

Abstract
Pharmacological probes for the melanocortin receptors have been utilized for studying various disease states including cancer, sexual function disorders, Alzheimer's disease, social disorders, cachexia, and obesity. This study focused on the design and synthesis of bivalent ligands to target melanocortin receptor homodimers. Lead ligands increased binding affinity by 14- to 25-fold and increased cAMP signaling potency by 3- to 5-fold compared to their monovalent counterparts. Unexpectedly, different bivalent ligands showed preferences for particular melanocortin receptor subtypes depending on the linker that connected the binding scaffolds suggesting structural differences between the various dimer subtypes. Homobivalent compound 12 (CJL-1-140) possessed a functional profile that was unique from its monovalent counterparts providing evidence of the discrete effects of bivalent ligands. Lead compound 7 (CJL-1-87) significantly decreased feeding in mice after intracerebroventricular administration. To the best of our knowledge, this is the first report of a melanocortin bivalent ligand's in vivo physiological effects.
Keywords: Melanotropin, peptide, GPCR dimers, homobivalent, homodimer, obesity, feeding
Introduction
The melanocortin receptor system is involved in various physiological functions including pigmentation,1-2 sexual behavior,3 blood pressure modulation,4 memory,5-7 and energy homeostasis.8-11 The system contains five Gαs protein-coupled receptor subtypes (MC1-5R) that stimulate the cAMP signal transduction pathway upon agonist binding.1, 12-17 Ligands targeting the melanocortin G protein-coupled receptors (GPCRs) have been utilized as probes or investigated as potential therapeutics for Alzheimer's disease,18-20 cancer targeting,21-25 sexual function,3, 26 social disorders,27-28 cachexia,29-33 and obesity.8, 34-36 Due to the wide range of pharmacological effects, the development of ligands with new scaffolds, unique functional properties, or more selective profiles are needed as in vitro and in vivo probes for the various melanocortin dependent functions.
Bivalent ligands have been shown to offer access to properties and pharmacological profiles which are unique from classic monovalent ligands. The growing acceptance of GPCR dimers as pharmacological targets has fostered the development of bivalent ligands to target them. There have been several reports establishing that all known subtypes of melanocortin receptors form homodimers.37-43 Competitive binding studies suggested that melanocortin receptors have two tandem binding sites, each with different binding properties which may indicate targetable homodimers.44-45 Bivalent ligands offer a potential avenue to target melanocortin GPCR dimers and investigate their functional effects both in vitro and in vivo. Since Portoghese and coworkers pioneered bivalent ligands targeting GPCRs,46 bivalent ligands have been developed for various GPCR systems including the opioid,46-50 serotonin,51-53 adenosine,54 cannabinoid,55-56 chemokine,57 dopamine,58 and melanocortin receptors.25, 59-69 Bivalent ligands have been demonstrated to have a variety of different pharmacological effects as compared to their monovalent counterparts including: increasing or decreasing binding affinity,52, 58, 64 positively or negatively changing functional responses,53-55, 59, 70 altering receptor subtype selectivity,47, 58 changing receptor trafficking,71-73 and creating tissue selectivity.50, 74 Due to these unique characteristics, bivalent ligands offer distinct advantages over the classical monomeric approach.
Employing the use of bivalent ligands has also been shown as a feasible route to avoid the undesirable side effects exhibited by classic monovalent ligands.49, 73-75 A previous study of heterobivalent ligands targeting the δ and μ opioid receptor heterodimers resulted in a ligand with 50-fold higher opioid agonist potency, but devoid of tolerance commonly seen with monovalent opioid ligands.49 Bivalent melanocortin ligands may, therefore, be able to circumvent undesirable side effects seen with classic monovalent ligands. For example, a melanocortin bivalent agonist ligand may be able to have reduced effects on erectile function,3 sexual behavior,3 and blood pressure4 while maintaining the desirable effect of decreasing food intake for an anti-obesity drug.8-10, 34, 36
Melanocortin bivalent ligands with various designs and linkers have resulted in increased binding affinity for human (h)MC4R expressed in HEK293 cells.60-69 There has not previously been a report of bivalent ligands' pharmacology using the cloned MC1R, MC3R, and MC5R cell lines. Studying bivalent ligands' effects at each receptor subtype is important for understanding ligand selectivity and for transitioning molecules to more complicated whole animal models that are expressing multiple receptor isoforms. Furthermore, there has been no investigation of bivalent ligands' effects on the cloned mouse receptors. Results obtained from the cloned mouse receptors would be advantageous to inform the use of melanocortin bivalent ligands in the developed mouse models8-11 and represent an important translational step in the development of bivalent ligands as pharmacological probes.
Reports of melanocortin bivalent ligand's effects on functional activity have been limited. The initial functional activity study showed effects on frog-melanophore cells and used an unoptimized linker.59 In this system, an agonist bivalent ligand increased agonist signal. They also reported a bivalent ligand based on an antagonist monovalent pharmacophore that interestingly became a full agonist at high concentrations.59 Another report described bivalent ligands having increased potency in cAMP accumulation assays in HEK293 cells expressing the hMC4R.25 To the best of our knowledge, these are the only two functional studies of bivalent ligands at the melanocortin receptors reported to date.25, 59 While these preliminary studies with frog melanophores and hMC4R cell lines illustrate the uniqueness and utility of melanocortin bivalent ligands, further functional studies at other receptor subtypes would help advance melanocortin bivalent ligands as potentially selective and potent pharmacological probes.
Given the lack of studies reporting bivalent ligands' effects at different melanocortin receptor subtypes, there has been little understanding of how different linkers and design strategies affect binding and functional selectivity between the different melanocortin receptor isoforms. One objective of this study was to investigate receptor subtype preference patterns by screening in parallel the different melanocortin receptors (the MC2R was excluded since it is reported to only be stimulated by ACTH)1 with bivalent ligands using different design strategies. Furthermore, in order for the bivalent ligands to be suitable in vivo functional probes for mouse studies, their effects must be characterized at the mouse melanocortin receptors otherwise interpretation of in vivo mouse studies would be confounding. The current study reports the design and synthesis of a library of agonist, partial agonist, and antagonist melanocortin homobivalent ligands which underwent in vitro binding and functional evaluation at the mouse (m)MC1R, mMC3R, mMC4R, and mMC5R subtypes. It also gives, to the best of our knowledge, the first in vivo functional evaluation of a melanocortin bivalent ligand.
Results and Discussion
Design
It is hypothesized that appropriately designed bivalent ligands could be used to target melanocortin receptor dimers, and that there may be differences in the receptor subtype homodimer pharmacological profiles. Our approach to target receptor homodimers was to create bivalent ligands comprised of two selected pharmacophore scaffolds connected with two different linkers (Figure 1). The previously reported tetrapeptides Ac-His-DPhe-Arg-Trp-NH2 76-77 and Ac-His-DNal(2′)-Arg-Trp-NH2 78 were selected as the scaffold templates to incorporate into the bivalent ligands. These tetrapeptides are based on His-Phe-Arg-Trp which is the minimal messaging sequence of the endogenous melanocortin hormones.76, 79-81 Truncation studies of the potent and enzymatically stable peptide NDP-MSH (Ac-Ser-Tyr-Ser-Nle-Glu-His-DPhe-Arg-Trp-Gly-Lys-Pro-Val-NH2) have previously shown the tetrapeptide Ac-His-DPhe-Arg-Trp-NH2 to be the most active fragment.76
Figure 1.
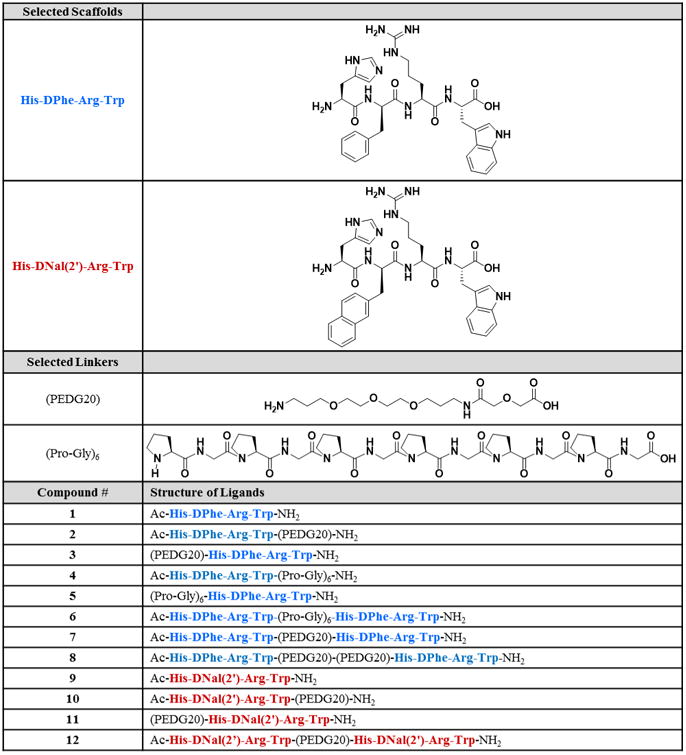
Design of ligands from selected scaffolds and linkers.
The Ac-His-DPhe-Arg-Trp-NH2 peptide was reported to have a high nanomolar to low micromolar binding affinity at the melanocortin receptors.77 Herein, it is postulated that the incorporation of the His-DPhe-Arg-Trp scaffold into bivalent ligands would retain the relatively potent agonist functional effects, but would have a lower binding affinity than if longer peptide scaffolds were incorporated. This is an important consideration in the design strategy presented, since bivalent ligands based off of low affinity scaffolds often allow easier detection of synergistic binding effects.60, 82-83 This allows for detection of larger increases in binding affinity which is characteristic of bivalent ligands targeting dimers.46, 63, 84 Incorporation of the tetrapeptide His-DPhe-Arg-Trp into bivalent ligands has already been reported to significantly increase binding at the hMC4R.64 The current design and experiments advance the field by examining the binding and functional effects of bivalent ligands based on this tetrapeptide with different linkers at the various melanocortin receptor subtypes. The previous report consisted of 14 atom, 19 atom, and 38 atom linkers separating the two His-DPhe-Arg-Trp scaffolds;64 the design herein consisted of 20 atom, 36 atoms, and 40 atom linkers connecting the same scaffolds. The small extensions in our design can significantly change activity, as bivalent ligands are quite sensitive to linker length.49, 74, 85-86 Single atom linker extensions previously resulted in noteworthy changes (>500-fold) in the in vivo potency in a series of bivalent ligands tested for antinociception.74 A two atom linker extension in a bivalent ligand previously increased potency by 1100-fold.86
In order to study the effects of antagonist and partial agonist based bivalent ligands, the DPhe in the agonist scaffold was substituted to a DNal(2′) to yield the His-DNal(2′)-Arg-Trp tetrapeptide scaffold that has previously been reported to be a mMC3R/mMC4R antagonist, with partial activity at the mMC3R, and full agonist activity at the mMC1R and mMC5R.78 To our knowledge, the binding affinity of this tetrapeptide has not been previously reported, but it was assumed that the binding affinity would be similar to Ac-His-DPhe-Arg-Trp-NH2 in that it would have a low enough affinity to detect synergistic binding modes when used in homobivalent ligands. Carrithers and Lerner reported that an antagonist monomer yielded an agonist bivalent ligand at high concentrations in a functional frog melanocyte dispersion assay.59 This result lead to the present hypotheses that bivalent ligands based on antagonist and partial agonist monomers may result in unique pharmacological profiles with general activity trends that could be exploited in future bivalent ligand design.
Both a polyethylene diamine diglycolyic acid (PEDG, but also referred to in the literature as PEGO or PEG2) and an alternating proline-glycine (Pro-Gly) linker system were selected for this study based upon previous work of Hruby and coworkers demonstrating these linker systems enhance binding affinity at the hMC4R.62-64, 87-88 It was hypothesized that different linker systems and lengths may have varying effects at the different receptor homodimer subtypes. The PEDG linker is flexible with good solubility. Both a 20 atom PEDG20 linker and a 40 atom PEDG20-PEDG20 linker have increased binding affinity at the hMC4R when joining seven residue analogs of the NDP-MSH scaffold.63 The Pro-Gly linker is a semi-rigid linker system with the Pro giving the linker rigidity and the Gly giving the linker flexibility. A linker system of 36 atoms based on six repeats of Pro-Gly has been shown to be an effective linker system for targeting the hMC4R.62-63, 88 The PEDG20, PEDG20-PEDG20, and (Pro-Gly)6 have previously been estimated to be 4-18 Å, 8-36 Å, and 10-20 Å, respectively.22, 63 The PEDG20 linker was selected for the Ac-His-DNal(2′)-Arg-Trp-NH2 series based on solubility, ease of synthesis, and preliminary functional results. Although the metabolic stability was not tested in the current study, it has previously been shown that adding polyethylene glycol to a peptide can increase metabolic stability.89-90 By incorporating the polyethylene glycol-like PEDG20 into our design, it may increase the likelihood of identifying a suitable in vivo probe.
Peptide synthesis
All compounds were synthesized on Rink-amide-MBHA resin using standard Fmoc-chemistry and solid phase synthesis methodology utilizing both a semi-automated synthesizer and a microwave synthesizer (Scheme 1).91-93 Similar to a strategy previously employed,62 the resin was split at various points in the synthesis to produce the desired linker controls. This strategy allowed the production of the desired control ligands (i.e. the tetrapeptides and the tetrapeptides with the linker attached) in route to the synthesis of the bivalent ligands. This strategy was also used to derivatize control ligands with the linker attached to the N-terminus for both DPhe and DNal(2′) compounds.
Scheme 1. Synthesis of bivalent ligands and control ligands.

