

Abstract
Bivalent ligands targeting putative melanocortin receptor dimers have been developed and characterized in vitro, however studies of their functional in vivo effects have been limited. The current report compares the effects of homobivalent ligand CJL-1-87, Ac-His-DPhe-Arg-Trp-PEDG20-His-DPhe-Arg-Trp-NH2, to monovalent ligand CJL-1-14, Ac-His-DPhe-Arg-Trp-NH2 on energy homeostasis in mice after central intracerebroventricular (ICV) administration into the lateral ventricle of the brain. Bivalent ligand CJL-1-87 had noteworthy advantages as an anti-obesity probe over CJL-1-14 in a fasting-refeeding in vivo paradigm. Treatment with CJL-1-87 significantly decreased food intake compared to CJL-1-14 or saline (50% less intake 2 to 8 hours after treatment). Furthermore, CJL-1-87 treatment decreased the respiratory exchange ratio (RER) without changing the energy expenditure indicating that fats were being burned as the primary fuel source. Additionally, CJL-1-87 treatment significantly lowered body fat mass percentage 6 hours after administration (p < 0.05) without changing the lean mass percentage. The bivalent ligand significantly decreased insulin, C-peptide, leptin, GIP, and resistin plasma levels compared to levels after CJL-1-14 or saline treatments. Alternatively, ghrelin plasma levels were significantly increased. Serum stability of CJL-1-87 and CJL-1-14 (T1/2 = 6.0 h and 16.8 h, respectively) was sufficient to permit physiological effects. The differences in binding affinity of CJL-1-14 compared to CJL-1-87 are speculated as a possible mechanism for the bivalent ligand’s unique effects. We also provide in vitro evidence for the formation of a MC3R-MC4R heterodimer complex, for the first time to our knowledge, that may be an unexploited neuronal molecular target. Regardless of the exact mechanism, the advantageous ability of CJL-1-87 compared to CJL-1-14 to increase in vitro binding affinity, increase the duration of action in spite of decreased serum stability, decrease in vivo food intake, decrease mice’s body fat percent, and differentially affect mouse hormone levels demonstrates the distinct characteristics achieved from the current melanocortin agonist bivalent design strategy.
Keywords: melanocortin homodimer, obesity, metabolic serum stability, melanotropin, MC3R-MC4R heterodimer, BRET, interleukin-6 (IL-6), magnetic resonance imagining (MRI), metabolic syndrome
TOC image
Introduction
Bivalent ligand design strategies have been utilized to develop novel ligands for various GPCR systems including the opioids,1–8 gonadotropin-releasing hormone receptor,9, 10 adenosine,11 cannabinoid,12, 13 serotonin,14–16 dopamine,17, 18 chemokine,6, 19 oxytocin,20 and melanocortin receptor systems.21–36 There has been increasing evidence that heterobivalent ligands featuring pharmacophores for two different receptors can be an efficacious targeting strategy for heterodimers and results in unique properties in vivo.6, 7, 37, 38 Reports of homobivalent ligands containing two pharmacophores for the same receptor have shown them to possess distinct characteristics in vitro compared to monovalent ligands. Reports of homobivalent ligands’ in vivo functions compared to their monovalent counterparts are sparser.20, 21, 39, 40 Because bivalent ligands can have unique functional characteristics compared to monovalent ligands that are not easily assayed in vitro (i.e. alterations of receptor trafficking41–43 or tissue selectivity1, 6), it is important to establish their in vivo functional significance. This in vivo data can be used to guide future bivalent drug design strategies and identify molecular probes for in vivo mechanism of action hypothesis driven research. A recent report demonstrated that homobivalent ligands targeting the oxytocin receptor system exhibited 40- and 100-fold greater potency in zebrafish and mice, respectively, establishing that a homobivalent design strategy can have noteworthy advantages in vivo.20
There have been several reports of bivalent ligands targeting the melanocortin receptor system.21–36 Many of these studies focus on the development of bivalent ligands for their use as high affinity imaging tools or targeting agents for melanoma.24–36 Given that chemical probes for the five melanocortin receptor subtypes (MC1-5R) have been utilized to study several diseases and disorders including Alzheimer’s disease, 44–46 sexual function disorders, 47, 48 social disorders, 49, 50 cachexia, 51–56 and obesity; 21, 57, 58 it is of interest to study the biological functions of bivalent ligands targeting these receptors. The limited reports of the in vitro functional effects of melanocortin homobivalent have all shown increased functional potency (up to 16-fold).21, 22, 24 Although melanocortin bivalent ligands have been used in vivo as imaging tools;34–36 to our knowledge no other laboratories have reported on the functional effects of melanocortin homobivalent ligands in vivo.21
