

Abstract
Atomic-force-microscopy (AFM)-based single-molecule force spectroscopy (SMFS) is a powerful yet accessible means to characterize mechanical properties of biomolecules. Historically, accessibility relies upon the nonspecific adhesion of biomolecules to a surface and a cantilever and, for proteins, the integration of the target protein into a polyprotein. However, this assay results in a low yield of high-quality data, defined as the complete unfolding of the polyprotein. Additionally, nonspecific surface adhesion hinders studies of α–helical proteins, which unfold at low forces and low extensions. Here, we overcame these limitations by merging two developments: (i) a polyprotein with versatile, genetically encoded short peptide tags functionalized via a mechanically robust Hydrazino-Pictet Spengler ligation and (ii) the efficient site-specific conjugation of biomolecules to PEG-coated surfaces. Heterobifunctional anchoring of this polyprotein construct and DNA via copper-free click chemistry to PEG-coated substrates and a strong but reversible streptavidin-biotin linkage to PEG-coated AFM tips enhanced data quality and throughput. For example, we achieved a 75-fold increase in the yield of high-quality data and repeatedly probed the same individual polyprotein to deduce its dynamic force spectrum in just 2 h. The broader utility of this polyprotein was demonstrated by measuring three diverse target proteins: an α-helical protein (calmodulin), a protein with internal cysteines (rubredoxin), and a computationally designed three-helix bundle (α3D). Indeed, at low loading rates, α3D represents the most mechanically labile protein yet characterized by AFM. Such efficient SMFS studies on a commercial AFM enable the rapid characterization of macromolecular folding over a broader range of proteins and a wider array of experimental conditions (pH, temperature, denaturants). Further, by integrating these enhancements with optical traps, we demonstrate how efficient bioconjugation to otherwise nonstick surfaces can benefit diverse single-molecule studies.
Keywords: Single-molecule force spectroscopy, atomic force microscopy, site-specific attachment, protein folding
TOC image
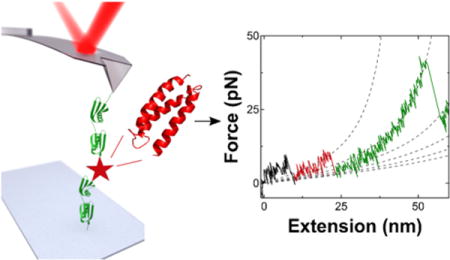
Introduction
Single-molecule force spectroscopy (SMFS) is a powerful tool to mechanically measure the dynamics and energetics of individual biomolecules folding and unfolding (proteins, RNA, and DNA).1,2 A common need in all SMFS assays is a simple and efficient method to site-specifically conjugate biomolecules to a surface and a force probe while avoiding nonspecific attachments that degrades data quality. Another ongoing need is to improve throughput.3 For instance, the vast majority of attempts to stretch a single molecule using traditional atomic force microscopy (AFM)-based SMFS techniques yield uninterpretable force-extension curves.4 Yet, it typically requires hundreds to thousands of unfolding events to deduce the zero force unfolding rate (k0) and the distance to the transition state (Δx‡) of a protein via dynamic force spectroscopy.4 As a result, AFM-based studies of protein folding would be advanced if one could collect high-quality data in a relatively high-throughput manner (~50–100 measurements/hr).3 Ideally, such success would be accompanied by sub-ms temporal resolution, so that short-lived protein-folding intermediates5,6 can be detected and instrument-induced artifacts minimized.7 However, like most AFM experiments,4 our data quality and throughput were limited by nonspecific attachment (Figure 1a–c).
Figure 1.
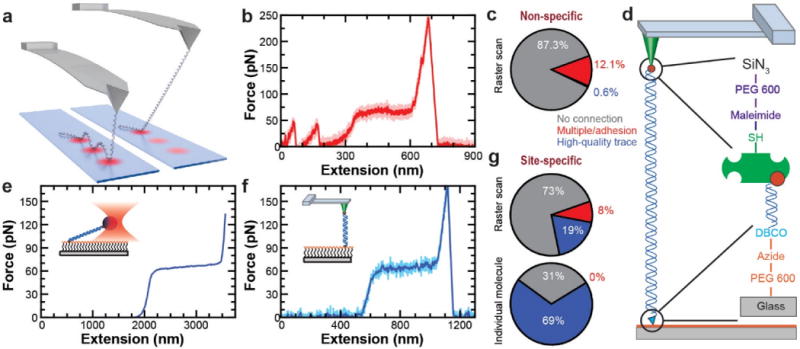
Site-specific chemistry improves data quality. (a) Schematic of experiment showing multiple nonspecific attachments o the DNA to the surface. (b) A typical force-extension curve (FEC) for a 700-nm-long DNA shows adhesion artifacts from multiple nonspecific attachments to the surface. The high-bandwidth trace (16.6 kHz, pink) was filtered to 333 Hz (red). (c) A pie chart quantifies the percentage assay efficiency when using nonspecific attachment by showing the fraction of high-quality FEC (blue), low quality FEC containing multiple attachments (red), and no molecular attachment (grey)(N = 1,278). (d) Schematic of the overall coupling scheme, illustrating the site-specific attachment of DNA to the surface using copper-free click chemistry and to the tip using streptavidin-biotin. PEG coating of both surfaces minimizes nonspecific adhesion. (e) A FEC for a 2-μm-long DNA molecule, site specifically anchored to a PEG-coated coverslip, stretched using an optically trapped streptavidin-coated bead. Data filtered to 500 Hz. (f) A FEC for a 650-nm-long DNA molecule stretched using a streptavidin-coated tip. High-bandwidth records (50 kHz, light blue) were filtered to 200 Hz (dark blue). (g) Pie charts quantify the fraction of high-quality FEC such as the one shown in (f) using the same color scheme as in (c). Top pie chart (“Raster scan”) quantifies a normal search strategy (i.e., scanning the AFM tip around the surface in a grid) (N = 202). Bottom pie chart (“Individual molecule”) shows repeated measurements (N = 218) at a single surface location where a single DNA molecule was located using a grid search.
