

Abstract
Solid-phase synthesis of antibacterial cyclohexapeptides including wollamides A, B and desotamide B has been developed. Briefly, the protected linear hexapeptides were assembled on 2-chlorotrityl chloride resin using standard Fmoc chemistry and diisopropylcarbodiimide/hydroxybenzotriazole coupling reagents, cleaved off-resin with hexafluoroisopropanol/dichloromethane to keep side-chain protecting groups intact, and cyclized in solution. Final global removal of all protecting groups using a cocktail of trifluoroacetic acid/triisopropylsilane/dichloromethane afforded the desired cyclic hexapeptides, which were characterized by 1H, 13C NMR, and HRMS. Subsequent investigation of macrocyclization parameters such as terminal residues, coupling reagents, and cyclization concentration revealed the optimized conditions for the synthesis of this class of cyclic hexapeptides.
Keywords: Cyclohexapeptide, Desotamide B, Macrocyclization, Macrolactamization, Solid-phase synthesis, Wollamide A, Wollamide B
Introduction
Desotamide B (1) (Fig. 1), a cyclic hexapeptide, was first described in 2014 by Ju and colleagues as a secondary metabolite from a deep-sea Streptomyces scopuliridis (SCSIO ZJ46).1 To date, this rare family of cyclic peptides also includes desotamides A,2 B-D,1 E-F,3 and G.4 In the same year, 1 was also reported by Capon and colleagues as a metabolite from a soil Streptomyces nov. sp. (MST-115088) along with two new cyclic hexapeptides, wollamides A (3) and B (2) (Fig. 1).3 The amino acid sequence and stereochemistry of these compounds were established by a combination of spectroscopic analysis and chemical degradation.3 Compounds 1 and 2 contain five amino acid residues in common: l-Asn, l-Val, d-Leu, l-Leu, and l-Trp. Since Gly occurs as the final amino acid in its sequence, 1 contains all proteinogenic residues except for d-Leu. Of much more biological interest are wollamides B (2) and A (3) that incorporate the nonproteinogenic and basic amino acid residue ornithine (Orn),5 which is also commonly found in other naturally occurring antimicrobial peptides such as daptomycin6 and bacitracin. Wollamide A (3) differs from 2 by the substitution of a single amino acid, l-Val, with l-allo-Ile. This unique residue is found in other bioactive molecules such as mar-formycins,7 and its biosynthetic origins have until recently been uncharacterized.8
Fig. 1.

Structures of desotamide B and wollamides A and B.
In addition to their rarity as secondary metabolites of Streptomyces, several members of this cyclic hexapeptide family have been shown to exhibit antibacterial activity.1,3 Desotamide B has been reported to have moderate Gram-positive antibacterial activity against S. aureus, S. pneumoniae,1 and B. subtilis.3 Notably, the basic d-Orn in wollamides A and B expanded the antibacterial spectrum to include mycobacteria (IC50s = 2.8 and 3.1 mM against M. bovis, respectively) that was not exhibited by desotamides A, B, E or F.3 In addition to the M. bovis activity reported for these d-Orn homologues, wollamides also displayed higher IC50 values (>30 μM) against mammalian cancer cell lines (SW620 and NCIH-460), indicating potential antibacterial and/or antimycobacterial selectivity.3
Desotamide B (1) and the wollamides (2 and 3) have been previously isolated in very low yields (<1%) from Streptomyces extract,3 which greatly hinders further antibacterial profiling and development. As natural products such as the wollamides can serve as promising antitubercular leads for medicinal chemistry efforts, an efficient synthetic route to these molecules is needed for subsequent structure-activity relationship studies. Very recently, synthesis and antitubercular evaluation of wollamide B and analogues were reported by Imming and colleagues.9 So far, no synthesis has been reported for wollamide A and desotamide B. Herein we report the synthesis and optimization studies toward 1–3 on solid support with solution phase macrocyclization.10
Results and discussion
Retrosynthetic strategy
Scheme 1 outlines a retrosynthetic methodology to obtain desotamide B(1), wollamide B (2),and wollamide A (3). To synthesize the target molecules, the constitutive amino acid residues with acid-labile protecting groups on their side chains were commercially available in their fluorenylmethyloxycarbonyl (Fmoc) protected forms. The first amino acid would be anchored on the highly acid-labile 2-chlorotrityl chloride (2-CTC) resin, a solid support that prevents diketopiperazine formation due to steric effect.11 Furthermore, 2-CTC resin is applicable to mild acidolytic conditions12 that would leave the boc and trt protecting groups of Trp, Asn, and Orn intact for the succeeding head-to-tail macrolactamization in solution. Finally, subsequent global deprotection of the side-chain protecting groups would afford the final products 1–3.