Synthetic scheme for synthesis of bivalent ligands and control ligands. (A) The synthesis of N-terminal linker controls and bivalent ligands by microwave synthesis. (B) Synthesis of C-terminal linker controls using a semi-automated synthesizer. (a) 20 % piperidine in DMF (b) Fmoc-NH-AA-COOH or Fmoc-NH-PEDG20-COOH, HBTU, DIEA in DMF. Repeat (a) and (b) to achieve desired sequence. When Fmoc-NH-PEDG20-COOH was incorporated, reaction was allowed to proceed for an extra hour at room temperature. (c) 75% acetic anhydride/ 25% pyridine. (d) Cleavage with 91% TFA, 3% EDT, 3% TIS, 3% water for 1.5-3 hours.
Peptides were cleaved off of the resin using a mixture of triisopropylsilane, 1,2-ethanedithiol, and water in trifluoroacetic acid (TFA). A cleavage time of 3 hours caused significant degradation of 3. Interestingly, the major degradation product had the same retention time by analytical RP-HPLC in acetonitrile and aqueous TFA (0.1%) (Figure 2A), but clear peak separation could be seen by analytical RP-HPLC in methanol and aqueous TFA (0.1%) (Figure 2B) illustrating the importance of using two diverse solvent systems (e.g. acetonitrile and methanol) when assessing compound purity. Mass spectrometry revealed the mass of the desired product (mass of 961.6) and the mass of the impurity (mass of 685.4). The impurity peak possessed the same mass and had similar retention times as the parent tetrapeptide Ac-His-DPhe-Arg-Trp-NH2 that could indicate the degradation of the linker. Co-injection of purified 1 with crude sample of 3 from the 3 hour cleavage resulted in increased relative intensity of the impurity peak demonstrating the similar retention times of 1 and the impurity (Figure 2C). A shorter cleavage time of 1.5 hours resulted in minimal degradation products of 3 as seen by analytical HPLC in methanol and aqueous TFA (0.1%) (Figure 2D). A shorter cleavage time was also used for 11 and minimal degradation was observed. The remaining peptides reported were synthesized with little difficulty. All final ligands were purified to greater than 95% pure and their mass was confirmed by ESI-MS. Further details can be found in the experimental section.
Figure 2.
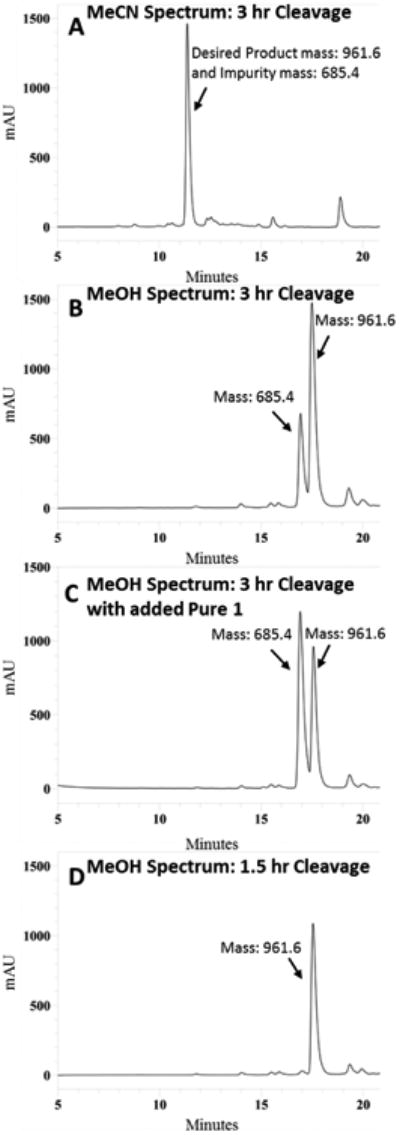
Crude RP-HPLC analytical chromatograms at 214 nm of 3 (mass of 961.6) in a gradient from 10% to 90% MeCN or MeOH in water containing 0.1 % trifluoroacetic acid at a flow rate of 1.5 mL/min over 35 minutes (5 to 20 minutes are shown) using an analytical Vydac C18 column (Vydac 218TP104). (A) Analytical HPLC trace in MeCN of crude peptide 3 after a three hour cleavage which shows only one major peak. A major impurity peak (mass of 685.4) is masked in this chromatogram. (B) Analytical HPLC trace in MeOH of crude peptide 3 after a three hour cleavage which identifies both the desired product and an impurity peak masked in MeCN chromatogram. (C) Co-injection of crude 3 from three hour cleavage with purified 1 (mass of 685.4) increases the intensity of the impurity peak demonstrating similar retention times. (D) A shorter cleavage time of 1.5 hours diminishes degradation product giving better crude peptide purity.
125I-NDP-MSH Competitive Binding Affinity Studies
The ligands' ability to competitively displace 125I-NDP-MSH was studied in HEK293 cells expressing the melanocortin receptors. The results are summarized in Table 1 and illustrated in Figure 3. The varying effects of the linker, pharmacophore, and receptor subtype are summarized below.
Figure 3.
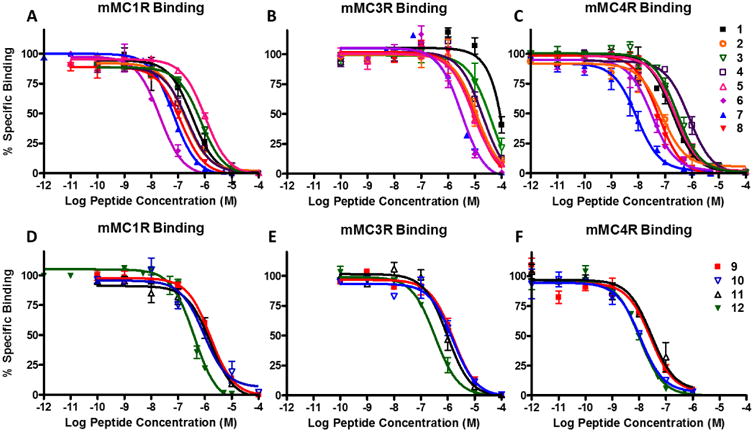
Illustrations of the competitive binding experiments at the mMC1R, mMC3R, and mMC4R. Top figures shows the
 based ligands. The bottom figures show the
based ligands. The bottom figures show the
 based ligands.
based ligands.
Linker effects
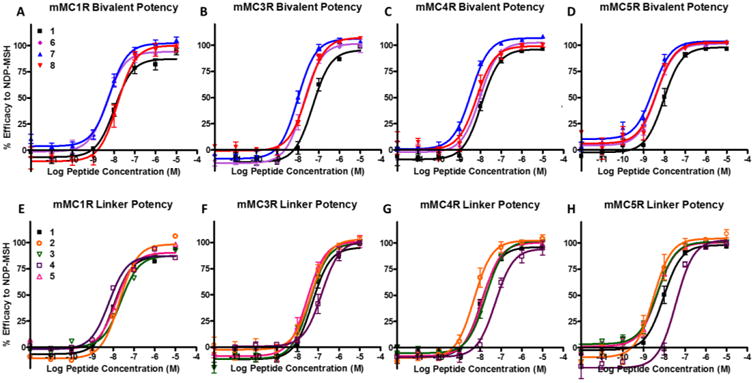
The addition of the linker to the monovalent tetrapeptide scaffold affected binding of the ligand depending on the type of linker, site of addition (N- or C-terminus), and receptor subtype. At the mMC1R, the linkers had minimal effects on binding (Figure 3A and D) and the difference between control ligand 1 or 9 and their corresponding linker control ligands are within experimental error. Changes less than 3-fold were considered to be within experimental error associated with the assay (in our hands). At the mMC3R, the addition of a linker to the tetrapeptides resulted in equal or increased binding affinity (Figure 3B and E). Most notably was the addition of the PEDG20 to the C-terminus and (Pro-Gly)6 to the N-terminus of the Ac-His-DPhe-Arg-Trp-NH2 scaffold in compounds 2 and 5. The addition of the linker resulted in increased mMC3R binding affinity of ca. 6- and 9-fold, respectively, as compared to its monovalent counterpart 1 (Table 1). At the mMC4R, the (Pro-Gly)6 linker reduced the binding affinity by 4-fold compared to 1 when added to the C-terminus in compound 4 (Figure 3C and F). In contrast, the PEDG20 linker when added to the C-terminus in 2 resulted in a 3-fold increased binding affinity compared to 1. All other linker control compounds resulted in less than 3-fold changes at the mMC4R compared to their monovalent counterpart.
Table 1. Summary of competitive binding experiments for compounds evaluated at the mouse melanocortin receptors.
| mMC1R | mMC3R | mMC4R | Selectivity | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (nM) | IC50 (nM) | IC50 (nM) | Ratios | ||||||||
| Compound Number | Compound Name | Mean ± SEM | n | Fold Diff. | Mean±SEM | n | Fold Diff.* | Mean±SEM | n | Fold Diff. | 1R:3R:4R |
| NDP-MSH | 0.31±0.08 | 6 | 4.18±0.61 | 14 | 1.09±0.12 | 19 | 1:13:4 | ||||
| 1 | 388±52 | 2 | 1 | 60% @100 μM | 2 | 1* | 214±55 | 6 | 1 | 1:374:1 | |
| 2 | CJL-5-35-4 | 178±4 | 2 | 2 | 13400±5100 | 2 | 6 | 83±14 | 4 | 3 | 2:162:1 |
| 3 | CJL-1-116 | 705±6 | 2 | 0.6 | 78% @100 μM | 3 | ND | 292±67 | 4 | 0.7 | 2:ND:1 |
| 4 | CJL-5-35-1 | 225±63 | 2 | 2 | 85% @100 μM | 2 | ND | 841±290 | 2 | 0.3 | 1:ND:4 |
| 5 | CJL-1-41 | 1090±110 | 3 | 0.4 | 8430±230 | 2 | 9 | 258±27 | 2 | 0.8 | 4:33:1 |
| 6 | CJL-1-31 | 27.5±2.2 | 2 | 14 | 3250±760 | 2 | 25 | 33±5.1 | 3 | 6 | 1:119:1 |
| 7 | CJL-1-87 | 68.6±5.3 | 3 | 6 | 3470±510 | 2 | 23 | 9.9±2.9 | 4 | 22 | 7:351:1 |
| 8 | CJL-5-72 | 131±18 | 2 | 3 | 10200±1300 | 2 | 8 | 54±10 | 2 | 4 | 2:190:1 |
|
| |||||||||||
| 9 | 1630±120 | 2 | 1 | 1430±190 | 2 | 1 | 26.0±3.4 | 2 | 1 | 63:55:1 | |
| 10 | CJL-5-35-5 | 997±190 | 2 | 2 | 1310±120 | 2 | 1 | 10.7±0.6 | 2 | 2 | 94:123:1 |
| 11 | CJL-1-132 | 1870±220 | 4 | 1 | 999±290 | 2 | 1 | 21.8±1.9 | 3 | 1 | 85:46:1 |
| 12 | CJL-1-140 | 430±40 | 3 | 4 | 350±80 | 5 | 4 | 10.4±0.03 | 2 | 2 | 41:34:1 |
The experimental compounds were used to displace 125I-NDP-MSH in a dose-response manner to calculate the IC50 values. % represent the amount of 125I-NDP-MSH signal reduction at 100 μM. The “n” column represents the number of independent experiments performed. The reported errors are the standard error of the mean (SEM). ND means not determined.
Fold difference for mMC3R was calculated based on an estimated IC50 of 80,000 nM for 1.
It is worth noting that there were changes in binding affinity when the linker was added to the C-terminus of the peptide, which has been seldom investigated when studying melanocortin bivalent ligands. The present SAR study demonstrated that the site of linker addition to either the C-terminus or N-terminus is an important consideration when designing bivalent ligands.
Bivalent Ligands
All His-DPhe-Arg-Trp based bivalent ligands had increased binding affinity (3- to 25-fold) compared to the parent tetrapeptide 1. The SAR of the His-DPhe-Arg-Trp based bivalent ligands at the different receptor subtypes was an intriguing finding. At the mMC1R, the most significantly enhanced compound was the (Pro-Gly)6 linked compound 6 (Figure 3A). Its binding affinity increased by 14-fold compared to monovalent ligand 1. The PEDG20 linked compounds 7 and 8 resulted in a 6- and 3-fold increased binding affinity, respectively. At the mMC3R, the (Pro-Gly)6 and PEDG20 linkers had the greatest effect (Figure 3B). It was observed that 6 and 7 possessed c.a. 25- and 23-fold increased binding affinity, respectively, compared to 1. Compound 8 resulted in an 8-fold increased binding affinity compared to 1. At the mMC4R, the PEDG20 linked bivalent ligand 7 increased binding affinity 22-fold as compared to the monovalent counterpart 1 (Figure 3C). Compounds 6 and 8 possessed 6- and 4-fold increased binding affinity, respectively, as compared to 1.
