Many categories, ranging from cell surface to organelle membranes, were highly enriched with low P values and high glycoprotein counts. We also clustered the glycoproteins with a relatively high synthesis rate (ks>0.03 h−1) according to molecular function. Interestingly, the highly enriched categories are receptor binding and transportation. The most highly enriched are those with glycosylation enzymatic activity, including transferase activity (transferring hexosyl groups) and hydrolase activity (hydrolyzing O-glycosyl compounds) (Fig. 4b)
Figure 4.
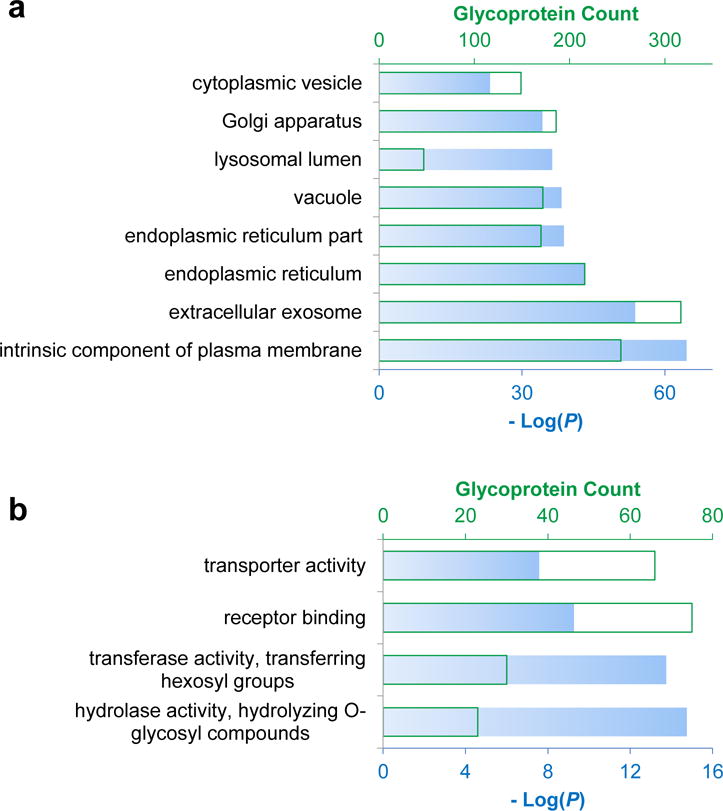
Clustering of (a) the quantified glycoproteins according to cellular compartment and (b) the glycoproteins with a relatively higher synthesis rate based on molecular function.
Here we quantified the synthesis rates of 83 CD glycoproteins (Table S6), and twelve of them also belong to the family of cell adhesion molecules (CAMs), which are listed in Table 1. Many of them are integrins, which are typically a group of important transmembrane receptors that participate in the interactions between cells and extracellular matrix. For instance, ITGB1 was confidently quantified in the current experiments based on 11 unique glycopeptides. It is known to be conjoining with integrin alpha subunits to form various cell-surface receptors, such as forming a laminin receptor with integrin alpha subunit 6 (ITGA6). The latter is also quantified with a synthesis rate of 0.0380 h−1. The four integrins (ITGB1, ITGB2, ITGAV, and ITGA6) quantified all have similar synthesis rates, ranging from 0.0377 h−1 to 0.0483 h−1, correlating well with the fact that they adjoin one another to form the receptor complex on the cell surface.
Table 1.
