

Summary
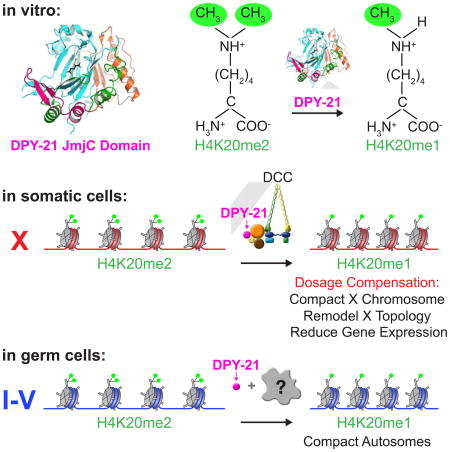
Chromatin modification and higher-order chromosome structure play key roles in gene regulation, but their functional interplay in controlling gene expression is elusive. We discovered the machinery and mechanism underlying the dynamic enrichment of histone modification H4K20me1 on hermaphrodite X chromosomes during C. elegans dosage compensation and demonstrated H4K20me1's pivotal role in regulating higher-order chromosome structure and X-chromosome-wide gene expression. Structure and activity of dosage-compensation-complex (DCC) subunit DPY-21 defined a Jumonji demethylase subfamily that converts H4K20me2 to H4K20me1 in worms and mammals. Selective inactivation of demethylase activity eliminated H4K20me1 enrichment in somatic cells, elevated X-linked gene expression, reduced X-chromosome compaction, and disrupted X-chromosome conformation by diminishing formation of topologically-associating domains (TADs). Unexpectedly, DPY−21 also associates with autosomes of germ cells in a DCC-independent manner to enrich H4K20me1 and trigger chromosome compaction. Our findings demonstrate the direct link between chromatin modification and higher-order chromosome structure in long-range regulation of gene expression.
Graphical abstract
Introduction
Gene regulation in multicellular organisms entails diverse strategies that operate over different distances. Some mechanisms act locally on individual genes, while others act over long range to co-regulate expression of numerous genes within sub-chromosomal domains (e.g. imprinted gene clusters, hox clusters) (Andrey et al., 2013; Plasschaert and Bartolomei, 2014; Williamson et al., 2012), or across entire chromosomes (e.g. X-chromosome dosage compensation) (da Rocha and Heard, 2017; Kuroda et al., 2016; Meyer, 2010; Strome et al., 2014). Post-translational modifications of histones and three-dimensional chromosome architecture are prime elements of long-range gene regulation, but their functional interplay in controlling gene expression has been elusive (Dekker and Mirny, 2016; Soshnev et al., 2016).
Here we dissect X-chromosome dosage compensation (DC) in C. elegans to determine the effect of chromatin modifications on higher-order chromosome structure during long-range gene regulation. DC is exemplary because it controls hundreds of genes simultaneously, it distinguishes X chromosomes from autosomes, and it discriminates between the sexes in modulating gene expression. The C. elegans dosage compensation complex (DCC), a ten subunit complex with five condensin subunits, binds to both hermaphrodite X chromosomes to reduce gene expression by half, thereby equalizing it with that of the single male X (Meyer, 2010). Moreover, the DCC remodels the topology of hermaphrodite X chromosomes into a spatial conformation distinct from that of autosomes or male X chromosomes by forming topologically associating domains (TADs) (Crane et al., 2015). During that process, the chromatin modification H4K20me1 becomes enriched on the dosage-compensated X chromosomes (Kramer et al., 2015; Liu et al., 2011; Vielle et al., 2012; Wells et al., 2012). Neither the role of this modification in chromosome-wide gene control nor its effect on X-chromosome structure has been determined.
H4K20me1 is also enriched on the inactive X of female mammals, revealing a common feature of diverse DC strategies (Kohlmaier et al., 2004). Its contribution to transcriptional silencing is unknown. In general, the role of H4K20me1 in gene regulation has remained a puzzle due to its context-dependent contribution to both gene activation and gene repression (Beck et al., 2012).
Although H4K20 methylation has been implicated in myriad nuclear functions including gene regulation, DNA replication and repair, mitotic chromosome condensation, and cell cycle control, the mechanisms that regulate different H4K20me states (H4K20me1/me2/me3) and transduce these states into properly executed nuclear functions are not well understood (Beck et al., 2012; Jorgensen et al., 2013; van Nuland and Gozani, 2016). In principle, H4K20me1 enrichment on C. elegans X chromosomes could occur by activating the methyltransferase (SET-1) that converts H4K20 to H4K20me1, by inhibiting the methyltransferase (SET−4) that converts H4K20me1 to H4K20me2/me3, by inhibiting the demethylase (JMJD−1.1/1.2) that converts H4K20me1 to H4K20, or by activating an unknown demethylase that converts H4K20me2 to H4K20me1. Although H4K20me2 is the predominant form of H4K20 in eukaryotic cells (Pesavento et al., 2008), only a neuron-specific H4K20me2 demethylase has been reported for any organism (Wang et al., 2015). No H4K20me2 demethylase has yet been identified that could regulate H4K20me1 levels during DC.
We pursued the machinery and mechanisms that catalyze H4K20me1 enrichment on X chromosomes and the role of H4K20me1 in higher-order chromosome structure and gene regulation. The key element in controlling H4K20me1 levels is a DCC subunit. Apart from five condensin subunits (DPY−26, DPY−27, DPY−28, MIX−1, CAPG−1), the DCC includes an XX-specific protein (SDC−2) that triggers assembly of the DCC onto X chromosomes, two proteins (SDC−3 and DPY−30) that aid SDC−2 in recruiting the DCC to X, and two proteins (SDC−1 and DPY−21) required for full DCC activity but not assembly (Meyer, 2010).
Through X-ray crystallography and biochemical assays, we found that DPY−21 defines a new subfamily of Jumonji C (JmjC) histone demethylases that converts H4K20me2 to H4K20me1 and is widely conserved from worms to mammals. Inactivation of JmjC activity in vivo abrogated H4K20me1 enrichment on X, reduced X-chromosome compaction, disrupted X-chromosome topology by weakening TAD boundaries, and elevated X-linked gene expression, demonstrating key roles for H4K20me1 in DC. Unexpectedly in germ cells, DPY−21 associates with autosomes, but not X chromosomes, in a DCC-independent manner to enrich H4K20me1 and facilitate chromosome compaction. Thus, DPY−21 is an adaptable chromatin regulator that is harnessed during development for distinct biological functions. In both somatic cells and germ cells, H4K20me1 enrichment modulates 3D chromosome architecture, demonstrating the direct impact of chromatin modification on establishing higher-order chromosome structure.
Results
DPY−21 is a Jumonji C (JmjC) domain-containing protein
Although none of the ten DCC subunits had a demethylase domain identified by primary sequence analysis, structure prediction programs (Methods) suggested homology between the C-terminal domain of DPY−21 and JmjC domain-containing lysine demethylases (KDMs), despite low (15%) sequence identity. JmjC KDMs are Fe2+ and α-ketoglutarate (α-KG) dependent dioxygenases that demethylate lysines in histone and non-histone proteins (Markolovic et al., 2016). To investigate this potential connection, we determined a 1.8 Å crystal structure of the C. elegans DPY−211210–1617 fragment that encompasses the putative JmjC domain (Table S1 and Methods). Strikingly, comparisons to known structures using DALI (Holm and Rosenstrom, 2010) revealed DPY−21 to be a JmjC domain-containing protein most similar to the JmjC KDMs (Z score 21.3–14.5 and root-mean-square deviation of 2.9 Å – 3.7 Å), strongly suggesting that DPY−21 indeed acts as a histone demethylase.
Similar to other JmjC KDMs, the DPY−211210–1617 structure includes a JmjC domain that is folded into a double-stranded β-helix (DSBH) and surrounded by a Jumonji N (JmjN) domain, a β-hairpin motif, and a mixed domain (Figure 1A) (Chen et al., 2006). As in all α-KG dioxygenases, the DPY−21 DSBH core bears facial triad residues (H1452, D1454, and H1593) that chelate Fe2+ and form an active site (Figures 1B and 1C). The electron density map revealed an unexpected active site density that was identified as an α-KG molecule, although no α-KG was added during purification or crystallization. α-KG coordinates Fe2+ in a bidentate manner, and a water molecule completes the octahedral coordination of Fe2+ (Figures 1B and 1C). The α-KG is further stabilized by hydrogen bonding to side chains of T1449 and Y1585 and also by hydrogen bonding to side chains of W1410, K1526, and S1603 via water molecules. In addition, α-KG forms van der Waals contacts with A1499, L1587, and F1595 (Figures 1B and 1C).
Figure 1. Structural and Functional Characterization of the DPY−21 JmjC Demethylase Domain.

(A) DPY-211210–1617 structure in complex with a-KG (black) and Fe2+ (orange) showing JmjC (cyan), JmjN (orange), b-hairpin (pink), and mixed (green) domains.
(B) Active site of DPY−211210–1617 showing JmjC domain residues (cyan) in complex with a-KG (black), Fe2+ (orange), and water molecules (red). Facial triad residues H1452 and D1454 (red letters) were changed to alanines for in vitro and in vivo studies. The electron density, 2Fo-Fc (mesh), contoured at 1.0 s above the mean is shown for Fe2+, a-KG, and water molecules.
(C) Schematic diagram of contacts made by a-KG with active site residues and water molecules. Contacts include electrostatic (grey dashed lines in Å) and hydrophobic (blue and red eyelashes) interactions.
(D) In vitro demethylase assay performed on bulk histones. DPY−211210–1641 and mROSBIN350–795 specifically demethylate H4K20me2 only in the presence a-KG, FeSO4, and ascorbate. Mutation of facial triad residues (H1452A D1454A) in DPY−211210–1641 abolished enzymatic activity.
(E) Mass spectrometry analysis of mROSBIN350–795 activity showed demethylation of an H4K20me2 peptide. Upper and lower panels show spectra in the absence and presence of a-KG, FeSO4, and ascorbate, respectively. See Figures S1, S2 and Table S1.
The DPY−21 C-terminal domain is similar (33% sequence identity) to C-terminal domains of metazoan proteins named round spermatid basic proteins 1 (ROSBIN) (Yonker and Meyer, 2003) (Figure S1B). Mouse ROSBIN, an essential protein with minimal biochemical characterization, is expressed in embryos and male gonads (Koscielny et al., 2014) (Takahashi et al., 2004). Structure-guided sequence comparison of DPY−21 with ROSBIN family members showed that all residues that interact with Fe2+ and α-KG are conserved (Figures S1A and S1B). Moreover, sequence conservation between these proteins (Figure S1A) extends to regions participating in histone peptide binding, as revealed by JmjC KDM structures (Markolovic et al., 2016). Extensive sequence conservation with DPY−21 suggests that ROSBINs may also be JmjC demethylases that share substrate specificity.
DPY−21 and mROSBIN are the first Jumonji C H4K20me2 demethylases
We performed in vitro histone demethylase assays to determine whether DPY−21 is a bona fide histone demethylase and to define its substrate specificity. We incubated a set of purified DPY−21 fragments with bulk histones and assayed H3 and H4 methylation marks by immunoblotting. DPY−211210–1641, which contains the core crystallized JmjC domain and the remaining 24 C-terminal residues, exhibited robust demethylase activity with histone H4 methyl substrates (Figure 1D), but not with histone H3 substrates (Figure S2A). DPY−211210–1641 demethylated H4K20me2 to H4K20me1 only in the presence of all cofactors (Figure 1D). Changes in facial triad residues H1452 and D1454 prevented demethylation of H4K20me2, confirming their critical role in demethylase activity.
Together these data establish DPY−21 as the first JmjC domain-containing demethylase with specificity for H4K20me2.
To assess the evolutionary conservation of this JmjC demethylase activity, we assayed a mouse ROSBIN fragment (mROSBIN350–795) that includes the JmjC domain (Figure S1B). Similar to DPY−211210–1641, mROSBIN350–795 displayed robust H4K20me2 demethylase activity only in the presence of cofactors (Figure 1D), demonstrating wide conservation of JmjC demethylase activity. mROSBIN350–795 was also inactive against histone H3 methyl substrates (Figure S2A), suggesting specificity for H4.
While the in vitro demethylase assay clearly demonstrated a reciprocal decrease in the level of H4K20me2 with an increase in H4K20me1, results for H4K20me3 were not compelling (Figure S2B). To further dissect substrate specificity, we assayed methylated H4K20 histone peptides. Mass spectrometry showed that mROSBIN350–795 demethylated the H4K20me2 peptide (Figure 1E) but not the H4K20me3 peptide (data not shown), while DPY−211210–1641 was not active against either peptide (data not shown), plausibly due to differences in the C termini (Figure S1B). Collectively, our in vitro results demonstrate that DPY−211210–1641 and mROSBIN350–795 are histone lysine demethylases, with a strong preference for H4K20me2 as the substrate.
DPY−21 is the founding member of a new JmjC subfamily, KDM9
Histone lysines are demethylated by two classes of demethylases that use different chemistry: flavin-dependent lysine-specific demethylases that belong to the KDM1 subfamily, and Fe2+ and α-KG dependent demethylases that belong to the JmjC family (Markolovic et al., 2016). JmjC demethylases are divided into subfamilies KDM2-KDM7 based on domain architecture and specificity toward histone methylation marks.
Very few demethylases of either class are known to act on methylated H4K20 substrates. For H4K20me2, only the neuron-specific demethylase LSD1n had been found prior to our work. LSDN1n, an isoform of LSD1, is a flavin-dependent monoamine oxidase that demethylates both H4K20me2 and H4K20me1 (Wang et al., 2015). The only known JmjC H4K20 demethylases (PHF2 and PHF8) have specificity for H4K20me3 and H4K20me1, respectively (Liu et al., 2010; Qi et al., 2010; Stender et al., 2012) and belong to the KDM7 subfamily. Both PHF2 and PHF8 also have an N-terminal PHD domain required for H4K20 demethylase activity in vitro, which occurs only in the context of nucleosomes.
In contrast, DPY−21 and mROSBIN are unique compared to PHF2 and PHF8 not only because of their H4K20me2 substrate specificity but also their architecture. DPY−21 and mROSBIN lack PHD and other chromatin-interacting domains. They only require the JmjC domain for substrate recognition, allowing them to demethylate H4K20me2 in vitro on histones and histone peptides. Based on our structural and biochemical results, we propose a new subfamily of JmjC lysine demethyases, the KDM9 subfamily, founded by Caenorhabditis DPY−21 and the evolutionarily related ROSBIN proteins. These enzymes are the first JmjC demethylases shown to demethylate H4K20me2.
The DPY−21 JmjC demethylase acts in vivo to enrich H4K20me1 on X chromosomes of somatic cells
To assess JmjC demethylase functions in vivo, we edited codons for catalytic residues in endogenous dpy−21 loci to generate mutant proteins with the following substitutions in the facial triad: H1452A [allele, dpy−21(y607 JmjC)], D1454A [allele, dpy−21(y618 JmjC)], or H1452A D1454A [allele, dpy−21(y622 JmjC)].
In wild-type XX hermaphrodites (Figures 2A and 2C), but not XO males (Figure S3B), H4K20me1 is enriched on X chromosomes relative to autosomes (see also (Vielle et al., 2012; Wells et al., 2012). Reciprocally, H4K20me2/me3 are depleted from X (Figures 2B and 2D) and (Vielle et al., 2012). In all three engineered dpy−21(JmjC) mutants, sex-specific enrichment of H4K20me1 was abolished from X chromosomes of XX adults (Figures 2A, 2C, and S3A) and embryos (Figures 3A and S3D), and H4K20me2/me3 levels were restored (Figures 2B, 2D, and S3C). Thus, H4K20me1 enrichment on X occurs by demethylation of H4K20me2 to H4K20me1.
Figure 2. DPY−21 Demethylase Enriches H4K20me1 on X in vivo.

(A), (B) Confocal images of gut nuclei from wild-type and dpy−21 mutants show H4K20me1 (A) and H4K20me2/me3 (B) levels on X vs. autosomes, with DPY−27 marking X.
(C), (D) Boxplots summarize H4K20me1 or H4K20me2/me3 levels on X relative to their average levels on all chromosomes of wild-type and mutant nuclei. Boxes, middle 50% of ratios. Center bars, medians. X chromosomes of dpy−21(null) and dpy−21(JmjC) mutants have reduced H4K20me1 (p < 10−14) and enhanced H4K20me2/me3 (p < 10−9) relative to wild-type. H4K20me1 enrichment (p = 0.1) and H4K20me2/me3 depletion (p = 0.2) are not different in sdc−1(null) vs. wild-type. P values, 2-sided Wilcoxon rank-sum test.