Scheme 1.

Retrosynthetic scheme.
Representative total synthesis of wollamide B
FMOC-SPPS (Scheme 2) of the resin-bound linear hexapeptide 9 commenced with the attachment of Fmoc-Asn(trt)-OH onto the 2-CTC resin with N,N-diisopropylethylamine (DIPEA) in dichloro-methane (DCM). Fmoc deprotection with 25% 4-methylpiperidine in N,N-dimethylformamide (DMF) exposed the N-terminal amine of Asn(trt) 4 that was coupled to Fmoc-Val-OH with hydroxybenzotriazole (HOBt) and N,N′-diisopropylcarbodiimide (DIC).13 This afforded the immobilized dipeptide 5 (Scheme 2). Sequence elongation with d-Leu, Leu, Trp(boc), and d-Orn(boc) in a C to N direction afforded the immobilized linear precursor 9. Mild acidolytic cleavage of the resin-bound hexapeptide 9 with 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) in DCM (1:4)12 yielded the side-chain protected linear precursor 10 in 55% overall yield. By HPLC and NMR analysis, the linear hexapeptide 10 was obtained in high purity and used directly for subsequent macrolactamization without further purification. Macrolactamization of 10 between the C-terminus l-Asn(trt) and the N-terminus d-Orn(boc) was facilitated by N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hex-afluorophosphate (HBTU) and DIPEA, affording the side-chain protected cyclic hexapeptide 11 after purification by flash column chromatography. Global deprotection of the N-boc and trityl groups was accomplished with a cocktail of trifluoroacetic acid (TFA) and the carbocation scavenger triisopropylsilane (TIPS)14 in DCM. Following purification with amine functionalized silica gel column chromatography, wollamide B (2) was isolated in 17% yield over 15 steps. The 1H and 13C NMR spectroscopic data (Fig. 2A and Supporting Information) of synthetic 2 were in agreement with those of the natural product.3
Scheme 2.

Solid-phase synthesis of wollamide B (2) on 2-CTC resin. Reagents and conditions: (a) Fmoc-Asn(trt)-OH, DIPEA, DCM, 3 h; (b) 25% 4-methylpiperidine in DMF; (c) Fmoc-Val-OH, DIC, HOBt, DMF/DCM 1:1, 4 h; (d) Fmoc-d-Leu-OH, DIC, HOBt, DMF/DCM 1:1, 4 h; (e) Fmoc-Leu-OH, DIC, HOBt, DMF/DCM 1:1, 4 h; (f) Fmoc-Trp(boc)-OH, DIC, HOBt, DMF/DCM 1:1, 4 h; (g) Fmoc-d-Orn(boc)-OH, DIC, HOBt, DMF/DCM 1:1, 4 h; (h) HFIP/DCM 1:4, 30 min; (i) HBTU, DIPEA, DMF, 30 min; (j) TFA/TIPS/DCM 50:5:45,30 min.
Fig. 2.