The synthesis rates of glycoproteins that are both CD and CAM molecules.
| CD Name | Gene Symbol | UniProt ID | Unique Glycopeptide Hits | Median Synthesis Rate (h−1) | Annotation |
|---|---|---|---|---|---|
| CD58 | CD58 | P19256 | 1 | 0.0552 | Lymphocyte function-associated antigen 3 |
| CD54 | ICAM1 | P05362 | 3 | 0.0538 | Intercellular adhesion molecule 1 |
| CD166 | ALCAM | Q13740 | 9 | 0.0517 | CD166 antigen |
| CD29 | ITGB1 | P05556 | 11 | 0.0483 | Integrin beta-1 |
| CD276 | CD276 | Q5ZPR3 | 4 | 0.0427 | CD276 antigen |
| CD51 | ITGAV | P06756 | 8 | 0.0412 | Integrin alpha-V |
| CD171 | L1CAM | P32004 | 6 | 0.0388 | Neural cell adhesion molecule L1 |
| CD49f | ITGA6 | P23229 | 5 | 0.0380 | Integrin alpha-6 |
| CD18 | ITGB2 | P05107 | 3 | 0.0377 | Integrin beta-2 |
| CD62l | SELL | P14151 | 1 | 0.0272 | L-selectin |
| CD275 | ICOSLG | O75144 | 3 | 0.0205 | ICOS ligand |
| CD155 | PVR | P15151 | 1 | 0.0046 | Poliovirus receptor |
Comparison of the Difference between the Synthesis and Degradation Rates
We then analyzed the difference between the synthesis and degradation rates for glycoproteins (Table S5), and the rate differences are plotted in Fig. 5. To ensure the analysis confidence, we only analyzed the glycoproteins with their synthesis/degradation rates accurately quantified (without “very slow” annotation) in at least two experiments, where 400 proteins fall into this category. Since cells were still growing throughout the time course, we anticipated that the majority of the proteins would have a faster synthesis rate than degradation rate. This was proved by the results (Fig. 5a) as 352 proteins (88%) had higher synthesis rates.
Figure 5.
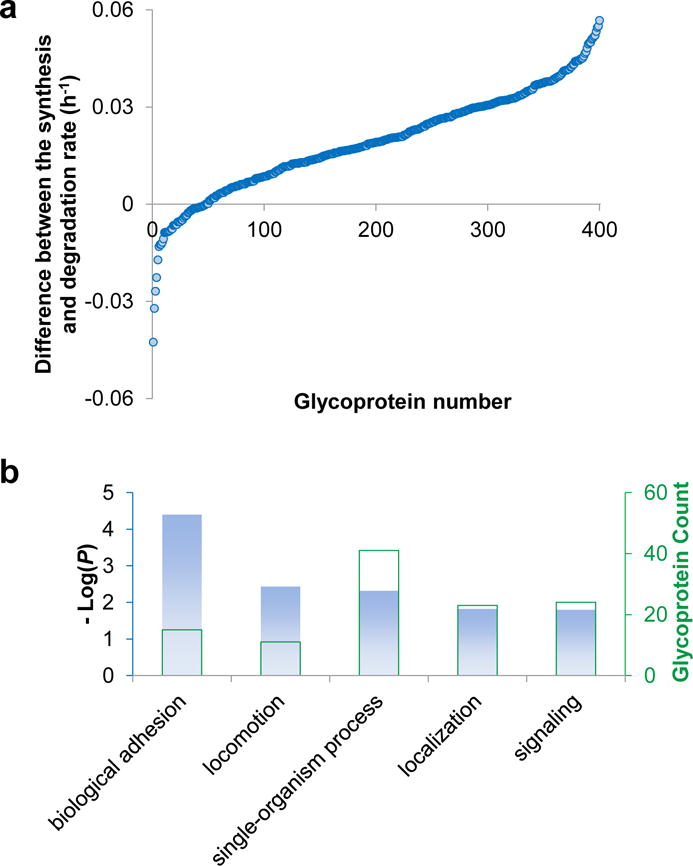
(a) The difference between the synthesis and degradation rates for 400 glycoproteins with both rates quantified; (b) The biological processes in which 48 glycoproteins with a lower synthesis rate are involved in.