We have previously reported melanocortin homobivalent ligands possessed increased binding affinity of 14- to 25-fold, and increased in vitro functional potency of 3- to 5-fold depending on the individual melanocortin receptor subtype compared to monovalent control counterparts.21 Specifically, compound CJL-1-87, Ac-His-DPhe-Arg-Trp-PEDG20-His-DPhe-Arg-Trp-NH2, was the most potent bivalent ligand at the melanocortin 3 receptor (MC3R) and melanocortin 4 receptor (MC4R). This compound consists of two monovalent agonist His-DPhe-Arg-Trp tetrapeptide scaffolds connected through a 20-atom polyethylene diamine diglycolyic acid linker (PEDG20). It had an increased binding affinity of 23- and 22- fold, and an increased functional potency of 5- and 4-fold compared to its monovalent counterpart CJL-1-14, Ac-His-DPhe-Arg-Trp-NH2, at the MC3R and MC4R, respectively. As predicted, administering CJL-1-87 directly into the brain dose-dependently decreased in food intake demonstrating its in vivo efficacy at the MC3R and/or MC4R.21
The MC3R and MC4R are centrally located and are implicated in the melanocortin system’s role in energy homeostasis.57, 59, 60 Agonist stimulation of the MC3R and MC4R decreases food intake and increases energy expenditure. Therefore, agonist ligands may be potential therapeutics for the treatment of obesity.57, 59–61 It is hypothesized that the distinct pharmacological profile of agonist homobivalent ligand CJL-1-87 may be advantageous in vivo compared to monovalent agonist ligands by enhancing desirable effects such as decreasing food intake and weight loss while hopefully minimizing undesirable side effects. However, given the lack of studies directly comparing the effects of melanocortin monovalent and bivalent ligands in vivo; exploratory in vivo studies are necessary to characterize the physiological profile of homobivalent ligands, assess their side effect profiles, and guide future design and in vitro SAR studies. Direct comparison of bivalent ligand CJL-1-87 to monovalent ligand CJL-1-14 may provide insight into the advantages and disadvantages of a melanocortin bivalent ligand design strategy in the development of anti-obesity therapeutics. These studies may also help further elucidate the complex melanocortin pharmacology within the brain and particularly the role of the MC3-MC4 melanocortin receptor homo- and heterodimerization.
Previously published literature pertinent to comparing CJL-1-87 to its monovalent counterpart was inconclusive as to whether there is any significant advantages or disadvantages of the bivalent ligand CJL-1-87 over the monovalent ligand CJL-1-14 in vivo. 21, 57 Thus it is necessary to perform direct head to head comparison studies in different experimental paradigms to draw definitive conclusions.21, 57 In the current study, evidence is provided that the melanocortin bivalent agonist ligand CJL-1-87 possesses distinct advantages in a mouse fasting-refeeding paradigm compared to its monovalent counterpart CJL-1-14 including: decreased in vivo food intake, reduced in vivo body fat percentage, and differentially effected mouse hormone levels. These data have led us to postulate two possible mechanisms to explain the differences in the in vivo pharmacology between CJL-1-14 and CJL-1-87. The first possible mechanism is that CJL-1-87 can compete more effectively with endogenous antagonist agouti-related peptide (AGRP) in the fasting state due to its increased binding affinity compared to CJL-1-14. The other possible mechanism is that CJL-1-87 is targeting melanocortin dimers. To test this hypothesis, bioluminescence resonance energy transfer (BRET) studies were performed and support the feasibility of MC3R-MC4R heterodimer formation in vitro. This heterodimer may be a novel neuronal target for treating metabolic disorders, but its physiological relevancy remains to be determined. Although the exact molecular mechanism accounting for the differences observed in vivo remains unclear, the current studies provide direct evidence that the ICV administration of bivalent ligand CJL-1-87 and monovalent control CJL-1-14 result in different effects on energy homeostasis in mice in the fasting-refeeding experimental paradigm.