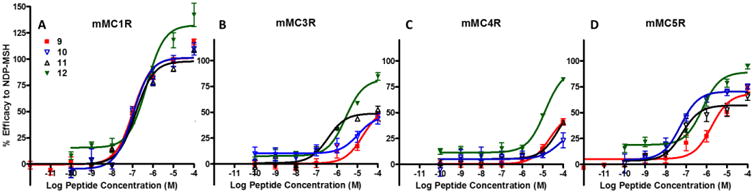
In the Ac-His-DNal(2′)-Arg-Trp-NH2 series, bivalent ligand 12 resulted in 4-fold increased binding affinity at both the mMC1R and mMC3R as compared to its monovalent counterpart 9 (Figure 3D and E). The binding affinity of compound 12 was within experimental error of the binding affinity of 9 at the mMC4R (Figure 3F). It was postulated that no increase in binding affinity was observed at the mMC4R because of the potent binding affinity of Ac-His-DNal(2′)-Arg-Trp-NH2 scaffold that potentially masked multivalent interactions.60, 82-83 The lower binding affinity of the Ac-His-DPhe-Arg-Trp-NH2 scaffolds allows easier detection of the enhancements in the binding affinity. As Kiessling and Lamanna explain, an “increase in apparent affinity of a multivalent display of middle-affinity epitopes quickly exceeds measurable binding constants and is indistinguishable from multivalent scaffolds of high-affinity ligands. In contrast, the increase in functional affinity between multivalent displays of weakly versus more weakly interacting epitopes falls within a range discernible by most biological systems.”83
The increased binding affinity (14- to 25-fold) of the lead bivalent ligands in the Ac-His-DPhe-Arg-Trp-NH2 based series support the hypothesis that these ligands are binding in a synergistic bivalent mode utilizing a second binding site. The second binding site could be either an auxiliary binding site on the same receptor or an orthosteric binding site on a neighboring receptor.83, 94 The use of Occam's razor directs us to the latter possibility since it has been observed that melanocortin receptors dimerize,37-43 and therefore, a neighboring orthosteric binding site should be readily accessible for synergistic binding versus an unknown auxiliary binding site for the His-DPhe-Arg-Trp pharmacophore.
In the proposed bivalent binding mode, the first binding interaction of one pharmacophore is postulated to tether the second pharmacophore to the receptor surface. If the linker has the correct properties (e.g. length, flexibility) to orientate the second pharmacophore into a tandem binding site (i.e. a GPCR dimer), the second binding interaction proceeds with lowered entropic cost (Figure 4).46, 94 Based on these results, the PEDG20-PEDG20 linker in compound 8 may be too long to tether the second pharmacophore in the correct location of the second binding site, and therefore, loses the entropic gains of the bivalent design reflected in the lower fold changes in binding affinity at all receptor subtypes.46
Figure 4.
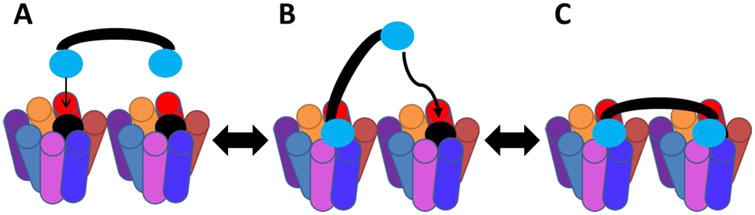
Proposed binding mode of the bivalent ligands. (A) First pharmacophore engages GPCR dimer or two neighboring binding sites. (B) The first binding event tethers the second pharmacophore in close proximity to the second binding site significantly increasing the likelihood of the second binding event. (C) The second pharmacophore binds with low entropic cost.
An interesting trend observed was the differential effects of the (Pro-Gly)6 and PEDG20 linkers at the melanocortin receptor subtypes. Compound 6 with the (Pro-Gly)6 linker resulted in the greatest fold increase in ligand binding affinity at the mMC1R (14-fold) and notable fold increase at the mMC3R (25-fold), but lower fold changes at the mMC4R (6-fold). Ligand 7 with the PEDG20 linker resulted in the greatest fold increase in binding affinity at the mMC4R (22-fold) and notable fold increase at the mMC3R (23-fold), but lower fold increase at the mMC1R (6-fold). In this study, it was identified that the mMC1R has a preference for the (Pro-Gly)6 linker, the mMC4R has preference for the PEDG20 linker, and the mMC3R bound well with both of the two linkers (Figure 5).
Figure 5.
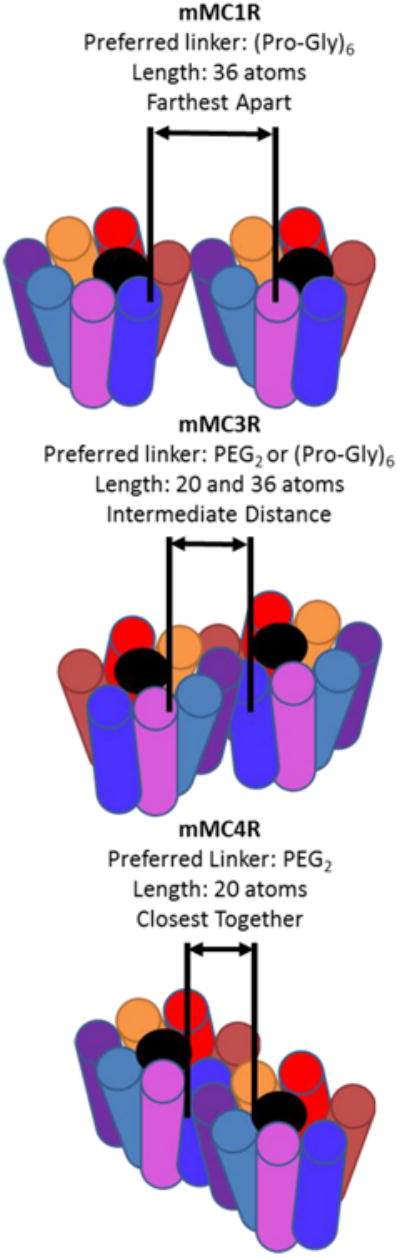
Postulated rationale for linker-dependent preferences at the different melanocortin homodimer subtypes. The different linker systems had varying effects on enhancing binding or functional responses depending on which receptor subtype was expressed. Since the linkers connect the same pharmacophore, it appears the difference are due to the linkers' physicochemical properties such as linker length. These differences suggest that there are differences between the various subtypes of melanocortin receptor dimers such as the distance between tandem binding sites (see text). The figure demonstrates how different distances between tandem binding sites would show preference for the different length linker systems.
The receptor subtype differences observed with compounds 6 and 7 are not due to the binding scaffold or the binding pocket since these remain constant when comparing the bivalent ligands to the monovalent counterparts at each receptor subtype. In addition, the tetrapeptide plus linker control ligands resulted in minimal increased binding affinity (<4-fold) at the mMC1R and mMC4R suggesting that the linker by itself is not the driving factor for the bivalent ligands increased activity. At the mMC3R, the linker control ligands did result in increased binding affinity (≤ 9-fold), but their affinities were lower than the affinities of the lead bivalent ligands and are likely not the primary driving factor for the increased mMC3R binding affinities of the bivalent ligands. Instead the bivalent ligand-receptor differences are conjectured to be due to differences in the physiochemical nature of the linker (e.g. flexibility, length, ect.) and how these may change the presentation of the pharmacophore to the tandem binding site.
Based upon these results it can be postulated that there are differences how the tandem binding sites of different melanocortin homodimer subtypes present themselves. For example, if the mMC1R homodimer had more distance between the two binding sites than a mMC4R homodimer, the (Pro-Gly)6 linker (36 atoms, ∼8-36 Å22, 63), that is hypothesized to be longer based on atom length than the PEDG20 linker (20 atoms, ∼4-18 Å22, 63), would favor the mMC1R homodimer and the PEDG20 would favor the mMC4R. To be consistent with the data, the mMC3R homodimer would have an intermediate distance between their two binding sites compared to the mMC1R and mMC4R homodimers, and therefore would show enhanced binding with both linker systems (Figure 5). It should be noted that the flexibility of the linkers makes prediction of their exact lengths in solution difficult, and this is just a hypothesis to explain the trends observed.
This idealized situation only accounts for the distance between tandemly arranged binding pockets and the linker's length. Other factors including the linker's other physiochemical properties, the two pharmacophores' orientations, and the binding pockets' accessibility may play a role in the binding affinity preferences observed. Nevertheless, these data suggest bivalent ligands could be exploited to achieve selectivity between the different melanocortin homodimers. This is, to our knowledge, the first indication of ligand preference patterns (albeit not selectivity) between the melanocortin homodimer-subtypes; however this phenomenon has been observed in several other bivalent ligand systems targeting GPCR systems.47, 58 For example, Kuhhorn and coworkers previously observed different linker systems connecting bivalent ligands resulted in varying dopamine receptor subtype specificity.58 In addition, Portoghese and coworkers synthesized bivalent ligands with different selectivity profiles for the μ, κ, and δ opioid receptors based on single glycyl unit linker extensions.47 Given these three examples of differential binding of bivalent ligands, it suggests that ligand selectivity between different receptor homodimer subtypes, as opposed to monomer orthosteric selectivity, may be a general phenomenon among GPCRs. However, more investigation into melanocortin receptor homodimerization (or higher-order oligomerization) will be necessary. The current study reports foundational work and results in novel chemical probes for future studies for both melanocortin GPCR homodimers and heterodimers.
Functional cAMP Accumulation Studies
The AlphaScreen® cAMP Assay Technology was utilized to examine the ligands ability to stimulate intracellular cAMP signaling in live HEK293 cells stably expressing the mMC1R, mMC3R, mMC4R, and mMC5R. Compounds which did not produce full activation at 100 μM (compared to maximal NDP-MSH signal) were analyzed for antagonist properties via a Schild analysis at the mMC3R and mMC4R.95 The results of the studies are summarized in Tables 2 and 3 as well as illustrated in Figures 6 and 7.
Table 2. Summary of functional experiments for
based compounds evaluated at the mouse melanocortin receptors.
| mMC1R | mMC3R | mMC4R | mMC5R | Selectivity | |||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | Agonist EC50 (nM) | Agonist EC50 (nM) | Agonist EC50 (nM) | Agonist EC50 (nM) | Ratios | ||||
| Mean±SEM | Fold Diff. | Mean±SEM | Fold Diff. | Mean±SEM | Fold Diff. | Mean±SEM | Fold Diff. | 1R: 3R:4R:5R | |
| NDP-MSH | 0.03±0.01* | 0.24±0.01* | 0.46±0.04* | 0.31±0.03* | 1:9:18:12 | ||||
| α-MSH | 0.15±0.05 | 0.76±0.05 | 4.0±0.9 | 0.59±0.03 | 1:5:28:4 | ||||
| Ɣ-MSH | 1090±300 | 34.6±4.0 | 869±66 | 35.1±18.7 | 31:1:25:1 | ||||
| 1 | 14.1±2.6* | 1 | 55.5±12.2* | 1 | 13.7±1.9* | 1 | 9.8±2.7* | 1 | 1:4:1:1 |
| 2 | 26.2±9.9 | 0.5 | 47.4±12.7 | 1 | 5.7±2.9 | 2 | 2.1±0.3 | 5 | 13:23:3:1 |
| 3 | 24.9±5.9* | 0.6 | 30.9±7.5* | 2 | 18.5±2.9* | 0.7 | 3.9±1.3* | 2 | 6:8:5:1 |
| 4 | 5.7±.7 | 2 | 107±66 | 0.5 | 74.1±14.1 | 0.2 | 52.4±10.3 | 0.2 | 1:19:13:9 |
| 5 | 16.4±4.7* | 0.9 | 27.2±5.2* | 2 | 14.5±2.7* | 0.9 | 4.9±1.2* | 2 | 3:6:3:1 |
| 6 | 4.4±0.6* | 3 | 20.2±4.0* | 3 | 9.2±1.0* | 1 | 3.8±0.9* | 2 | 1:5:2:1 |
| 7 | 6.4±1.5* | 2 | 10.1±2.5* | 5 | 3.6±0.5* | 4 | 3.1±0.6* | 3 | 1:3:1:1 |
| 8 | 12.6±3.8 | 1 | 22.7±3.7 | 2 | 7.3±2.1 | 2 | 4.0±0.2 | 2 | 3:6:2:1 |
AlphaScreen® assays were performed to determine relative potency of compounds to induce cAMP signaling. The reported errors are the standard error of the mean (SEM) determined from at least three independent experiments. Changes less than 3-fold were considered to be within the inherent experimental assay error.
denotes six or more independent experiments.
Table 3. Summary of functional experiments for
based compounds evaluated at the mouse melanocortin receptors.
| mMC1R | mMC3R | mMC4R | mMC5R | ||||
|---|---|---|---|---|---|---|---|
| Compound | Agonist (EC50) | Agonist (EC50) | Antagonist (pA2) | Agonist (EC50) | Antagonist (pA2) | Agonist (EC50) | |
| Mean±SEM | Fold | Mean±SEM | Mean±SEM | Mean±SEM | Mean±SEM | Mean±SEM | |
| 9 | 98.4±32.2* | 1 | 45% at 100 μM | 6.04±0.09 | 40% at 100 μM | 8.09±0.04 | 75% at 100 μM |
| 10 | 112±19 | 0.9 | 40% at 100 μM* | 6.14±0.06 | 20% at 100 μM* | 8.39±0.08 | PA, 70% at 100 μM |
| 11 | 139±17* | 0.7 | PA, 50% at 100 μM* | 6.07±0.18 | 40% at 100 μM* | 7.97±0.29 | PA, 65% at 100 μM* |
| 12 | 563±142* | 0.2 | 80% at 100 μM* | 6.08±0.11 | 85% at 100 μM* | 7.68±0.37 | 786±185* |
AlphaScreen® assays were performed to determine relative potency of compounds to induce cAMP signaling. The reported errors are the standard error of the mean (SEM) of at least three independent experiments. Antagonist activity was evaluated at only the mMC3R and mMC4R with all compounds based on the His-DNal(2′)-Arg-Trp pharmacophore. The pA2 values were calculated by a Schild analysis in which NDP-MSH was in a standard dose response (10-12 to 10-6) and three doses of antagonist were used to shift the agonist dose response. PA indicates partial agonist activity was observed. Percentage (%) indicate the amount of activity relative to maximal NDP-MSH response was observed at 100 μM.
denotes six or more independent experiments. A bar graph representation of the partial functional responses and the associated error can be seen in the supplemental information.
Figure 6.
Illustrations of the in vitro functional pharmacology at the mMC1R, mMC3R, mMC4R, and mMC5R of the
based ligands. Top figures show the bivalent ligands compared to the control peptide 1. The bottom figures show the effects of the linkers plus pharmacophore compared to control peptide 1.