(E) DPY−21 tethering to ectopic rex−1 DCC binding sites inserted on chromosome V caused JmjC-dependent H4K20me1 enrichment in gut nuclei. Scale bars (A), (B), (E), 2 μm.
(F) Quantification of DPY−27-marked rex insertions with HK20me1 enrichment from confocal images of strains in (E).
(G) Spike-in corrected H4K20me1 enrichment on X (red) vs. autosomes (blue) in ChIP-seq assays reveals significant decrease in H4K20me1 on X in dpy−21(JmjC) or dpy−21(null) vs. wild-type embryos. Average enrichment of two biological replicates is shown for each genotype with error bars indicating SD.
(H), (I) ChIP-seq profiles show spike-in corrected H4K20me1 enrichment in representative regions of chr. X (H) and chr. I (I) in wild-type and dpy−21(JmjC) or dpy−21(null) mutant embryos.
(J) Average spike-in corrected H4K20me1 enrichment plotted across transcription start sites (TSS) defined by GRO-CAP and GRO-seq (Kruesi et al., 2013) or across the middle of intergenic regions on X (red) and autosomes (blue). Similar decreases in H4K20me1 were found on all regions of X in dpy−21 mutants vs. wild-type. n, number of X or A regions in each set of related plots. See Figure S3.
Figure 3. Recruitment of DPY−21 to X Chromosomes is Cell-cycle dependent and Coincident with H4K20me1 Enrichment.

(A) Nuclei from wild-type and dpy−21(y607 JmjC) XX embryos of different ages stained with DAPI and H4K20me1, SDC−3, and DPY−21 antibodies. DPY−21 localizes to X later in development than other DCC subunits, coincident with H4K20me1 enrichment on X. DPY−21 localized to X with similar timing in dpy−21(JmjC) and wild-type embryos. Yellow arrows, foci of SDC−3, DPY−21, or H4K20me1 on X. Red arrows, diffuse nuclear localization of H4K20me1 or DPY−21. Scale bar, 2 μm.
(B) Individual nuclei of a wild-type 376-cell embryo reveal cell-cycle dependent association of DPY−21 on X during interphase but not mitosis, unlike other DCC subunits. Scale bar, 1 μm. See Figures S3 and S4.
To examine the genome-wide distribution of H4K20me1 at high resolution in wild-type and dpy-21 mutant embryos, we performed ChIP-seq analysis using C. tropicalis embryos as a spike-in control to normalize read enrichment (Figures 2G-2J and S3G). Consistent with immunofluorescence experiments, H4K20me1 was enriched on X compared to autosomes in wild-type embryos. Enrichment was observed in both genic and intergenic regions (Figure 2J). Moreover, the H4K20me1 level on X was reduced by 2-fold in both dpy−21(y607 JmjC) and dpy−21(e428 null) mutants, making it comparable to that on autosomes (Figures 2G-2J and S3G). In contrast, autosomal levels of H4K20me1 were not significantly changed in dpy−21(JmjC) or dpy−21(null) mutants (Figures 2G-2J an S3G). Thus, DPY−21 functions in vivo as a chromosome-wide histone demethylase to enrich H4K20me1 on X.
Our findings predict that tethering of DPY−21 to an ectopic site on an autosome via its association with the DCC should cause JmjC-dependent H4K20me1 enrichment at that site. Indeed, inserting 72 copies of DCC binding site rex−1 in a site on chromosome V was sufficient to induce site-specific enrichment of H4K20me1 that was abolished in dpy−21(JmjC) and dpy−21(null) mutants (Figures 2E and 2F). This result confirms that the DCC targets DPY−21 to chromosomes to perform demethylation.
DPY−21 binding to X chromosomes coincides with enrichment of H4K20me1 on X
H4K20me1 enrichment on X in XX embryos occurs long after initial recruitment of DCC subunits to X, which starts around the 30–40-cell stage of embryogenesis with the synthesis of SDC−2, the hermaphrodite-specific trigger of DCC binding to X. H4K20me1 enrichment was not evident before the 200-cell stage and became reliably detectable in most cells around the 300–350-cell stage (Figure 3A) (Vielle et al., 2012; Wells et al., 2012).
To assess the timing of DPY−21 binding to X relative to DCC binding and H4K20me1 enrichment, we performed immunofluorescence experiments using two different DPY−21-specific reagents. We used an antibody against the N-terminal region of DPY−21 to monitor DPY−21 in a wild-type strain and an antibody against 3×FLAG to monitor DPY−21 in a strain with 3×FLAG inserted at the 5′ end of the endogenous dpy−21 gene. The 3×FLAG tag had no adverse effects on DPY−21's DC functions (Methods). In both cases, DPY−21 was not detected on X before the 200-cell stage and was only reliably found on X in most cells around the 300–350-cell stage, the same timing as X enrichment of H4K20me1, and long after binding of other DCC subunits (Figures 3A and S4A). The timing of DPY−21 binding is consistent with its demethylase activity causing H4K20me1 enrichment and our prior finding that DPY−21 's association with the DCC is not as stable as that of other subunits (Yonker and Meyer, 2003).
Catalytically inactive DPY−21 proteins exhibited the same timing in X binding as active DPY−21 (Figures 3A, S4B, S4C). Furthermore, western blots revealed no reduction in DPY−21 levels in dpy−21 (y607 JmjC) vs. wild-type embryos (Figures S3E and S3F). Hence, loss of H4K20me1 enrichment on X in JmjC mutants was not due to defective DPY−21 localization or protein degradation. Finally, mRNA levels of all other DCC subunits were not changed in JmjC mutants (GEO files), and X binding was not visibly affected (Figures 2A, 3A, S3A, S3D, S4B, S4C), indicating that loss of H4K20me1 enrichment did not result from defective functioning of other DCC subunits.
Cell-cycle-dependent activities of DPY−21
Unlike other DCC subunits, DPY−21's association with X is cell-cycle dependent. DPY-21 colocalizes with X during interphase but appears diffusely distributed in mitosis (Figures 3B and S4A). In contrast, other DCC subunits remain on X throughout the cell cycle. Coincident with this dynamic DPY−21 behavior, H4K20me1 is not enriched on X during mitosis (Figure 3B). Instead, all mitotic chromosomes have uniformly high levels of H4K20me1 (Figure 4A). In dpy−21(JmjC) mutants, the inactive protein exhibits the same dynamic behavior (Figures S4B and S4C), and H4K20me1 remains enriched on all mitotic chromosomes (Figure S4C), showing that DPY−21 does not control H4K20 methylation during mitosis. Thus, the selective enrichment of H4K20me1 on X is reestablished by DPY−21 during interphase of each cell cycle. The factors that regulate this dynamic behavior are not known, but DPY−21 is phosphorylated at T565, S776, and T1329 (Zielinska et al., 2009), suggesting that phosphorylation might contribute to its regulation.
Figure 4. Depletion of Histone Methyl Transferase set−1 Causes Severe Mitotic Chromosome Segregation Defects.

(A) Confocal images of a wild-type 8-cell embryo stained with DAPI and H4K20me1 antibody. H4K20me1 is significantly elevated in mitotic vs. interphase (arrows) nuclei and is distributed across all chromosomes.
(B), (C) Live images of set−1(RNAi) embryos show severe mitotic defects. (B) Defective mitotic chromosome segregation caused embryonic lethality. The time series of chromosome segregation starts with 2-cell wild-type or set−1(RNAi) embryos expressing Histone::GFP. Both wild-type cells completed a round of division in 25 minutes. The set−1(RNAi) embryo arrested in mitosis and died. Arrow, two polar bodies. (C) Live image of an older set−1(RNAi) embryo that received less set−1 RNA than in (B) shows mitotic segregation defects in nearly all cells, including chromatin bridges and micronuclei (arrows). Enlargements of chromatin bridges are shown. Scale bars (A-C), 5 μm. See Table S2.
Mutations that inactivate JmjC activity disrupt dosage compensation
dpy−21(JmjC) mutations enabled us to selectively eliminate H4K20me1 enrichment on X and thereby assess whether H4K20me1 plays a direct role in X-chromosome gene regulation or instead occurs as a secondary consequence of the DC process. Prior studies reduced H4K20me1 levels globally throughout the cell cycle via RNAi to the set−1 methyltransferase gene or blocked its progression to H4K20me2/me3 via mutation of the set−4 methyltransferase gene. Both conditions caused genome-wide changes in gene expression (Kramer et al., 2015; Vielle et al., 2012; Wells et al., 2012), thus complicating interpretations about H4K20me1's role.
In studies asking whether set−1(RNAi) enhances the XX-specific lethality caused by weak DC mutations, we (Table S2) and others (Vielle et al., 2012; Wells et al., 2012) found XX lethality to be enhanced. However, we also found that enhanced lethality is neither sex-specific nor specific to the disruption of DC (Table S2). The sex-independent effect is likely due to both genome-wide changes in mRNA levels (Figure 5C and (Kramer et al., 2015) and H4K20me1's role in mitosis. Supporting the latter, we found that strong depletion of SET−1 caused severe mitotic chromosome defects, resulting in arrested embryonic development (Figure 4B and Table S2), and weaker depletion caused chromosome bridges and milder chromosome segregation defects, permitting longer embryonic development (Figure 4C). These results compromise any conclusion that synergistic lethality is an indicator of H4K20me1's role in DC.
Figure 5. The DPY−21 Demethylase Regulates X-Chromosome Gene Expression.

(A) Cartoon of DCC regulation in different genotypes. In XX embryos, xol−1 is repressed, allowing SDC−2 to trigger DCC assembly on X and reduce gene expression by half (light blue). In XO animals, xol−1 is active and represses sdc−2, thereby blocking DCC binding and promoting full X expression (dark blue). In xol−1 XO mutants, SDC−2 triggers DCC binding to X, causing reduced X expression (light blue) and death. Most dpy−21(JmjC); xol−1 XO animals are viable because dpy−21(JmjC) mutations reduce activity of the X-bound DCC (grey) and partially restore X expression (medium blue) (Table S3A).
(B) Rescue of xol−1 XO lethality by dpy−21(JmjC) / dpy−21(null) heterozygous mutations shows that JmjC mutations disrupt DC less severely than a dpy−21(null) mutation (Table S4).
(C) Cumulative plots show the distribution of expression changes for genes on X (red) and autosomes (blue) in mutant vs. wild-type embryos as assayed by RNA-seq. X-axis represents the log2 fold change in expression. X chromosomes have elevated expression compared to autosomes in all mutants except set−1(RNAi). P values, 1-sided Wilcoxon rank-sum tests. See Tables S3, S4, S5, and S6.
To assess the role of H4K20me1 in DC, we first asked whether dpy−21(JmjC) mutations suppress the XO-specific lethality caused by xol−1 mutations, which inappropriately activate the DCC in males, causing reduced X expression and death (Figure 5A) (Miller et al., 1988; Rhind et al., 1995). If JmjC activity contributes to reducing X expression during DC, JmjC mutations should suppress the xol−1 XO-specific lethality. Indeed, all three homozygous JmjC mutations suppressed the lethality (40–56%), showing that JmjC activity, and hence H4K20me1 enrichment, is important for DC (Table S3A).
Because suppression by dpy−21(JmjC) mutations was less than that by a dpy−21(null) mutation (76%, p < 0.01, chi-square test) (Table S3A), we compared the viability of dpy−21(JmjC/null); xol−1 XO animals with dpy−21(null/null); xol−1 XO animals under conditions that enabled us to detect all rescued XO animals regardles of their sex (Figure 5B and Table S4). The goal was to determine the relative contribution of the JmjC domain to DPY−21's overall functions in DC. We found that 62–72% of JmjC/null XO animals were viable compared to 93% of null/null animals. These results confirmed the importance of DPY−21 demethylase activity in DC and suggested that DPY−21 has additional roles in DC. Supporting this idea, dpy−21(null) mutant hermaphrodites are less viable and have more developmental abnormalities than dpy−21(JmjC) mutants (Table S3B).
The role of the DPY−21 demethylase in enriching H4K20me1 on X, and thereby reducing X expression, predicts that blocking H4K20me2/me3 production via a set−4 mutation should prevent the rescue of xol−1 XO males by dpy−21(JmjC) mutations. This prediction was met (Table S5), providing strong genetic evidence that production of H4K20me2/me3 on X is an intermediate step in the enrichment of H4K20me1 on X.
H4K20me1 regulates X-chromosome gene expression
To assay more directly the effect of dpy−21(JmjC) mutations on X-chromosome gene expression, we performed RNA-seq on dpy−21(JmjC), dpy−21(null), and wild-type XX embryos and compared changes in gene expression on X and autosomes (Figures 5C). For all three dpy−21(JmjC) mutants, X-chromosome gene expression was significantly elevated relative to that of all autosomes (Figure 5C). The relative elevation in X expression was greater In dpy−21(null) mutants, consistent with their more severe mutant phenotypes and greater suppression of xol−1 XO lethality (Figure 5C). Thus, the DPY−21 demethylase, and hence X enrichment of H4K20me1, helps repress X-linked gene expression in vivo.
Malignant brain tumor repeat (MBT) proteins associate with H4K20me1 nucleosomes and repress gene expression (Blanchard et al., 2014; Trojer et al., 2007). Hence, we tested whether the MBT repeat proteins of C. elegans (Harrison et al., 2007) contribute to DC. Simultaneous knockout of the two MBT repeat genes mbtr−1 and lin−61 failed to suppress xol−1 XO lethality (Table S6), making it unlikely that MBT repeat proteins play a significant role in X-chromosome repression.
H4K20me1 modulates X-chromosome conformation in somatic cells
Cytological studies showed that DCC binding increases X-chromosome compaction in interphase nuclei of somatic cells (Lau et al., 2014). Genome-wide chromosome conformation capture (Hi-C) studies showed the DCC remodels X-chromosome structure by inducing topologically associating domains (TADs) using its highest-affinity binding sites (rex sites) to mediate long-range chromatin interactions (Crane et al., 2015). We asked whether the DPY-21 demethylase contributes to X compaction and to the DCC-driven remodeling of X-chromosome topology.
To assess compaction, we determined the fraction of total chromosome volume occupied by X in gut nuclei of wild-type and dpy−21(JmjC) mutant XX adults (Figure 6A). Loss of demethylase activity caused a significant increase (∼30%, p < 10−6, 2-sided Wilcoxon rank-sum test) in X volume in all three dpy−21(JmjC) mutants (Figure 6A). An even greater increase in X volume (∼55%, p < 10−9) was found in dpy−21(null) mutants, suggesting additional roles for DPY−21 in X organization. In contrast, X volume was not significantly changed in sdc−1(null) mutants (p = 0.2), which have normal H4K20me1 enrichment on X, but modest DC defects like dpy−21(JmjC) mutants. Thus, H4K20me1 plays a direct role in X-chromosome compaction, and loss of compaction is not a byproduct of DC defects.
Figure 6. DPY−21 Demethylase Activity Modulates Spatial Organization of X Chromosomes.

(A) Boxplots showing ratios of X volume relative to the total volume of chromatin in gut nuclei of different genotypes reveal that demethylase activity promotes X compaction. n, number of nuclei assayed.
(B) TAD boundary formation occurs prior to DPY−21 binding to X. Box plots show the distribution of Pearson's correlation coefficients between FISH probes within (red) or across (blue) the rex-14 TAD boundary in embryos of different cell number. Boxes, middle 50% of coefficients. Center bars, median (m) coefficients. n, total number of nuclei. Asterisks of same color specify data sets compared using 1-sided Wilcoxon rank-sum test.
(C), (D), (F), (G) Heatmaps of Hi-C data binned at 50 kb resolution show chromatin interaction frequencies on chr. X or chr. I in wild-type and dpy-21(JmjC) mutant embryos.
(E), (H) Z-score difference heatmaps at 50 kb resolution show chromatin interactions that are increased (orange-red) and decreased (blue) on X or I in dpy−21(JmjC) mutant vs. wild-type embryos.
(I), (J) Insulation plots for chr. X or chr. I of wild-type (grey) or dpy−21(JmjC) mutant (blue) embryos and insulation difference plots (red). Black bars, location of TAD boundaries in wild-type embryos. Red dots, DCC-dependent boundaries greatly diminished or eliminated upon DCC depletion (Crane et al, 2015.)
(K) Box plots compare insulation changes for different classes of TAD boundaries. In dpy−21(JmjC) mutants, DCC-dependent TAD boundaries on X (red) have greater insulation changes than DCC-independent boundaries on X (blue) (p < 10−3) or autosomal boundaries (black) (p < 10−4). P values, Wilcoxon ranked-sum test.