An important early advance to increasing data quality for protein studies was the head-to-tail linking of identical or similar protein domains—called a polyprotein—that gives rise to a distinct mechanical fingerprint for a single molecule: a saw-tooth-like force-extension curve.8–10 Target proteins inserted into such polyproteins can then be reliably identified by changes in the mechanical fingerprint.11 Polyprotein-based assays commonly use nonspecific attachment to the surface and the AFM tip.4,8 This assay is easy to implement, but uncontrolled attachment of the tip along the polyprotein backbone and surface adhesion lead to a very low percentage of high-quality records, defined as the full unfolding of all the domains in the polyprotein.4 Moreover, random attachment along the backbone leads to lateral shifts in the mechanical fingerprint along the extension axis, which needs to be corrected.12 In addition, surface adhesion hinders studying proteins with high αhelical content because such proteins tend to unfold at low forces (5–20 pN) and extensions.13 As a result, recent studies detailing the unfolding pathway of mechanically labile proteins have relied upon optical traps.14–16 However, modern commercial AFMs offer significantly enhanced ease-of-use compared to custom-built optical traps and can achieve sub-pN force stability when using cantilevers with their gold coating removed.17 Hence, there is an opportunity to apply AFM to rapidly characterize diverse targets, particularly mechanically labile α-helical proteins, by developing an efficient means to site specifically anchor a polyprotein to otherwise non-stick surfaces.
Multiple groups have developed site-specific anchoring of polyproteins with a focus on cysteine9,10,18 and proteinmediated linkages.19–24 Introducing terminal cysteines into polyproteins offers the advantage of covalent attachment to gold-coated9,10,22 or maleimide-functionalized18 surfaces. However, many proteins contain internal cysteines, some of which are essential to their function. Larger protein-mediated tags have also shown great success (e.g., Halo-tags,20,22 SpyCatcher/SpyTag,21 cohesin-dockerin,23 and Strep-tag II/Strep-Tactin24), but they increase the size and complexity of the polyprotein and require anchoring of specialized cognate ligands/proteins to a surface. Further, when using a fully covalently linked construct, repeated unfolding and rupture of the macromolecules leads to an unwanted protein coat at the apex of the tip that will progressively reduce the rate of attachment. Recently, a novel polyprotein construct combined covalent anchoring in series with a strong, but non-covalent, cohesin-dockerin protein bond.25 When integrated with a microfluidic platform, this construct enabled screening multiple protein variants by SMFS with a single cantilever for improved force precision.23 Notwithstanding the success of this platform, it requires a substantial investment in developing expertise in microfluidics and protein-ligand constructs when trying to study new proteins by traditional AFM techniques.
Here, we initially address the general issue of surface conjugation for SMFS by developing a robust and simple-to-implement protocol to site specifically anchor DNA and proteins to nonstick surfaces. While the anchoring to glass coverslips was covalent, we used a strong but reversible bond to AFM tips. Such reversibility preserves tip functionality over days to weeks by avoiding the buildup of an undesired macromolecular coating on the tip’s apex that inhibits subsequent attachment. Specifically, we used copper-free click chemistry26 to anchor both DNA and proteins to a PEG-coated coverslip and used a streptavidin-biotin bond for tip attachment (Figure 1d). Our surface-conjugation scheme generalizes to other SMFS platforms. To demonstrate this applicability, we used it to overstretch DNA27,28 with both an optical trap and an AFM (Figure 1e,f). Importantly, the AFM data showed up to a ~100-fold increase in the rate of acquiring high-quality DNA data (Figure 1g, lower panel), compared to nonspecific attachment (Figure 1c).
To apply this coupling scheme to proteins, we also developed a new polyprotein construct, minimizing its size and complexity by using either a terminal-cysteine and/or a short genetically encoded aldehyde tag29 (Figure 2). By merging this new polyprotein with our enhanced site-specific conjugation strategy, we achieved a ~75-fold increase in the rate of acquiring of high-quality protein unfolding data over the traditional non-specific coupling scheme. Our polyprotein’s broader utility was demonstrated by measuring three diverse target proteins: an α-helical protein (calmodulin), a protein with internal cysteines (rubredoxin), and a computationally designed three-helix bundle (α3D). Indeed, at low loading rates, α3D represents the most mechanically labile protein yet characterized by AFM, highlighting the sensitivity of this versatile polyprotein-based assay.
Figure 2.

Overview of two schemes for polyprotein functionalization and surface attachment based upon (a) a directional and (b) a dual-aldehyde construct. The polyprotein consisted of two repeats of two NuG2 (green circles) flanking the protein of interest (blue star). To generate an aldehyde, a 6-amino-acid tag (LCTPSR, purple rectangle) was introduced into the DNA coding for the polyprotein (not shown). After expression and purification, the polyprotein was reacted overnight (o/n) with formylglycine-generating enzyme (FGE), which converts the cysteine only in the short tag into a formylglycine (fGly), an aldehyde-containing amino acid. Next, the aldehyde at one or both ends of the polyprotein was functionalized with a HIPS-based reagent. In both constructs, one end was labelled with a biotin and, in the right column, the other end with a DBCO, a copperless click chemistry reagent that reacts with azide (N3) moieties. If desired, the directional construct could be directly linked to a maleimide functionalized surface using a terminal cysteine or subsequently DBCO-labelled for linkage to an azide-functionalized surface. Note that while the dualaldehyde construct only yields a fraction of polyproteins with both DBCO and biotin labels, only such heterobifunctionally labeled proteins are efficiently stretched between an azide-functionalized surface and a streptavidin-coated tip.
Methods
Expression and purification of double aldehyde NuG2 polyprotein
As shown in Figure 2, our polyprotein construct contains four repeats of NuG2, a fast-folding variant of the GB1 domain, that has been repeatedly characterized by AFM.30,31 To achieve site-specific conjugation, the polyprotein contained a short (6-amino-acid) polypeptide tag (LCTPSR) where the cysteine is converted to an aldehyde by formylglycine-generating enzyme (FGE). Aldehydes provide excellent chemical orthogonality to cysteine-mediated labeling and access to a wide variety of commercially available reagents, including fluorophores for single-molecule studies.32 To increase efficiency, we performed the conversion in vitro after protein purification. Details of the FGE purification and other details including inserting a protein of interest into the polyprotein, expressing the directional construct for proteins that do not contain solvent-exposed cysteines, and synthesis of Hydrazino-Pictet Spengler (HIPS) reagents, which provide a mechanically stable linkage at neutral pH are presented in the Supporting Information.
With the same coupling chemistry at each end of the protein, differential labeling arises stochastically by mixing equal molar ratios of biotinand DBCO-based reagents. Specifically, we used HIPS-biotin and HIPS dibenzocyclooctyne (DBCO) incubated at a 10X molar excess to the protein for 3 days at 37 °C. Note, shorter incubations are possible, particularly when only using the HIPS biotin reagent in the context of the directional construct. Excess reagent was removed by gel filtration chromatography and peak fractions were concentrated to 0.5–1 mg/mL.