Interestingly, 48 proteins had lower synthesis rates, and they mostly participated in the biological processes of adhesion, locomotion, localization, and signaling (Fig. 5b). We reasoned that at the end of the time course, as the cells were approaching a static state due to high confluence, these biological processes were not supposed to robustly continue, and thus the related glycoproteins were down-regulated. Another possible explanation arises from the current approach to calculate these two rates. The absolute glycoprotein degradation rate was calculated based on the abundance changes of heavy glycoproteins, and the contribution from heavy protein synthesis was negligible since heavy isotopic lysine and arginine were not supplied during the 48 h time course. However, light glycoproteins were used to calculate the synthesis rates while they were synthesized and degraded simultaneously. Since the starting light protein abundance was extremely low, we neglected the contribution from light protein degradation, but this contribution accumulated throughout the whole time course. Therefore, the real protein synthesis rates are likely slightly higher than the rates obtained in this work.
CONCLUSIONS
Evolution has endowed cells the ability to synthesize proteins in a conservative and low-risk pattern.53 In this study, we integrated pulse-chase SILAC, chemical enrichment of glycopeptides, and multiplex proteomics to simultaneously investigate the glycoprotein synthesis and degradation rates in human cells on a large scale. Rigorous criteria were applied for glycopeptide filtering, and 3,390 unique heavy and light glycopeptides with site-specific information led to the simultaneous quantitation of the degradation and/or synthesis rates of many glycoproteins. We quantified the synthesis rates of 847 N-glycoproteins and the degradation rates of 704 N-glycoproteins, and demonstrated this method to be reproducible based on the results from the biological triplicate experiments. The glycoproteins related to binding, transportation, and enzyme activity were determined to have higher synthesis rates in the cells under the current culture conditions. The majority of the quantified glycoproteins were synthesized faster than degraded due to the cell growth. In combination with pulse-chase SILAC and glycopeptide enrichment, we can simultaneously quantify the synthesis and degradation rates of glycoproteins. This method can be extensively applied to investigate glycoprotein dynamics, which will aid in a better understanding of glycoprotein functions and the molecular mechanisms of biological events.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (R01GM118803).
Footnotes
SUPPORTING INFORMATION
This material is available free of charge via the Internet at http://pubs.acs.org. Unique light glycopeptides identified in the three experiments (Table S1); Unique heavy glycopeptides identified in the three experiments (Table S2); The synthesis rates of the quantified N-glycoproteins in the three experiments (Table S3); The degradation rates of the quantified N-glycoproteins in the three experiments (Table S4); Glycoproteins with both degradation and synthesis rates quantified (Table S5); The synthesis rates of the 83 quantified CD molecules (Table S6); An example of glycopeptide identification and quantification (Figure S1); Reproducibility evaluation of the heavy glycopeptides/glycoproteins (Figure S2); The overlap of the glycoproteins with degradation and synthesis rates quantified (Figure S3).
References
- 1.Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler EM. Essentials of glycobiology. 2nd. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2008. [PubMed] [Google Scholar]