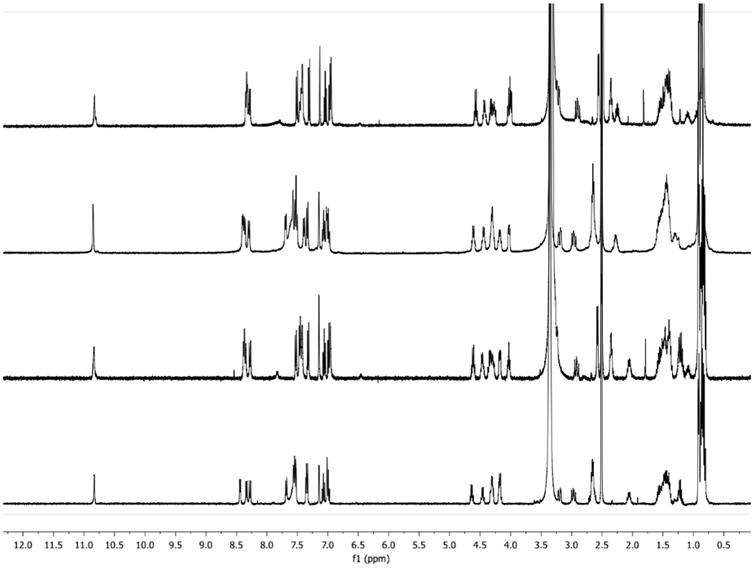
1H NMR spectra for synthesized wollamides. (A) The free base form of wollamide B (2); (B) The TFA salt form of wollamide B (2); (C) The free base form of wollamide A (3); (D) The TFA salt form of wollamide A (3). Samples for 1H NMR were dissolved in DMSO-d6 and spectra were collected on a Bruker AVANCE DRX-400 MHz NMR spectrometer (Bruker, Billerica, MA, USA) at 298 K.
Next, in an effort to improve the yield of cyclohexapeptides and streamline the purification process following final global deprotection, we found that, instead of running a column chromatography, the lipophilic byproducts from the boc and trt deprotection could be simply removed by precipitation and decanting using trituration with ethyl acetate and hexanes. After lyophilization of the resulting solid, the 1H NMR spectrum of the TFA salt form of wollamide B showed some notable differences compared to that of the free base form of wollamide B (Fig. 2A and B). In the literature, the 1H NMR spectra differences between the free base and its salt form were well documented.15–18 To our surprise, the 1H NMR spectrum of the TFA salt form of synthetic wollamide B showed striking similarity to that of recently reported free base form of wollamide B, and both spectra were recorded at 400 MHz in DMSO-d6.9 The free base and TFA salt forms of synthetic wollamide B (2) were also supported by their IR and 19F NMR spectra (Fig. 3). To further confirm, the TFA salt form of 2 was then converted to its free base form by washing with NaHCO3 solution,19 and the 1H NMR spectrum of the resulting free base form was consistent with that of the free base form isolated using the base-modified silica purification (Supporting Information).
Fig. 3.
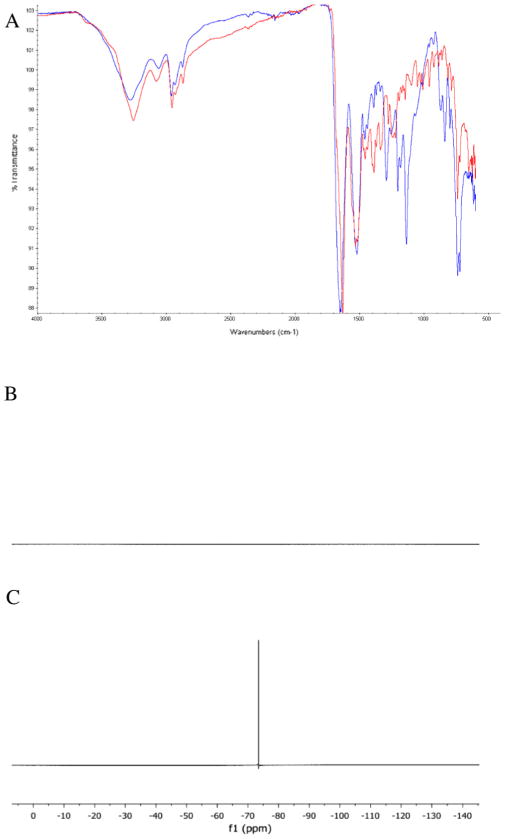
The IR and 19F NMR spectra of wollamide B (2). (A) The overlay IR spectra of wollamide B, red refers to the free base form, and blue refers to the TFA salt form; (B) The 19F NMR of the free base form of wollamide B; (C) The 19F NMR of the TFA salt form of wollamide B, showing the 19F signal at −73.47 ppm.