Coverslip functionalization and bioconjugation
An overview of this process is shown in Figure 3A. Prior to this process, the glass coverslips were first cleaned with a series of solutions [acetone, ethanol, and ethanolic KOH (3 M)] and then sequentially rinsed twice in ultrapure water. We dried the slides with a gentle stream of dry nitrogen (N2). We then irradiated the coverslips with UV light for 30 min (PSD-UV8, Novascan Technologies, Inc.) or 60 min (BondWand, Electro-Lite Corporation) to increase the efficiency of the silane coupling. Next, the coverslips were immersed in a 0.15 mg/mL solution of silane-PEG-azide (MWPEG = 600, PG2-AZSL-600, Nanocs Inc.) dissolved in toluene in a glass-capped 50-mL beaker for 3 h at 60 °C, while stirring the solution at 400 rpm with a Teflon coated stir bar. We then immediately immersed the functionalized surfaces for ~30 s sequentially in toluene, isopropanol, and ultrapure water held in individual 200-mL beakers and then gently dried them using a stream of N2. A rapid transfer between silane-PEG-azide and toluene is critical to prevent drying and the resulting PEG agglomeration. Azide-functionalized surfaces were stored dry at 4 °C in sealable, 1″ wafer holders (e.g., H22-101-0615, Entegris) and remained functional for up to 1 month, with optimal results in the first 2 weeks.
Figure 3.
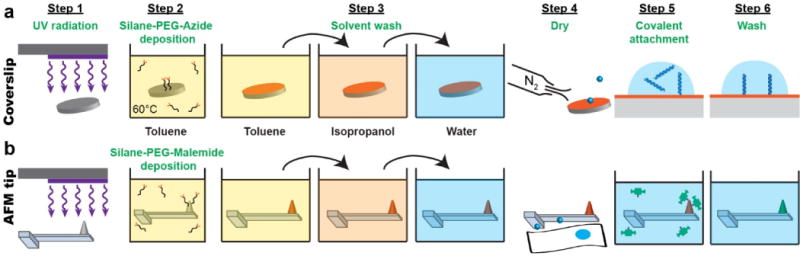
Efficient six-step protocol for functionalization of glass coverslips (a) and silicon-nitride tips (b) using hetero-bifunctional PEG. Tips and coverslips were activated using UV radiation and then silane-PEG functionalized in toluene at elevated temperature resulting in azide-derivatized coverslips and maleimide-derivatized AFM tips. After a series of solvent washes, the surfaces were dried. Biomolecules were covalently bound to coverslips using copper-free click chemistry, while thiol-modified streptavidin was covalently attached to maleimide-coated AFM tips. Finally, coverslips and tips were washed and stored in buffer at 4 °C until use.
The next step was to covalently attach the biomolecule (DNA or protein) to these functionalized surfaces. To do so, we deposited 20 μL of either 650-nm-long DNA (1–20 ng/μL) or our polyprotein construct (50–300 ng/μL) onto the azide-functionalized coverslip. This coverslip was then sealed in a wafer holder contained in a simple humidity chamber for 4–24 h at 4 °C. To remove unbound molecules after this incubation, samples were rinsed 15 times with 1 mL of PBS [140 mM NaCl, 3 mM KCl, and 10 mM phosphate buffer (pH 7.4), unless otherwise noted].
Functionalizing AFM tips with streptavidin
An overview of this process is shown in Figure 3B. Prior to this functionalization, we first chemically stripped the gold and the underlying chromium layer from long, soft cantilevers [L = 100 μm; k = 6 pN/nm BioLever (Olympus)] to improve force stability.17 The cantilevers were first sequentially immersed in toluene, isopropanol, and ultrapure water in individual 100-mL beakers and finally dried by gently pressing the chip against a Kimwipe. Next, individual cantilevers were sequentially immersed in gold etchant (Type TFA, Transene) in a 10-mL beaker for 30 s, ultrapure water in a 500-mL beaker for 40 s, chromium etchant (Cr Etchant, Transene) in a 10-mL beaker for 30 s, and then a second 500-mL beaker of ultrapure water for 40 s. The cantilevers were then dried by gently pressing the chip—not the cantilever—against a Kimwipe to wick away excess water.
To prepare the cantilevers for silanization, we irradiated them with UV light for 30 min (PSD-UV8, Novascan Technologies, Inc.) or 60 min (BondWand, Electro-Lite Corporation), depending on the source. The resulting cantilevers were immediately immersed in a 0.15 mg/mL solution of silane-PEG-maleimide (MWPEG = 600; PG2-MLSL600, Nanocs Inc.) in toluene at 60 °C in a glass-capped 50mL beaker for 3 h. As with the coverslips, the cantilevers were sequentially rinsed in toluene, isopropanol, and ultrapure water and finally dried.
We simplified streptavidin attachment to the cantilever by using a commercially available variant of streptavidin that was thiol-modified (SAVT, Protein Mods LLC). Specifically, immediately after drying, we immersed the maleimide-functionalized cantilever tips for 3 h at room temperature in 50 μL of 200 μg/mL modified streptavidin dissolved in PBS (pH 6.75) supplemented with 1 mM TCEP [Tris(2-carboxyethyl)phosphine]. We used a lower pH to reduce maleimide hydrolysis while staying in the optimal pH range (6.5–7.5) for maleimide-thiol reactions.33 As with the coverslips, the coated cantilevers were then washed by rinsing the cantilevers in 3 separate 10-ml beakers for 30 s each in PBS (pH 7.4) and a subsequent immersion for 5 min in a 300-mL beaker of PBS to remove unreacted streptavidin. Streptavidin-coated cantilevers were then stored at 4 °C in PBS using a sealed wafer holder within humidity chambers for up to 2 weeks. We note that this same protocol has been used with other cantilevers for SMFS, specifically, focused-ion-beam modified (FIB) BioLever Mini34 and FIB-modified BioLever Fasts.35
AFM assay and analysis
Our assay was designed to efficiently stretch only proteins and DNA that are heterobifunctionally labelled with DBCO and biotin. Yet, at best, 50% of individual dual-aldehyde polyproteins will be terminally labelled with DBCO and biotin while 25% will be doubly labelled with DBCO and 25% doubly labelled with biotin. Doubly labelled DBCO protein attaches efficiently to the surface but is unlikely to make a connection to the streptavidin-coated tip for three reasons: (i) it lacks biotin for site-specific attachment (Figure S1), (ii) both labelled ends are likely to be anchored to the azide surface via the DBCO moiety, and (iii) non-specific adhesion is suppressed by the low contact force (100 pN) along with the tip’s PEG coating. Similarly, for protein doubly labelled with biotin, the protein lacks the chemical moiety to site specifically bind to the azide-PEG-coated surfaces and therefore should be efficiently removed during subsequent washing of these relatively non-stick surfaces.