- 2.Dennis JW, Granovsky M, Warren CE. Bioessays. 1999;21:412–421. doi: 10.1002/(SICI)1521-1878(199905)21:5<412::AID-BIES8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 3.Maverakis E, Kim K, Shimoda M, Gershwin ME, Patel F, Wilken R, Raychaudhuri S, Ruhaak LR, Lebrilla CB. J Autoimmun. 2015;57:1–13. doi: 10.1016/j.jaut.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lehle L, Strahl S, Tanner W. Angew Chem-Int Edit. 2006;45:6802–6818. doi: 10.1002/anie.200601645. [DOI] [PubMed] [Google Scholar]
- 5.Sola RJ, Griebenow K. J Pharm Sci-US. 2009;98:1223–1245. doi: 10.1002/jps.21504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang H, Li XJ, Martin DB, Aebersold R. Nat Biotechnol. 2003;21:660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- 7.Zhu ZK, Desaire H. In: Annual review of analytical chemistry. Cooks RG, Pemberton JE, editors. Vol. 8. Vol. 8. Annual Reviews; Palo Alto: 2015. pp. 463–483. [DOI] [PubMed] [Google Scholar]
- 8.Yang Y, Liu F, Franc V, Halim LA, Schellekens H, Heck AJR. Nat Commun. 2016;7:10. doi: 10.1038/ncomms13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei X, Dulberger C, Li LJ. Anal Chem. 2010;82:6329–6333. doi: 10.1021/ac1004844. [DOI] [PubMed] [Google Scholar]
- 10.Breidenbach MA, Palaniappan KK, Pitcher AA, Bertozzi CR. Mol Cell Proteomics. 2012;11:10. doi: 10.1074/mcp.M111.015339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khatri K, Klein JA, Zaia J. Anal Bioanal Chem. 2017;409:607–618. doi: 10.1007/s00216-016-9970-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loziuk PL, Hecht ES, Muddiman DC. Anal Bioanal Chem. 2017;409:487–497. doi: 10.1007/s00216-016-9776-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trinidad JC, Barkan DT, Gulledge BF, Thalhammer A, Sali A, Schoepfer R, Burlingame AL. Mol Cell Proteomics. 2012;11:215–229. doi: 10.1074/mcp.O112.018366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alfaro JF, Gong CX, Monroe ME, Aldrich JT, Clauss TRW, Purvine SO, Wang ZH, Camp DG, Shabanowitz J, Stanley P, Hart GW, Hunt DF, Yang F, Smith RD. Proc Natl Acad Sci U S A. 2012;109:7280–7285. doi: 10.1073/pnas.1200425109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khidekel N, Ficarro SB, Clark PM, Bryan MC, Swaney DL, Rexach JE, Sun YE, Coon JJ, Peters EC, Hsieh-Wilson LC. Nat Chem Biol. 2007;3:339–348. doi: 10.1038/nchembio881. [DOI] [PubMed] [Google Scholar]
- 16.Chen WX, Smeekens JM, Wu RH. Chem Sci. 2015;6:4681–4689. doi: 10.1039/c5sc01124h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banazadeh A, Veillon L, Wooding KM, Zabet-moghaddam M, Mechref Y. Electrophoresis. 2017;38:162–189. doi: 10.1002/elps.201600357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaunitz S, Nagy G, Pohl NLB, Noyotny MV. Anal Chem. 2017;89:389–413. doi: 10.1021/acs.analchem.6b04343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chandler KB, Costello CE. Electrophoresis. 2016;37:1407–1419. doi: 10.1002/elps.201500552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan ZJ, Yin HD, Nie S, Lin ZX, Zhu JH, Ruffin MT, Anderson MA, Simone DM, Lubman DM. J Proteome Res. 2015;14:1968–1978. doi: 10.1021/acs.jproteome.5b00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng JN, Xiao HP, Wu RH. Angew Chem Int Ed. 2017;56:7107–7111. doi: 10.1002/anie.201702191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu T, Qian WJ, Gritsenko MA, Camp DG, Monroe ME, Moore RJ, Smith RD. J Proteome Res. 2005;4:2070–2080. doi: 10.1021/pr0502065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L, Aryal UK, Dai ZY, Mason AC, Monroe ME, Tian ZX, Zhou JY, Su D, Weitz KK, Liu T, Camp DG, Smith RD, Baker SE, Qian WJ. J Proteome Res. 2012;11:143–156. doi: 10.1021/pr200916k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Storr SJ, Royle L, Chapman CJ, Hamid UMA, Robertson JF, Murray A, Dwek RA, Rudd PM. Glycobiology. 2008;18:456–462. doi: 10.1093/glycob/cwn022. [DOI] [PubMed] [Google Scholar]