Figure 7.
Illustrations of the in vitro functional agonist pharmacology at the mMC1R, mMC3R, mMC4R, and mMC5R of the
based ligands.
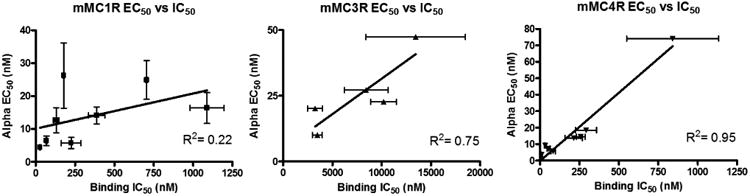
As anticipated, compounds with greater binding affinity tended to have greater functional potency. However, plotting the EC50 values versus IC50 values of His-DPhe-Arg-Trp ligands suggested that binding affinity does not correlate linearly to function at the mMC1R and mMC3R with R2 values of 0.22 and 0.75, respectively (Figure 8). At the mMC4R, it does appear to correlate linearly (R2 value of 0.95) such that a ligand's EC50 potency is approximately 10-fold more potent compared to its IC50 binding affinity. Although some of the poor correlation could be due to inherent experimental error, these data show that binding affinity and functional potency do not necessarily correlate within receptor isoforms highlighting the importance of studying both a ligand's binding affinity and functional potency in complementary assays. A more detailed description of the functional activity with the linker control compounds and the bivalent compounds follows.
Figure 8.
Correlation of IC50 (nM) vs EC50 (nM) at the different receptor subtypes for
based ligands. The mMC4R had a relatively linear correlation between receptor activation and ligand binding. At the mMC1R there appears to be relatively little correlation. The lack of correlation stresses the importance of studying ligands' binding affinity and functional effects in complementary assays. Data is shown as mean ± SEM.
Linker effects
There were three situations in which the addition of a linker to the tetrapeptide scaffold resulted in a noteworthy changes in agonist activity. Changes less than 3-fold were considered to be within the intrinsic experimental error associated with this functional assay (in our hands). The attachment of the (Pro-Gly)6 linker to the C-terminus in 4 decreased the potency of the Ac-His-DPhe-Arg-Trp-NH2 by 5-fold at the mMC4R and mMC5R (Table 2, Figure 6G and H ). In contrast, at the mMC5R the attachment of PEDG20 to the C-terminus in 2 resulted in 5-fold increased functional potency (Table 2, Figure 6H). These data indicated that the C-terminus of His-DPhe-Arg-Trp is amendable to changes, but sensitive to modifications. These findings reinforced the importance of including C-terminal linker controls in bivalent ligand studies.
Bivalent ligands
At the mMC1R, the bivalent ligand 6, based off of the His-DPhe-Arg-Trp scaffold, resulted in 3-fold increased agonist potency as compared to its monovalent counterpart 1 (Figure 6A). At the mMC3R, ligands 7 and 6 (that possessed 23- and 25-fold increased binding affinity) resulted in 5- and 3-fold increased agonist potency, respectively, as compared to 1 (Figure 6B). At the mMC4R, bivalent ligand 7 resulted in 4-fold increased potency as compared to the parent tetrapeptide 1 (Figure 6C). At the mMC5R, bivalent ligand 7 resulted in a 3-fold increased potency as compared to the monovalent control 1 (Figure 6D). However, this data was confounded by the 5-fold increased potency of linker control compound 2 compared to 1 (Figure 6H). It is therefore hard to interpret whether increased potency at this receptor subtype is due to the addition of the linker or because of the bivalent design.
At the mMC1R, bivalent ligand 12, derived from the His-DNal(2′)-Arg-Trp tetrapeptide, decreased agonist potency 5-fold as compared to the monovalent counterpart 9 (Figure 7A). This is the only bivalent ligand that displayed a decreased potency; an unanticipated result since the binding affinity of this ligand was increased as compared to its monovalent counterpart 9. At the mMC3R, ligand 12 increased receptor efficacy relative to NDP-MSH, resulting in an 80% maximal signal at 100 μM compared to monovalent ligand's 45% signal (Figure 7B). The ligand was analyzed via a Schild analysis for antagonist activity but showed no change in antagonist potency as compared to the monovalent ligand 9 (Table 3). At the mMC4R, bivalent ligand 12 showed increased receptor efficacy showing 85% maximal signal at 100 μM relative to NDP-MSH compared 40% maximal signal by the monovalent ligand 9 (Figure 7C). The ligand was also analyzed by a Schild analysis, but showed minimal change in antagonist potency compared to the monovalent ligand 9 (Table 3). At the mMC5R, ligand 12 displayed full agonist pharmacology (EC50 = 790 nM) and increased receptor efficacy as compared to 9 that showed 75% agonist signal at 100 μM relative to NDP-MSH (Figure 7D).
An interesting pattern with the bivalent ligands based on the His-DNal(2′)-Arg-Trp scaffold was observed: compound 12 increased efficacy at the mMC3R, mMC4R, and mMC5R as compared to the monovalent control ligand 9. This trend observed of a monovalent scaffold that possessed relatively low agonist efficacy and potent antagonism being converted to a bivalent ligand showing increased agonist efficacy at high concentrations was previously seen by Carrithers and Lerner.59 They observed this trend in a frog skin melanocyte dispersion assay with a bivalent ligand that consisted of the nonapeptide antagonist scaffold Met-Pro-DPhe-Arg-DTrp-Phe-Lys-Pro-Val96 linked with a polylysine linker.59 The current study extends this finding by showing a similar trend at the individually cloned mMC3R, mMC4R, and mMC5R subtypes. Interestingly, bivalent ligand 12 also showed decreased agonist potency at the mMC1R despite an increased binding affinity. The ligand had a receptor functional profile different than the original monovalent ligand 9 in which its agonist potency increased at the mMC3R, mMC4R, and mMC5R yet decreased at the mMC1R. Since the region of the ligand which purportedly binds the receptor is not changing from the monovalent ligand, the pharmacology of 12 is a result of joining the two scaffolds.
This SAR is unique from classical monovalent ligand SAR and demonstrates that bivalent ligands can create unique pharmacologies. The conversion observed at the mMC3R and mMC4R of monovalent scaffold with relatively low agonist efficacy and potent antagonism to a bivalent ligand with increased agonist efficacy, yet similar antagonism, may be just one potential functional consequence of bivalent compounds targeting the melanocortin receptors. Furthermore, the observation that 12 possessed increased binding affinity at the mMC1R but decreased functional potency once again emphasizes the importance of studying both the binding and function of bivalent ligands.
It should be noted that the fold increases observed in functional potency (3- to 5-fold) were not as pronounced as the fold increases observed in binding affinity (14- to 25-fold). There are several possibilities for why the increased binding affinities did not translate to larger increases in functional potency. First, similar to discussion above about the smaller fold increases observed with Ac-His-DNal(2′)-Arg-Trp-NH2 based compounds' binding affinity, the Ac-His-DPhe-Arg-Trp-NH2 scaffold's potent nanomolar agonist efficacy could be masking functional increases. Second, binding affinity is a molecular recognition event whereas functional potency is a signaling transduction event and is dependent on a conformational change of the receptor. The tethered bivalent ligands could cause a change in the orientation of the second pharmacophore in the second binding pocket in which it can still bind to the receptor, but it does not activate the cAMP signal transduction pathway as effectively. Third, the bivalent ligand could be binding in an auxiliary binding site that has minimal functional effects. Fourth, the lower fold changes in functional potency could be a result of asymmetric signaling of the two receptors present in the dimer. It could be postulated that the first binding event activates cAMP signal transduction through the first receptor, but the second binding event does not activate the cAMP pathway. The second binding could result in no conformational change of the second receptor, or a different conformational change that results in biased signaling through a different pathway (e.g. β-arrestin recruitment). This would result in a lack of increase in functional activity in spite of increased bivalent ligand binding. This type of asymmetry in GPCR dimers was previously observed by Han and coworkers.97 They observed that an agonist binding a single dopamine D2 receptor resulted in maximal functional activation, while an agonist binding the second receptor in the GPRC dimer blunted signaling.97 Further investigation into these possible mechanisms is necessary.
In Vivo ICV Administration Studies
Although there have been reports of melanocortin bivalent ligands as in vivo imaging tools,98-100 to the best of our knowledge, the in vivo functional effects of bivalent ligands have not been reported. Given the unique characteristics of bivalent ligands compared to their monovalent counterparts that were not explicitly tested (e.g. altering receptor trafficking71-73 and creating tissue selectivity50), it is important to demonstrate that the in vitro pharmacology translated to the in vivo pharmacology.
In order to better understand the functional effects of melanocortin bivalent ligands and their potential as in vivo probes, the intracerebroventricular (ICV) administration of bivalent ligand 7 was performed in mice. The central administration of melanocortin ligands has previously been used to study their effect on the centrally located MC3R and MC4R. Specifically, ICV administration of melanocortin agonists has been reported to decrease food intake and antagonists to increase food intake.8, 36, 101 Compound 7 was selected for study since it showed the greatest potency and binding affinity at the mMC3R and mMC4R.
Treatment of mice with 7 was well tolerated and resulted in a dose dependent decrease in food intake after ICV administration as anticipated for an agonist compound (Figure 9, Supp. Info. Figure 1). A significant decrease in food intake was observed in male mice 2, 4, 6, and 8 hours after 5 nmols of 7 was administered (Figure 9). No significant effect in male mice was seen for later time points (24-72 hours) (Supp. Info. Figure 1C). No significant difference were observed in female mice at 2, 4, 6, and 8 hour time points (Supp. Info. Figure 9B). A significant decrease in food intake was observed 24 hours post-treatment in female mice (p<0.05), but no significant effect was observed at the 48 and 72 hour time points (Supp. Info. Figure 1D). No significant effect on body weight was observed in either female or male mice.
Figure 9.
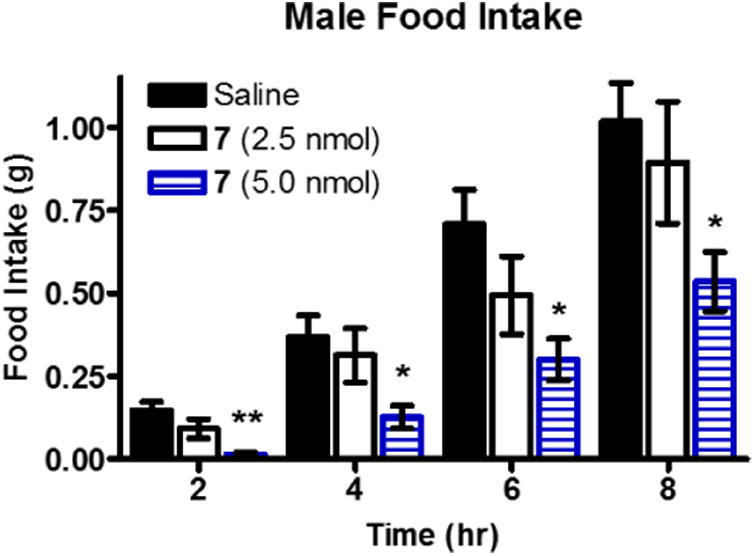
Cumulative food intake following intracerebroventricular administration of either saline (n=16) or 7 in saline (n=8) in male wild type mice. Data is shown as mean ± SEM. Data was analyzed using the PRISM program (v4.0; GraphPad Inc.) by a one-way ANOVA followed by a Bonferroni post test in order to compare individual doses to saline administration. *p<0.05, ** p=0.01.
These data are consistent with the in vitro data that 7 acts as a melanocortin agonist at the centrally expressed melanocortin receptors. The monovalent counterpart, Ac-His-DPhe-Arg-Trp-NH2, was previously shown to decrease food intake 3, 4, 5, 6, 16, and 24 hours after administration of a 2 nmol dose.36 An interesting observation between the two studies is the longer lasting effect previously reported with Ac-His-DPhe-Arg-Trp-NH2 at 24 hours which was not observed with 7 in male mice in the current study. This may indicate a difference in the physiological effects of the compound in vivo and may be due to any number of reasons including increased receptor desensitization, increased compensation pathways, changes in hormone signaling, or increased metabolic degradation of the compound. Additional experiments would need to be performed to establish an explanation for the differences observed.
The previous study showed that Ac-His-DPhe-Arg-Trp-NH2 at the 2.0 nmol dose had similar results to 7 at the 5 nmol dose at 4 and 6 hours after administration. These results would indicate that the in vitro functional results (as opposed to binding results) are more indicative of the in vivo effects in the current nocturnal satiated feeding paradigm. However, there are key differences between the current study and the previous study that should be mentioned. Firstly, the food intake after saline administration in the previous study was significantly lower than in the current study at the 6 and 24 hour time points (p<0.05). Secondly, different mouse chow was used. The previous study utilized Harlan Teklad 8604 containing 4% fat and 3.30 kcal/g digestible energy, whereas, Harlan Teklad 2018 containing 6.2 % fat and 3.1 kcal/g digestible energy was used in the current study. This difference in chow may have been responsible for the differences in saline food intake, since a mouse eating Harlan Teklad 8604 would need to consume less chow to achieve the same caloric intake. There were also varying environment factors including the animal facilities, the lab staff, frequency of measurements, and type of nesting material. Subtle changes in experimental conditions have been shown to result in differences in animal behavior.102-105 Given these factors, a direct head to head comparison study of the monovalent 1 and the bivalent ligand 7 would be necessary to draw more definitive conclusions.