(L) Cumulative distributions of Z-score differences between dpy−21(JmjC) mutant and wild-type embryos for Hi-C interactions at specified distances on X (left) or all autosomes (right) showed an increase in short-range interactions on X (< 1 Mb apart) and a decrease in long-range interactions on X (> 1 Mb) in JmjC mutants. Medians of Z-score differences for each distance range are shown. See Figures S5, S6 and Table S7.
To assess the impact of H4K20me1 on the remodeling of X topology, we first examined the relative timing of TAD boundary formation and DPY−21 binding. In embryos of different ages, we imaged pairs of fluorescent probes that tile adjacent 500 kb regions within a TAD or flanking a DCC-dependent TAD boundary (Crane et al., 2015) (Figure 6B). In 80–120-cell embryos, less overlap occurred between probes flanking the rex−14 TAD boundary than between probes within the TAD, indicating boundary formation had begun. The boundary was well formed in 180–250-cell embryos (Figure 6B), before DPY−21 was reliably detected on X (Figure 3). Thus, demethylase activity is not essential to initiate formation of DCC-dependent TAD boundaries.
To determine the effect of H4K20me1 enrichment on X-chromosome topology at higher resolution, we performed in-situ Hi-C (Figures 6C–6H, S5A-S5C, Table S7) (Crane et al., 2015; Rao et al., 2014). X chromosomes were organized into TADs in both wild-type and JmjC-mutant embryos (Figures 6C and 6D), but TADs were less distinct in JmjC mutants (Figures 6D and 6E).
To quantify changes in TAD boundaries, we calulated insulation profiles for X and autosomes (Figures 6I and 6J). An insulation score reflects the aggregate of interactions across each interval, and local minima in insulation profiles denote TAD boundaries. On X chromosomes of both wild-type and JmjC-mutant embryos, TAD boundaries formed at the same 17 locations, consistent with TAD formation starting prior to DPY−21 binding. However, the difference in X insulation profiles showed an increase in insulation, and thus a reduction in boundary strength, at all eight DCC-dependent TAD boundaries (Figures 6I and 6K) in JmjC mutants. In contrast, the nine DCC-independent TAD boundaries on X and the boundaries on autosomes were not significantly weakened. These FISH and Hi-C results suggest a two-tier model for TAD formation during DC: the DCC initiates TAD formation at specific locations prior to DPY−21 recruitment. DPY−21 then binds to X and enriches H4K20me1, which strengthens TAD boundaries.
To further define the role of H4K20me1 in X-chromosome remodeling, we compared genomic-distance-normalized Z-scores between wild-type and JmjC-mutant embryos (Figures 6E, 6H, and S5A). We found distance-dependent changes in interactions across the entire X (Figures 6E and 6L left). In JmjC mutants, interactions between sites within 1 Mb increased significantly (p < 10−15, 2-sided KS test), while interactions between sites more than 1 Mb apart decreased significantly (p < 10−15). Notably, long-range interactions among the DCC-dependent TAD boundaries and the 25 highest-affinity rex sites key for TAD boundary formation were significantly reduced (Figure S5D), consistent with reduced TAD boundary strength. In contrast, interactions on autosomes exhibited modest changes in the opposite direction (Figures 6H, 6L right, and S5A), consistent with X chromosomes having a different pattern of distance-dependent chromatin interaction decay than autosomes in JmjC mutants (Figure S5E). These findings show that H4K20me1 promotes long-range interactions across X that enhance the integrity of TADs.
We verified the role of demethylase activity in X-chromosome conformation and compaction using FISH to examine the spatial separation between pairs of genomic loci in wild-type and JmjC-mutant embryos. Based on Hi-C data (Crane et al., 2015), we chose several pairs of rex and non-rex sites that were separated by more than 1 Mb and had a comparable decrease in interaction frequency upon DCC depletion. FISH utilized 30–40 kb probes on embryos with more than 300 cells.
In dpy−21(JmjC) mutants, we found a significant increase in the spatial separation of all three pairs of non-rex sites (Figure S6A), consistent with the decrease in Hi-C interaction frequencies and in chromosome compaction. Equivalently spaced sites on chromosome I showed no change in spatial separation (Figure S6C). However, the spatial separation of rex sites was not changed by either the dpy−21(JmjC) or dpy−21(null) mutation, but was increased by the sdc−2(null) mutation (Figure S6B). The discrepancy between FISH and Hi-C at these rex sites likely reflects a difference in sensitivity of the two methods for detecting changes in chromosome structure (Giorgetti and Heard, 2016).
Our combined results show that H4K20me1 enrichment contributes significantly to X-chromosome remodeling during DC. At least two different mechanisms contribute to X-chromosome structure: a demethylase-independent mechanism that initiates TAD formation and a demethylase-dependent mechanism that compacts X chromosomes and strengthens TADs.
The DPY−21 demethylase selectively enriches H4K20me1 and compacts autosomes in germ cells via a DCC-independent mechanism
H4K20me1 exhibits a dynamic pattern of chromatin enrichment in germ cells (Vielle et al., 2012) (Figures 7A, S7A-S7C), prompting the question of whether DPY−21 also regulates H4K20me1 in the gonad, where the DCC does not function. H4K20me1 levels are high on all mitotic chromosomes and become reduced as nuclei enter meiosis (Figure 7A). By early pachytene, H4K20me1 is enriched only on autosomes and is absent from X chromosomes of both XX and XO germ cells (Figures 7C, S7B, and S7C). Autosomal enrichment persists through mid-late pachytene.
Figure 7. DPY−21 Demethylase Acts Independently of the DCC in Germ Cells to Enrich H4K20me1 and Compact Autosomes.

(A), (B) Maximum intensity projections of wild-type (A) and dpy−21(y607 JmjC) mutant (B) XX gonads stained with DAPI and H4K20me1 antibodies. Demethylase inactivation does not reduce H4K20me1 levels in mitotic nuclei but prevents H4K20me1 accumulation in meiotic nuclei. Arrows, somatic sheath cells. Scale bar, 10 μm.
(C) Pachytene nuclei from dpy−21(5′-FLAG) and dpy−21(5′-FLAG JmjC) XX and XO gonads stained with antibodies to H4K20me1 and X-specific marker HIM−8. H4K20me1 is selectively enriched on autosomes in pachytene nuclei of both sexes in the dpy−21(5′-FLAG) strain but absent from X (top). H4K20me1 is absent from all pachytene chromosomes of dpy−21(JmjC) mutant XX and XO gonads (bottom).
(D) Pachytene nuclei from dpy−21(5′-FLAG) and dpy−21(5′-FLAG JmjC) XX gonads stained with FLAG and HIM-8 antibodies. DPY-21 localizes to autosomes but not X, consistent with its demethylation of only autosomes. Scale bars (C), (D), 2 μm.
(E) DPY-21 demethylase is required for full compaction of autosomes in germ cells. (Top) Pachytene nucleus from a wild-type gonad stained with a FISH probe to chr. I (blue) and antibodies to HIM-8 (red) and axis protein HTP-3 (green). The 3D traces of X and I (yellow) from wild-type and dpy-21(JmjC) gonads were used to straighten the chromosomes. (Middle) Computationally straightened X and I with average axis lengths and SEMs. (Bottom) Box plots show that the ratio of I:X axis lengths is significantly lower in wild-type vs. dpy-21(JmjC) pachytene nuclei (p = 1.7x10-8, 2-sided Wilcoxon rank-sum test).
See Figure S7.
To determine the function of DPY−21 JmjC activity in germ cells, we examined H4K20me1 localization in dpy−21(JmjC) XX and XO mutant gonads. H4K20me1 levels remained high on mitotic chromosomes of mutant gonads (Figures 7B and S7A), but H4K20me1 was absent from all chromosomes of pachytene nuclei (Figures 7B and 7C). Thus, demethylase activity is essential for the autosomal enrichment of H4K20me1 during germ cell meiosis but is not required for H4K20me1 enrichment during germ cell mitosis.
These results lead to three predictions about DPY−21 localization in the germline: DPY−21 should localize to all autosomes of pachytene nuclei but not to X chromosomes of pachytene nuclei or to any mitotic chromosomes. All three predictions were met, indicating that DPY−21 is a direct regulator of H4K20me1 during germ cell meiosis (Figures 7D, S7A-S7C). Because the DCC does not assemble or function in germlines of XX or XO animals, DPY−21 must operate via a DCC-independent mechanism. DPY−21's selective localization to autosomes during meiosis must occur through different partners that either recruit it to autosomes or repel it from X.
The association of DPY−21 with autosomes in germ cells raised the question of whether H4K20me1 enrichment plays a general role in regulating chromosome compaction across cell types (Figure 7E). A change in meiotic chromosome compaction should alter the length of the chromosome axis. We measured and compared axis lengths of chromosomes X and I in pachytene nuclei of wild-type and dpy−21(JmjC) mutant gonads by tracing the axes in three dimensions and computationally straightening them. X axis length was not different in dpy−21(JmjC) vs. wild-type nuclei, as predicted by the lack of DPY−21 on X, but chromosome 1 axis length was extended in the mutant. The ratio of I:X lengths increased significantly from 1.4 in wild-type nuclei to 1.7 in dpy−21(JmjC) nuclei (p = 1.7 × 10−8). Thus, higher-order structure of different chromosomes can be regulated in somatic cells versus germ cells by selective localization of DPY−21, and hence H4K20me1 enrichment. DPY−21 is an adaptable chromatin regulator that controls distinct biological functions in development.
Discussion
The function of chromatin modification in establishing higher-order chromosome structure during gene regulation has been elusive. Here we determined the mechanism underlying the dynamic control of histone modification H4K20me1 throughout the cell cycle and demonstrated the H4K20me1-driven remodeling of X-chromosome architecture and repression of X-chromosome gene expression.
Our structural and biochemical analysis of DCC subunit DPY−21 revealed a novel, widely conserved subfamily of JmjC demethylases (KDM9) that catalyzes the demethylation of H4K20me2 to H4K20me1 in worms and mammals. DPY−21 is the first known H4K20me2 demethylase with pan-cellular activity. DPY−21 lacks a recognizable DNA binding domain to confer target specificity, but it is recruited to diverse genomic locations by different partners to catalyze H4K20me2 demethylation. In somatic cells, the DCC, a condensin complex, targets the demethylase to X chromosomes. DCC binding and spreading distributes demethylase activity, enhancing H4K20me1 chromosome-wide.
Thus, the DCC acts as an “eraser” of a prominent histone post-translational modification, promoting H4K20me1 enrichment on X by activating an H4K20me2 demethylase. DPY−21's role in chromatin modification further illustrates how dosage compensation evolved by co-opting conserved machinery used in other biological processes for the new task of fine-tuning X-chromosome expression. Moreover, our results suggest that other condensin and SMC complexes could act as scaffolds to recruit proteins with chromosome-modifying activities.
Chromatin modifications can recruit specialized proteins to regulate the structure of nucleosome arrays and gene expression. In flies and mammals, malignant brain tumor repeat (MBT) proteins associate with nucleosomes enriched in H4K20me1 or H4K20me2, compact the chromatin fiber, and help repress transcription (Blanchard et al., 2014; Trojer et al., 2007). Knockout of worm MBT proteins failed to disrupt DC, implying that if H4K20me1 binding proteins modulate X-chromosome conformation and gene repression, they will define a new class of H4K20me1-specific histone “readers”. Alternatively, H4K20me1 may control nucleosome or chromatin folding directly or antagonize other chromatin modifying activities.
While histone modifications can alter inter-nucleosomal interactions that govern the compaction state of a chromatin fiber (Francis et al., 2004; Kalashnikova et al., 2013; Lu et al., 2008), the potential impact of histone modifications on higher-order chromosome organization beyond chromatin-fiber compaction is not well understood. Only recently has super-resolution imaging shown that chromatin domains enriched in H3K27me3 and polycomb proteins adopt unique folded states (Boettiger et al., 2016). Once the polycomb repressive complex 1 is recruited to H3K27me3-enriched chromatin, it generates chromatin domains 20–140 kb in size that are distinct from previously defined TADs (Kundu et al., 2017). Our results show that H4K20me1 controls chromosome structure over a larger length scale. H4K20me1 modulates the formation of TADs (≥ 1 Mb) by enhancing long-range DNA interactions across X.
Our combined data support a two-tier model for the DCC-dependent remodeling of X-chromosome topology. In the first stage, the condensin DCC initiates TAD boundary formation via a demethylase-independent mechanism that promotes long-range interactions between the highest affinity DCC binding sites (rex sites) prior to DPY−21's assembly onto X. In the second stage, DPY−21 demethylase catalyzes enrichment of H4K20me1 on X, thereby enhancing long-range DNA interactions across X. X-chromosome compaction is generally increased, and TAD boundaries are strengthened by the elevation of long-range rex interactions critical for TAD formation.
Unexpectedly, higher-order chromosome structure is also regulated in germ cells by DPY−21. In meiotic nuclei of both sexes, DPY−21 is localized to autosomes via a DCC-independent mechanism. The targeted H4K20me1 enrichment then condenses the lengths of autosomal axes, providing broad evidence for H4K20me1's regulation of higher-order chromosome structure. Axis expansion can alter crossover frequency and distribution (Mets and Meyer, 2009), raising the possibility that autosomal H4K20me1 might have consequences for crossover recombination as well as gene expression.
DPY−21's DCC-independent activity is relevant for recent findings that DPY−21 modulates nematode growth and metabolism through the TORC2 pathway (Webster et al., 2013) and entry into the quiescent dauer state through the insulin-signaling pathway (Delaney et al., 2017; Dumas et al., 2013). Evidence in both cases suggests at least partial DCC-independence for DPY−21's functions. Determining the effect of JmjC-specific mutations on both pathways will be key for distinguishing the contribution of demethylase activity from general DCC functions. Moreover, because metabolism is altered in both biological contexts and α-KG is a necessary DPY−21 co-factor and a product of the Krebs cycle, the worm's metabolic state, rather than the DCC, could influence DPY−21's functions (Chin et al., 2014).
Relevance of our studies for mammalian development is underscored by the enrichment of H4K20me1 on the inactive female X chromosome (Kohlmaier et al., 2004). Mechanisms underlying the H4K20me1 enrichment and its role in chromosome silencing are not known. The long non-coding RNA XIST, the trigger of mammalian X inactivation, induces accumulation of H4K20me1 on X, but X inactivation per se is not required for H4K20me1 deposition, suggesting that H4K20me1 might contribute to establishing inactivation. Consistent with this idea, knockout of the H4K20me1 methyltransferase causes decondensation of X (Oda et al., 2009). Our discoveries offer new directions for unraveling the regulation and function of H4K20me1 in X-chromosome inactivation and other long-range mechanisms of gene control.
Star-Methods
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Barbara J. Meyer (bjmeyer@berkeley.edu).
Experimental Model and Subject Details
C. elegans and C. tropicalis Strains
Strains were cultured on NGM agar plates with OP50 bacteria grown in Luria Broth (LB) except as indicated for RNAi and Hi-C experiments. The hcp−6(mr17ts) strain was grown at 15°C; all other strains were grown at 20°C.
E. coli Strains
Rosetta 2 (DE3) pLysS was grown in LB media with 50 μg/ml carbenicillin and 34 μg/ml chloramphenicol. The HT115 (DE3) strain used for RNAi experiments was grown first in LB media with 50 μg/ml carbenicillin and then on NGM agar plates with 1 mM IPTG and 100 μg/ml carbenicillin. The HB101 strain used for nematode culture in Hi-C experiments was grown on NGM agar plates.
Method Details
Naming of the DPY−21-ROSBIN Subfamily of JmjC Demethylases
Even though the JmjC demethylases discovered before DPY−21 are only divided into subfamilies KDM2-KDM7, the name KDM9 was chosen for the new DPY-21-ROSBIN subfamily, because the name KDM8 is currently the HGNC symbol for JMJD5, which demethylates H3K36.
Structure Prediction
Structure predictions were made using the Phyre2 (Kelley et al., 2015) web server for the complete DPY−21 amino acid sequence (1–1641) and Robetta (Kim et al., 2004) for DPY−21 (655–1641).