AFM experiments were performed on a commercial AFM (Cypher ES, Asylum Research). Cantilevers were calibrated using standard protocols.36 Specifically, we calibrated the stiffness using the thermal method36 far from the surface and deduced sensitivity by pressing the cantilever into hard contact. On average, the cantilevers used in this work had k ≈ 8 pN/nm. As is typical in AFM-based SMFS, a force-extension curve acquisition was initiated by bringing the cantilever into hard contact with the surface (v = 1 μm/s). However, to reduce non-specific attachment, the tip was pushed into the surface for a brief period (~1–2 s) at 100 pN, approximately 10-fold less force than is generally used to promote non-specific attachment.4 We then retracted at 50–3,800 nm/s while digitizing at 50 kHz. For presentation purposes, these high-bandwidth records were boxcar averaged to the indicated bandwidth.
The sample surface was typically probed with a raster scan. Specifically, we moved the AFM tip in a grid pattern, with locations separated by several microns, and probed each location 10 times. This standard scheme was further optimized by repeatedly probing the same location when a molecule was detected. The repeated probing of the same individual molecule was discontinued after ~10 consecutive attempts failed to yield a connection.
Force was determined by cantilever deflection after scaling for the sensitivity and stiffness of each cantilever. Extension was deduced from the motion of the sample minus the deflection of the cantilever. We determined the loading rate (pN/s) for each rupture observed in a forceextension curve by fitting a line to the force-versus-time curve in the immediate vicinity of a rupture.
Results and Discussion
Efficient coupling protocol and highly stable functionalized sample surfaces and AFM tips
An efficient and reproducible method to site-specifically anchor various biomolecules between the AFM tip and the surface is necessary to achieve improved throughput for AFM-based SMFS. Development of such schemes has been pursued by multiple groups.9,10,18–24 Despite these important advances, several issues remain. First, the surface-coupling protocol is generally time and labor intensive. For instance, the widely used protocol for thiol-maleimide chemistry takes on average ~8 h to prepare surfaces for biomolecular conjugation.18 Second, maleimide moieties rapidly hydrolyze under aqueous conditions, leading to higher sample-tosample variability and lower coupling efficiency. Third, such samples should ideally be used on the same day. Fourth, protein-mediated covalent-coupling schemes20–23 have several downsides: they generally require the expression of additional proteins and are difficult to extend to nucleic acids. Finally, when using a fully covalent coupling schemes,20–22 there is undefined partitioning of the protein between the tip and the surface after rupture.
To address these limitations, we developed an efficient, heterobifunctional anchoring of biomolecules via copperfree click chemistry to a PEG-coated substrate and a strong but reversible streptavidin-biotin linkage to a PEG-coated AFM tip (Figure 1d). Our rapid protocol to functionalize surfaces and AFM tips reduced surface functionalization time from ~8 h to ~3.5 h (Figure 3). Simplicity was achieved, in part, by using a commercially available heterobifunctional PEG with a silane moiety for coupling to glass or silicon-nitride surfaces and an orthogonal chemical-crosslinking moiety for attachment to biomolecules. Our functionalized substrates and tips were stable over weeks and yielded high-quality data over multiple experimental runs. Specifically, azide-coated surfaces worked for up to one month in SMFS assays when stored dry at 4 °C. Streptavidin-coated AFM tips were reused for up to two weeks to measure thousands of DNA molecules over multiple samples. We also reused protein and DNA samples for up to 3 and 9 days, respectively, with a gradual decrease in attachment rate. Overall, the long shelf life of functionalized surfaces and protein-coated samples and tips significantly accelerates the application of SMFS by reducing the time and labor dedicated to daily sample preparation.
Data quality and quantity improves for DNA and proteins when using site-specific attachment to non-stick surfaces
To test our DBCO and biotin conjugation scheme, we mechanically stretched DNA, a common biophysical assay.37 The PEG coating on both the coverslip and the tip led to significantly less adhesion artifacts (Figure 1f) compared to traditional nonspecific coupling (Figure 1c) that is still widely used in AFM-based SMFS.4 More quantitatively, we observed a ~30-fold improvement in the efficiency of measuring high-quality records of DNA stretching (Figure 1e,g) in comparison to traditional nonspecific attachment (Figure 1c) when probing a different location after each attempt (i.e., raster scanning). Indeed, even though the tip was pressed into the surface at ~10-fold less force (~100 pN) than typically used to initiate nonspecific attachment (~1,000 pN),4 the percentage of high-quality records is higher than that for detecting any molecular attachment with nonspecific coupling. This high coupling efficiency at low force together with accompanying control experiments (Figure S1) demonstrates the site-specific nature of the attachment of the streptavidin-coated AFM tip to the biotinylated DNA. Importantly, the combination of a covalent, site-specific anchoring at one end and a strong but reversible linkage at the other deterministically left the target molecule on the sample surface and positioned to be remeasured after returning the AFM tip to the surface. As a result, the yield for acquiring high-quality records from a single individual DNA molecule at a single location increased further to 69%, a ~100-fold improvement over our results using nonspecific adhesion.
Next, we demonstrated improvements in the rate of acquiring high-quality protein records by incorporating our site-specific bioconjugation scheme with our co-developed polyprotein construct (Figure 2). Specifically, we first deposited the polyprotein consisting of four repeats of NuG2 [i.e., (NuG2)4] with variable chemical functionalizations at a fixed concentration (0.3 mg/ml) (Figure 4). Next, the tip was gently (100 pN) pressed into the surface for 1 s and then retracted at a constant velocity (400 nm/s), changing locations every third attempt. When using nonspecific adhesion, the vast majority (85.5%) of attempts yielded no attachment (Figure 4b). Less often, we observed a trace showing a single rupture or pulling on multiple proteins in parallel (Figure 4c) or a trace showing incomplete unfolding of a single polyprotein (Figure 4d), at 10.8% and 3.5% respectively. The desired high-quality data, the full unfolding of the polyprotein (Figure 4e), was observed least frequently (0.2%). As summarized in Figure 4f, the sequential introduction of site-specific surface coupling and tip attachment led to an increased yield of high-quality data. In particular, we achieved a ~75-fold increase in the rate of acquiring high-quality data when using the dual-aldehyde construct in conjunction with an azide-functionalized surface and a streptavidin-coated tip. Optimization of protein deposition concentration is likely to further increase this yield.
Figure 4.