- 25.Thaysen-Andersen M, Packer NH, Schulz BL. Mol Cell Proteomics. 2016;15:1773–1790. doi: 10.1074/mcp.O115.057638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Plomp R, Bondt A, de Haan N, Rombouts Y, Wuhrer M. Mol Cell Proteomics. 2016;15:2217–2228. doi: 10.1074/mcp.O116.058503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kurcon T, Liu ZY, Paradkar AV, Vaiana CA, Koppolu S, Agrawal P, Mahal LK. Proc Natl Acad Sci USA. 2015;112:7327–7332. doi: 10.1073/pnas.1502076112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alvarez-Manilla G, Warren NL, Atwood J, Orlando R, Dalton S, Pierce M. J Proteome Res. 2010;9:2062–2075. doi: 10.1021/pr8007489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zielinska DF, Gnad F, Wisniewski JR, Mann M. Cell. 2010;141:897–907. doi: 10.1016/j.cell.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 30.Wollscheid B, Bausch-Fluck D, Henderson C, O’Brien R, Bibel M, Schiess R, Aebersold R, Watts JD. Nat Biotechnol. 2009;27:378–386. doi: 10.1038/nbt.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woo CM, Iavarone AT, Spiciarich DR, Palaniappan KK, Bertozzi CR. Nat Methods. 2015;12:561–567. doi: 10.1038/nmeth.3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun SS, Shah P, Eshghi ST, Yang WM, Trikannad N, Yang S, Chen LJ, Aiyetan P, Hoti N, Zhang Z, Chan DW, Zhang H. Nat Biotechnol. 2016;34:84–88. doi: 10.1038/nbt.3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeng Y, Ramya TNC, Dirksen A, Dawson PE, Paulson JC. Nat Methods. 2009;6:207–209. doi: 10.1038/nmeth.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiao HP, Wu RH. Anal Chem. 2017;89:3656–3663. doi: 10.1021/acs.analchem.6b05064. [DOI] [PubMed] [Google Scholar]
- 35.Wang XS, Yuan ZF, Fan J, Karch KR, Ball LE, Denu JM, Garcia BA. Mol Cell Proteomics. 2016;15:2462–2475. doi: 10.1074/mcp.O115.049627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fierro-Monti I, Racle J, Hernandez C, Waridel P, Hatzimanikatis V, Quadroni M. PLOS One. 2013;8 doi: 10.1371/journal.pone.0080423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hinkson IV, Elias JE. Trends Cell Biol. 2011;21:293–303. doi: 10.1016/j.tcb.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 38.Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Hamon C. Anal Chem. 2003;75:1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 39.Chen WX, Smeekens JM, Wu RH. Mol Cell Proteomics. 2014;13:1563–1572. doi: 10.1074/mcp.M113.036251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaji H, Saito H, Yamauchi Y, Shinkawa T, Taoka M, Hirabayashi J, Kasai K, Takahashi N, Isobe T. Nat Biotechnol. 2003;21:667–672. doi: 10.1038/nbt829. [DOI] [PubMed] [Google Scholar]
- 41.Chen WX, Smeekens JM, Wu RH. J Proteome Res. 2014;13:1466–1473. doi: 10.1021/pr401000c. [DOI] [PubMed] [Google Scholar]
- 42.Eng JK, McCormack AL, Yates JR. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 43.Elias JE, Gygi SP. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 44.Kall L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. Nat Methods. 2007;4:923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 45.Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausolei SA, Villen J, Haas W, Sowa ME, Gygi SP. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. Nat Biotechnol. 2006;24:1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 47.Xiao HP, Smeekens JM, Wu RH. Analyst. 2016;141:3737–3745. doi: 10.1039/c6an00144k. [DOI] [PubMed] [Google Scholar]
- 48.Xiao HP, Tang GX, Wu RH. Anal Chem. 2016;88:3324–3332. doi: 10.1021/acs.analchem.5b04871. [DOI] [PubMed] [Google Scholar]
- 49.Xiao HP, Wu RH. Chem Sci. 2017;8:268–277. doi: 10.1039/c6sc01814a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 51.Chen WX, Smeekens JM, Wu RH. Chem Sci. 2016;7:1393–1400. doi: 10.1039/c5sc03826j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang DW, Sherman BT, Lempicki RA. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 53.Orozco M. Chem Soc Rev. 2014;43:5051–5066. doi: 10.1039/c3cs60474h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.