Expression and Purification of DPY−211210–1617
A DNA fragment encoding the C-terminal part of C. elegans DPY−21 (residues 1210–1617; UniProtKB Q9GRZ3) was cloned into the 2RRT vector, a pET StrepII-StrepII TEV LIC cloning vector (gift from Scott Gradia, MacroLab, Addgene plasmid #55215) to create a C- terminal DPY−21 fusion protein with an N-terminal double Strep tag followed by a TEV-cleavage site. The resulting plasmid was transformed into the Escherichia coli Rosetta 2 (DE3) pLysS (Novagen) strain and cultured overnight at 37°C in 50 ml LB media supplemented with 50 μg/ml carbenicillin and 34 μg/ml chloramphenicol. The overnight culture was used to inoculate 2×YT media supplemented with 2 % (w/v) dextrose, 50 μg/ml carbenicillin and 34 μg/ml chloramphenicol (15 ml of overnight culture per L of media). The bacterial cultures grew at 37°C to a n OD600 ∼0.7. Flasks were then placed in an ice bath for 30 min and protein production was induced with 0.5 mM IPTG. Expression of the fusion protein was allowed to continue overnight at 18°C. Cells were collected by centrifugation (7000 × g, 20 min, 4°C) and frozen at −80°C (total yield ∼3.4 g/L).
To purify the DPY−21 recombinant protein, frozen cell pellets (10 g) were re-suspended in 70 ml of lysis buffer (100 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.5 mM TCEP-NaOH (pH 4.0), final pH 8.0) supplemented with 1 mM PMSF and protease inhibitor mix (Calbiochem). Cells were disrupted by sonication (Qsonica Q500 with a 1/2” probe, 45% amplitude), and the cell debris and insoluble material were removed by centrifugation (30000 × g, 45 min, 4°C). The recombinant DPY−21 was purified by Strep-affinity, anion-exchange, and size-exclusion chromatography on an ÄKTA pure 25 L system (GE Healthcare). All purification steps were performed at 4°C. The supernatant was loaded onto a 1 ml StrepTrap HP column (GE Healthcare), washed with 20 column volumes of lysis buffer, and eluted with lysis buffer supplemented with 2.5 mM d-desthiobiotin (Sigma-Aldrich). The elution peak was collected and concentrated to ∼3 ml. The buffer was exchanged to buffer A (50 mM Tris-HCl (pH 8.0), 50 mM NaCl, 5% (v/v) glycerol, 0.5 mM TCEP-NaOH (pH 4.0), final pH 8.0). The concentrated sample was loaded onto a 1 ml HiTrap Q HP column (GE Healthcare), washed with 10 columns of buffer A, and eluted with a 50 to 700 mM NaCl gradient in buffer A. DPY−21 fractions eluted in the range of 11–29 mS/cm. The eluted DPY−21 peak was concentrated (Amicon Ultracel, 30 kDa, Millipore) and loaded onto a Superose 6 HR 10/30 column (GE Healthcare) pre-equilibrated in buffer containing 50 mM HEPES-NaOH (pH 7.5), 500 mM NaCl, 5% (v/v) glycerol and 0.5 mM TCEP-NaOH (pH 4.0), final pH 7.5. Fractions containing DPY−21 protein (as determined by A280 and SDS-PAGE) were pooled, concentrated to 10 mg/ml and stored at –80°C. The typical yield of purified protein was 0.6 mg/L of media.
Crystallization and Structure Determination
Frozen aliquots of purified DPY−21 were thawed on ice, and the buffer was exchanged to 50 mM HEPES-NaOH (pH 7.5), 200 mM NaCl, 0.5 mM TCEP-NaOH (pH 4.0), final pH 7.5. Protein was concentrated (Amicon Ultracel, 30 kDa, Millipore) to 5 mg/ml and used in crystallization experiments set up by the Mosquito Crystal robot (TTP Labtech). Crystals were grown at 18°C by vapor diffusion in hanging drops containing 200 nl DPY−21 (5 mg/ml) and 200 nl well solution [100 mM DL-malic acid (pH 5.6) and 25% (w/v) PEG 550 MME]. Crystals were cryo-protected using 100 mM DL-malic acid (pH 5.6) (Hampton Research), 26% (w/v) PEG 550 MME (Hampton Research) and 16% (v/v) glycerol and flash cooled in liquid nitrogen.
X-ray diffraction data were collected at the Lawrence Berkeley National Laboratory Advanced Light Source (Beamline 8.3.1), and processed using the XDS (Kabsch, 2010) and AIMLESS software (Evans, 2011). Indexed crystals belonged to the orthorhombic space group C 2 2 21 and contained a single molecule of DPY−21 in the asymmetric unit. Excitation scans revealed the presence of Fe, and experimental phase information was collected from a native crystal using a combination of iron and sulfur single-wavelength anomalous dispersion. Iterative data sets were collected at a minimal accessible wavelength (∼7235 eV, 1.7324 Å). Strong anomalous signal and clear phase solution were obtained after ∼65× multiplicity. Twenty-one sulfur sites and a single Fe site were identified with Hyss in PHENIX (Adams et al., 2002), and SOLVE/RESOLVE (Terwilliger, 1999) was used to extend phases to the native DPY−21 set to ∼1.8 Å. Model building was completed in Coot (Emsley and Cowtan, 2004), and the structure was refined with PHENIX. X-ray data for refinement were extended according to an I/σ resolution cutoff of ∼2.0. Statistics of data collection, phasing and refinement are provided in Table S1. Figures were prepared using PyMOL (Schrodinger, LLC), LigPlot+ (Laskowski and Swindells, 2011), ESPript 3.0 (Robert and Gouet, 2014), and ProtSkin (www.mcgnmr.mcgill.ca/ProtSkin) programs.
Immunoblotting-Based Demethylase Assay
cDNAs encoding wild-type DPY−211210–1641, mutant DPY−211210–1641 (H1452A D1454A), and wild-type mouse ROSBIN350–795 (cDNA clone GenBank BC094667.1 obtained from Dharmacon, MMM1013-202731745) were cloned into the 2RRT vector as described for DPY-211210–1617. All three proteins were expressed and purified as described for DPY−211210–1617 with the following modification: proteins were purified using a single chromatography step over a StrepTrap HP column. Purified proteins (13 μM) were incubated with 30 μg of calf thymus type II-A histones (Sigma-Aldrich) in 50 mM HEPES-NaOH (pH 7.5), 300 mM NaCl, 50 μM (NH4)2•FeSO4•6H20 (Sigma-Aldrich), 1 mM oc-ketoglutaric acid (Sigma-Aldrich), and 1 mM L-ascorbic acid (Sigma-Aldrich) for 4 h at 22°C. Samples were separated by SDS-PAGE and immunoblotted using specific antibodies. Western blots were analyzed using ImageJ software (Schneider et al., 2012). The following primary and secondary antibodies were used in Western blots: mouse monoclonal anti-H4K20me1 (Active Motif), rabbit polyclonal anti-H4K20me2 (Abcam), rabbit monoclonal anti-H4K20me3 (Abcam), mouse monoclonal anti-H4 (Abcam), rabbit polyclonal anti-H3K4me1 (Abcam), rabbit polyclonal anti-H3K4me2 (EMD Millipore), rabbit polyclonal anti-H3K4me3 (Abcam), rabbit polyclonal anti-H3K9me1 (Abcam), mouse monoclonal anti-H3K9me2 (Abcam), rabbit polyclonal anti-H3K9me3 (EMD Millipore), rabbit polyclonal anti-H3K27me1 (EMD Millipore), rabbit polyclonal anti-H3K27me2 (Abcam), mouse monoclonal anti-H3K27me3 (Abcam), rabbit polyclonal anti-H3K36me1 (Abcam), rabbit polyclonal anti-H3K36me2 (Abcam), rabbit polyclonal anti-H3K36me3 (Abcam), rabbit polyclonal anti-H3K79me1 (Abcam), rabbit polyclonal anti-H3K79me2 (Abcam), rabbit polyclonal anti-H3K79me3 (Abcam), donkey anti-rabbit HRP (Jackson ImmunoResearch), and donkey anti-mouse HRP (Jackson ImmunoResearch).
Dot Blot Analysis of Antibody Specificity Against H4K20me3
Different amounts (0.1 μg and 1 μg) of a recombinant H4K20 protein with a specific methylated lysine analog (MLA) [(H4K20me1, Active Motif), (H4K20me2, Active Motif), and (H4K20me3, Active Motif)] were spotted in duplicate onto nitrocellulose membranes and immunoblotted separately with six different anti-H4K20me3 antibodies: rabbit monoclonal ab177190 at 1.2 μg/ml (Abcam), mouse monoclonal ab78517 at 1 μg/ml (Abcam), mouse monoclonal 39671 at 1 μg/ml (Active Motif), rabbit polyclonal ab9053 at 1 μg/ml (Abcam), rabbit polyclonal 07-463 at 2 μg/ml (EMD Millipore), and rabbit polyclonal C15410207 at 1 μg/ml (Diagenode) . All antibodies were diluted in blocking buffer containing milk (5% milk in 1 × TBS supplemented with 0.1% v/v Tween-20). Secondary antibodies donkey anti-rabbit HRP (Jackson ImmunoResearch) and donkey anti-mouse HRP (Jackson ImmunoResearch) were used at 0.8 μg/ml concentration and diluted in blocking buffer containing milk. The nitrocellulose membrane was incubated in WesternBright Sirius ECL solution (Advansta Corporation) for 3 min and the chemiluminescence signal was acquired using FluorChem HD2 (ProteinSimple) software. The dot blot intensities were quantified using ImageJ software. Under the experimental conditions described, only two of six antibodies showed a strong preference for H4K20me3 over H4K20me2: rabbit monoclonal ab177190 (Abcam) (Figure S2C) and mouse monoclonal 39671 (Active Motif, data not shown). The remaining four antibodies also recognized H4K20me2 to varying degrees, making them less specific as anti-H4K20me3 antibodies (see example for ab78517 in Figure S2D).
Mass Spectrometry-Based Demethylase Assay
Demethylase assays were performed using 13 μM enzyme (DPY−211210–1641 or ROSBIN350–795), 30 μM of peptide in 50 mM HEPES-NaOH (pH 7.5), 300 mM NaCl, 50 μM (NH4)2•FeSO4•6H20 (Sigma-Aldrich), 1 mM oc-ketoglutaric acid (Sigma-Aldrich), and 1 mM L-ascorbic acid (Sigma-Aldrich) for 4 h at 22°C. Demethylation of substrate peptides was assessed by electrospray ionization - Fourier transform ion cyclotron resonance mass spectrometry (9.4T) on a Bruker Apex III instrument after desalting samples on a Zip-tip. The peptide used in the assay was H4K20me2 [AKRHR-K(me2)-VLRDNI].
Genome Editing
C. elegans strains encoding altered DPY−21 proteins having single or double amino acid substitutions were made using TALENS and single-stranded oligonucleotide repair templates to edit the endogenous dpy−21 locus (Lo et al., 2013). TY5686 dpy−21(y607 JmjC) encodes DPY−21 H1452A (codon 1452 changed to GCC from CAC), TY5693 dpy−21(y618 JmjC) encodes DPY−21 D1454A (codon 1454 changed to GCT from GAT), and TY5695 dpy−21(y622 JmjC) encodes DPY−21 H1452A D1454A (codon 1452 changed to GCC and codon 1454 to GCT).
The following primers were used to amplify the wild-type and edited dpy−21 genomic regions: kb445 and kb442r. dpy−21 mutant strains were identified by DNA sequence analysis of PCR fragments. dpy−21 mutants were outcrossed five times against a strain carrying the closely linked unc−76(e911) mutation to remove extraneous DNA changes introduced during the editing process.
A 5′ 3×FLAG tag was introduced into the endogenous dpy−21 locus of the N2 strain or the endogenous locus of the dpy−21(y607 JmjC) strain (H1452A) by CRISPR/Cas9 using rol−6 as a co-conversion marker (Farboud and Meyer, 2015; Paix et al., 2015). The 5′ 3×FLAG tag was inserted after codon 5 (ACT) using double stranded repair template kb596ds. Primers used to amplify the dpy−21 region with the 5′ 3×FLAG insertion are kb552 and kb559r. The dpy−21 FLAG-tagged strains were outcrossed 4 times against an N2 strain to remove potential DNA changes introduced during the editing process.
To assess whether addition of the 3×FLAG tag to the N-terminus of the DPY−21 protein had any deleterious effects on DPY−21 function in dosage compensation, we used a very stringent genetic test to assay for dosage compensation defects. dpy−21(y692 5′-FLAG) V males were mated to unc−42 dpy−21(e428) V; lon−2 xol−1(y9) X hermaphrodites, and cross progeny were examined for the appearance of Lon non-Unc XO animals [(dpy−21(y692 5′-FLAG) / unc−42 dpy−21(e428 null); lon−2 xol−1 XO] and non-Unc non-Lon XX animals dpy−21(y692 5′-FLAG) / unc−42 dpy−21(e428); lon-2 xol-1/++ XX. Appearance of any Lon non-Unc XO animals would indicate that dpy-21(y692 5′-FLAG) failed to complement the dpy-21(e428 null) allele because DPY-21 function was compromised by the 5′-FLAG tag. xol-1 mutations kill XO animals because they permit the DCC to load inappropriately onto the male X chromosome and cause reduced X-linked gene expression. Disruption of dpy-21 function suppresses the xol-1 XO-specific lethality (Miller et al., 1988). We found zero Lon non-Unc XO progeny from multiple crosses in which at least 1000 cross progeny non-Unc non-Lon XX animals were identified, indicating that the 5′-FLAG tag on the DPY-21 protein causes no obvious deleterious effects on dosage compensation.
Quantification of Progeny from Matings on Feeding RNAi
Hermaphrodites were cultured on NGM agar plates seeded with OP50 bacteria grown in LB medium. L1 hermaphrodites were picked onto feeding RNAi plates (NGM agar with 1mM IPTG and 100 μg/ml carbenicillin) seeded with gene-specific RNAi bacteria and allowed to mature. Their progeny were grown to the L4 stage on RNAi feeding plates and then used for matings. Individual L4 hermaphrodites were placed onto new RNAi plates and mated with 6-10 males of the same genotype that had also been grown for two generations on RNAi plates as male-hermaphrodite mating stocks. Matings were transferred daily to new RNAi plates. Progeny counts began with the first plates that had male progeny. The set-1 RNAi construct was from the Ahringer feeding library (Kamath et al., 2003).
Generating Males for Mating RNAi Experiments
Males used in the male-hermaphrodite matings on RNAi plates were first generated by heat shocking hermaphrodites at 37°C for 1 hr. The rare males were used to generate male-hermaphrodite mating stocks that were maintained for each genotype on NGM plates seeded with OP50.
sdc−2 RNAi
sdc−2(y93) L4 hermaphrodites were grown on RNAi plates seeded with bacteria carrying an sdc−2 RNAi construct from the Ahringer feeding library (Kamath et al., 2003). Their progeny were first scored for embryos and then viable adults. The same protocol was used for control animals grown on bacteria carrying the empty L4440 feeding RNAi vector.
set−1 dsRNA for RNAi Injection
The set−1 clone from the Ahringer RNAi library was PCR amplified using T7 primers to make a DNA template for RNA transcription. PCR product (1 μg) was then transcribed using the MegaScript T7 Transcription Kit (Invitrogen) in a 4-6 hr incubation. Single-stranded RNAs were allowed to anneal, and double-stranded RNA at 1 mg/ml was injected into hermaphrodite gonads.
Immunofluorescence
The following antibodies were used for immunofluorescence: Rabbit polyclonal anti-DPY-27 (rb699) (Chuang et al., 1994), rat polyclonal anti-SDC-3 (PEM4A) (Crane et al., 2015) mouse monoclonal anti-H4K20me1 (Active Motif), rabbit polyclonal anti-H4K20me1 (Abcam), rabbit polyclonal anti-H4K20me2 (Abcam), rabbit monoclonal anti-H4K20me3 (Abcam), mouse monoclonal anti-H4K20me3 (Active Motif), mouse monoclonal anti-H4K20me3 (Abcam), rabbit polyclonal anti-DPY-21 (Novus Biologicals, raised against amino acids 482-581), mouse monoclonal anti-FLAG M2 antibody (Sigma-Aldrich), mouse monoclonal anti-Nuclear Pore Complex Mab414 (Abcam), rat anti-HIM-8 antisera (A. Dernburg lab) and guinea pig anti-HTP-3 antisera (A. Dernburg lab).