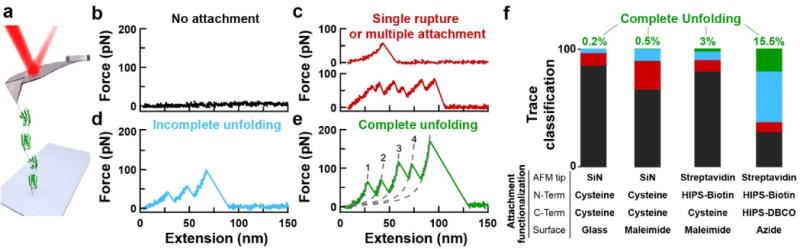
Improved single-molecule force spectroscopy. (a) A schematic of the experiment shows a polyprotein with four NuG2 domains being stretched between the surface and an AFM tip. (b–e) Force-extension curves (FECs) typically show one of four classes of mechanical fingerprints ranging from no attachment to full unfolding of the polyprotein. In panel e, the segments of the FEC between domain ruptures are well described by a worm-like-chain model (dashed lines). FEC data smoothed to 1 kHz. (f) Bar graphs characterizing the fraction of records with no unfolding (black), traces showing a single unfolding event or pulling on multiple proteins in parallel (red), incomplete unfolding of a single polypeptide (blue), and complete unfolding (green). Sitespecific attachment to the functionalized, PEG-coated surface used either a maleimide-thiol reaction or a reaction between dibenzocyclooctyne (DBCO), a copperless-click-chemistry reagent, and an azide-derivatized surface. Hydrazino-Pictet-Spengler (HIPS) reagents labelled genetically encoded aldehydes with biotin and DBCO. Going from left to right, the bar graphs were based on 595, 595, 1237, and 898 individual stretching attempts. SiN, silicon nitride.
HIPS chemistry provides mechanically robust covalent coupling
In comparison to the longevity of our tips in DNA assays, the yield of high-quality protein data showed a decrease in efficiency after 2.5 h (~300 pulls) when we functionalized the aldehyde-labelled polyprotein with commercial oxyamine (and hydrazide) reagents instead of the HIPS-based reagents (Figure S2). We attributed this decline to mechanically induced degradation of the oxime linkage. Rupture of this linkage, in turn, reduced the activity of the streptavidin-coated tip by occupying the accessible biotin-binding sites and/or developing an unwanted protein coat at the apex of the tip. We therefore developed HIPS-based reagents, which significantly improved stability at neutral pH.38 We describe the synthesis of such reagents (see Supporting Information), which are now also commercially available from a third party (Click Chemistry Tools). With these reagents, hundreds of individual polyproteins can be mechanically unfolded by the same individual tip (Figure S2), with only minimal loss in the activity of the tip over days. We therefore conclude that HIPSbased functionalization of the aldehyde is mechanically robust.
Dynamic force spectra of a single, individual polyprotein
Heterogeneity in the folding and unfolding of individual RNA molecules has been observed by single-molecule fluorescence.39,40 However, in traditional AFM studies, dynamic force spectra are derived from a large number of rupture forces from a multitude of different molecules, masking any such heterogeneity. To enable looking for such heterogeneity, we first wanted to establish the necessary experimental capability to derive a full dynamic spectrum from a single individual polyprotein.
To do so, we used our (NuG2)4 polyprotein (Figure 5a) that contained a biotin at one end and a DBCO at the other. As before, the assay was initiated by pressing the tip into the surface at a low force (~100 pN) and then retracting it at constant velocity (v = 50–1,000 nm/s). We again achieved a high percentage of force-extension records (Figure 5b) that showed minimal surface adhesion and all four protein-unfolding events. For instance, in a set of 200 sequential attempts, six sequential force-extension curves contained the full polyprotein unfolding (Figure S3), and ~14% of the ~200 attempts yielded full unfolding. This success rate is very high by AFM standards, albeit lower than our results with DNA (Figure 1g), presumably due to the polyprotein’s much smaller radius of gyration compared to a 650-nm-long DNA molecule. Analysis of the force-extension curves yielded a change in contour length (ΔL = 17.4 nm) that matched the literature values for NuG2 (ΔL ≈ 17.6 nm).30,31 We then repeated the measurement at different pulling velocities to acquire a dynamic force spectrum (Figure 5c) at one surface location (Figure 5d) for a total of 357 unfolding events in 2 h.
Figure 5.
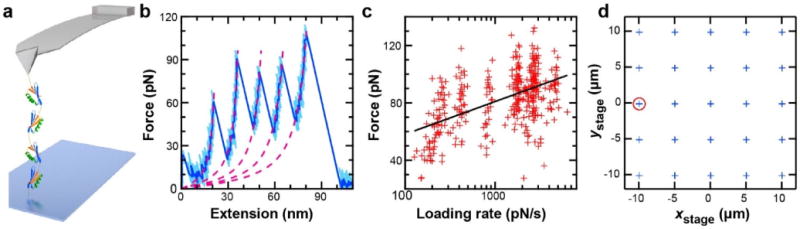
Dynamic force spectroscopy of a single individual polyprotein. (a) Schematic of experiment showing a polyprotein of four repeats of NuG2 [i.e., (NuG2)4] covalently bound to a PEG-coated surface and reversibly bound to a streptavidin-coated cantilever. (b) A force-extension curve shows the full unfolding of (NuG2)4 and minimal adhesion at small extensions (<10 nm). Highbandwidth data (50 kHz, light blue) were filtered to 200 Hz (dark blue). Magenta dashed lines represent worm-like chain fits. (c) A dynamic force spectrum for NuG2 generated from one individual polyprotein where each data point (red; N = 357) represents the rupture of an individual NuG2 domain. Bell-Evans analysis of this data (black line) yielded values for the zero-force off-rate (ko = 0.03 s−1) and distance to the transition (Δx‡ = 4.2 Å). (d) Surface locations (xstage and ystage, blue crosses) probed during polyprotein experiment. All data for (c) were measured from one location (red circle) with no indication of multiple attachments (Figure S4).
We interpret this data set as a single individual polyprotein being repeatedly unfolded for the following three reasons: (i) displacements away from a particular location (Figure 5d, red circle) led to the absence of attachment; (ii) none of the force-extension curves over the 2-h period showed any signature of multiple proteins being attached to the tip in parallel (Figure S4); and (iii) the purposely low probability (8 %) of achieving molecular attachment at a particular location in this experiment argues for a very low probability (0.6 %) of stretching multiple different molecules at one location.41
To compare our data with existing results, we analyzed the rupture force vs loading rate using a Bell-Evans model,42 since the Bell-Evans analysis is often used in AFMbased SMFS and has been applied to NuG2 recently.31 Analysis of a single individual polyprotein yielded ko = 0.03 ± 0.02 s−1 and Δx‡ = 4.2 ± 0.4 Å, where ko is the off-rate at zero force, and Δx‡ is the distance to the unfolding-transition state. These parameters are in quantitative agreement with the recently reported values (0.04 s−1, and 4.2 Å).31 Hence, the finite strength of the streptavidin-biotin bond did not lead to an altered result, at least for NuG2, which is considered a mechanically robust protein.30 Rather, this strong but reversible attachment of the polyprotein to the tip in conjunction with its covalent anchoring to an otherwise nonstick surface significantly accelerated the acquisition of high-quality AFM-based SMFS data and thereby enables probing for kinetic differences between individual proteins and, more generally, acquiring large, high-quality data sets rapidly.