Results and Discussion
In Vitro Mouse Serum Stability Assays
In order to assess the metabolic stability of the peptides, in vitro mouse serum stability assays were performed (Figure 1, Table 1). As reported previously, α-MSH was rapidly degraded with less than 1% intact peptide remaining following a 6 h incubation (Supplemental Table 1).62–64 It had a half-life of 0.9 h which is comparable with previous literature reports in rat serum.62 Compound CJL-1-14, Ac-His-DPhe-Arg-Trp-NH2, was the most stable peptide in mouse serum and had a half-life of 16.8 h. After incubation in mouse serum for 72 hours 12% of the Ac-His-DPhe-Arg-Trp-NH2 peptide remained intact. The next most stable peptides were the linker control peptides CJL-5-35-4 and CJL-1-116 that have a PEDG20 linker added to the N-terminus or C-terminus to Ac-His-DPhe-Arg-Trp-NH2. They possessed half-lives of 12.5 h and 10.6 h, respectively. This supports the hypothesis that the PEDG20 linker is not rapidly degraded in mouse serum. The PEDG20 based bivalent ligand CJL-1-87 had similar stability to NDP-MSH, the enzymatically stable analog of α-MSH.63, 64 Compound CJL-1-87 and NDP-MSH had half-lives of 6.0 and 5.1 h, respectively. Considering NDP-MSH is currently approved for clinical use in the European Union,65 the similar half-lives of CJL-1-87 and NDP-MSH suggest that CJL-1-87 would have a reasonable metabolic stability for in vivo applications.
Figure 1.
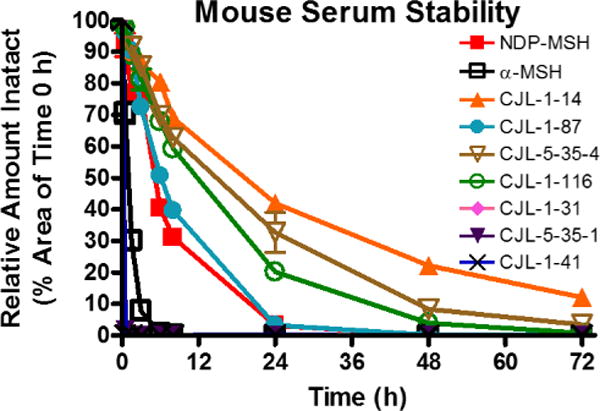
In vitro serum stability of bivalent ligands and control ligands. Ligands (10 µM) were incubated in mouse serum and monitored for degradation of the parent molecule by LC-ESI+-MS/MS. The PEDG20 based compounds were relatively metabolically stable, whereas (Pro-Gly)6 based compounds were rapidly degraded.
Table 1.
Sequence information of compounds used in the serum stability assays and their half-lives. Half-lives were calculated from two technical replicates of the in vitro serum stability assays (see Figure 1).