C. elegans embryos were obtained by dissecting gravid adult worms in 8 μl of egg buffer (25 mM HEPES-NaOH (pH 7.3), 118 mM NaCl, 48 mM KCl, 2 mM CaCl2, 2 mM MgCl2) on polylysine coated slides. A coverslip was placed on top of the dissected worms, and the slides were then frozen on a dry ice block for at least 10 min. Coverslips were cracked off, and the samples were dehydrated for 10 min in 95% ethanol pre-cooled to -20°C. The samples were then fixed for 10 min in a humid chamber with 1% (w/v) paraformaldehyde (Sigma-Aldrich) freshly made in egg buffer. After fixation, slides were washed 3 times for 10 min with 1× PBS-T (0.5% Triton X-100 in 1× PBS, 1 mM EDTA) at room temperature. Excess 1× PBS-T was then removed and 50 μl of primary antibodies at 1:200 dilutions in PBS-T were applied. Samples were incubated overnight in a humid chamber. Slides were washed 3 times for 10 min with 1× PBS-T at room temperature and then incubated in secondary antibodies, Alexa-Fluor-488 donkey anti-mouse (Jackson ImmunoResearch), Alexa-Fluor-594 donkey anti-rabbit (Jackson ImmunoResearch), Alexa-Fluor-647 donkey anti-rat (Jackson ImmunoResearch) at 1:250 dilution for 6 h to overnight. Slides were then washed 3 times for 10 min with 1× PBS-T at room temperature and then mounted with ProLong Diamond antifade mountant (Life Technologies) with DAPI (1 μg/ml).
To stain C. elegans gut cells and gonads, adult worms were dissected in 4 μl of egg buffer on an 18 mm × 18 mm coverslip, and 4 μl of 2% (w/v) paraformaldehyde in egg buffer was added to the sample. The coverslip was flipped onto a poly-lysine coated glass slide Fisherbrand Superfrost Plus, Fisher Scientific 12-550-15) and incubated in a humid chamber for 10 min to fix the sample. After fixation, the slides were frozen on a dry ice block for at least 10 min, and the coverslips were removed. The slides were immersed for 1 min in methanol pre-cooled to -20°C (for gut staining) or for 10 min in ethanol pre-cooled to -20°C (for gona d staining) and washed at room temperature with PBS-T. The slides were then stained with antibodies as described above.
We evaluated the performance of four antibodies against H4K20me2/me3 in immunofluorescence experiments. Of the antibodies tested, only the mouse monoclonal antibody ab78517, which detected both H4K20me2 and H4K20me3 in immunoblotting assays (Figure S2D), resulted in strong, reliable nuclear signals. In contrast, antibodies that preferentially recognized H4K20me2 (ab9052) or H4K20me3 (ab177190 and Active Motif 39671) by immunoblotting failed to produce detectable signals in embryos older than the 100-cell stage or in adult somatic tissues. Therefore, we used ab78517 to assess the level of H4K20me2/me3 on X chromosomes in vivo.
FISH
To assess chromosome structure changes in dpy-21 mutants, FISH probes targeting multiple X and autosomal regions were made using the following fosmids (BioScience LifeSciences): WRM0618bA10 (chromosome X, 8.86-8.9 Mb, Pair 2); WRM0634cA04 (chromosome X,10.46-10.5 Mb, Pair 2); WRM061dF08 (chromosome X, 13.41-13.44 Mb, Pair 3); WRM0613bE04 (chromosome X, 12.31-13.35 Mb, Pair 3); WRM0632bH06 (chromosome X, 3.13-3.17 Mb, Pair 1); WRM0617dE07 (chromosome X, 4.09-4.12 Mb, Pair 1); WRM0610cG11 (chromosome X, rex-14, 8.01-8.04 Mb); WRM0631aB04 (chromosome X, rex-47, 9.46-9.49 Mb); WRM0626cG08 (chromosome X, rex-23, 4.19-4.22 Mb); WRM0638aF07 (chromosome X, rex-32, 3.01-3.05 Mb); WRM0627cE11 (chromosome I, 4.51-4.54 Mb); WRM0641bD11 (chromosome I, 3.3-3.33 Mb). The probes were labeled with Alexa−488 or Alexa−594 using FISH Tag DNA Kit (Life Technologies).
FISH probes were used to assess the spatial proximity between 2 pairs of rex sites and 3 pairs of non-rex sites that exhibit DCC-dependent Hi-C interactions and 1 pair of autosomal sites that exhibit DCC-independent Hi-C interactions. Pairwise combinations of probes are listed below, with WTObs indicating the Hi-C observed interaction frequency in wild-type embryos and DiffObs indicating the difference between wild-type and dosage compensation mutant (sdc−2 (y93, RNAi)) Hi-C observed interaction frequency (Crane et al., 2015): rex−32/rex−23, chromosome X, 3.01-3.05 Mb and 4.19-4.22 Mb, WTObs = 1.615, DiffObs = 0.310; rex−14/47, chromosome X, 8.01-8.04 Mb and 9.46-9.49 Mb, WTObs = 2.031, DiffObs = 1.085; Pair 1, chromosome X, 3.13-3.17 Mb and 4.09-4.12 Mb, WTObs = 1.608, DiffObs = 0.310; Pair 2, chromosome X, 8.86-8.9 Mb and 10.46-10.5 Mb, WTObs = 1.558, DiffObs = 0.812; Pair 3, chromosome X, 12.31-13.35 Mb and 13.41-13.44 Mb, WTObs = 2.137, DiffObs = 1.023; Pair 4, chromosome I, 3.3-3.33 Mb and 4.51-4.54 Mb, WTObs = 1.981, DiffObs = 0.074.
C. elegans embryos were obtained by dissecting gravid N2, dpy−21(y607 JmjC), dpy−21(e428 null) or szT1/sdc−2(y74) unc−3(e151) worms. FISH was performed on embryos as described previously (Crane et al., 2015). Following FISH, the embryos were stained with rabbit DPY−27 antibody (rb699), mouse anti-Nuclear Pore Complex antibody (Mab414) at 1:200 dilution and Alexa-Fluor−555 goat anti-rabbit antibody (Life Technologies) and Cy5 goat anti-mouse antibody (Jackson ImmunoResearch). For N2, dpy−21(y607 JmjC) and dpy−21(e428 null) strains, X-specific DPY−27 staining was used to identify XX embryos. Absence of DPY−27 staining on X was used to identify homozygous sdc−2(y74) unc−3(e151) XX embryos. Nuclear pore staining was used together with DAPI to segment nuclei and serve as a positive staining control.
Microscopy and Image Analysis
To image embryos, intestinal nuclei, and gonads, confocal image stacks with a 50 nm or 51.5 nm XY pixel size and a 83.9 nm Z step size were acquired on a Leica TCS SP8 WLL microscope using a ×63, 1.4 NA objective lens and HyD detectors. Image deconvolution with theoretical point spread function was performed using Huygens Professional Software (Scientific Volume Imaging, The Netherlands). For images in Figures 3B and S4, representative nuclei at specific cell cycle stages were manually cut out from images of entire embryos using edit polygon and cut mask functions of the Priism software (Chen et al., 1996).
To measure the enrichment of H4K20me1 and the depletion of H4K20me2/3 on X, individual intestinal nuclei were manually cut out from deconvolved image stacks based on DAPI staining using the edit polygon and cut mask functions of the Priism software. The DPY−27 and DAPI channels were blurred using the 3D Filter Function to make the signals continuous. The channels were then thresholded using three times the standard deviation of signal intensity (for DPY-27) or using the basic thresholding function in Priism (for DAPI) to create binary masks for X-chromosome territories and for all chromosomes, respectively. The fold enrichment of H4K20me1 or H4K20me2/3 on X (Figures 2C and 2D) is the ratio between (average intensity of H4K20me1 or H4K20me2/3 signal in the X-chromosome mask) and (average intensity of H4K20me1 or H4K20me2/3 in the all-chromosomes mask), as calculated using a custom python script.
Similarly, to assess whether the demethylase activity contributes to X compaction (Figure 6A), we calculated the ratio between the volume of X chromosomes and the total volume of all combined chromosomes in gut nuclei of wild-type and mutant strains using masks as described above.
Box plots were generated using R to compare the ratio of H4K20me1 or H4K20me2/3 on X versus all chromosomes in different genotypes, as well as the ratio of X-chromosome volume versus total chromosome volume. The lower and higher whiskers of the box plots represent the lowest datum still within 1.5 IQR (interquartile range) of the first quartile and the highest datum still within 1.5 IQR of the third quartile.
To measure the 3D distances between FISH foci, we used custom software as described previously (Crane et al., 2015). DAPI and nuclear pore complex signals were blurred using the 3D Filter Function in Prism to make the nuclear signal continuous and thereby permit nuclei to be segmented accurately. The find points function was then used to count and record the local centre of mass (LCOM) of each nucleus and each FISH spot in x, y, and z using user-defined threshold values. After counting the number of nuclei, embryos at 300–400 cell stage were selected for further analysis. The software then assigned FISH spots to appropriate nuclei, and quantified the 3D distance between all combinations of red and green FISH spots within the same nucleus. To eliminate the bias caused by inclusion of distances between FISH spots on different chromosomes, we used only the shortest calculated distances between red and green probes in each nucleus for X-linked loci in XX embryos and for autosomal loci in all embryos. The distributions of distances in different genotypes were compared using the Wilcoxon rank-sum test function in R.
To quantify the length of chromosome axes in the gonad, we traced chromosomes I and X in 3D based on HTP−3 staining as described previously (Mets and Meyer, 2009). A FISH probe targeting a 500 kb genomic region on chromosome I (4.6 Mb to 5.1 Mb (Crane et al., 2015)), and HIM−8 signal were used to mark chromosome I and X, respectively.
We observed a progressive increase in nuclear size and chromosome length during pachytene. We quantified ChrX and ChrI axis lengths all along the pachytene region and observed that the increase in chromosome lengths was the most significant between early- and mid-pachytene and plateaued at late-pachytene, with the latest quarter of pachytene exhibiting minimal variation in chromosome lengths. Therefore, we restricted the meiotic chromosome length comparison between wild-type and dpy−21(JmjC) mutants to nuclei in the last quarter of pachytene.
Live Imaging of Embryos
The strain EG4601 (oxIs279 [pie−1p::GFP::H2B + unc−119(+)]II; unc−119(ed3) III) was used to image the effect of set−1(RNAi) on mitosis in young embryos. EG4601 carries an integrated copy of a gene encoding histone H2B fused with GFP driven by the pie−1 promoter. Live imaging was performed on a Leica SP8 WLL with x63 oil lens. A gravid hermaphrodite was dissected in water 24–30 hr after injection with double-stranded set−1 RNA to release the post-fertilization embryos. Young embryos were collected with a capillary pipet and placed on a 2% agar pad on a slide. A 22x22 mm No 1.5 coverslip was placed on the agar pad, and the space was filled with water before sealing the edges of the coverslip with melted Vaseline. Scanning was done at 0.1 mm XY pixel size and 0.3 mm Z step size. Between 15 to 25 Z sections were taken at each time point. The time interval for each Z stack was set at 5–15 min. The maximum number of sections during the time course was limited to 3,000 to allow 100% of EG4601 embryos to survive and hatch by the next day. All imaged embryos were incubated at room temperature on the slide for 24 hr to check whether they had hatched. The high pixel value noise from the detectors was marked by hand and filled in with the average of neighboring pixels using Priism. Data stacks were Gaussian filtered in 3D to reduce the noise, and movies and images were generated in Priism.
Growth of set−1(RNAi) and Control RNAi Nematodes for RNA-seq
HT115 DE3 bacteria carrying either the empty RNAi feeding library vector L4440 or the L4440 vector with the set−1 coding region were grown overnight to saturation in 5 ml cultures of LB with 100 μg/ml ampicillin. For both bacterial strains, 2 ml of saturated culture were seeded into each of 3 separate 1 L volumes of LB with 100 μg/ml ampicillin. The 1 L cultures were grown for 4–6 h, induced with a final concentration of 4 mM IPTG for 2 h at 37°C to produce dsRNA, and grown to saturation. Bacteria were concentrated by centrifugation at 4300 × g for 10 min, resuspended in a 1:1 (w/v) ratio with LB, and seeded (2 ml) onto 9 cm RNAi plates (NGM agar with 1 mM IPTG and 100 μg/ml carbenicillin). Plates were allowed to dry overnight at 25°C. Synchronized populations of starved N2 L1s were placed onto two separate sets of plates with L4440 vector bacteria and three separate sets of plates with set−1(RNAi) bacteria and allowed to grow to adulthood. The resulting F0 gravid hermaphrodite populations were bleached separately, and the resulting starved L1s were transferred to independent sets of freshly prepared RNAi plates. The F1s were grown to adulthood and then bleached separately to obtain independent populations of embryos. mRNA extracted from these independently isolated sets of embryos was used for making RNA-seq libraries as described below. This set−1(RNAi) treatment caused ∼95% embryonic lethality.
RNA-seq Library Preparation
Mixed-stage embryos from each biological replicate of wild-type (N2) (7 total replicates), dpy−21(y607 JmjC) (3), dpy−21(y618 JmjC) (3), dpy−21(y622 JmjC) (3), dpy−21(e428 null) (7), set−4(n4600) (3), set−1(RNAi) (3), and N2 on L4440 (2) were isolated following the procedures above and frozen at −80°C in 1X M9 buffer. RNA was extrac ted as described previously (Crane et al., 2015). Libraries were prepared from 10 μg of total RNA. PolyA+ RNA was purified using the Dynabeads mRNA purification kit (Ambion) and fragmented using Fragmentation Reagent (Ambion). First strand cDNA was synthesized from polyA+ RNA using the SuperScript III Reverse Transcriptase Kit (Life Technologies) with random primers (Life Technologies). Second strand cDNA synthesis was performed using Second Strand Synthesis buffer, DNA Pol I, and RNase H (Life Technologies, 10812014, 18010025, 18021014). cDNA libraries were prepared for sequencing using the mRNA TruSeq protocol (Illumina).
Gene Expression Analysis
RNA-seq libraries were sequenced with Illumina's HiSeq 2000 platform. Reads were required to have passed the CASAVA 1.8 quality filtering to be considered further. To remove and trim reads containing the adapter barcodes, we used cutadapt version 0.9.5 (https://cutadapt.readthedocs.org/) (Martin, 2011). Reads were aligned to the WS220 transcriptome using GSNAP version 2012-01-11 (Wu and Nacu, 2010). Uniquely mapping reads were assigned to genes using HTSeq version 0.5.4p3 using the union mode (Anders et al., 2015). DESeq was used to calculate normalization factors and gene expression fold change and for significance testing (Anders and Huber, 2010).
Western Blot Analysis
Synchronized populations of N2 and dpy-21 (y607 JmjC) were grown on NGM agar plates with HB101 bacteria. Mixed-stage embryos were harvested from gravid hermaphrodites by bleaching, resuspended in homogenization buffer (50 mM HEPES-NaOH [pH 7.6], 140 mM KCl, 1 mM EDTA, 10% (v/v) glycerol, 0.5% (v/v) NP-40, 1 mM PMSF, 5 mM DTT, protease inhibitor cocktail II [Calbiochem]), and frozen in liquid nitrogen. Embryos were disrupted using Covaris S2 (10% duty cycle, power level 5, 200 cycles/ burst) for total of 7 min (60 sec ON, 7 cycles), and the cell debris was removed by centrifugation (16100 × g, 30 min, 4°C). Total protein concentration was det ermined using a BCA assay compared to a BSA standard (Thermo Fisher). Identical protein amounts (45 μg) for both strains were separated by SDS-PAGE and immunoblotted using rabbit polyclonal anti-DPY-21 (Novus Biologicals) and mouse monoclonal anti-α-tubulin (Sigma-Aldrich) antibodies. Western blots were analyzed using Image Lab software (Bio-Rad Laboratories, Inc.).
ChIP-seq and Spike-in Analysis
Mixed-stage C. elegans embryos from two biological replicates of wild-type (N2), dpy-21(y607 JmjC) and dpy-21(e428 null) were isolated as described above. As the spike-in control, C. tropicalis (TY5752) embryos were mixed with each C. elegans sample at 1: 6.5 weight ratio. Embryos were fixed for 15 min at room temperature with 2% (w/v) formaldehyde prepared by diluting 16% (w/v) formaldehyde (Polysciences) in 1 × M9 buffer. ChIP-seq was then performed as described previously (Kruesi et al., 2013) using H4K20me1 antibody (Abcam ab9051). For each sample, 6 μg of H4K20me1 antibody was used for 2 mg embryo extract. Single-end 50-bp sequencing was performed for ChIP and input libraries for each replicate using the Illumina HiSeq-4000 platform. To analyze the ChIP-seq data, a hybrid reference genome was built by combining the C. elegans genome version WS230 and the largest 70 contigs in the C. tropicalis genome version WS230. Single end reads from ChIP and input libraries were then mapped to the hybrid genome using Bowtie2 version 1.1.1 (Langmead et al., 2009), allowing up to two mismatches in the seed region and only retaining reads uniquely mapped to the hybrid genome. The reads mapped to the C. elegans genome were then extracted and sorted using SAMtools version 0.1.19 (Li et al., 2009). The H4K20me1 read coverage in the C. elegans genome was calculated by normalizing the read number in each 50 bp bin to the total read number using the bamCoverage function in deepTools version 2.5.0.1 (Ramirez et al., 2014). The H4K20me1 ChIP-seq enrichment profile of each sample was generated by calculating the ratios between read coverage scores in each ChIP dataset and corresponding input dataset using the bwCompare function in deepTools.