The current report advances the field by indicating that melanocortin bivalent ligands are suitable to probe for melanocortin effects in vivo. In the current study, there does not appear to be a dramatic advantage between monovalent ligand 1 and bivalent ligand 7. However, only one experimental paradigm of food intake was evaluated. Since bivalent ligands show increased binding affinity compared to their monovalent counterparts, they may be beneficial in conditions that the ligand is competing with the natural antagonist AGRP for binding such as in a fasting state. The bivalent ligands developed currently may also be exploited as imaging tools as described previously,21-22, 98-100 but for the centrally located receptors. This would be especially useful if bivalent ligands featuring selective scaffolds for different melanocortin receptor subtypes were used for imaging the isoforms' locations in the brain. Bivalent ligands are also uniquely poised to study melanocortin receptor dimerization in vivo. The use of 7 to decrease food intake in mice demonstrates its utility as a probe for metabolic diseases such as obesity. Additionally, the current study's finding that melanocortin bivalent are well tolerated and functionally active in vivo indicates that they will also be useful probes for other disease states in which the melanocortin system plays a role including Alzheimer's disease,18-19 sexual function,3, 26 and social disorders.27-28
Conclusion
Melanocortin bivalent ligands can be exploited to increase receptor binding affinity and functional potency. This report identified a preference of the receptor subtypes for different bivalent ligand linkers indicating differences in the homodimer subtypes. Compound 7 resulted in significant decreased feeding in vivo upon ICV administration which was consistent with its agonist in vitro pharmacology. This foundational work can be applied to the various fields in which melanocortin ligands are currently under investigation as pharmacological probes and potential therapeutics. Specifically, our in vivo results indicate bivalent ligands' utility in studying melanocortin-dependent metabolic disease states. It also serves as a foundation for the development of melanocortin bivalent ligands as functional pharmacological probes for melanocortin receptor homodimers and heterodimers.
Experimental
Peptide Synthesis
Peptides were synthesized using standard fluorenyl-9-methoxycarbonyl (Fmoc) methodology to protect the elongating peptide chain.92 Couplings were performed in a CEM Discover SPS microwave peptide synthesizer except for the last two residues of 2, 4, and 10 which were performed on a semi-automated synthesizer (LabTech,Louisville, KY). The 4-(2′,4′-dimethoxyphenyl-Fmoc-aminomethyl)phenoxyacetyl-MBHA resin[Rink-amide-MBHA (200-400 mesh), 0.35-0.37 meq/g substitution], 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), and Fmoc-protected amino acids[Fmoc-Pro, Fmoc-Gly, Fmoc-His(Trt), Fmoc-DPhe, Fmoc-Arg(Pbf), Fmoc-Trp(Boc), andFmoc-DNal(2′)] were purchased from Peptides International (Louisville, KY, USA). The O-(N-Fmoc-3-aminopropyl)-O'-(N-diglycolyl-3-aminopropyl)-diethyleneglycol [Fmoc-NH-(PEG)2-COOH (20atoms) or Fmoc-NH2-PEDG20-COOH] was purchased from Novobiochem® EMDMillipore Corp (Billerica, MA, USA). The N,N-diisopropylethylamine (DIEA), triisopropylsilane (TIS), 1,2-ethanedithiol (EDT), piperidine, pyridine, and trifluoroacetic acid (TFA) were purchased from Sigma-Aldrich (St. Louis, MO). Acetonitrile (MeCN), N,N-dimethylformamide (DMF), acetic anhydride, dichloromethane (DCM), and methanol (MeOH) were purchased from FischerScientific. All reagents were ACS grade or better and were used without further purification.
Peptides were assembled on the Rink-amide-MBHA resin in a fritted polypropylene reaction vessel (25 mL CEM reaction vessel). After resin was swelled in DCM for at least one hour, it was continually mixed by bubbling nitrogen. From here a two-step cycle of deprotection with 20% piperidine in DMF, then amide coupling with the Fmoc-amino acid, HBTU, and DIEA was repeated until the final peptide was synthesized on resin. All deprotection or coupling reagents were removed by 3-5 washes of DMF between steps. A Kaiser/ninhydrin test was utilized after each deprotection or coupling step to indicated the presence or lack of a free primary amine.106 For Pro residues, the presence or lack of a free secondary amine was indicated by a chloranil test.93, 107 Deprotection was achieved in a twostep process. An initial two minute deprotection was performed outside of the microwave. The deprotection solution was removed by vacuum. A second aliquot of 20% piperidine was added and further deprotection was assisted by microwave heating (75°C, 30 W, 4 min).
Microwave assisted amide coupling was achieved by addition of 3.1-fold excess Fmoc-protected amino acids (5.1-fold for Arg) and 3-fold excess of HBTU (5-fold for Arg) dissolved in DMF added to the deprotected elongating peptide on the resin. The coupling reaction was initiated via addition of 5-fold excess of DIEA (7-fold for Arg) and the reaction was heated in the microwave synthesizer (75°C, 30 W or 50°C, 30 W for His) for five minutes (10 min for Arg). Fmoc-NH-(PEDG20)-COOH was incorporated into the peptide using the standard microwave protocol except it was allowed to cool for at least one hour post coupling to ensure the reaction went to completion.
The semi-automated synthesizer couplings were achieved by splitting dry resin with the Fmoc protected elongating peptide chain (∼0.2 nmols) into a 16-well Teflon reaction block. The resin and peptide swelled for two hours in DCM, and then were deprotected by addition of 20% piperidine in DMF for two minutes. The deprotection mixture was removed, then a second aliquot of 20% piperidine was added and mixed for 18 minutes. Following a positive Kaiser test, the coupling was initiated by addition of reagents as described above. The reaction was then mixed for at least two hours at room temperature.
All N-terminal acetylated peptides were acetylated on resin after the final Fmoc deprotection by addition of 3:1 mixture of acetic anhydride to pyridine and were mixed with bubbling nitrogen for 30 minutes at room temperature. After syntheses were completed, all peptides were washed with DCM at least 3 times and dried overnight in a desiccator. Simultaneous side chain deprotection and resin cleavage was accomplished via addition of 8 mL of a cleavage cocktail (91% TFA, 3% EDT, 3% TIS, 3% water) for 1.5-3 hours. The crude peptides and cleavage solution were filtered into a pre-weighed 50 mL conical tube. The cleaved resin was rinsed with an additional 2 mL of cleavage cocktail to remove residual peptides. Peptides were precipitated using cold (4°C) anhydrous diethyl ether. The turbid mixture was vortexed and centrifuged at 4°C and 4000 RPMs for 4 minutes (Sorval Super T21 high-speed centrifuge swinging bucket rotor). The supernatant was decanted leaving the crude peptide pellet. This was then washed with cold (4°C) diethyl ether and centrifuged. This process was repeated at least 3 times until no thiol aroma was present and the peptide was dried overnight in a desiccator.
A 5-20 mg sample of crude peptide was purified by RP-HPLC using a Shimadzu chromatography system with a photodiode array detector and a semipreparative RP HPLC C18 bonded silica column (Vydac 218TP1010, 1 cm × 25 cm). The solvent system for purification was either MeCN or MeOH in water with 0.1% TFA. After purified fractions were collected, peptides were concentrated in vacuo and lyophilized. A purity of greater than 95% was confirmed by RP-HPLC in two diverse solvent systems (10% MeCN in 0.1 % TFA/water and a gradient to 90% MeCN over 35 min; and 10% MeOH in 0.1 % TFA/water and a gradient to 90% MeOH over 35 minutes). The correct molecular mass was confirmed by ESI-MS (Table 4) (University of Minnesota Department of Chemistry Mass Spectrometry Laboratory).
Table 4. Analytical data for peptides synthesized in this study.

|
HPLC k′ = (peptide retention time - solvent retention time) / solvent retention time. System 1 is a 10% to 90% gradient of acetonitrile in water containing 0.1% trifluoroacetic acid over 35 minutes at a flow rate of 1.5 mL/min, and system 2 is the same gradient with methanol replacing acetonitrile. Product purity is determined using solvent system which showed the least purity and integrating the area under the curves of the chromatograms collected at 214 nm. Mass observed was calculated from the M+1 or (M+2)/2 peak.
Cell Culture
HEK293 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% newborn calf serum (NCS), and 1% penicillin/streptomycin in humidified atmosphere of 95% air and 5% CO2 at 37°C. Stable cell lines were generated with wild type mMC1R, mMC4R, mMC5R, and mMC3R-Flag DNA in pCDNA3 expression vector (20 μg) using the calcium phosphate transfection method.108 Stable populations were generated using G418 selection (0.7-1.0 mg/mL) and used in bioassays unless indicated otherwise. Experimental ligands were dissolved to a 10-2 M stock in DMSO and stored at -20°C. Subsequent dilutions were performed in the stated assay buffer to achieve the final concentration in the well. The ligands were assayed as TFA salts.
125I-NDP-MSH Competitive Binding Affinity Studies
125I-NDP-MSH was purchased from Dr. Robert Speth, Nova Southeastern University (specific activity: 2175 Ci/mmol). HEK293 cells stably expressing the mMC1R and mMC4R were maintained as described above. Binding experiments on the mMC3R were performed on transiently transfected HEK293 cells. Transfection was performed two days prior to binding experiment in 10 cm plates using FuGene6 transfection reagent (15 μL/plate; Promega), Opti-Mem medium (1.7 mL/ plate; Invitrogen), and mMC3R-Flag DNA (3.33 μg/plate). One or two days preceding the experiment, cells were plated into 12-well tissue culture plates (Corning Life Sciences, Cat. # 353043) and grown to 90-100 % confluency. On the day of the assay, media was aspirated and the cells were treated with a freshly diluted aliquot of non-labeled compound at the concentration being tested (ranging from 10-12 to 10-4 M as appropriate) in assay buffer (DMEM and 0.1% BSA) and a constant amount of 125I- NDP-MSH (100,000 cpm/well). Cells were incubated at 37° C for one hour. After which, media was gently aspirated and cells were washed gently once with assay buffer.
Buffer was gently aspirated and cells were lysed with NaOH (500 μL; 0.1M) and Triton X-100 (500 μL; 1%) for at least 10 minutes. Cell lysate was transferred to 12×75 mm polystyrene tubes and radioactivity was quantified on WIZARD2 Automatic Gamma Counter (PerkinElmer). Experiments were performed with duplicate data points and repeated in at least two independent experiments. Each experiment included unlabeled NDP-MSH as a positive control. Non-specific values were obtained using a 10-6 M unlabeled NDP-MSH. Data was analyzed using the PRISM program (v4.0; GraphPad Inc.). Dose-response curves and IC50 values were generated and analyzed by a nonlinear regression method. The standard deviation (SD) was derived from the IC50 values from at least two independent experiments.
AlphaScreen® cAMP Functional Bioassay
The AlphaScreen® cAMP technology (PerkinElmer Life Sciences, Cat #6760625M) was utilized to measure cAMP signaling after ligand stimulation in HEK293 cells stably expressing the mMC1R, mMC3R, mMC4R, and mMC5R. The AlphaScreen® assay was performed as described by manufacturer, and it is described briefly below.
Cells were 70-95% confluent on the day of the assay. Cells were removed from 10 cm plates with Gibco® Versene solution. Cells were pelleted by centrifugation (Sorvall Super T21 high speed centrifuge, swinging bucket rotor) at 800 rpm for five minutes. Media was aspirated and cells were resuspended in Dulbecco's phosphate buffered saline solution (DPBS 1X [-] without calcium and magnesium chloride, Gibco ® Cat # 14190-144). A 10 μL aliquot was removed for cell counting. After addition of Trypan blue dye solution (BioRad) to the cell aliquot (1:1 by volume), cells were counted manually using a hemocytometer. Cells were again pelleted at 800 rpm for 5 minutes and DPBS was gently aspirated. The pelleted cells were then resuspended in a solution of freshly made stimulation buffer (Hank's Balanced Salt Solution [HBSS 10X [-] sodium bicarbonate] and [-] phenol red, Gibco®], 0.5 mM isobutylmethylxanthine [IBMX], 5 mM HEPES buffer solution [1M, Gibco®], 0.1% bovine serum albumin [BSA] in Milli-Q water, pH=7.4) and anti-cAMP acceptor beads (1.0 unit per 5 μL, AlphaScreen®). At this point 5 μL of cell/acceptor bead solution was added manually to each well of a 384 well microplate (OptiPlate-384; PerkinElmer). Final concentrations were 10,000 cells/well and 1.0 Unit anti-cAMP acceptor beads/well. The cells were then stimulated with 5 μL of ligand diluted in stimulation buffer to achieve their final concentrations in the well ranging from 10-13 to 10-4 M. Cells were incubated in a dark laboratory drawer at room temperature for two hours.
A biotinylated cAMP/streptavidin donor bead working solution was made by adding biotinylated cAMP (1 Unit/well, AlphaScreen®) and streptavidin donor beads (1 Unit/well, AlphaScreen®) to a lysis buffer (10% Tween-20, 5 mM HEPES buffer solution [1M, Gibco®], 0.1% bovine serum albumin [BSA] in Milli-Q water, pH=7.4) one to two hours before addition. After the two hour stimulation period, 15 μL of the biotinylated cAMP/ Streptavidin donor bead working solution was added to each well in the dark or under green light and mixed well by pipetting up and down. The cells were then incubated for another two hours at room temperature in a dark drawer. After this final incubation, the plate was read on an EnSpire™ Alpha plate reader using a pre-normalized assay protocol set by the manufacturer. Assays were performed with duplicate data points and repeated in at least three independent experiments. Each plate contained a control ligand dose response (NDP-MSH, α-MSH, or Ɣ2-MSH), a 10-4 M forskolin positive control, and a no ligand assay buffer negative control.
Data was analyzed using the PRISM program (v4.0; GraphPad Inc.). Dose response curves and potency EC50 values (concentration which caused 50% receptor activation) were generated and analyzed by a nonlinear regression method. Because the AlphaScreen® assay is a competition assay and to be consistent with functional data being represented as an increasing response with increasing concentration, a transformation was carried out for illustration purposes to normalize data to control compounds and flip data curves. A detailed explanation of the transformation can be found in the supplemental materials.