With the successful total synthesis of wollamide B (2), the method was applied to the synthesis of its close structural analogues, desotamide B (1) and wollamide A (3). The synthesis of 1 and 3 proceeded in the same manner as 2, but with Gly as the last residue and l-allo-Ile as the second residue in the linear hexapeptide assembly, respectively. Again, following purification with the base-modified silica gel column chromatography, the 1H NMR spectrum of synthetic desotamide B (1) was also in agreement with that of the isolated natural product.1,3 However, in the case of wollamide A (3), column purification with amine functionalized silica gel resulted in a product (Fig. 2C) that had notable 1H NMR differences (e.g., in the regions of 4–4.75 ppm and 8.25–8.5 ppm) from isolated natural product.3 The TFA salt form of wollamide A was then prepared as previously done for wollamide B to determine if this was the cause of the observed chemical shift discrepancies.20 Indeed, we found that the 1H and 13C NMR spectra (Fig. 2D and Supporting Information) of the TFA salt form of synthetic wollamide A were in good agreement with those of the natural product,3 revealing the reported natural wollamide A was most likely isolated in its TFA salt form, not the free base form as shown in the paper. The presence of TFA in natural sample may have been introduced during the preparative HPLC purification process when using 0.01% TFA in water and methanol in mobile phases.3
Effect of amino acid residues at the C and N terminals on cyclization efficiency
After the successful total synthesis of the target cyclohexapeptides, additional optimization studies were performed to facilitate future construction of cyclic hexapeptide libraries. In the synthesis of cyclic peptides, strategies often focused on points of macrolactamization and/or coupling reagents for successful synthesis.21 To evaluate different disconnection sites of macrocyclization, the linear hexapeptides of wollamide B were synthesized in parallel starting with a different amino acid in the wollamide B sequence; this produced six linear precursors (10, 12–16) (Table 1), followed by cyclization to afford protected wollamide B precursor 11.
Table 1.
Optimization and evaluation on cyclization efficiency of linear hexapeptide precursors with different amino acid sequence.
| |||||
|---|---|---|---|---|---|
|
| |||||
| Entry | Linear hexapeptide substrates HO-AA1AA2AA3AA4AA5AA6-NH2 | Linear yield (%) | tR (min)a | Linear purity (%)b | Cyclic yield (%)c |
| 1 | HO-Asn(trt)-Val-d-Leu-Leu-Trp(boc)-d-Orn(boc)-NH2 (10) | 55 | 6.93 | 100 (100) | 72 |
| 2 | HO-Val-d-Leu-Leu-Trp(boc)-d-Orn(boc)-Asn(trt)-NH2 (12) | 49 | 6.92 | 98.2 (98.6) | 65d |
| 3 | HO-d-Leu-Leu-Trp(boc)-d-Orn(boc)-Asn(trt)-Val-NH2 (13) | 57 | 6.87 | 98.9 (98.6) | 63 |
| 4 | HO-Leu-Trp(boc)-d-Orn(boc)-Asn(trt)-Val-d-Leu-NH2 (14) | 58 | 6.89/6.90 | 98.7 (99.7) | 77 |
| 5 | HO-Trp(boc)-d-Orn(boc)-Asn(trt)-Val-d-Leu-Leu-NH2 (15) | 53 | 6.90 | 99.6 (98.0) | 65 |
| 6 | HO-d-Orn(boc)-Asn(trt)-Val-d-Leu-Leu-Trp(boc)-NH2 (16) | 52 | 6.85 | 99.5 (99.6) | 68 |
HPLC retention time.
Determined by HPLC analysis at 254 and 220 nm, respectively.
Yield refers to isolated peptides after silica gel column chromatography.
Desired product 11 (41%) and C-terminal d-Val epimer 18 (24%).