To directly compare the H4K20me1 levels between experimental replicates and different genotypes, a spike-in normalization factor was calculated for each ChIP-seq dataset. The number of reads mapping to C. elegans and C. tropicalis genomes in ChIP and corresponding input data were calculated. The normalization factor was then calculated as (C. elegans ChIP / C. tropicalis ChIP) / (C. elegans Input / C. tropicalis Input). To correct the H4K20me1 ChIP-seq enrichment with spike-in, we multiplied the read coverage for each ChIP dataset by the normalized factor and then compared it with the corresponding input. The spike-in corrected enrichment ratios for total number of reads mapped to X chromosomes or all autosomes were calculated, and the average ratios of two replicates were plotted in Figure 2G. The genome-wide spike-in corrected log2 enrichment profiles were generated using the bwCompare function in deepTools.
To examine the enrichment of H4K20me1 across TSSs or intergenic regions on X and autosomes in different genotypes, the enrichment scores of H4K20me1 in 50 bp bins across the TSSs defined by GRO-Cap and GRO-seq (Kruesi et al., 2013) or across the middle of intergenic regions were extracted using the computeMatrix function in deepTools and then averaged and plotted using R. The expected nucleosome-free region was evident when H4K20me1 reads were plotted against the true TSSs. Intergenic regions were defined as genomic regions that do not overlap with any genes, including protein-coding genes and non-coding RNAs, from C. elegans genome annotation version WS245.
in situ Hi-C
Embryos were collected and nuclei were isolated as in (Crane et al., 2015), and in situ Hi-C was performed based on a previously published protocol (Rao et al., 2014), with some modifications. Nuclei (1.5 × 108) were resuspended in 50 μl of 0.5% (w/v) SDS and incubated at 62°C for 5–10 min. SDS was quenched as in the in situ Hi-C protocol of (Rao et al., 2014). Then 25 μl of 10× DpnII Buffer and 2 μl (100 U) of DpnII (NEB R0543M) were added, and chromatin was digested overnight at 37°C with rotation. DpnII was inactivated, restr iction fragment overhangs were filled in, and ligation was performed as in (Rao et al., 2014), except that the following recipe was used for the ligation master mix: 13 μl 1 U/μl T4 DNA Ligase (Life Technologies 15224-090), 240 μl 5X T4 DNA ligase buffer, 100 μl 10% Triton X−100, 12 μl 10mg/ml Bovine Serum Albumin, 535μl water. DNA was purified as in (Rao et al., 2014) and sheared in a 130 μl microTUBE (Covaris, 520045) using an S2 Covaris (Covaris 520045) for 55 sec with duty cycle 10%, intensity 4, and 200 cycles/burst. 300-500bp fragments were selected as in (Rao et al., 2014), except that 40 μl of AMPure XP beads were used for the second size selection step. Biotinylated fragments were pulled down according to (Rao et al., 2014) and Dynabeads were washed in 100 μl 1X End-It DNA End-Repair Kit (Lucigen, ER81050) End-Repair buffer. Beads were resuspended in 100 μl of End-Repair Mix (10 μl 10X End-Repair buffer, 10 μl 2.4mM dNTP mix, 10 μl 10mM ATP, 2 μl End-Repair enzyme mix, 68 μl water) and incubated at room temperature for 45 min with gentle rotation. A-tailing and adapter ligation were performed as in (Rao et al., 2014) using 2 μl NEXTflex DNA Barcode (Bioo Scientific, 514101). To avoid PCR inhibition, the beads were divided between seven 50 μl PCRs and amplified for 4–6 cycles using Phusion polymerase (Thermo Fischer Scientific, F−530L) and NEXTflex Primer Mix (Bioo Scientific, 514107–48). The final library was purified according to (Rao et al., 2014). To verify successful Hi-C, 1 μl of the library was cloned using a Zero Blunt TOPO PCR cloning kit (Life Technologies, 450245), and 20–50 colonies were amplified and sequenced. If at least 50% of the fragments contained informative ligations, the library was sequenced using 100 bp paired end reads on a HiSeq4000 platform. A step-by-step version of this protocol is available on our website (http://mcb.berkeley.edu/labs/meyer/in_situ_Hi_C_protocol.pdf).
Hi-C Data Analysis
Read Mapping, Filtering and Iterative Correction
Reads were mapped to genome assembly ce11 using Bowtie2 version 2.2.4 (Langmead and Salzberg, 2012), binned at 50 kb, and iteratively corrected as previously described (Imakaev et al., 2012) using scripts derived from those available at https://bitbucket.org/mirnylab/hiclib. Briefly, after alignment, the paired reads were removed if both sides aligned to the same DpnII restriction fragment. These ‘same fragment reads’ include the read pairs in which the reads face in opposite directions (self circles), face toward each other (dangling ends) or were uniquely aligned to the same strand (error pairs). The read pairs from neighboring fragments that face toward each other and are separated by less than 500 bp (extra dangling ends) were also removed. To further improve the data quality, additional filtering steps were performed to remove read pairs corresponding to repeated instances of the same DNA molecule (duplicates), fragments starting within 5 bp from the DpnII site (fragments starting near restriction sites), fragments longer than 100 kb or shorter than 100 bp (extremely large and small fragments) and top 0.5% of fragments with the greatest number of reads (most frequently detected fragments). The mapping results and numbers of different categories of DNA molecules in the libraries are summarized in Table S8. Binning and iterative correction were then performed on the filtered data.
Insulation Analysis and TAD Boundary Calling
To quantitatively identify TADs and examine the strength of TAD boundaries, we calculated the insulation scores from Hi-C interaction data binned at 50 kb and performed boundary calling using a previously described method (Crane et al., 2015).
Calculating Z Scores and Scaling of the Contact Probability
To correct for experimental variation in the overall decay of chromatin contacts with genomic distance, we transformed the Hi-C interaction data into Z-scores as described before (Crane et al., 2015). To calculate the scaling of the contact probability, we plotted contact probability as a function of genomic separation and used the contact probability between 200 kb and 2 Mb to calculate the scaling factor for chromosome X, combined autosomes, and combined all chromosomes in wild-type and dpy−21(y607 JmjC) mutant replicates.
3D Plots
We made 3D plots to examine the changes in interactions between specific classes of genomic loci in wild-type vs. dpy−21(y607 JmjC) mutant embryos. We created 10x10 bin submatrices of wild-type or mutant Hi-C Z-scores around interactions between pairs of top 25 rex and predicted rex sites (Crane et al., 2015), DCC-dependent TAD boundaries, or DCC-independent TAD boundaries on chromosome X. Only interactions within 2.5 Mb were included. The submatrices were then averaged and plotted in 3D using R.
Quantification and Statistical Analysis
P values for statistical comparison between datasets were calculated using the 1-sided or 2-sided Wilcoxon rank-sum test in R or the 2-sided KS test. P values are provided in the figures or figure legends.
Data and Software Availability
The RNA-seq, ChIP-seq and Hi-C data sets generated in this paper have been deposited in the NCBI GEO under the accession number GSE84581. The DPY−21 crystal structure data generated in this paper have been deposited in the PDB under ID code 5UQD.
Supplementary Material
Figure S1. Evolutionary Conservation of the DPY−211210–1617 JmjC Domain, Related to Figure 1
(A) Front and back views of the DPY−211210–1617 structure colored according to residue conservation with ROSBIN proteins as shown in panel (B). The green color gradient indicates the degree of conservation (from 0 to 100% identity, light to dark, respectively). a-KG is shown in a stick representation (black carbon atoms). Arrows point to regions involved in histone peptide binding in other JmjC KDMs structures: region 1 (strands β6 and β7), region 2 (loop preceding the first DSBH strand β11), and region 3 (helical region composed of η3, a8, η4, and α9).
(B) Sequence alignment of the C-terminal domain of Caenorhabditis elegans DPY−21 (UniProtKB Q9GRZ3) with ROSBIN sequences from Drosophila melanogaster (UniProtKB B7YZX6, defined as Uncharacterized protein, isoform B), Danio rerio (UniProtKB A0A0G2KVT3), Mus musculus (UniProtKB Q80T69), Homo sapiens (UniProtKB Q5VWQ0), and Gallus gallus (UniProtKB F1NFM8). The alignment was generated using Clustal Omega and online ESPript 3.0 server (Robert and Gouet, 2014). The sequence is colored according to residue conservation: black background, conserved; bold black letters, highly similar; regular letters, non-conserved. Residues involved in Fe2+ binding are boxed in purple. Residues surrounding a-KG are boxed in red. Alanines replaced H1452 (purple star) in dpy−21(y607 JmjC), D1454 (purple triangle) in dpy−21(y618 JmjC), and both H1452 and D1454 in dpy−21(y622 JmjC). Secondary structure elements for DPY−21 are given above the alignment (α; α-helix, β; β-strand, and η; 310 helix) and color-coded for the different domains: JmjN (orange), β-hairpin (pink), mixed domain (green) and JmjC domain (cyan). Dashed lines represent regions of DPY−21 that are disordered and not built in the DPY−211210–1617 structure.
Figure S2. in vitro Assays for Demethylase Activity, Related to Figure 1
(A) Assays for demethylase specificity to histone H3 methylation marks. DPY−211210–1641 WT, DPY-211210–1641 mutant (H1452A D1454A), and mouse ROSBIN350–795 WT were incubated with calf thymus bulk histones with and without components (a-ketoglutarate, FeSO4, and ascorbate). Demethylase activity was evaluated by immunoblotting with specific antibodies against several H3 methylation marks. None of the assayed H3 marks showed changes.
(B) Assays for demethylase specificity to H4K20me3 were performed as in (A), using an H4K20me3 antibody (ab177190) for immunoblotting. Ambiguity exists as to whether the decrease in H4K20me3 level was due to true H4K20me3 demethylase activity or more likely to a combination of imperfect antibody specificity and low abundance of H4K20me3 in vivo. Of total H4 histones in human cell lines, 80% are H4K20me2, and only 2% are H4K20me3 (Pesavento et al., 2008). The anti-H4K20me3 monoclonal antibody ab177190 used also recognizes H4K20me2 at a lower level
(C,D). Non-specific antibody binding to H4K20me2 may have contributed to the observed H4K20me3 signal. (C), (D) Dot-blot analysis of antibody specificity for H4K20me3. (C) Rabbit monoclonal anti-H4K20me3 antibody (ab177190) showed a preference for H4K20me3 over H4K20me2. Detection of H4K20me2 was evident only after a long exposure (second blot).
(D) Using the same experimental conditions as in (A) mouse monoclonal anti-H4K20me3 antibody (ab78517) recognized both H4K20me2 and H4K20me3 to a similar extent. Antibody ab78517 was used for immunofluorescence experiments in Figure 2B and Figure S3C. The ab177190 antibody had an unusably weak signal in immunofluorescence experiments.
Figure S3. The DPY−21 JmjC Demethylase Enriches H4K20me1 on X in Adult Gut Cells and During Embryonic Development, Related to Figures 2 and 3
(A), (C) Confocal images of gut nuclei in XX animals (single Z sections) show levels of H4K20me1 (A) and H4K20me2/me3 (C) on X compared to autosomes. DPY−27 marks X chromosomes. dpy−21(y607 JmjC) and dpy−21(y622 JmjC) mutations eliminate H4K20me1 enrichment on X and restore H4K20me2/me3 levels. In contrast, an sdc−1(null) mutation, which causes simililarly weak dosage compensation defects as a dpy−21(null) mutation, had no effect on H4K20 methylation status (also Figures 2C and 2D), in contrast to prior reports of others (Vielle et al., 2012; Wells et al., 2012).
(B) Confocal images of a representative intestinal XO nucleus. The absence of SDC−3 staining signal indicates that the DCC is not bound to X. H4K20me1 is not enriched in any region of the nucleus. Scale bars in (A-C), 2 μm.
(D) Confocal images of representative nuclei from embryos > 300-cell stage of different genotypes. H4K20me1 enrichment on X is eliminated by dpy−21(e428 null), dpy−21(y607 JmjC), dpy−21(y618 JmjC) and dpy−21(y622 JmjC) mutations, but is not affected by the sdc−1(y415) mutation. In set−4(n4600) mutants and set−4(n4600); dpy−21(y607 JmjC) mutants, the H4K20me1 enrichment on X relative to autosomes is also lost due to the global elevation of the H4K20me1 level. Yellow arrows show foci of SDC−3 or H4K20me1 concentrated on X. Red arrows show diffuse nuclear localization of H4K20me1. Scale bar, 2 μm.
(E) Western blot of DPY−21 and α-tubulin in wild-type and dpy−21(y607 JmjC) embryos.
(F) Histogram showing quantification of western blot signal in (E) reveals no reduction in DPY−21 levels in dpy−21(y607 JmjC) vs. wild-type embryos, indicating that the JmjC amino acid substitution H1452A reduces catalytic activity but not protein abundance. Values represent the average of three protein bands +/- SEM.
(G) H4K20me1 enrichment on X (light red) vs. autosomes (light blue) in two biological ChIP-seq replicates (rep) of each genotype before the spike-in correction.
(H) H4K20me1 enrichment on X (red) vs. autosomes (blue) in the same two biological ChIP-seq replicates as in (G) after the spike-in correction reveals significant decrease in H4K20me1 on X in dpy−21(JmjC) or dpy−21(null) mutant vs. wild-type embryos.
Figure S4. Cell-cycle Dependent Localization of DPY−21 to X in Wild-type and dpy−21(JmjC) Mutant Embryos. Related to Figure 3.
(A) Immunofluorescence of 3×FLAG-tagged DPY-21 confirmed the cell-cycle dependent recruitment of DPY−21 to X chromosomes. Enlargements of individual nuclei at different stages of the cell cycle from a confocal image of a dpy-21(5′-FLAG) XX embryo at 277-cell stage stained with DAPI and antibodies against SDC-3, DPY−27 and FLAG. 3×FLAG-tagged DPY−21 colocalizes with SDC−3 and DPY−27 on X during interphase but dissociates from X during mitosis, while SDC−3 and DPY−27 remain on X throughout the cell cycle. Scale bar, 1 μm.
(B) Immunofluorescence of the 3×FLAG-tagged dpy−21(JmjC) mutant confirmed that the JmjC demethylase mutation does not affect the recruitment of DPY−21 to X chromosomes in interphase nuclei. Enlargements of individual nuclei at different stages of the cell cycle from confocal images of a dpy−21(y720 5′-FLAG, y607 JmjC) XX embryo at the 396-cell stage co-stained with DAPI and antibodies against SDC−3, DPY−27, and FLAG. Scale bar, 1 μm.
(C) Immunofluorescence of the dpy−21(JmjC) using DPY−21 antibodies also showed that the JmjC mutation does not affect the cell-cycle dependent recruitment of DPY−21 to X chromosomes of interphase nuclei but eliminates H4K20me1 enrichment. Individual nuclei at different stages of the cell cycle from the dpy-21(y607 JmjC) mutant 335-cell embryo shown in Figure 3 stained with DAPI and antibodies to H4K20me1, SDC−3, and DPY−21. In interphase, DPY−21 and SDC−3 colocalize on X. During mitosis, DPY−21 dissociates from X, while SDC−3 remains on X. The JmjC mutation eliminates H4K20me1 enrichment on X during interphase, but does not affect the H4K20me1 level during mitosis. Scale bar, 1 μm.
Figure S5. DPY-21 Demethylase Activity Modulates X-Chromosome Topology, Related to Figure 6
(A) Subtraction heatmaps from Hi-C data binned at 50 kb resolution depict the Z-score differences between dpy−21(y607JmjC) mutant and wild-type embryos for chromosomes II, III, IV, V and X. X exhibits more significant changes in chromatin interactions compared to autosomes, with interactions within 1 Mb on X exhibiting an overall increase (orange-red) and interactions greater than 1 Mb apart on X exhibiting an overall decrease (blue).