Compounds which showed partial receptor activation at 100 μM at the mMC3R or mMC4R were analyzed for antagonist properties using a Schild regression analysis. Ligands were tested in a dose dependent manner to inhibit NDP-MSH agonist receptor stimulation and the pA2 values were calculated [pA2=-log(Ki)].95 The standard error of the mean (SEM) was derived from the potency values and pA2 from at least three independent experiments.
Animal Studies
All studies were conducted in accordance with the guidelines set up by the Institutional Animal Care and Use Committee (IUCUC) of the University of Minnesota. Wildtype male and female 129/Sv×C57BL/6J hybrid mice were derived from in house breeding colony.11, 36 Mice were individually housed to protect cannula assembly in standard polycarbonate conventional cages provided by the University of Minnesota's Research Animal Resources (RAR). Weekly cage changes were conducted by lab research staff. Mice were housed in a temperature-controlled room (23°-25°C) and maintained on a 12-h light/dark cycle (lights off at 11:00 am). Mice had ad libitum access to standard chow (Harlan Teklad 2018 Diet: 18.6% crude protein, 6.2% crude fat, 3.5% crude fiber, with energy density of 3.1 kcal/g) and tap water. Mice were 8 weeks of age at the beginning of the experiment.
Cannulation surgery and placement validation
Mice were anesthetized with mixture of ketamine (100 mg/kg) and xylazine (5 mg/kg) administered intraperitoneal (IP) and placed in a stereotaxic apparatus (David Kopf Instruments). Mice were given a dose of flunixin meglumine (FluMegluine, Clipper Distribution Company) for post-surgery analgesic. A 26-gauge cannula (PlasticsOne, Roanoke, VA) was inserted into the lateral cerebral ventricle at coordinates 1.0 mm lateral and 0.46 mm posterior to bregma and 2.3 mm ventral to the skull.109 The cannula was secured using n-butyl cyanoacrylate glue (3M Vetbond) and dental cement (Jet Dental and Fleck's Zinc). All cannulation surgeries took place during one week. During post-operative care, mice were administered 0.5 mL of 0.9% saline (Hospira, Lake Forrest, IL) subcutaneously. Mice were allowed to recover for seven days following surgery.
Cannula placement was verified using 2.5 μg human PYY3-36 (Bachem) as described by Marsh et al. 101 In these cannula placement validation experiments, each mouse was administered hPYY and saline on different days separated by a washout period of at least four days. Food intake and mouse weight was measured at 2, 4, and 6 hours post-injection. A mouse with a validated properly placed cannula consumed at least 0.75 g more food four hours after hPYY administration than after saline administration. Validated mice on average ate 0.42 ±0.07 g of food four hours after saline administration and 1.79 ± 0.13 g of food four hours after hPYY administration.
Study design
All intracerebroventricular (ICV) administration experiments were a crossover design (Supp. Table 1) and standard chow was provided ab libitum. Mice were housed in RAR supplied conventional mouse cages during experimentation. Compound 7 was dissolved to a stock solution of 10 nmols/μL in saline (0.9% sodium chloride, Hospira Inc., Lake Forest, IL). The day of compound administration, an aliquot of stock solution was diluted to concentrations of 1.0 nmol, 2.5 nmol and 5.0 nmol in 3 μL of saline. Desired experimental dose (3 μL) or saline vehicle control (3 μL) was administered via ICV injection two hours before lights out (t=0 hr). Food intake and mouse weight was manually measured at T = 0, 2, 4, 6, 8, 24, 48, and 72 hours post-injection. This paradigm measures nocturnal food consumption of free feeding mice. It was chosen because it causes minimal disruption of normal feeding patterns and homeostasis, yet is sensitive to subtle changes that can be masked in fasting paradigms.110 It is also a sensitive way to measure the effects of compounds which induce satiation which might only reduce the size of an initial meal (e.g. two hour time point measurements), but no other.110 Mice were given 6-7 days recovery between treatments to reestablish pretreatment body weight and feeding patterns. Data was analyzed using the PRISM program (v4.0; GraphPad Inc.) by a one-way ANOVA followed by a Bonferroni post test in order to compare individual doses to saline administration at each time point.
Data Analysis
All data analysis was analyzed using the PRISM program (v4.0; GraphPad Inc.). Statistical significance is considered if p<0.05. Data analysis is discussed in more detail at the end of each assays' experimental.
Supplementary Material
Acknowledgments
We would like to thank Skye Doering, Stacey Wilber, Katlyn Fleming, Dr. Mark Ericson, Dr. Srinivasa Tala, and Dr. Anamika Singh their insightful dialogue and guidance while preparing this manuscript. This work has been supported by NIH Grant R01DK091906.
Funding Sources: This work has been supported by NIH Grant R01DK091906 (CHL).
Abbreviations
- MC1R
melanocortin-1 receptor
- MC3R
melanocortin-3 receptor
- MC4R
melanocortin-4 receptor
- MC5R
melanocortin-5 receptor
- MSH
melanocyte-stimulating hormone
- NDP-MSH
[Nle4,DPhe7]-α-MSH
- cAMP
cyclic adenosine monophosphate
- HEK293
human embryonic kidney 293
- RP-HPLC
reverse-phase high-pressure liquid chromatography
- ESI-MS
electron-spray ionization mass spectrometry
- MeCN
acetonitrile
- MeOH
methanol
- PEDG20
19-amino-5-oxo-3,10,13,-16-tetraoxa-6-azanonadecan-1-oic acid
- SAR
structure-activity relationship
- DNal(2′)
D-(2-Naphthyl)alanine
Footnotes
Supporting Information. AlphaScreen® data normalization, Latin-Square (crossover) feeding paradigm, Female and Male Mice ICV Feeding (0-72 hours), Graphical representation of partial functional receptor activation.
Author Contributions: The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- 1.Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science. 1992;257:1248–1251. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- 2.Lerner AB, McGuire JS. Effect of alpha- and betamelanocyte stimulating hormones on the skin colour of man. Nature. 1961;189:176–179. doi: 10.1038/189176a0. [DOI] [PubMed] [Google Scholar]
- 3.Van der Ploeg LH, Martin WJ, Howard AD, Nargund RP, Austin CP, Guan X, Drisko J, Cashen D, Sebhat I, Patchett AA, Figueroa DJ, DiLella AG, Connolly BM, Weinberg DH, Tan CP, Palyha OC, Pong SS, MacNeil T, Rosenblum C, Vongs A, Tang R, Yu H, Sailer AW, Fong TM, Huang C, Tota MR, Chang RS, Stearns R, Tamvakopoulos C, Christ G, Drazen DL, Spar BD, Nelson RJ, MacIntyre DE. A role for the melanocortin 4 receptor in sexual function. Proc Natl Acad Sci U S A. 2002;99:11381–11386. doi: 10.1073/pnas.172378699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenfield JR, Miller JW, Keogh JM, Henning E, Satterwhite JH, Cameron GS, Astruc B, Mayer JP, Brage S, See TC, Lomas DJ, O'Rahilly S, Farooqi IS. Modulation of blood pressure by central melanocortinergic pathways. N Engl J Med. 2009;360:44–52. doi: 10.1056/NEJMoa0803085. [DOI] [PubMed] [Google Scholar]
- 5.Gonzalez PV, Schioth HB, Lasaga M, Scimonelli TN. Memory impairment induced by IL-1beta is reversed by alpha-MSH through central melanocortin-4 receptors. Brain Behav Immun. 2009;23:817–822. doi: 10.1016/j.bbi.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Beckwith BE, Sandman CA, Hothersall D, Kastin AJ. Influence of neonatal injections of Alpha-MSH on learning, memory and attention in rats. Physiol Behav. 1977;18:63–71. doi: 10.1016/0031-9384(77)90095-6. [DOI] [PubMed] [Google Scholar]
- 7.Sandman CA, George JM, Nolan JD, Vanriezen H, Kastin AJ. Enhancement of attention in man with ACTH-MSH 4-10. Physiol Behav. 1975;15:427–431. doi: 10.1016/0031-9384(75)90209-7. [DOI] [PubMed] [Google Scholar]
- 8.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 9.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 10.Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao L, Metzger JM, Strack AM, Camacho RE, Mellin TN, Nunes CN, Min W, Fisher J, Gopal-Truter S, MacIntyre DE, Chen HY, Van der Ploeg LH. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat Genet. 2000;26:97–102. doi: 10.1038/79254. [DOI] [PubMed] [Google Scholar]
- 11.Haskell-Luevano C, Schaub JW, Andreasen A, Haskell KR, Moore MC, Koerper LM, Rouzaud F, Baker HV, Millard WJ, Walter G, Litherland SA, Xiang Z. Voluntary exercise prevents the obese and diabetic metabolic syndrome of the melanocortin-4 receptor knockout mouse. FASEB J. 2009;23:642–655. doi: 10.1096/fj.08-109686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chhajlani V, Wikberg JES. Molecular cloning and expression of the human melanocyte stimulating hormone receptor cDNA. FEBS Lett. 1992;309:417–420. doi: 10.1016/0014-5793(92)80820-7. [DOI] [PubMed] [Google Scholar]
- 13.Gantz I, Konda Y, Tashiro T, Shimoto Y, Miwa H, Munzert G, Watson SJ, Delvalle J, Yamada T. Molecular cloning of a novel melanocortin receptor. J Biol Chem. 1993;268:8246–8250. [PubMed] [Google Scholar]
- 14.Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ, Delvalle J, Yamada T. Molecular cloning, expression, and gene localization of a 4th melanocortin receptor. J Biol Chem. 1993;268:15174–15179. [PubMed] [Google Scholar]
- 15.Gantz I, Tashiro T, Barcroft C, Konda Y, Shimoto Y, Miwa H, Glover T, Munzert G, Yamada T. Localization of the genes encoding the melanocortin-2 (adrenocorticotropic hormone) and melanocortin-3 receptors to chromosomes 18p11.2 and 20q13.2-Q13.3 by fluorescence in-situ hybridization. Genomics. 1993;18:166–167. doi: 10.1006/geno.1993.1448. [DOI] [PubMed] [Google Scholar]
- 16.Mountjoy KG, Mortrud MT, Low MJ, Simerly RB, Cone RD. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol Endocrinol. 1994;8:1298–1308. doi: 10.1210/mend.8.10.7854347. [DOI] [PubMed] [Google Scholar]
- 17.Roselli-Rehfuss L, Mountjoy KG, Robbins LS, Mortrud MT, Low MJ, Tatro JB, Entwistle ML, Simerly RB, Cone RD. Identification of a receptor for gamma melanotropin and other proopiomelanocortin peptides in the hypothalamus and limbic system. Proc Natl Acad Sci U S A. 1993;90:8856–8860. doi: 10.1073/pnas.90.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giuliani D, Galantucci M, Neri L, Canalini F, Calevro A, Bitto A, Ottani A, Vandini E, Sena P, Sandrini M, Squadrito F, Zaffe D, Guarini S. Melanocortins protect against brain damage and counteract cognitive decline in a transgenic mouse model of moderate Alzheimers disease. Eur J Pharmacol. 2014;740:144–150. doi: 10.1016/j.ejphar.2014.06.063. [DOI] [PubMed] [Google Scholar]
- 19.Giuliani D, Bitto A, Galantucci M, Zaffe D, Ottani A, Irrera N, Neri L, Cavallini GM, Altavilla D, Botticelli AR, Squadrito F, Guarini S. Melanocortins protect against progression of Alzheimer's disease in triple-transgenic mice by targeting multiple pathophysiological pathways. Neurobiol Aging. 2014;35:537–547. doi: 10.1016/j.neurobiolaging.2013.08.030. [DOI] [PubMed] [Google Scholar]
- 20.Giuliani D, Neri L, Canalini F, Calevro A, Ottani A, Vandini E, Sena P, Zaffe D, Guarini S. NDP-alpha-MSH induces intense neurogenesis and cognitive recovery in Alzheimer transgenic mice through activation of melanocortin MC4 receptors. Mol Cell Neurosci. 2015;67:13–21. doi: 10.1016/j.mcn.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 21.Xu LP, Josan JS, Vagner J, Caplan MR, Hruby VJ, Mash EA, Lynch RM, Morse DL, Gillies RJ. Heterobivalent ligands target cell-surface receptor combinations in vivo. Proc Natl Acad Sci U S A. 2012;109:21295–21300. doi: 10.1073/pnas.1211762109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Josan JS, Handl HL, Sankaranarayanan R, Xu L, Lynch RM, Vagner J, Mash EA, Hruby VJ, Gillies RJ. Cell-specific targeting by heterobivalent ligands. Bioconjugate Chem. 2011;22:1270–1278. doi: 10.1021/bc1004284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barkey NM, Tafreshi NK, Josan JS, De Silva CR, Sill KN, Hruby VJ, Gillies RJ, Morse DL, Vagner J. Development of melanoma-targeted polymer micelles by conjugation of a melanocortin 1 receptor (MC1R) specific ligand. J Med Chem. 2011;54:8078–8084. doi: 10.1021/jm201226w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barkey NM, Preihs C, Cornnell HH, Martinez G, Carie A, Vagner J, Xu LP, Lloyd MC, Lynch VM, Hruby VJ, Sessler JL, Sill KN, Gillies RJ, Morse DL. Development and in vivo quantitative magnetic resonance imaging of polymer micelles targeted to the melanocortin 1 receptor. J Med Chem. 2013;56:6330–6338. doi: 10.1021/jm4005576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brabez N, Lynch RM, Xu LP, Gillies RJ, Chassaing G, Lavielle S, Hruby VJ. Design, synthesis, and biological studies of efficient multivalent melanotropin ligands: Tools toward melanoma diagnosis and treatment. J Med Chem. 2011;54:7375–7384. doi: 10.1021/jm2009937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uckert S, Bannowsky A, Albrecht K, Kuczyk MA. Melanocortin receptor agonists in the treatment of male and female sexual dysfunctions: results from basic research and clinical studies. Expert Opin Inv Drug. 2014;23:1477–1483. doi: 10.1517/13543784.2014.934805. [DOI] [PubMed] [Google Scholar]