From the results summarized in Table 1, it was found that the overall linear yields of 10 and 12–16 ranged from 49% to 58%, with the lowest linear yield (49%) resulted when l-Val was anchored to the 2-CTC resin (entry 2, Table 1). For the cyclization with HBTU, all linear precursors were cyclized in 30 min (<5% linear hexapeptide detected by HPLC), except for 12 that required 1 h to complete the cyclization. Additionally, all the linear hexapeptide substrates 10 and 13–16 cyclized smoothly to give 11 with moderate to good isolated yields (63–77%) after column purification. However, in the case of 12 (entry 2, Table 1; Scheme 3), two products with the same mass and the same C18 HPLC retention time closely eluted during normal phase silica gel column purification. The slower eluting compound with a lower Rf value was confirmed as the desired product 11, and its 1H NMR spectrum was consistent with that of the sole cyclic product 11 from all the other linear precursors (10, 13– 16) (Supporting Information). The faster eluting compound was later identified as the C-terminal d-Val epimer 18, which was confirmed by an independent synthetic route from the d-Val incorporated linear peptide [HO-Asn(trt)-d-Val-d-Leu-Leu-Trp(boc)-d-Orn (boc)-NH2] (17) (Scheme 3 and Supporting Information).22,23 Subsequent cyclization of 17 resulted in a single product that had an identical 1H NMR spectrum (Supporting Information) with the epimer byproduct 18 from the cyclization of 12 (entry 2, Table 1). In this study, only 12 resulted in partial loss (24%) of C-terminal stereointegrity and detectable C-terminal epimerization as determined by isolation after column purification. In the comparative analysis of these cases, it was also found that the cyclization reaction of 12 with terminal l-Asn and l-Val residues proceeded notably slower than all the other cyclizations with HBTU; the increased reaction time of its activated linear peptide intermediate as well as conformational and/or steric factors may have reduced its cyclization efficiency, providing opportunity for the formation of the C-terminal epimer.21,23,24
Scheme 3.
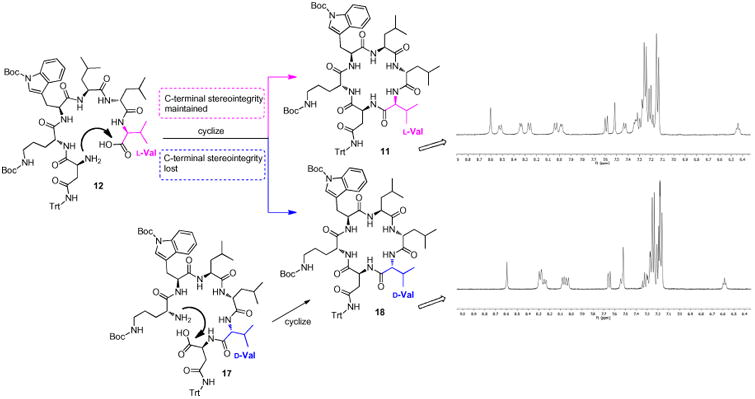
Cyclization of 12 using HBTU and DIPEA to give the desired product 11 and the C-terminal d-Val epimer 18, which was confirmed through an independent synthetic route from 17. For clarity, only partial 1H NMR spectra of 11 and 18 are shown.
Moreover, on the basis of this study, it was observed that the linear hexapeptide substrate 14 with terminal l- and d-Leu residues underwent efficient cyclization to afford the highest yield (77%, entry 4, Table 1) without detectable epimerization, thereby offering an alternate linear order to the synthesis of wollamide B.
Effect of coupling reagents and concentration on cyclization efficiency
In the synthesis of 2, cyclization was facilitated by HBTU, a uronium reagent.25 To evaluate the effect of different coupling reagents on cyclization efficiency, several other coupling reagents including O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU), (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC)/DIC, and 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM) were also assessed in this study (Table 2).25 TBTU is in the same class of uronium reagents as HBTU, differing only in its counterion. PyBOP belongs to a class of phosphonium reagents and EDC/DIC belong to the more economical carbodiimide class of reagents. In general, the carbodiimides also require the use of a racemization suppressant (e.g., HOBt), which was not needed for the other coupling reagents. DMTMM is often classified as a miscellaneous reagent based on its structure.25
Table 2.