(B) Scatter plots depict the decay of overall chromatin interaction frequencies with increasing genomic distance in wild-type (left) and dpy−21(y607 JmjC) (right) mutant embryos. Red lines indicate the fitted loess curve for interaction frequency.
(C) Scatter plots show the correlation of chromatin interaction frequencies between wild-type and dpy−21(y607JmjC) mutant Hi-C replicates. Normalized interactions between 50 kb bins for each of the replicates compared were plotted on the X and Y axis. The bins close to the diagonal (y = × ± 1) were excluded. Pearson's correlation coefficients (R) for the pairwise comparisons are shown. Replicates of the same genotype exhibit higher correlation coefficients than replicates of different genotypes (also see Table S8).
(D) Three-dimensional profiles of average Hi-C interaction frequencies (Z-scores) in 50 kb bins around pairs of top 25 rex sites, DCC-dependent boundaries on X, and DCC-independent boundaries on X in wild-type or dpy−21(y607 JmjC) mutant embryos. Profiles are centered at 0. Interactions between rex sites and DCC-dependent boundaries are reduced in mutants.
(E) Scaling of intra-chromosomal contact probability with genomic distance for chromosome X (red) and autosomes (blue) in wild-type and dpy−21(y607 JmjC) mutant embryos. Scaling factors were calculated using average contact probability between 200 kb and 2 Mb (shaded region, fit showed as dotted lines). X and autosomes exhibit similar scaling factors in wild-type embryos (−0.62 and −0.59) but slightly different scaling factors in mutants (−0.76 and −0.67).
Figure S6. DPY−21 Demethylase Activity Is Required for X-Chromosome Compaction, Related to Figure 6
(A)-(C) Distribution of distances between FISH probes in embryos of different genotypes shown as cumulative plots. A shift to the right indicates increased distances and hence less compaction. Locations of probes on chromosome X or chromosome I are depicted above the plots. Shown for each probe pair are the genomic separation (red text) and number of embryos (in parentheses) and nuclei assayed (at bottom of plots). P values were calculated using the 2-sided Wilcoxon rank-sum test.
(A) Three pairs of non-rex loci located in different regions of the X chromosome show similarly increased distances in dpy−21(y607 JmjC) and dpy−21(e428 null) mutants that were less than the increases in sdc−2(null) mutants, which lack DCC binding to X. Pair 1 probes are within a TAD. Pair 2 probes are in adjacent TADs delineated by the rex−47 TAD boundary. Pair 3 probes are in adjacent TADs.
(B) Distances between two pairs of rex sites show no increase in dpy−21 mutants but a significant increase in sdc-2 mutants.
(C) Distances between two loci on chromosome I are not responsive to perturbations of dosage compensation.
Figure S7. Dynamic Localization of DPY−21 and H4K20me1 During Meiotic Progression, Related to Figure 7
(A) Confocal images of metaphase chromosomes from the mitotic zones of dpy−21(5′-FLAG) and dpy−21(y720 5′-FLAG, y607 JmjC) gonads co-stained with H4K20me1 and FLAG antibodies. H4K20me1 is highly enriched on all chromosomes, but DPY-21 is expressed at a low level and does not localize to chromosomes. Scale bar, 1 μm.
(B), (C) Confocal images of different regions of dpy-21(5′-FLAG) gonads show distinct patterns of DPY-21 and H4K20me1 staining.
(B) Transition zone – pachytene regions from two gonads stained with antibodies against FLAG-tagged DPY-21 (top) or H4K20me1 (bottom). Arrows mark the entry into pachytene. As meiotic cells transition into pachytene, DPY-21 staining becomes stronger and DPY-21's association with autosomes becomes evident. Meanwhile, the overall level of H4K20me1 decreases and H4K20me1 becomes selectively enriched on autosomes. The abrupt changes of DPY-21 and H4K20me1 patterns indicate the switch from a mitosis-specific, DPY-21-independent mode to a meiosis-specific, DPY-21−dependent mode of H4K20me1 regulation.
(C) Pachytene regions from two gonads stained with antibodies against FLAG-tagged DPY−21 (top) or H4K20me1 (bottom). In pachytene, both DPY−21 and H4K20me1 selectively localize to autosomes and are absent from X. Arrows mark several representative X chromosomes in different nuclei. Scale bars in B and C, 10 μm.
Highlights.
H4K20me2 JmjC demethylase subfamily revealed by DPY-21 structure and activity
In somatic cells, DPY-21 enriches H4K20me1 on X chromosomes to repress gene expression
H4K20me1 enrichment controls the higher-order structure of X chromosomes
In germ cells, DPY-21 enriches H4K20me1 on autosomes to promote chromosome compaction
In Brief.
A histone demethylase regulates 3D chromosome structure and gene expression by modulating the dynamic enrichment of H4K20me1.
Acknowledgments
We thank D. Fujimori, D. Minor, T. Cline, E. Ralston, and M. Marletta for general discussions, J. Dekker for Hi-C discussions, D. Stalford for figures, T. Cline, J. Berger, B. Farboud, and N. Fuda for manuscript comments, and J. Doudna for sharing equipment and expertise. We used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 OD018174 Instrumentation Grant. Huygens software was licensed to the CNR Biological Imaging Facility at UC Berkeley. Research was supported in part by Miller Institute funds and NIGMS grant R01 GM030702 to B.J.M. and NIGMS grant F32 GM100647 to B.S.W. B.J.M. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Author Contributions: K.B., Q.B., S.U., E.A., P.K., B.S.W., D.S.K., and C.P. conducted experiments and analyzed data. K.B., Q.B., and E.A. edited the manuscript. B.J.M. guided the study, analyzed data, and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrey G, Montavon T, Mascrez B, Gonzalez F, Noordermeer D, Leleu M, Trono D, Spitz F, Duboule D. A switch between topological domains underlies HoxD genes collinearity in mouse limbs. Science. 2013;340:1234167. doi: 10.1126/science.1234167. [DOI] [PubMed] [Google Scholar]
- Beck DB, Oda H, Shen SS, Reinberg D. PR-Set7 and H4K20me1: at the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. Genes Dev. 2012;26:325–337. doi: 10.1101/gad.177444.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard DP, Georlette D, Antoszewski L, Botchan MR. Chromatin reader L(3)mbt requires the Myb-MuvB/DREAM transcriptional regulatory complex for chromosomal recruitment. Proc Natl Acad Sci U S A. 2014;111:E4234–4243. doi: 10.1073/pnas.1416321111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettiger AN, Bintu B, Moffitt JR, Wang S, Beliveau BJ, Fudenberg G, Imakaev M, Mirny LA, Wu CT, Zhuang X. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature. 2016;529:418–422. doi: 10.1038/nature16496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Hughes DD, Chan TA, Sedat JW, Agard DA. IVE (Image Visualization Environment): a software platform for all three-dimensional microscopy applications. J Struct Biol. 1996;116:56–60. doi: 10.1006/jsbi.1996.0010. [DOI] [PubMed] [Google Scholar]
- Chen Z, Zang J, Whetstine J, Hong X, Davrazou F, Kutateladze TG, Simpson M, Mao Q, Pan CH, Dai S, et al. Structural insights into histone demethylation by JMJD2 family members. Cell. 2006;125:691–702. doi: 10.1016/j.cell.2006.04.024. [DOI] [PubMed] [Google Scholar]
- Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, Deng G, Diep S, Lomenick B, Meli VS, Monsalve GC, et al. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature. 2014;510:397–401. doi: 10.1038/nature13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang PT, Albertson DG, Meyer BJ. DPY-27: a chromosome condensation protein homolog that regulates C. elegans dosage compensation through association with the X chromosome. Cell. 1994;79:459–474. doi: 10.1016/0092-8674(94)90255-0. [DOI] [PubMed] [Google Scholar]
- Crane E, Bian Q, McCord RP, Lajoie BR, Wheeler BS, Ralston EJ, Uzawa S, Dekker J, Meyer BJ. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature. 2015;523:240–244. doi: 10.1038/nature14450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Rocha ST, Heard E. Novel players in X inactivation: insights into Xist-mediated gene silencing and chromosome conformation. Nat Struct Mol Biol. 2017;24:197–204. doi: 10.1038/nsmb.3370. [DOI] [PubMed] [Google Scholar]
- Dekker J, Mirny L. The 3D Genome as Moderator of Chromosomal Communication. Cell. 2016;164:1110–1121. doi: 10.1016/j.cell.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney CE, Chen AT, Graniel JV, Dumas KJ, Hu PJ. A histone H4 lysine 20 methyltransferase couples environmental cues to sensory neuron control of developmental plasticity. Development. 2017 doi: 10.1242/dev.145722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas KJ, Delaney CE, Flibotte S, Moerman DG, Csankovszki G, Hu PJ. Unexpected role for dosage compensation in the control of dauer arrest, insulin-like signaling, and FoxO transcription factor activity in Caenorhabditis elegans. Genetics. 2013;194:619–629. doi: 10.1534/genetics.113.149948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Evans PR. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr D Biol Crystallogr. 2011;67:282–292. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farboud B, Meyer BJ. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design. Genetics. 2015;199:959–971. doi: 10.1534/genetics.115.175166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis NJ, Kingston RE, Woodcock CL. Chromatin compaction by a polycomb group protein complex. Science. 2004;306:1574–1577. doi: 10.1126/science.1100576. [DOI] [PubMed] [Google Scholar]
- Giorgetti L, Heard E. Closing the loop: 3C versus DNA FISH. Genome Biol. 2016;17:215. doi: 10.1186/s13059-016-1081-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison MM, Lu X, Horvitz HR. LIN-61, one of two Caenorhabditis elegans malignant-brain-tumor-repeat-containing proteins, acts with the DRM and NuRD-like protein complexes in vulval development but not in certain other biological processes. Genetics. 2007;176:255–271. doi: 10.1534/genetics.106.069633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J, Mirny LA. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Methods. 2012;9:999–1003. doi: 10.1038/nmeth.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen S, Schotta G, Sorensen CS. Histone H4 lysine 20 methylation: key player in epigenetic regulation of genomic integrity. Nucleic Acids Res. 2013;41:2797–2806. doi: 10.1093/nar/gkt012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalashnikova AA, Porter-Goff ME, Muthurajan UM, Luger K, Hansen JC. The role of the nucleosome acidic patch in modulating higher order chromatin structure. Journal of the Royal Society, Interface. 2013;10:20121022. doi: 10.1098/rsif.2012.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004;32:W526–531. doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmaier A, Savarese F, Lachner M, Martens J, Jenuwein T, Wutz A. A chromosomal memory triggered by Xist regulates histone methylation in X inactivation. PLoS Biol. 2004;2:E171. doi: 10.1371/journal.pbio.0020171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koscielny G, Yaikhom G, Iyer V, Meehan TF, Morgan H, Atienza-Herrero J, Blake A, Chen CK, Easty R, Di Fenza A, et al. The International Mouse Phenotyping Consortium Web Portal, a unified point of access for knockout mice and related phenotyping data. Nucleic Acids Res. 2014;42:D802–809. doi: 10.1093/nar/gkt977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer M, Kranz AL, Su A, Winterkorn LH, Albritton SE, Ercan S. Developmental Dynamics of X-Chromosome Dosage Compensation by the DCC and H4K20me1 in C. elegans. PLoS Genet. 2015;11:e1005698. doi: 10.1371/journal.pgen.1005698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruesi WS, Core LJ, Waters CT, Lis JT, Meyer BJ. Condensin controls recruitment of RNA polymerase II to achieve nematode X-chromosome dosage compensation. eLife. 2013;2:e00808. doi: 10.7554/eLife.00808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu S, Ji F, Sunwoo H, Jain G, Lee JT, Sadreyev RI, Dekker J, Kingston RE. Polycomb Repressive Complex 1 Generates Discrete Compacted Domains that Change during Differentiation. Mol Cell. 2017;65:432, 446 e435. doi: 10.1016/j.molcel.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda MI, Hilfiker A, Lucchesi JC. Dosage Compensation in Drosophila-a Model for the Coordinate Regulation of Transcription. Genetics. 2016;204:435–450. doi: 10.1534/genetics.115.185108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnel C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- Lau AC, Nabeshima K, Csankovszki G. The C. elegans dosage compensation complex mediates interphase X chromosome compaction. Epigenetics Chromatin. 2014;7:31. doi: 10.1186/1756-8935-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Rechtsteiner A, Egelhofer TA, Vielle A, Latorre I, Cheung MS, Ercan S, Ikegami K, Jensen M, Kolasinska-Zwierz P, et al. Broad chromosomal domains of histone modification patterns in C. elegans. Genome Res. 2011;21:227–236. doi: 10.1101/gr.115519.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Tanasa B, Tyurina OV, Zhou TY, Gassmann R, Liu WT, Ohgi KA, Benner C, Garcia-Bassets I, Aggarwal AK, et al. PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature. 2010;466:508–512. doi: 10.1038/nature09272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo TW, Pickle CS, Lin S, Ralston EJ, Gurling M, Schartner CM, Bian Q, Doudna JA, Meyer BJ. Precise and heritable genome editing in evolutionarily diverse nematodes using TALENs and CRISPR/Cas9 to engineer insertions and deletions. Genetics. 2013;195:331–348. doi: 10.1534/genetics.113.155382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Simon MD, Chodaparambil JV, Hansen JC, Shokat KM, Luger K. The effect of H3K79 dimethylation and H4K20 trimethylation on nucleosome and chromatin structure. Nat Struct Mol Biol. 2008;15:1122–1124. doi: 10.1038/nsmb.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markolovic S, Leissing TM, Chowdhury R, Wilkins SE, Lu X, Schofield CJ. Structure-function relationships of human JmjC oxygenases-demethylases versus hydroxylases. Curr Opin Struct Biol. 2016;41:62–72. doi: 10.1016/j.sbi.2016.05.013. [DOI] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet journal. 2011;17:10–12. [Google Scholar]
- Mets DG, Meyer BJ. Condensins regulate meiotic DNA break distribution, thus crossover frequency, by controlling chromosome structure. Cell. 2009;139:73–86. doi: 10.1016/j.cell.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer BJ. Targeting X chromosomes for repression. Curr Opin Genet Dev. 2010;20:179–189. doi: 10.1016/j.gde.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LM, Plenefisch JD, Casson LP, Meyer BJ. xol-1: a gene that controls the male modes of both sex determination and X chromosome dosage compensation in C. elegans. Cell. 1988;55:167–183. doi: 10.1016/0092-8674(88)90019-0. [DOI] [PubMed] [Google Scholar]
- Oda H, Okamoto I, Murphy N, Chu J, Price SM, Shen MM, Torres-Padilla ME, Heard E, Reinberg D. Monomethylation of histone H4-lysine 20 is involved in chromosome structure and stability and is essential for mouse development. Mol Cell Biol. 2009;29:2278–2295. doi: 10.1128/MCB.01768-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paix A, Folkmann A, Rasoloson D, Seydoux G. High efficiency, homology-directed genome editing in Caenorhabditis elegans using CRISPR-Cas9 ribonucleoprotein complexes. Genetics. 2015;201:47–54. doi: 10.1534/genetics.115.179382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesavento JJ, Yang H, Kelleher NL, Mizzen CA. Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle. Mol Cell Biol. 2008;28:468–486. doi: 10.1128/MCB.01517-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plasschaert RN, Bartolomei MS. Genomic imprinting in development, growth behavior and stem cells. Development. 2014;141:1805–1813. doi: 10.1242/dev.101428. in. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi HH, Sarkissian M, Hu GQ, Wang Z, Bhattacharjee A, Gordon DB, Gonzales M, Lan F, Ongusaha PP, Huarte M, et al. Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature. 2010;466:503–507. doi: 10.1038/nature09261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Dundar F, Diehl S, Gruning BA, Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42:W187–191. doi: 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhind NR, Miller LM, Kopczynski JB, Meyer BJ. xol-1 acts as an early switch in the C. elegans male/hermaphrodite decision. Cell. 1995;80:71–82. doi: 10.1016/0092-8674(95)90452-2. [DOI] [PubMed] [Google Scholar]
- Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soshnev AA, Josefowicz SZ, Allis CD. Greater Than the Sum of Parts: Complexity of the Dynamic Epigenome. Mol Cell. 2016;62:681–694. doi: 10.1016/j.molcel.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stender JD, Pascual G, Liu W, Kaikkonen MU, Do K, Spann NJ, Boutros M, Perrimon N, Rosenfeld MG, Glass CK. Control of proinflammatory gene programs by regulated trimethylation and demethylation of histone H4K20. Mol Cell. 2012;48:28–38. doi: 10.1016/j.molcel.2012.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strome S, Kelly WG, Ercan S, Lieb JD. Regulation of the X Chromosomes in Caenorhabditis elegans. Cold Spring Harb Perspect Biol. 2014;6:a018366–a018366. doi: 10.1101/cshperspect.a018366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Tanaka H, Iguchi N, Kitamura K, Chen Y, Maekawa M, Nishimura H, Ohta H, Miyagawa Y, Matsumiya K, et al. Rosbin: a novel homeobox-like protein gene expressed exclusively in round spermatids. Biol Reprod. 2004;70:1485–1492. doi: 10.1095/biolreprod.103.026096. [DOI] [PubMed] [Google Scholar]
- Tan FJ, Fire AZ, Hill RB. Regulation of apoptosis by C. elegans CED-9 in the absence of the C-terminal transmembrane domain. Cell Death Differ. 2007;14:1925–1935. doi: 10.1038/sj.cdd.4402215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC. Reciprocal-space solvent flattening. Acta Crystallogr D Biol Crystallogr. 1999;55:1863–1871. doi: 10.1107/S0907444999010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojer P, Li G, Sims RJ, 3rd, Vaquero A, Kalakonda N, Boccuni P, Lee D, Erdjument-Bromage H, Tempst P, Nimer SD, et al. L3MBTL1, a histone-methylation-dependent chromatin lock. Cell. 2007;129:915–928. doi: 10.1016/j.cell.2007.03.048. [DOI] [PubMed] [Google Scholar]
- van Nuland R, Gozani O. Histone H4 Lysine 20 (H4K20) Methylation, Expanding the Signaling Potential of the Proteome One Methyl Moiety at a Time. Mol Cell Proteomics. 2016;15:755–764. doi: 10.1074/mcp.R115.054742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vielle A, Lang J, Dong Y, Ercan S, Kotwaliwale C, Rechtsteiner A, Appert A, Chen QB, Dose A, Egelhofer T, et al. H4K20me1 contributes to downregulation of X-linked genes for C. elegans dosage compensation. PLoS Genet. 2012;8:e1002933. doi: 10.1371/journal.pgen.1002933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Telese F, Tan Y, Li W, Jin C, He X, Basnet H, Ma Q, Merkurjev D, Zhu X, et al. LSD1n is an H4K20 demethylase regulating memory formation via transcriptional elongation control. Nat Neurosci. 2015;18:1256–1264. doi: 10.1038/nn.4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster CM, Wu L, Douglas D, Soukas AA. A non-canonical role for the C. elegans dosage compensation complex in growth and metabolic regulation downstream of TOR complex 2. Development. 2013;140:3601–3612. doi: 10.1242/dev.094292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells MB, Snyder MJ, Custer LM, Csankovszki G. Caenorhabditis elegans dosage compensation regulates histone H4 chromatin state on X chromosomes. Mol Cell Biol. 2012;32:1710–1719. doi: 10.1128/MCB.06546-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson I, Eskeland R, Lettice LA, Hill AE, Boyle S, Grimes GR, Hill RE, Bickmore WA. Anterior-posterior differences in HoxD chromatin topology in limb development. Development. 2012;139:3157–3167. doi: 10.1242/dev.081174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonker SA, Meyer BJ. Recruitment of C. elegans dosage compensation proteins for gene-specific versus chromosome-wide repression. Development. 2003;130:6519–6532. doi: 10.1242/dev.00886. [DOI] [PubMed] [Google Scholar]
- Zielinska DF, Gnad F, Jedrusik-Bode M, Wisniewski JR, Mann M. Caenorhabditis elegans has a phosphoproteome atypical for metazoans that is enriched in developmental and sex determination proteins. J Proteome Res. 2009;8:4039–4049. doi: 10.1021/pr900384k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Evolutionary Conservation of the DPY−211210–1617 JmjC Domain, Related to Figure 1
(A) Front and back views of the DPY−211210–1617 structure colored according to residue conservation with ROSBIN proteins as shown in panel (B). The green color gradient indicates the degree of conservation (from 0 to 100% identity, light to dark, respectively). a-KG is shown in a stick representation (black carbon atoms). Arrows point to regions involved in histone peptide binding in other JmjC KDMs structures: region 1 (strands β6 and β7), region 2 (loop preceding the first DSBH strand β11), and region 3 (helical region composed of η3, a8, η4, and α9).