- 27.Penagarikano O, Lazaro MT, Lu XH, Gordon A, Dong HM, Lam HA, Peles E, Maidment NT, Murphy NP, Yang XW, Golshani P, Geschwind DH. Exogenous and evoked oxytocin restores social behavior in the Cntnap2 mouse model of autism. Sci Transl Med. 2015;7 doi: 10.1126/scitranslmed.3010257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barrett CE, Modi ME, Zhang BC, Walum H, Inoue K, Young LJ. Neonatal melanocortin receptor agonist treatment reduces play fighting and promotes adult attachment in prairie voles in a sex-dependent manner. Neuropharmacology. 2014;85:357–366. doi: 10.1016/j.neuropharm.2014.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joppa MA, Ling N, Chen C, Gogas KR, Foster AC, Markison S. Central administration of peptide and small molecule MC4 receptor antagonists induce hyperphagia in mice and attenuate cytokine-induced anorexia. Peptides. 2005;26:2294–2301. doi: 10.1016/j.peptides.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 30.Deboer MD, Marks DL. Cachexia: lessons from melanocortin antagonism. Trends Endocrinol Metab. 2006;17:199–204. doi: 10.1016/j.tem.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 31.DeBoer MD, Marks DL. Therapy insight: Use of melanocortin antagonists in the treatment of cachexia in chronic disease. Nat Clin Pract Endocrinol Metab. 2006;2:459–466. doi: 10.1038/ncpendmet0221. [DOI] [PubMed] [Google Scholar]
- 32.Doering SR, Todorovic A, Haskell-Luevano C. Melanocortin antagonist tetrapeptides with minimal agonist activity at the mouse melanocortin-3 receptor. ACS Med Chem Lett. 2015;6:123–127. doi: 10.1021/ml500340z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ericson MD, Wilczynski A, Sorensen NB, Xiang Z, Haskell-Luevano C. Discovery of a beta-hairpin octapeptide, c[Pro-Arg-Phe-Phe-Dap-Ala-Phe-DPro], mimetic of agouti-related protein(87-132) [AGRP(87-132)] with equipotent mouse melanocortin-4 receptor (mMC4R) antagonist pharmacology. J Med Chem. 2015 doi: 10.1021/acs.jmedchem.5b00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh A, Dirain ML, Wilczynski A, Chen C, Gosnell BA, Levine AS, Edison AS, Haskell-Luevano C. Synthesis, biophysical, and pharmacological evaluation of the melanocortin agonist AST3-88: modifications of peptide backbone at Trp 7 position lead to a potent, selective, and stable ligand of the melanocortin 4 receptor (MC4R) ACS Chem Neurosci. 2014;5:1020–1031. doi: 10.1021/cn5000953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haslach EM, Huang H, Dirain M, Debevec G, Geer P, Santos RG, Giulianotti MA, Pinilla C, Appel JR, Doering SR, Walters MA, Houghten RA, Haskell-Luevano C. Identification of tetrapeptides from a mixture based positional scanning library that can restore nM full agonist function of the L106P, I69T, I102S, A219V, C271Y, and C271R human melanocortin-4 polymorphic receptors (hMC4Rs) J Med Chem. 2014;57:4615–4628. doi: 10.1021/jm500064t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Irani BG, Xiang Z, Yarandi HN, Holder JR, Moore MC, Bauzo RM, Proneth B, Shaw AM, Millard WJ, Chambers JB, Benoit SC, Clegg DJ, Haskell-Luevano C. Implication of the melanocortin-3 receptor in the regulation of food intake. Eur J Pharmacol. 2011;660:80–87. doi: 10.1016/j.ejphar.2010.10.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zanna PT, Sanchez-Laorden BL, Perez-Oliva AB, Turpin MC, Herraiz C, Jimenez-Cervantes C, Garcia-Borron JC. Mechanism of dimerization of the human melanocortin 1 receptor. Biochem Biophys Res Commun. 2008;368:211–216. doi: 10.1016/j.bbrc.2008.01.060. [DOI] [PubMed] [Google Scholar]
- 38.Mandrika I, Petrovska R, Wikberg J. Melanocortin receptors form constitutive homo-and heterodimers. Biochem Biophys Res Commun. 2005;326:349–354. doi: 10.1016/j.bbrc.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 39.Sebag JA, Hinkle PM. Opposite effects of the melanocortin-2 (MC2) receptor accessory protein MRAP on MC2 and MC5 receptor dimerization and trafficking. J Biol Chem. 2009;284:22641–22648. doi: 10.1074/jbc.M109.022400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Piechowski CL, Rediger A, Lagemann C, Muhlhaus J, Muller A, Pratzka J, Tarnow P, Gruters A, Krude H, Kleinau G, Biebermann H. Inhibition of melanocortin-4 receptor dimerization by substitutions in intracellular loop 2. J Mol Endocrinol. 2013;51:109–118. doi: 10.1530/JME-13-0061. [DOI] [PubMed] [Google Scholar]
- 41.Rediger A, Piechowski CL, Habegger K, Gruters A, Krude H, Tschop MH, Kleinau G, Biebermann H. MC4R dimerization in the paraventricular nucleus and GHSR/MC3R heterodimerization in the arcuate nucleus: Is there relevance for body weight regulation? Neuroendocrinology. 2012;95:277–288. doi: 10.1159/000334903. [DOI] [PubMed] [Google Scholar]
- 42.Nickolls SA, Maki RA. Dimerization of the melanocortin 4 receptor: A study using bioluminescence resonance energy transfer. Peptides. 2006;27:380–387. doi: 10.1016/j.peptides.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 43.Biebermann H, Krude H, Elsner A, Chubanov V, Gudermann T, Gruters A. Autosomal-dominant mode of inheritance of a melanocortin-4 receptor mutation in a patient with severe early-onset obesity is due to a dominant-negative effect caused by receptor dimerization. Diabetes. 2003;52:2984–2988. doi: 10.2337/diabetes.52.12.2984. [DOI] [PubMed] [Google Scholar]
- 44.Kopanchuk S, Veiksina S, Petrovska R, Mutule I, Szardenings M, Rinken A, Wikberg JE. Co-operative regulation of ligand binding to melanocortin receptor subtypes: Evidence for interacting binding sites. Eur J Pharmacol. 2005;512:85–95. doi: 10.1016/j.ejphar.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 45.Kopanchuk S, Veiksina S, Mutulis F, Mutule I, Yahorava S, Mandrika I, Petrovska R, Rinken A, Wikberg JE. Kinetic evidence for tandemly arranged ligand binding sites in melanocortin 4 receptor complexes. Neurochem Int. 2006;49:533–542. doi: 10.1016/j.neuint.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 46.Portoghese PS, Ronsisvalle G, Larson DL, Yim CB, Sayre LM, Takemori AE. Opioid agonist and antagonist bivalent ligands as receptor probes. Life Sci. 1982;31:1283–1286. doi: 10.1016/0024-3205(82)90362-9. [DOI] [PubMed] [Google Scholar]
- 47.Portoghese PS, Larson DL, Sayre LM, Yim CB, Ronsisvalle G, Tam SW, Takemori AE. Opioid agonist and antagonist bivalent ligands: The relationship between spacer length and selectivity at multiple opioid receptors. J Med Chem. 1986;29:1855–1861. doi: 10.1021/jm00160a010. [DOI] [PubMed] [Google Scholar]
- 48.Portoghese PS, Nagase H, Lipkowski AW, Larson DL, Takemori AE. Binaltorphimine-related bivalent ligands and their kappa-opioid receptor antagonist selectivity. J Med Chem. 1988;31:836–841. doi: 10.1021/jm00399a026. [DOI] [PubMed] [Google Scholar]
- 49.Daniels DJ, Lenard NR, Etienne CL, Law PY, Roerig SC, Portoghese PS. Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc Natl Acad Sci U S A. 2005;102:19208–19213. doi: 10.1073/pnas.0506627102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhushan RG, Sharma SK, Xie ZH, Daniels DJ, Portoghese PS. A bivalent ligand (KDN-21) reveals spinal delta and kappa opioid receptors are organized as heterodimers that give rise to delta(1) and kappa(2) phenotypes. Selective targeting of delta-kappa heterodimers. J Med Chem. 2004;47:2969–2972. doi: 10.1021/jm0342358. [DOI] [PubMed] [Google Scholar]
- 51.Russo O, Berthouze M, Giner M, Soulier JL, Rivail L, Sicsic S, Lezoualc'h F, Jockers R, Berque-Bestel I. Synthesis of specific bivalent probes that functionally interact with 5-HT(4) receptor dimers. J Med Chem. 2007;50:4482–4492. doi: 10.1021/jm070552t. [DOI] [PubMed] [Google Scholar]
- 52.Singh N, Hazari PP, Prakash S, Chuttani K, Khurana H, Chandra H, Mishra AK. A homodimeric bivalent radioligand derived from 1-(2-methoxyphenyl) piperazine with high affinity for in vivo 5-HT1A receptor imaging. MedChemComm. 2012;3:814–823. [Google Scholar]
- 53.Soulier JL, Russo O, Giner M, Rivail L, Berthouze M, Ongeri S, Maigret B, Fischmeister R, Lezoualc'h F, Sicsic S, Berque-Bestel I. Design and synthesis of specific probes for human 5-HT4 receptor dimerization studies. J Med Chem. 2005;48:6220–6228. doi: 10.1021/jm050234z. [DOI] [PubMed] [Google Scholar]
- 54.Karellas P, McNaughton M, Baker SP, Scammells PJ. Synthesis of bivalent beta(2)-adrenergic and adenosine A(1) receptor ligands. J Med Chem. 2008;51:6128–6137. doi: 10.1021/jm800613s. [DOI] [PubMed] [Google Scholar]
- 55.Nimczick M, Pemp D, Darras FH, Chen X, Heilmann J, Decker M. Synthesis and biological evaluation of bivalent cannabinoid receptor ligands based on hCB(2)R selective benzimidazoles reveal unexpected intrinsic properties. Bioorg Med Chem. 2014;22:3938–3946. doi: 10.1016/j.bmc.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 56.Huang G, Pemp D, Stadtmuller P, Nimczick M, Heilmann J, Decker M. Design, synthesis and in vitro evaluation of novel uni- and bivalent ligands for the cannabinoid receptor type 1 with variation of spacer length and structure. Bioorg Med Chem Lett. 2014;24:4209–4214. doi: 10.1016/j.bmcl.2014.07.038. [DOI] [PubMed] [Google Scholar]
- 57.Xu Y, Duggineni S, Espitia S, Richman DD, An J, Huang Z. A synthetic bivalent ligand of CXCR4 inhibits HIV infection. Biochem Biophys Res Commun. 2013;435:646–650. doi: 10.1016/j.bbrc.2013.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kuhhorn J, Hubner H, Gmeiner P. Bivalent dopamine D2 receptor ligands: Synthesis and binding properties. J Med Chem. 2011;54:4896–4903. doi: 10.1021/jm2004859. [DOI] [PubMed] [Google Scholar]
- 59.Carrithers MD, Lerner MR. Synthesis and characterization of bivalent peptide ligands targeted to G-protein-coupled receptors. Chem Biol. 1996;3:537–542. doi: 10.1016/s1074-5521(96)90144-1. [DOI] [PubMed] [Google Scholar]
- 60.Alleti R, Vagner J, Dehigaspitiya DC, Moberg VE, Elshan NG, Tafreshi NK, Brabez N, Weber CS, Lynch RM, Hruby VJ, Gillies RJ, Morse DL, Mash EA. Synthesis and characterization of time-resolved fluorescence probes for evaluation of competitive binding to melanocortin receptors. Bioorg Med Chem. 2013;21:5029–5038. doi: 10.1016/j.bmc.2013.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Elshan NGRD, Jayasundera T, Anglin BL, Weber CS, Lynch RM, Mash EA. Trigonal scaffolds for multivalent targeting of melanocortin receptors. Org Biomol Chem. 2015;13:1778–1791. doi: 10.1039/c4ob02094d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fernandes SM, Lee YS, Gillies RJ, Hruby VJ. Synthesis and evaluation of bivalent ligands for binding to the human melanocortin-4 receptor. Bioorg Med Chem. 2014;22:6360–6365. doi: 10.1016/j.bmc.2014.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Handl HL, Sankaranarayanan R, Josan JS, Vagner J, Mash EA, Gillies RJ, Hruby VJ. Synthesis and evaluation of bivalent NDP-alpha-MSH(7) peptide ligands for binding to the human melanocortin receptor 4 (hMC4R) Bioconjugate Chem. 2007;18:1101–1109. doi: 10.1021/bc0603642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vagner J, Handl HL, Gillies RJ, Hruby VJ. Novel targeting strategy based on multimeric ligands for drug delivery and molecular imaging: Homooligomers of alpha-MSH. Bioorg Med Chem Lett. 2004;14:211–215. doi: 10.1016/j.bmcl.2003.09.079. [DOI] [PubMed] [Google Scholar]
