

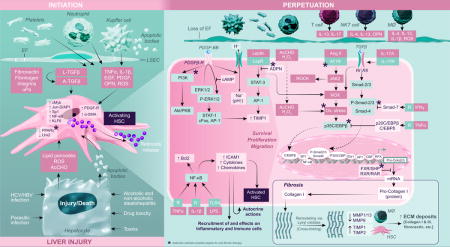
Liver fibrosis is a primary response to injury of etiologies including metabolic abnormalities, viral (hepatitis C and B virus [HCV/HBV]) and other infections, toxins and drugs. Chronic exposure to the injury agent(s) can cause fibrosis to progress to irreversible cirrhosis. Although effective therapies for HCV-induced fibrosis are now available, the new challenge is the alarming increase in liver fibrosis/cirrhosis due to non-alcoholic and alcoholic steatohepatitis. A clear understanding of the mechanisms of hepatic fibrosis/cirrhosis is critical.
Regardless of etiology, hepatic stellate cells (HSCs) are the major contributors to fibrosis/cirrhosis. Bone marrow-derived fibrocytes, epithelial-to-mesenchymal transition and portal fibroblasts (Pfb) are other sources, but their contribution is much lower. Pfb are important in the early stages of the disease, but HSCs are predominantly responsible for the progression of the disease, even for biliary fibrosis.1,2
Physiologically quiescent, HSCs become rapidly activated and transdifferentiate into proliferative, contractile and fibrogenic myofibroblast-like cells during liver injury. The process (known as “Initiation”) involves the loss of lipid droplets that contain retinoids, peroxisome proliferator-activated receptor (PPARγ) and LIM homodimer protein (Lhx2), and increased expression of platelet-derived growth factor (PDGF)-receptor, Kruppel-like factor 6 (KLF6) and α-smooth muscle actin (SMA).3 Apoptotic bodies derived from damaged or injured hepatocytes and reactive oxygen species (ROS), acetaldehyde and lipid peroxidation products are strong initiating signals for HSC activation.3 Kupffer cells (KC) also phagocytose the hepatocyte-apoptotic bodies. Mediators (ROS, TNFα, PDGF, 1L-1β, osteopontin, EGF, TGFβ) produced by KC, platelets, neutrophils and infiltrating immune/inflammatory cells as well as sinusoidal endothelial cells and cholangiocytes all induce HSC activation/proliferation.
The “Initiation” of HSC activation overlaps and continues with the “Perpetuation” phase involving signals of survival, migration, proliferation, fibrogenesis and retention of activated phenotype as the injury stimulus persists. PDGF is a powerful mediator of HSC chemotaxis and proliferation. PDGF stimulates Na+/H+ antiporter activity, and activates cFos, AP-1 and STAT-1 via ERK1/2-MAPK and P13K-PKB/Akt signaling, the critical pathways of HSC proliferation/migration. The abnormal extracellular matrix (ECM) is also a potent stimulus for migration/proliferation of activated HSCs (aHSCs) by their expression of integrins and signaling via Rho and Rac GTP-binding proteins.
The aHSCs produce and deposit increasing amounts of type I and III collagen and fibronectin in the ECM, a process compounded by an imbalance in matrix metalloproteinases ([MMPs], produced by macrophages) and predominantly, tissue inhibitor of metalloproteinases ([TIMPs], produced by HSCs).4 The most potent fibrogenic cytokine, TGFβ1 produced by several cells including aHSCs, is released in an inactive form and activated via proteolysis by fibronectin, urokinase-type plasminogen activator (uPa) and integrins. It’s binding to TGFβ-RII (increased by IL-175) promotes heterodimerization with TGFβ-RI, followed by Smad signaling-induced procollagen mRNA expression.6 Decreased expression of PPARγ, which inhibits Smad-3 expression, allows fibrogenesis to proceed.6,7 IFNγ and TGFβ1 can stimulate expression of Smad-7 that inhibits Smad activity.6 Os-teopontin, bFGF and CTGF are other cytokines that promote ECM synthesis by HSCs.3,6,7 Oxidative stress induced by various stimuli, including Ang II, in aHSCs also induces collagen I synthesis through the transcription factor p35C/EBP, which can be antagonized by TN-Fα-induced expression of p20C/EBP.3,6,8 The JAK2 activation by Ang II also instigates RhoA/Rho-kinase (ROCK) pathway coupled to the migration and contraction of HSCs. Endothelin-1 and thrombin are other major factors of HSC contraction, contributing to portal hypertension.