Notwithstanding our present success using a streptavidin-biotin linkage to study NuG2 (which builds upon earlier success in studying a high-force structural transition in polysaccharides43), this linkage is not widely used in AFM in comparison to its ubiquitous use in optical-trapping and magnetic-tweezer-based assays.1 In part, this difference arises because of the historically labor intensive process for labelling AFM tips and that AFM has been used to study proteins that are more mechanically robust than NuG2, such as titin’s IG domain.10,13 In such studies, the streptavidin-biotin bond can dissociate before a polyprotein completely unfolds, decreasing throughput.
In our assay, a simple path to increasing the strength of the linkage between the biomolecule under study and the AFM tip is to use substrates containing multiple, closely spaced biotins, a common practice in torsionally constrained DNA44,45 and DNA overstretching assays.46,47 More generally, our short genetically encoded aldehyde tags provide an orthogonal chemical moiety to incorporate future advances into our polyprotein construct. Indeed, as better ligands are introduced, a HIPS-based version of that ligand can be developed. These stronger but still reversible ligands could then be immediately incorporated to further advance the utility and throughput of AFM-based SMFS.
A versatile platform for studying diverse globular proteins
To demonstrate the versatility of our technology, we tested three different proteins: (i) calmodulin, (ii) rubredoxin, an iron-binding protein containing four internal cysteines, and (iii) α3D, a computationally designed three helix bundle48 not previously characterized by SMFS. The plasmid underlying the polyprotein construct enables efficient insertion of new protein sequences through a multiple cloning site amenable to both typical restriction digest cloning and sticky end cloning,49 a method that does not rely on digestion of the DNA insert. All three proteins expressed well within the dual-aldehyde construct and yielded high-quality SMFS data with a mechanical fingerprint matching the size of the inserted protein (Figure 6a–c). As expected, the rupture forces for calmodulin and rubredoxin agreed with previous studies.50,51
Figure 6.
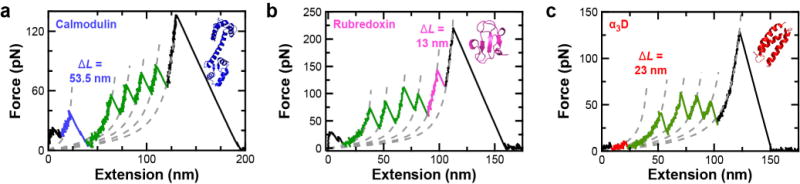
Efficient SMFS studies of diverse proteins. (a–c) FECs show the complete unfolding of a polyprotein containing calmodulin, rubredoxin and α3D, respectively. Segments of the FEC between domain ruptures are well described by a worm-like chain model (grey dashed lines). The contour length (ΔL) increases associated with each protein agree with the number of unfolded amino acids and the known structure. FEC data smoothed to 1 kHz.
Given the unfolding of proteins with high α-helical content generally occurs at low forces and low extensions, it is often masked by surface adhesion and the typical force noise in AFM of 5–10 pN.1 Thus, it is noteworthy that minimal surface adhesion was observed in the force-extension curves at low extensions, even when α3D unfolded at less than 12 pN (Figure 7a). In part, this success arises from integrating site-specific anchoring to PEG-coated surfaces with improved force stability achieved by removing the metallic coating from the cantilevers.17 Additionally, the traces obtained were exceptionally consistent, requiring minimal lateral shifts. For example, we generated a heat map, a standard AFM analysis, by overlaying 11 individual force-extension curves without any lateral offset (Figure 7b). Importantly, the insertion of mechanically labile target proteins (e.g., α3D and calmodulin) into the polyprotein resulted in similarly high efficiencies of the complete unfolding of the full construct (Figure S5). Thus, we acquired high quality data of diverse mechanically labile proteins, while still maintaining excellent data throughput.
Figure 7.
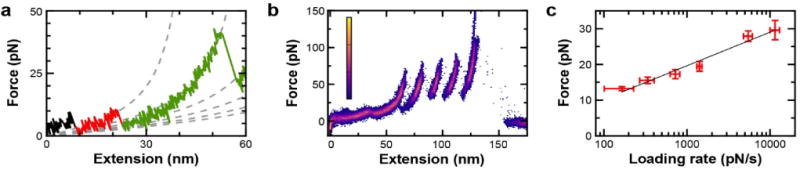
Dynamic force spectrum for α3D. (a) A force-extension curve showing an expanded view of α3D unfolding. (b) A heat map of 11 traces of α3D generated with no lateral offset, a commonly applied correction in AFM-based SMFS. (c) A dynamic force spectrum for α3D shows the mean rupture force as a function of loading rate. Errors represent the standard error of the mean. Analysis of this data with a Bell-Evans model (black line) yielded the distance to the transition and the zero-force off-rate (k0 = 2.0 ± 0.7 s−1). FEC data smoothed to 1 kHz.
Characterizing the free-energy landscape of mechanically labile proteins with AFM
We leveraged our increased efficiency and high sensitivity at low force to mechanically characterize α3D, obtaining its dynamic force spectrum (Figure 7c). To do so, the rupture force was measured as a function of loading rate, a process that can often take days to weeks. In contrast, we acquired a spectrum of a mechanically fragile protein in just ~24 h of instrument time, despite only analyzing force-extension curves that exhibited the full unfolding of the polyprotein (i.e., α3D plus four NuG2 ruptures followed by tip detachment). Further acceleration could be achieved if we optimized the data acquisition protocol to acquire hundreds of records from individual molecules and averaged over multiple molecules. However, in this first SFMS study of α3D, we chose a more traditional approach that incorporated raster scanning of the tip across the protein-coated surface.
We analyzed the α3D dynamic force spectrum using the Bell-Evans model,4 which is the standard for AFM-based SMFS. For α3D, this analysis yielded Δx‡ = 1.0 ± 0.1 nm, and k0 = 2.0 ± 0.7 s−1. Hence, as expected for an all-α-helical protein, α3D unfolds at low force and is mechanically compliant (large Δx‡) relative to proteins that contain β-sheet structures,13 including NuG2 (Δx‡ = 0.42 nm).30,31 Indeed, the mean rupture force of α3D at 165 pN/s (v = 50 nm/s) is less than that of calmodulin,50 making it arguably the mechanically weakest protein studied to date by AFM.