- 65.Vagner J, Handl HL, Monguchi Y, Jana U, Begay LJ, Mash EA, Hruby VJ, Gillies RJ. Rigid linkers for bioactive peptides. Bioconjug Chem. 2006;17:1545–1550. doi: 10.1021/bc060154p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bowen ME, Monguchi Y, Sankaranarayanan R, Vagner J, Begay LJ, Xu L, Jagadish B, Hruby VJ, Gillies RJ, Mash EA. Design, synthesis, and validation of a branched flexible linker for bioactive peptides. J Org Chem. 2007;72:1675–1680. doi: 10.1021/jo062276g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jagadish B, Sankaranarayanan R, Xu L, Richards R, Vagner J, Hruby VJ, Gillies RJ, Mash EA. Squalene-derived flexible linkers for bioactive peptides. Bioorg Med Chem Lett. 2007;17:3310–3313. doi: 10.1016/j.bmcl.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dehigaspitiya DC, Navath S, Weber CS, Lynch RM, Mash EA. Synthesis and bioactivity of MSH4 oligomers prepared by an A + B strategy. Tetrahedron Lett. 2015;56:3060–3065. doi: 10.1016/j.tetlet.2014.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dehigaspitiya DC, Anglin BL, Smith KR, Weber CS, Lynch RM, Mash EA. Linear scaffolds for multivalent targeting of melanocortin receptors. Org Biomol Chem. 2015 doi: 10.1039/c5ob01779c. [DOI] [PubMed] [Google Scholar]
- 70.Jorg M, May LT, Mak FS, Lee KC, Miller ND, Scammells PJ, Capuano B. Synthesis and pharmacological evaluation of dual acting ligands targeting the adenosine A2A and dopamine D2 receptors for the potential treatment of Parkinson's disease. J Med Chem. 2015;58:718–738. doi: 10.1021/jm501254d. [DOI] [PubMed] [Google Scholar]
- 71.Harikumar KG, Akgun E, Portoghese PS, Miller LJ. Modulation of cell surface expression of nonactivated cholecystokinin receptors using bivalent ligand-induced internalization. J Med Chem. 2010;53:2836–2842. doi: 10.1021/jm100135g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yekkirala AS, Kalyuzhny AE, Portoghese PS. An immunocytochemical-derived correlate for evaluating the bridging of heteromeric mu-delta opioid protomers by bivalent ligands. ACS Chem Biol. 2013;8:1412–1416. doi: 10.1021/cb400113d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Le Naour M, Akgun E, Yekkirala A, Lunzer MM, Powers MD, Kalyuzhny AE, Portoghese PS. Bivalent ligands that target mu opioid (MOP) and cannabinoid1 (CB1) receptors are potent analgesics devoid of tolerance. J Med Chem. 2013;56:5505–5513. doi: 10.1021/jm4005219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Akgün E, Javed MI, Lunzer MM, Powers MD, Sham YY, Watanabe Y, Portoghese PS. Inhibition of inflammatory and neuropathic pain by targeting a mu opioid receptor/chemokine receptor5 heteromer (MOR-CCR5) J Med Chem. 2015;58:8647–8657. doi: 10.1021/acs.jmedchem.5b01245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Smeester BA, Lunzer MM, Akgun E, Beitz AJ, Portoghese PS. Targeting putative mu opioid/metabotropic glutamate receptor-5 heteromers produces potent antinociception in a chronic murine bone cancer model. Eur J Pharmacol. 2014;743:48–52. doi: 10.1016/j.ejphar.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Haskell-Luevano C, Holder JR, Monck EK, Bauzo RM. Characterization of melanocortin NDP-MSH agonist peptide fragments at the mouse central and peripheral melanocortin receptors. J Med Chem. 2001;44:2247–2252. doi: 10.1021/jm010061n. [DOI] [PubMed] [Google Scholar]
- 77.Haskell-Luevano C, Hendrata S, North C, Sawyer TK, Hadley ME, Hruby VJ, Dickinson C, Gantz I. Discovery of prototype peptidomimetic agonists at the human melanocortin receptors MC1R and MC4R. J Med Chem. 1997;40:2133–2139. doi: 10.1021/jm960840h. [DOI] [PubMed] [Google Scholar]
- 78.Holder JR, Bauzo RM, Xiang Z, Haskell-Luevano C. Structure-activity relationships of the melanocortin tetrapeptide Ac-His-DPhe-Arg-Trp-NH(2) at the mouse melanocortin receptors: Part 2 modifications at the Phe position. J Med Chem. 2002;45:3073–3081. doi: 10.1021/jm010524p. [DOI] [PubMed] [Google Scholar]
- 79.Castrucci AM, Hadley ME, Sawyer TK, Wilkes BC, al-Obeidi F, Staples DJ, de Vaux AE, Dym O, Hintz MF, Riehm JP, et al. Alpha-melanotropin: the minimal active sequence in the lizard skin bioassay. Gen Comp Endocrinol. 1989;73:157–163. doi: 10.1016/0016-6480(89)90066-x. [DOI] [PubMed] [Google Scholar]
- 80.Haskell-Luevano C, Sawyer TK, Hendrata S, North C, Panahinia L, Stum M, Staples DJ, Castrucci AM, Hadley MF, Hruby VJ. Truncation studies of alpha-melanotropin peptides identify tripeptide analogues exhibiting prolonged agonist bioactivity. Peptides. 1996;17:995–1002. doi: 10.1016/0196-9781(96)00141-6. [DOI] [PubMed] [Google Scholar]
- 81.Hruby VJ, Wilkes BC, Hadley ME, Alobeidi F, Sawyer TK, Staples DJ, Devaux AE, Dym O, Castrucci AMD, Hintz MF, Riehm JP, Rao KR. Alpha-melanotropin: The minimal active sequence in the frog-skin bioassay. J Med Chem. 1987;30:2126–2130. doi: 10.1021/jm00394a033. [DOI] [PubMed] [Google Scholar]
- 82.Carlson CB, Mowery P, Owen RM, Dykhuizen EC, Kiessling LL. Selective tumor cell targeting using low-affinity, multivalent interactions. ACS Chem Biol. 2007;2:119–127. doi: 10.1021/cb6003788. [DOI] [PubMed] [Google Scholar]
- 83.Kiessling LL, Lamanna AC. Multivalency in biological systems. NATO Sci Ser, II. 2003;129:345–357. [Google Scholar]
- 84.Hiller C, Kuhhorn J, Gmeiner P. Class A G-protein-coupled receptor (GPCR) dimers and bivalent ligands. J Med Chem. 2013;56:6542–6559. doi: 10.1021/jm4004335. [DOI] [PubMed] [Google Scholar]
- 85.Zhang SJ, Yekkirala A, Tang Y, Portoghese PS. A bivalent ligand (KMN-21) antagonist for mu/kappa heterodimeric opioid receptors. Bioorg Med Chem Lett. 2009;19:6978–6980. doi: 10.1016/j.bmcl.2009.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Akgun E, Javed MI, Lunzer MM, Smeester BA, Beitz AJ, Portoghese PS. Ligands that interact with putative MOR-mGluR5 heteromer in mice with inflammatory pain produce potent antinociception. Proc Natl Acad Sci U S A. 2013;110:11595–11599. doi: 10.1073/pnas.1305461110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vagner J, Xu LP, Handl HL, Josan JS, Morse DL, Mash EA, Gillies RJ, Hruby VJ. Heterobivalent ligands crosslink multiple cell-surface receptors: The human melanocortin-4 and delta-opioid receptors. Angew Chem, Int Edit. 2008;47:1685–1688. doi: 10.1002/anie.200702770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Josan JS, Vagner J, Handl HL, Sankaranarayanan R, Gillies RJ, Hruby VJ. Solid-phase synthesis of heterobivalent ligands targeted to melanocortin and cholecystokinin receptors. Int J Pept Res Ther. 2008;14:293–300. doi: 10.1007/s10989-008-9150-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kostelnik KB, Els-Heindl S, Kloting N, Baumann S, von Bergen M, Beck-Sickinger AG. High metabolic in vivo stability and bioavailability of a palmitoylated ghrelin receptor ligand assessed by mass spectrometry. Biorg Med Chem. 2015;23:3925–3932. doi: 10.1016/j.bmc.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 90.Veronese FM, Pasut G. PEGylation, successful approach to drug delivery. Drug Discovery Today. 2005;10:1451–1458. doi: 10.1016/S1359-6446(05)03575-0. [DOI] [PubMed] [Google Scholar]
- 91.Carpino LA, Han GY. 9-Fluorenylmethoxycarbonyl function, a new base-sensitive amino-protecting group. J Am Chem Soc. 1970;92:5748–5749. [Google Scholar]
- 92.Carpino LA, Han GY. 9-Fluorenylmethoxycarbonyl amino-protecting group. J Org Chem. 1972;37:3404–3409. [Google Scholar]
- 93.Stewart JM, Young JD. Solid Phase Peptide Synthesis. 2nd. Pierce Chemical Co.; Rockford, Il: 1984. [Google Scholar]
- 94.Kiessling LL, Gestwicki JE, Strong LE. Synthetic multivalent ligands in the exploration of cell-surface interactions. Curr Opin Chem Biol. 2000;4:696–703. doi: 10.1016/s1367-5931(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 95.Schild HO. pA, a new scale for the measurement of drug antagonism. Br J Pharmacol Chemother. 1947;2:189–206. doi: 10.1111/j.1476-5381.1947.tb00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jayawickreme CK, Quillan JM, Graminski GF, Lerner MR. Discovery and structure-function analysis of alpha-melanocyte-stimulating hormone antagonists. J Biol Chem. 1994;269:29846–29854. [PubMed] [Google Scholar]
- 97.Han Y, Moreira IS, Urizar E, Weinstein H, Javitch JA. Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nat Chem Biol. 2009;5:688–695. doi: 10.1038/nchembio.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bapst JP, Froidevaux S, Calame M, Tanner H, Eberle AN. Dimeric DOTA-alpha-melanocyte-stimulating hormone analogs: synthesis and in vivo characteristics of radiopeptides with high in vitro activity. J Recept Signal Transduction Res. 2007;27:383–409. doi: 10.1080/10799890701723528. [DOI] [PubMed] [Google Scholar]
- 99.Bagutti C, Stolz B, Albert R, Bruns C, Pless J, Eberle AN. [111In]-DTPA-labeled analogues of alpha-melanocyte-stimulating hormone for melanoma targeting: receptor binding in vitro and in vivo. Int J Cancer. 1994;58:749–755. doi: 10.1002/ijc.2910580521. [DOI] [PubMed] [Google Scholar]
- 100.Morais M, Raposinho PD, Oliveira MC, Correia JDG, Santos I. Evaluation of novel Tc-99m(I)-labeled homobivalent alpha-melanocyte-stimulating hormone analogs for melanocortin-1 receptor targeting. J Biol Inorg Chem. 2012;17:491–505. doi: 10.1007/s00775-011-0871-y. [DOI] [PubMed] [Google Scholar]
- 101.Marsh DJ, Hollopeter G, Huszar D, Laufer R, Yagaloff KA, Fisher SL, Burn P, Palmiter RD. Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nat Genet. 1999;21:119–122. doi: 10.1038/5070. [DOI] [PubMed] [Google Scholar]
- 102.Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, Wren AM, Brynes AE, Low MJ, Ghatei MA, Cone RD, Bloom SR. Gut hormone PYY3-36 physiologically inhibits food intake. Nature. 2002;418:650–654. doi: 10.1038/nature00887. [DOI] [PubMed] [Google Scholar]
- 103.Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, Wren AM, Brynes AE, Low MJ, Ghatei MA, Cone RD, Bloom SR. Does gut hormone PYY3-36 decrease food intake in rodents? Reply. Nature. 2004;430 doi: 10.1038/nature00887. [DOI] [PubMed] [Google Scholar]
- 104.Tschop M, Castaneda TR, Joost HG, Thone-Reineke C, Ortmann S, Klaus S, Hagan MM, Chandler PC, Oswald KD, Benoit SC, Seeley RJ, Kinzig KP, Moran TH, Beck-Sickinger AG, Koglin N, Rodgers RJ, Blundell JE, Ishii Y, Beattie AH, Holch P, Allison DB, Raun K, Madsen K, Wulff BS, Stidsen CE, Birringer M, Kreuzer OJ, Schindler M, Arndt K, Rudolf K, Mark M, Deng XY, Withcomb DC, Halem H, Taylor J, Dong J, Datta R, Culler M, Craney S, Flora D, Smiley D, Heiman ML. Physiology: Does gut hormone PYY3-36 decrease food intake in rodents? Nature. 2004;430 doi: 10.1038/nature02665. [DOI] [PubMed] [Google Scholar]
- 105.Sorge RE, Martin LJ, Isbester KA, Sotocinal SG, Rosen S, Tuttle AH, Wieskopf JS, Acland EL, Dokova A, Kadoura B, Leger P, Mapplebeck JC, McPhail M, Delaney A, Wigerblad G, Schumann AP, Quinn T, Frasnelli J, Svensson CI, Sternberg WF, Mogil JS. Olfactory exposure to males, including men, causes stress and related analgesia in rodents. Nature methods. 2014;11:629–632. doi: 10.1038/nmeth.2935. [DOI] [PubMed] [Google Scholar]
- 106.Kaiser E, Colescott RL, Bossinger CD, Cook PI. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal Biochem. 1970;34:595–598. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- 107.Christensen T. Qualitative test for monitoring coupling completeness in solid-phase peptide-synthesis using chloranil. Acta Chem Scand. 1979;33:763–766. [Google Scholar]
- 108.Chen CA, Okayama H. Calcium phosphate-mediated gene transfer: a highly efficient transfection system for stably transforming cells with plasmid DNA. Biotechniques. 1988;6:632–638. [PubMed] [Google Scholar]
- 109.Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. Academic Press; San Diego: 1997. pp. 10–22. [Google Scholar]
- 110.Ellacott KL, Morton GJ, Woods SC, Tso P, Schwartz MW. Assessment of feeding behavior in laboratory mice. Cell Metab. 2010;12:10–17. doi: 10.1016/j.cmet.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.