(B) Sequence alignment of the C-terminal domain of Caenorhabditis elegans DPY−21 (UniProtKB Q9GRZ3) with ROSBIN sequences from Drosophila melanogaster (UniProtKB B7YZX6, defined as Uncharacterized protein, isoform B), Danio rerio (UniProtKB A0A0G2KVT3), Mus musculus (UniProtKB Q80T69), Homo sapiens (UniProtKB Q5VWQ0), and Gallus gallus (UniProtKB F1NFM8). The alignment was generated using Clustal Omega and online ESPript 3.0 server (Robert and Gouet, 2014). The sequence is colored according to residue conservation: black background, conserved; bold black letters, highly similar; regular letters, non-conserved. Residues involved in Fe2+ binding are boxed in purple. Residues surrounding a-KG are boxed in red. Alanines replaced H1452 (purple star) in dpy−21(y607 JmjC), D1454 (purple triangle) in dpy−21(y618 JmjC), and both H1452 and D1454 in dpy−21(y622 JmjC). Secondary structure elements for DPY−21 are given above the alignment (α; α-helix, β; β-strand, and η; 310 helix) and color-coded for the different domains: JmjN (orange), β-hairpin (pink), mixed domain (green) and JmjC domain (cyan). Dashed lines represent regions of DPY−21 that are disordered and not built in the DPY−211210–1617 structure.
Figure S2. in vitro Assays for Demethylase Activity, Related to Figure 1
(A) Assays for demethylase specificity to histone H3 methylation marks. DPY−211210–1641 WT, DPY-211210–1641 mutant (H1452A D1454A), and mouse ROSBIN350–795 WT were incubated with calf thymus bulk histones with and without components (a-ketoglutarate, FeSO4, and ascorbate). Demethylase activity was evaluated by immunoblotting with specific antibodies against several H3 methylation marks. None of the assayed H3 marks showed changes.
(B) Assays for demethylase specificity to H4K20me3 were performed as in (A), using an H4K20me3 antibody (ab177190) for immunoblotting. Ambiguity exists as to whether the decrease in H4K20me3 level was due to true H4K20me3 demethylase activity or more likely to a combination of imperfect antibody specificity and low abundance of H4K20me3 in vivo. Of total H4 histones in human cell lines, 80% are H4K20me2, and only 2% are H4K20me3 (Pesavento et al., 2008). The anti-H4K20me3 monoclonal antibody ab177190 used also recognizes H4K20me2 at a lower level
(C,D). Non-specific antibody binding to H4K20me2 may have contributed to the observed H4K20me3 signal. (C), (D) Dot-blot analysis of antibody specificity for H4K20me3. (C) Rabbit monoclonal anti-H4K20me3 antibody (ab177190) showed a preference for H4K20me3 over H4K20me2. Detection of H4K20me2 was evident only after a long exposure (second blot).
(D) Using the same experimental conditions as in (A) mouse monoclonal anti-H4K20me3 antibody (ab78517) recognized both H4K20me2 and H4K20me3 to a similar extent. Antibody ab78517 was used for immunofluorescence experiments in Figure 2B and Figure S3C. The ab177190 antibody had an unusably weak signal in immunofluorescence experiments.
Figure S3. The DPY−21 JmjC Demethylase Enriches H4K20me1 on X in Adult Gut Cells and During Embryonic Development, Related to Figures 2 and 3
(A), (C) Confocal images of gut nuclei in XX animals (single Z sections) show levels of H4K20me1 (A) and H4K20me2/me3 (C) on X compared to autosomes. DPY−27 marks X chromosomes. dpy−21(y607 JmjC) and dpy−21(y622 JmjC) mutations eliminate H4K20me1 enrichment on X and restore H4K20me2/me3 levels. In contrast, an sdc−1(null) mutation, which causes simililarly weak dosage compensation defects as a dpy−21(null) mutation, had no effect on H4K20 methylation status (also Figures 2C and 2D), in contrast to prior reports of others (Vielle et al., 2012; Wells et al., 2012).
(B) Confocal images of a representative intestinal XO nucleus. The absence of SDC−3 staining signal indicates that the DCC is not bound to X. H4K20me1 is not enriched in any region of the nucleus. Scale bars in (A-C), 2 μm.
(D) Confocal images of representative nuclei from embryos > 300-cell stage of different genotypes. H4K20me1 enrichment on X is eliminated by dpy−21(e428 null), dpy−21(y607 JmjC), dpy−21(y618 JmjC) and dpy−21(y622 JmjC) mutations, but is not affected by the sdc−1(y415) mutation. In set−4(n4600) mutants and set−4(n4600); dpy−21(y607 JmjC) mutants, the H4K20me1 enrichment on X relative to autosomes is also lost due to the global elevation of the H4K20me1 level. Yellow arrows show foci of SDC−3 or H4K20me1 concentrated on X. Red arrows show diffuse nuclear localization of H4K20me1. Scale bar, 2 μm.
(E) Western blot of DPY−21 and α-tubulin in wild-type and dpy−21(y607 JmjC) embryos.
(F) Histogram showing quantification of western blot signal in (E) reveals no reduction in DPY−21 levels in dpy−21(y607 JmjC) vs. wild-type embryos, indicating that the JmjC amino acid substitution H1452A reduces catalytic activity but not protein abundance. Values represent the average of three protein bands +/- SEM.
(G) H4K20me1 enrichment on X (light red) vs. autosomes (light blue) in two biological ChIP-seq replicates (rep) of each genotype before the spike-in correction.
(H) H4K20me1 enrichment on X (red) vs. autosomes (blue) in the same two biological ChIP-seq replicates as in (G) after the spike-in correction reveals significant decrease in H4K20me1 on X in dpy−21(JmjC) or dpy−21(null) mutant vs. wild-type embryos.
Figure S4. Cell-cycle Dependent Localization of DPY−21 to X in Wild-type and dpy−21(JmjC) Mutant Embryos. Related to Figure 3.
(A) Immunofluorescence of 3×FLAG-tagged DPY-21 confirmed the cell-cycle dependent recruitment of DPY−21 to X chromosomes. Enlargements of individual nuclei at different stages of the cell cycle from a confocal image of a dpy-21(5′-FLAG) XX embryo at 277-cell stage stained with DAPI and antibodies against SDC-3, DPY−27 and FLAG. 3×FLAG-tagged DPY−21 colocalizes with SDC−3 and DPY−27 on X during interphase but dissociates from X during mitosis, while SDC−3 and DPY−27 remain on X throughout the cell cycle. Scale bar, 1 μm.
(B) Immunofluorescence of the 3×FLAG-tagged dpy−21(JmjC) mutant confirmed that the JmjC demethylase mutation does not affect the recruitment of DPY−21 to X chromosomes in interphase nuclei. Enlargements of individual nuclei at different stages of the cell cycle from confocal images of a dpy−21(y720 5′-FLAG, y607 JmjC) XX embryo at the 396-cell stage co-stained with DAPI and antibodies against SDC−3, DPY−27, and FLAG. Scale bar, 1 μm.
(C) Immunofluorescence of the dpy−21(JmjC) using DPY−21 antibodies also showed that the JmjC mutation does not affect the cell-cycle dependent recruitment of DPY−21 to X chromosomes of interphase nuclei but eliminates H4K20me1 enrichment. Individual nuclei at different stages of the cell cycle from the dpy-21(y607 JmjC) mutant 335-cell embryo shown in Figure 3 stained with DAPI and antibodies to H4K20me1, SDC−3, and DPY−21. In interphase, DPY−21 and SDC−3 colocalize on X. During mitosis, DPY−21 dissociates from X, while SDC−3 remains on X. The JmjC mutation eliminates H4K20me1 enrichment on X during interphase, but does not affect the H4K20me1 level during mitosis. Scale bar, 1 μm.
Figure S5. DPY-21 Demethylase Activity Modulates X-Chromosome Topology, Related to Figure 6
(A) Subtraction heatmaps from Hi-C data binned at 50 kb resolution depict the Z-score differences between dpy−21(y607JmjC) mutant and wild-type embryos for chromosomes II, III, IV, V and X. X exhibits more significant changes in chromatin interactions compared to autosomes, with interactions within 1 Mb on X exhibiting an overall increase (orange-red) and interactions greater than 1 Mb apart on X exhibiting an overall decrease (blue).
(B) Scatter plots depict the decay of overall chromatin interaction frequencies with increasing genomic distance in wild-type (left) and dpy−21(y607 JmjC) (right) mutant embryos. Red lines indicate the fitted loess curve for interaction frequency.
(C) Scatter plots show the correlation of chromatin interaction frequencies between wild-type and dpy−21(y607JmjC) mutant Hi-C replicates. Normalized interactions between 50 kb bins for each of the replicates compared were plotted on the X and Y axis. The bins close to the diagonal (y = × ± 1) were excluded. Pearson's correlation coefficients (R) for the pairwise comparisons are shown. Replicates of the same genotype exhibit higher correlation coefficients than replicates of different genotypes (also see Table S8).
(D) Three-dimensional profiles of average Hi-C interaction frequencies (Z-scores) in 50 kb bins around pairs of top 25 rex sites, DCC-dependent boundaries on X, and DCC-independent boundaries on X in wild-type or dpy−21(y607 JmjC) mutant embryos. Profiles are centered at 0. Interactions between rex sites and DCC-dependent boundaries are reduced in mutants.
(E) Scaling of intra-chromosomal contact probability with genomic distance for chromosome X (red) and autosomes (blue) in wild-type and dpy−21(y607 JmjC) mutant embryos. Scaling factors were calculated using average contact probability between 200 kb and 2 Mb (shaded region, fit showed as dotted lines). X and autosomes exhibit similar scaling factors in wild-type embryos (−0.62 and −0.59) but slightly different scaling factors in mutants (−0.76 and −0.67).
Figure S6. DPY−21 Demethylase Activity Is Required for X-Chromosome Compaction, Related to Figure 6
(A)-(C) Distribution of distances between FISH probes in embryos of different genotypes shown as cumulative plots. A shift to the right indicates increased distances and hence less compaction. Locations of probes on chromosome X or chromosome I are depicted above the plots. Shown for each probe pair are the genomic separation (red text) and number of embryos (in parentheses) and nuclei assayed (at bottom of plots). P values were calculated using the 2-sided Wilcoxon rank-sum test.
(A) Three pairs of non-rex loci located in different regions of the X chromosome show similarly increased distances in dpy−21(y607 JmjC) and dpy−21(e428 null) mutants that were less than the increases in sdc−2(null) mutants, which lack DCC binding to X. Pair 1 probes are within a TAD. Pair 2 probes are in adjacent TADs delineated by the rex−47 TAD boundary. Pair 3 probes are in adjacent TADs.
(B) Distances between two pairs of rex sites show no increase in dpy−21 mutants but a significant increase in sdc-2 mutants.
(C) Distances between two loci on chromosome I are not responsive to perturbations of dosage compensation.
Figure S7. Dynamic Localization of DPY−21 and H4K20me1 During Meiotic Progression, Related to Figure 7
(A) Confocal images of metaphase chromosomes from the mitotic zones of dpy−21(5′-FLAG) and dpy−21(y720 5′-FLAG, y607 JmjC) gonads co-stained with H4K20me1 and FLAG antibodies. H4K20me1 is highly enriched on all chromosomes, but DPY-21 is expressed at a low level and does not localize to chromosomes. Scale bar, 1 μm.
(B), (C) Confocal images of different regions of dpy-21(5′-FLAG) gonads show distinct patterns of DPY-21 and H4K20me1 staining.
(B) Transition zone – pachytene regions from two gonads stained with antibodies against FLAG-tagged DPY-21 (top) or H4K20me1 (bottom). Arrows mark the entry into pachytene. As meiotic cells transition into pachytene, DPY-21 staining becomes stronger and DPY-21's association with autosomes becomes evident. Meanwhile, the overall level of H4K20me1 decreases and H4K20me1 becomes selectively enriched on autosomes. The abrupt changes of DPY-21 and H4K20me1 patterns indicate the switch from a mitosis-specific, DPY-21-independent mode to a meiosis-specific, DPY-21−dependent mode of H4K20me1 regulation.
(C) Pachytene regions from two gonads stained with antibodies against FLAG-tagged DPY−21 (top) or H4K20me1 (bottom). In pachytene, both DPY−21 and H4K20me1 selectively localize to autosomes and are absent from X. Arrows mark several representative X chromosomes in different nuclei. Scale bars in B and C, 10 μm.