Survey of cyclization conditions for protected wollamide B linear precursor 10 to 11.
| Entry | Cyclization conditions (equiv) | Conc. (M) | Reaction time (h) | Yielda (%) |
|---|---|---|---|---|
| 1 | HBTU/DIPEA (1.02, 3) | 0.001 | 0.5 | 72 |
| 2 | HBTU/DIPEA (1.02, 3) | 0.01 | 0.5 | 73 |
| 3 | HBTU/DIPEA (1.02, 3) | 0.05 | 0.5 | 71 |
| 4 | HBTU/DIPEA (1.02, 3) | 0.10 | 0.5 | 66 |
| 5 | TBTU/DIPEA (1.02, 3) | 0.001 | 0.5 | 61 |
| 6 | DMTMM/DIPEA (3, 3) | 0.001 | 0.5 | 59 |
| 7 | PyBOP/DIPEA (1.5, 3) | 0.001 | 0.5 | 68 |
| 8 | EDC-HCl/HOBt/DIPEA (1.5, 1.5, 3) | 0.001 | 54 | 43 |
| 9 | DIC/HOBt/DIPEA (3, 3, 3) | 0.001 | 1 week | 30 |
Isolated yields after silica gel column chromatography.
In the cyclization of 10 at 0.001 M concentration, we found that HBTU/DIPEA and PyBOP/DIPEA (entry 1 and 7, Table 2) resulted in the highest yields (72% and 68%, respectively). TBTU and DMTMM also cyclized the linear peptide in 30 min, but slightly lower yields (59–61%) were obtained after column purification. In contrast, the carbodiimide reagents (entry 8 and 9, Table 2) gave the lowest yields (30–43%). In addition, EDC/HOBt/DIPEA required 54 h for the starting material to be <5% as monitored by HPLC. Cyclization of 10 with DIC/HOBt/DIPEA proceeded even slower than with EDC, and the reaction was stopped after 1 week to afford 11 in 30% yield.
In the macrocyclization of linear peptides, concentration often plays an important role in the formation of desired macrocycles and minimization of unwanted oligomer and polymer side products.26 Therefore, it is generally recommended that the macrocyclization of linear peptides be performed under dilute submillimolar concentrations.23 While the initial cyclization of 10 was performed at 0.001 M using HBTU/DIPEA, cyclization was also carried out at 0.01, 0.05, and 0.1 M concentrations (entry 2–4, Table 2). At all concentrations evaluated, the mono-meric cyclohexapeptide 11 was obtained as the only product with comparable yields (66–73%), which will facilitate future scale-up and analogue synthesis. These results suggest that the linear peptide 10 from l-Asn to d-Orn order may have its reactive C- and N-terminus in close spatial proximity due to the secondary structural characteristics as determined by its amino acid residue order.23,27,28
Conclusions
In summary, we have reported the total synthesis and optimization studies of wollamides A, B and desotamide B by a combination of solid-phase peptide synthesis and solution-phase macrolactamization. Notably, the 1H NMR spectra of the free base and TFA salt forms of wollamides A and B revealed striking differences, especially in the regions of 4–4.75 ppm and 8.25–8.5 ppm. While the 1H NMR spectrum of the free base form of wollamide B is in agreement with that of the natural product, interestingly, the 1H NMR spectrum of the TFA salt form of wollamide A is consistent with that of reported natural wollamide A. This is the first report describing and clarifying the 1H NMR discrepancies between the free base and TFA salt forms of cyclohexapeptide wollamides. The cyclization studies of different linear hexapeptide substrates revealed that 14 with terminal l- and d-Leu residues underwent efficient cyclization to afford the highest yield (77%). Moreover, C-terminal epimerization was detected when cyclizing between the N-terminus l-Asn and C-terminus l-Val. Antibacterial activities and structure-activity relationships of these hexapeptides and structural analogues will be reported in due course.
Supplementary Material
Acknowledgments
This work was supported in part by the NIH grant (R15AI092315). We also thank Justin Reinicke for his assistance with the HRMS data.
Footnotes
Supplementary data: Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.tetlet.2017.05.084.