Conclusions
In summary, to accelerate the acquisition of high-quality SMFS data by AFM, we co-developed a new polyprotein construct featuring a versatile and mechanically robust HIPS chemistry in parallel with a conjugation protocol to site-specifically anchor biomolecules to otherwise nonstick surfaces in an accessible and significantly simplified manner. We then used protein and DNA molecules that were labelled at one end with DBCO, a copper-free click chemistry reagent, and at the other end with biotin. The DBCO label enabled us to leverage the efficiency and bioorthogonality of click chemistry while avoiding the detrimental effects of copper ions on many biomolecules.26 The biotin label provided a simple and accessible means to strongly but reversibly attach the DNA or polyprotein to a streptavidin coated tip, avoiding an undesired macromolecular coating at the apex of a tip that can decrease yield over time.
Importantly, this reversible linkage allowed hundreds to thousands of individual molecules to be probed and reprobed by a single cantilever over days to weeks, improving throughput and precision. Overall, we achieved a 75fold increase in the yield of high-quality protein-unfolding data in comparison to traditional AFM-based assays that rely upon non-specific attachment. Importantly, our advances in site-specific coupling are not limited to AFMbased SMFS, but should benefit other SMFS modalities. Indeed, we demonstrated such broader utility by overstretching DNA, a relatively high-force transition27,28 using both an AFM and an optical trap. Additionally, our labelling scheme using a cysteine and a 6-amino-acid tag can be extended to internal sites within a protein to control the directionality of the applied force, similar to prior work using a dual cysteine system,52 but with the added benefit of orthogonal coupling chemistry. More broadly, the increased rate of acquiring high-quality data demonstrated here should facilitate rapid collection of large data sets, enabling more complete investigation of macromolecular folding over a range of physiochemical conditions (pH, temperature, denaturant).
Supplementary Material
Acknowledgments
We thank Lyle Uyetake for preparing DNA, Mark Kastantin for technical discussions, Xinghua Shi for an FGE-encoding plasmid, and the Central Analytical Laboratory and Mass Spectrometry Facility at the University of Colorado at Boulder. This work was supported by a National Institute of Health (NIH) Molecular Biophysics Training Grant awarded to M.A.L. and S.R.O. (T32 GM-065103), National Research Council Fellowship to R.W. & D.T.E, the National Science Foundation (DBI-135398 to T.T.P.; Phys-1125844), NIH (R01 AI080709 to M.C.S.), NIH (S10 RR026641 to the Mass Spectrometry Facility), a Butcher Grant (to T.T.P.), and NIST. T.T.P. is a staff member of NIST’s Quantum Physics Division.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Plasmids are available for distribution from Addgene, plasmid IDs 80159 (heterobifunctional) and 80163 (dual-aldehyde).
Cloning methods, HIPS reagents synthetic schemes, and control experiment comparing oxyamine and HIPS stability to force (PDF)
ORCID
Marcelo Sousa: 0000-0002-0242-3619
Thomas Perkins: 0000-0003-4826-9490
Notes
David Rabuka is an employee of Catalent Biologics, which has partnered with Clickchem Tools for the distribution of reagents.
References
- 1.Neuman KC, Nagy A. Nat Methods. 2008;5:491. doi: 10.1038/nmeth.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greenleaf WJ, Woodside MT, Block SM. Annu Rev Biophys Biomol. 2007;36:171. doi: 10.1146/annurev.biophys.36.101106.101451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alsteens D, Tay S, Muller DJ. Nat Methods. 2015;12:45. doi: 10.1038/nmeth.3216. [DOI] [PubMed] [Google Scholar]
- 4.Bornschlogl T, Rief M. Methods Mol Biol. 2011;783:233. doi: 10.1007/978-1-61779-282-3_13. [DOI] [PubMed] [Google Scholar]
- 5.Yu H, Liu X, Neupane K, Gupta AN, Brigley AM, Solanki A, Sosova I, Woodside MT. Proc Natl Acad Sci USA. 2012;109:5283. doi: 10.1073/pnas.1107736109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu H, Siewny MG, Edwards DT, Sanders AW, Perkins TT. Science. 2017;355:945. doi: 10.1126/science.aah7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cossio P, Hummer G, Szabo A. Proc Natl Acad Sci USA. 2015;112:14248. doi: 10.1073/pnas.1519633112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carrion-Vazquez M, Oberhauser AF, Fisher TE, Marszalek PE, Li H, Fernandez JM. Prog Biophys Mol Biol. 2000;74:63. doi: 10.1016/s0079-6107(00)00017-1. [DOI] [PubMed] [Google Scholar]
- 9.Carrion-Vazquez M, Oberhauser AF, Fowler SB, Marszalek PE, Broedel SE, Clarke J, Fernandez JM. Proc Natl Acad Sci USA. 1999;96:3694. doi: 10.1073/pnas.96.7.3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rief M, Gautel M, Oesterhelt F, Fernandez JM, Gaub HE. Science. 1997;276:1109. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 11.Dietz H, Rief M. Proc Natl Acad Sci USA. 2004;101:16192. doi: 10.1073/pnas.0404549101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bosshart PD, Frederix PL, Engel A. Biophys J. 2012;102:2202. doi: 10.1016/j.bpj.2012.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffmann T, Dougan L. Chem Soc Rev. 2012;41:4781. doi: 10.1039/c2cs35033e. [DOI] [PubMed] [Google Scholar]
- 14.Stigler J, Ziegler F, Gieseke A, Gebhardt JCM, Rief M. Science. 2011;334:512. doi: 10.1126/science.1207598. [DOI] [PubMed] [Google Scholar]