Many factors including TNFα, IL-1β, LPS promote survival of aHSCs by stimulating NF-κB and downregulating pro-apoptotic genes. aHSCs themselves produce numerous cytokines, chemokines and growth mediators, LPS being an important stimulus.9,10 In addition to exerting autocrine effects, HSC-derived cytokines/chemokines recruit and activate inflammatory and immune cells, and react to the mediators released by them, perpetuating fibrogenesis.
Obviously, hepatic fibrosis is a complex phenomenon orchestrated by a multitude of cells, mediators and signaling pathways all converging on HSC activation, proliferation, migration and ECM deposition. Targeting the key pathways (indicated by asterisks on the snapshot) will be critical to develop effective therapies for fibrosis.
Acknowledgments
Financial support
VA Merit Review Award (11O1BX001174) and Department of Defense grant (W81XWH14-PRMRP-11RA).
Footnotes
Mediator of HSC proliferation: PDGF, EGF, VEGF, Ang II, bFGF
Factors that induce migration: PDGF, VEGF, Ang1, Fibronectin, Ang II, CXCR3-L, CXCR4-L, CCL2
Conflict of interest
The author declared he does not have anything to disclose regarding funding or conflict of interest with respect to this manuscript.
Author contribution
CRG is responsible for the content of this manuscript.
References
- 1.Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823. doi: 10.1038/ncomms3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kayama Y, Wang P, Brenner DA, Kisseleva T. In: Stellate cells in health and disease. Gandhi CR, Pinzani M, editors. Elsevier Press; 2015. pp. 87–106. [Google Scholar]
- 3.Puche JE, Saiman Y, Friedman SL. Hepatic stellate cells and liver fibrosis. Compr Physiol. 2013;3:1473–1492. doi: 10.1002/cphy.c120035. [DOI] [PubMed] [Google Scholar]
- 4.Campana L, Iredale J. Matrix metalloproteinases and their inhibitors. In: Gandhi CR, Pinzani M, editors. Stellate cells in health and disease. Elsevier Press; 2015. pp. 107–124. [Google Scholar]
- 5.Fabre T, Kared H, Friedman SL, Shoukry NH. IL-17A enhances the expression of profibrotic genes through upregulation of the TGF-β receptor on hepatic stellate cells in a JNK-dependent manner. J Immunol. 2014;193:3925–3933. doi: 10.4049/jimmunol.1400861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mann J, Mann DA. Transcriptional regulation of hepatic stellate cells. Adv Drug Deliv Rev. 2009;61:497–512. doi: 10.1016/j.addr.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 7.Marra F, Caligiuri A. Cytokine production and signaling in stellate cells. In: Gandhi CR, Pinzani M, editors. Stellate cells in health and disease. Elsevier Press; 2015. pp. 63–86. [Google Scholar]
- 8.Granzow M, Schierwagen R, Klein S, Kowallick B, Huss S, Linhart M, et al. Angiotensin-II type 1 receptor-mediated Janus kinase 2 activation induces liver fibrosis. Hepatology. 2014;60:334–348. doi: 10.1002/hep.27117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 10.Harvey SAK, Dangi A, Tandon A, Gandhi CR. The transcriptomic response of rat hepatic stellate cells to endotoxin: Implications for hepatic inflammation and immune regulation. PLoS One. 2013;8:e82159. doi: 10.1371/journal.pone.0082159. [DOI] [PMC free article] [PubMed] [Google Scholar]