References
- 1.Song Y, Li Q, Liu X, et al. J Nat Prod. 2014;77:1937. doi: 10.1021/np500399v. [DOI] [PubMed] [Google Scholar]
- 2.Miao S, Anstee MR, LaMarco K, Matthew J, Huang LHT, Brasseur MM. J Nat Prod. 1997;60:858. [Google Scholar]
- 3.Khalil ZG, Salim AA, Lacey E, Blumenthal A, Capon RJ. Org Lett. 2014;16:5120. doi: 10.1021/ol502472c. [DOI] [PubMed] [Google Scholar]
- 4.Li Q, Song Y, Qin X, Zhang X, Sun A, Ju J. J Nat Prod. 2015;78:944. doi: 10.1021/acs.jnatprod.5b00009. [DOI] [PubMed] [Google Scholar]
- 5.Groβ A, Hashimoto C, Sticht H, Eichler J. Front Bioeng Biotechnol. 2016;3 doi: 10.3389/fbioe.2015.00211. Article 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steenbergen JN, Alder J, Thorne GM, Tally FP. J Antimicrob Chemother. 2005;55:283. doi: 10.1093/jac/dkh546. [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Wang B, Li H, et al. Org Lett. 2015;17:1509. doi: 10.1021/acs.orglett.5b00389. [DOI] [PubMed] [Google Scholar]
- 8.Li Q, Qin X, Liu J, et al. J Am Chem Soc. 2016;138:408. doi: 10.1021/jacs.5b11380. [DOI] [PubMed] [Google Scholar]
- 9.Asfaw H, Laqua K, Walkowska AM, et al. PLoS One. 2017;12:e0176088. doi: 10.1371/journal.pone.0176088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsutsumi LS, Sun D. Part of this work has been presented at the following meeting. American Chemical Society National Meeting; Philadelphia, PA, USA. August 21–25; ORGN-158. [Google Scholar]
- 11.Chatzi KBO, Gatos D, Stavropoulos G. Int J Pept Protein Res. 1991;37:513. [PubMed] [Google Scholar]
- 12.Bollhagen R, Schmiedberger M, Barlos K, Grell E. Chem Commun. 1994;2559 [Google Scholar]
- 13.Sun D, Lee RE. J Comb Chem. 2007;9:370. doi: 10.1021/cc060154w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pearson DA, Blanchette M, Baker ML, Guindon CA. Tetrahedron Lett. 1989;30:2739. [Google Scholar]
- 15.Calvert MB, Sperry J. Chem Commun. 2015;51:6202. doi: 10.1039/c5cc00380f. [DOI] [PubMed] [Google Scholar]
- 16.Kumpulainen ETT, Koskinen AMP, Rissanen K. Org Lett. 2007;9:5043. doi: 10.1021/ol7022856. [DOI] [PubMed] [Google Scholar]
- 17.Davis RA, Baron PS, Neve JE, Cullinane C. Tetrahedron Lett. 2009;50:880. [Google Scholar]
- 18.Berry Y, Bremner JB, Davis A, Samosorn S. Nat Prod Res. 2006;20:479. doi: 10.1080/14786410500463270. [DOI] [PubMed] [Google Scholar]
- 19.Gibbons JB, Gligorich KM, Welm BE, Looper RE. Org Lett. 2012;14:4734. doi: 10.1021/ol3019242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Satisfactory elemental analyses were obtained for the TFA salt forms of synthetic wollamides B (2) and A (3), revealing a stoichiometry of 1.35 and 1.20 TFA per molecule, respectively.
- 21.Humphrey JM, Chamberlin AR. Chem Rev. 1997;97:2243. doi: 10.1021/cr950005s. [DOI] [PubMed] [Google Scholar]
- 22.Kaur H, Harris PW, Little PJ, Brimble MA. Org Lett. 2015;17:492. doi: 10.1021/ol503507g. [DOI] [PubMed] [Google Scholar]
- 23.White CJ, Yudin AK. Nat Chem. 2011;3:509. doi: 10.1038/nchem.1062. [DOI] [PubMed] [Google Scholar]
- 24.Ehrlich A, Heyne HU, Winter R, et al. J Org Chem. 1996;61:8831. doi: 10.1021/jo951108d. [DOI] [PubMed] [Google Scholar]
- 25.Han SY, Kim YA. Tetrahedron. 2004;60:2447. [Google Scholar]
- 26.Londregan AT, Farley KA, Limberakis C, Mullins PB, Piotrowski DW. Org Lett. 2012;14:2890. doi: 10.1021/ol301173m. [DOI] [PubMed] [Google Scholar]
- 27.Blankenstein J, Zhu J. Eur J Org Chem. 2005;2005:1949. [Google Scholar]
- 28.Martí-Centelles V, Pandey MD, Burguete MI, Luis SV. Chem Rev. 2015;115:8736. doi: 10.1021/acs.chemrev.5b00056. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.