- 15.Jahn M, Buchner J, Hugel T, Rief M. Proc Natl Acad Sci USA. 2016;113:1232. doi: 10.1073/pnas.1518827113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cecconi C, Shank EA, Bustamante C, Marqusee S. Science. 2005;309:2057. doi: 10.1126/science.1116702. [DOI] [PubMed] [Google Scholar]
- 17.Churnside AB, Sullan RM, Nguyen DM, Case SO, Bull MS, King GM, Perkins TT. Nano Lett. 2012;12:3557. doi: 10.1021/nl301166w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zimmermann JL, Nicolaus T, Neuert G, Blank K. Nat Protoc. 2010;5:975. doi: 10.1038/nprot.2010.49. [DOI] [PubMed] [Google Scholar]
- 19.Kufer SK, Dietz H, Albrecht C, Blank K, Kardinal A, Rief M, Gaub HE. Eur Biophys J. 2005;35:72. doi: 10.1007/s00249-005-0010-1. [DOI] [PubMed] [Google Scholar]
- 20.Taniguchi Y, Kawakami M. Langmuir. 2010;26:10433. doi: 10.1021/la101658a. [DOI] [PubMed] [Google Scholar]
- 21.Zakeri B, Fierer JO, Celik E, Chittock EC, Schwarz-Linek U, Moy VT, Howarth M. Proc Nat Acad Sci USA. 2012;109:E690. doi: 10.1073/pnas.1115485109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Popa I, Berkovich R, Alegre-Cebollada J, Badilla CL, Rivas-Pardo JA, Taniguchi Y, Kawakami M, Fernandez JM. J Am Chem Soc. 2013;135:12762. doi: 10.1021/ja4056382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Otten M, Ott W, Jobst MA, Milles LF, Verdorfer T, Pippig DA, Nash MA, Gaub HE. Nat Methods. 2014;11:1127. doi: 10.1038/nmeth.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baumann F, Bauer MS, Milles LF, Alexandrovich A, Gaub HE, Pippig DA. Nat Nanotechnol. 2016;11:89. doi: 10.1038/nnano.2015.231. [DOI] [PubMed] [Google Scholar]
- 25.Stahl SW, Nash MA, Fried DB, Slutzki M, Barak Y, Bayer EA, Gaub HE. Proc Natl Acad Sci USA. 2012;109:20431. doi: 10.1073/pnas.1211929109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Proc Natl Acad Sci USA. 2007;104:16793. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith SB, Cui Y, Bustamante C. Science. 1996;271:795. doi: 10.1126/science.271.5250.795. [DOI] [PubMed] [Google Scholar]
- 28.Cluzel P, Lebrun A, Heller C, Lavery R, Viovy JL, Chatenay D, Caron F. Science. 1996;271:792. doi: 10.1126/science.271.5250.792. [DOI] [PubMed] [Google Scholar]
- 29.Wu P, Shui W, Carlson BL, Hu N, Rabuka D, Lee J, Bertozzi CR. Proc Natl Acad Sci USA. 2009;106:3000. doi: 10.1073/pnas.0807820106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao Y, Balamurali MM, Sharma D, Li HB. Proc Natl Acad Sci USA. 2007;104:15677. doi: 10.1073/pnas.0705367104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He C, Hu C, Hu X, Xiao A, Perkins TT, Li H. Angew Chem Int Edit. 2015;54:9921. doi: 10.1002/anie.201502938. [DOI] [PubMed] [Google Scholar]
- 32.Shi X, Jung Y, Lin LJ, Liu C, Wu C, Cann IK, Ha T. Nat Methods. 2012;9:499. doi: 10.1038/nmeth.1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hermanson GT. Bioconjugate Techniques. Academic Press; San Diego: p. 1996. [Google Scholar]
- 34.Bull MS, Sullan RM, Li H, Perkins TT. ACS Nano. 2014;8:4984. doi: 10.1021/nn5010588. [DOI] [PubMed] [Google Scholar]
- 35.Edwards DT, Faulk JK, Sanders AW, Bull MS, Walder R, LeBlanc MA, Sousa MC, Perkins TT. Nano Lett. 2015;15:7091. doi: 10.1021/acs.nanolett.5b03166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sader JE, Chon JWM, Mulvaney P. Rev Sci Instrum. 1999;70:3967. [Google Scholar]
- 37.Bustamante C, Bryant Z, Smith SB. Nature. 2003;421:423. doi: 10.1038/nature01405. [DOI] [PubMed] [Google Scholar]
- 38.Agarwal P, Kudirka R, Albers AE, Barfield RM, de Hart GW, Drake PM, Jones LC, Rabuka D. Bioconjugate Chem. 2013;24:846. doi: 10.1021/bc400042a. [DOI] [PubMed] [Google Scholar]
- 39.Tan E, Wilson TJ, Nahas MK, Clegg RM, Lilley DM, Ha T. Proc Natl Acad Sci U S A. 2003;100:9308. doi: 10.1073/pnas.1233536100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okumus B, Wilson TJ, Lilley DM, Ha T. Biophys J. 2004;87:2798. doi: 10.1529/biophysj.104.045971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Block SM, Goldstein LS, Schnapp BJ. Nature. 1990;348:348. doi: 10.1038/348348a0. [DOI] [PubMed] [Google Scholar]
- 42.Evans E, Ritchie K. Biophys J. 1997;72:1541. doi: 10.1016/S0006-3495(97)78802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rief M, Oesterhelt F, Heymann B, Gaub HE. Science. 1997;275:1295. doi: 10.1126/science.275.5304.1295. [DOI] [PubMed] [Google Scholar]
- 44.Strick TR, Allemand JF, Bensimon D, Bensimon A, Croquette V. Science. 1996;271:1835. doi: 10.1126/science.271.5257.1835. [DOI] [PubMed] [Google Scholar]
- 45.Deufel C, Forth S, Simmons CR, Dejgosha S, Wang MD. Nat Methods. 2007;4:223. doi: 10.1038/nmeth1013. [DOI] [PubMed] [Google Scholar]
- 46.Paik DH, Perkins TT. J Am Chem Soc. 2011;133:3219. doi: 10.1021/ja108952v. [DOI] [PubMed] [Google Scholar]
- 47.Williams MC, Wenner JR, Rouzina I, Bloomfield VA. Biophys J. 2001;80:1932. doi: 10.1016/S0006-3495(01)76163-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu Y, Alonso DO, Maki K, Huang CY, Lahr SJ, Daggett V, Roder H, DeGrado WF, Gai F. Proc Natl Acad Sci USA. 2003;100:15486. doi: 10.1073/pnas.2136623100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zeng G. Biotechniques. 1998;25:206. doi: 10.2144/98252bm05. [DOI] [PubMed] [Google Scholar]
- 50.Junker JP, Ziegler F, Rief M. Science. 2009;323:633. doi: 10.1126/science.1166191. [DOI] [PubMed] [Google Scholar]
- 51.Zheng P, Takayama SJ, Mauk AG, Li H. J Am Chem Soc. 2013;135:7992. doi: 10.1021/ja402150q. [DOI] [PubMed] [Google Scholar]
- 52.Dietz H, Bertz M, Schlierf M, Berkemeier F, Bornschlogl T, Junker JP, Rief M. Nat Protoc. 2006;1:80. doi: 10.1038/nprot.2006.12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.