| Analog | Sequence | Half-Life (h) |
|---|---|---|
| NDP-MSH | Ac-Ser-Try-Ser-Nle-Glu-His-DPhe-Arg-Trp-Gly-Lys-Pro-Val-NH2 | 5.1 |
| α-MSH | Ac-Ser-Try-Ser-Met-Glu-His-Phe-Arg-Trp-Gly-Lys-Pro-Val-NH2 | 0.9 |
| CJL-1-14 | Ac-His-DPhe-Arg-Trp-NH2 | 16.8 |
| CJL-1-87 | Ac-His-DPhe-Arg-Trp-(PEDG20)-His-DPhe-Arg-Trp-NH2 | 6.0 |
| CJL-5-35-4 | Ac-His-DPhe-Arg-Trp-(PEDG20)-NH2 | 12.5 |
| CJL-1-116 | (PEDG20)-His-DPhe-Arg-Trp-NH2 | 10.6 |
| CJL-1-31 | Ac-His-DPhe-Arg-Trp-(Pro-Gly)6-His-DPhe-Arg-Trp-NH2 | 0.1 |
| CJL-5-35-1 | Ac-His-DPhe-Arg-Trp-(Pro-Gly)6-NH2 | 0.1 |
| CJL-1-41 | (Pro-Gly)6-His-DPhe-Arg-Trp-NH2 | 0.1 |
The current results demonstrate that the bivalent ligand CJL-1-87 is metabolized faster by the serum proteases present in the mouse serum than its monovalent counterpart CJL-1-14. This implies that the bivalent ligand CJL-1-87 would be at a disadvantage to the monovalent ligand CJL-1-14 if in vivo metabolic stability is desired. In order to interpret the data completely, it must be noted that it would be necessary to metabolically inactivate both pharmacophores of CJL-1-87 before it was rendered completely pharmacologically inactive. For example, if the N-terminal His-DPhe-Arg-Trp was cleaved by serum proteases, the C-terminal His-DPhe-Arg-Trp would retain activity at the melanocortin receptors similar to that of CJL-1-116 (cAMP signaling EC50 = 31 nM and 19 nM at the mMC3R and mMC4R, respectively) until the C-terminal His-DPhe-Arg-Trp was metabolized.21 In the current study, only the loss of fully intact peptide was monitored to give an indication of metabolic stability. Further studies into the rate of functional inactivation of the peptides will be necessary to determine how fast pharmacological activity is lost, but is outside the scope of the current study.
The previously reported bivalent ligands that were synthesized using a (Pro-Gly)6 linker were included in the current study for comparison of the linker design for future in vivo applications.21 The linker control ligands with the tetrapeptide and the (Pro-Gly)6, CJL-5-35-1 and CJL-1-41, and the bivalent ligand CJL-1-31 were rapidly degraded (Figure 1, Table 1). There was ≤ 2% intact peptide of CJL-1-41, CJL-1-31 and CJL-5-35-1 remaining after 0.5 h (Supplemental Table 1). It is unclear currently whether the loss of intact peptide results in loss of functional melanocortin activity or if the linker is merely being degraded rapidly while the active His-DPhe-Arg-Trp pharmacophore remains intact. Nevertheless, the current results validate the previous hypothesis that although a Pro-Gly based linker may be useful for determining in vitro pharmacology,21 its use is limited for in vivo applications to study bivalent design strategies unless rapid degradation is desired. It should also be noted that if a Pro-Gly linker system is used for in vitro applications in any bivalent design strategy for any receptor system, the supplementation of assay buffer with serums [i.e. fetal bovine serum (FBS), newborn calf serum (NCS)] may result in rapid degradation of the linker system.
Beyond the useful data obtained about the bivalent design strategy and metabolic stability, the current data demonstrates for the first time that the Ac-His-DPhe-Arg-Trp-NH2 is more stable in mouse serum than NDP-MSH (Figure 1, Table 1). This suggests that Ac-His-DPhe-Arg-Trp-NH2 is less susceptible to common proteases found in mouse serum and supports further research into the tetrapeptide scaffold.
The Effect of ICV Administration of CJL-1-14 versus CJL-1-87 on Mouse Energy Homeostasis
A direct head to head in vivo crossover experimental paradigm (Figure 2A) was designed to compare 5 nmol monovalent CJL-1-14 to 5 nmol bivalent CJL-1-87 utilizing the TSE Phenotypic metabolic cages configured to measure food intake, water intake, changes in CO2 and O2, and beam break activity. In a nocturnal satiated paradigm in which compound was administered ICV two hours before lights out and food is available ad libitum, no significant differences were observed in food intake, water intake, energy expenditure, respiratory exchange ratio (RER) values, or activity between CJL-1-14 and CJL-1-87. The observation of approximately equal food intake in this paradigm was consistent with the previous reports of food intake after CJL-1-14 and CJL-1-87 treatment in the literature, and no further experiments utilizing this experimental paradigm were performed.21, 57 However, significant differences between bivalent ligand CJL-1-87 and monovalent ligand CJL-1-14 were observed on food intake and RER values when utilizing a fasting-refeeding paradigm (Figure 2B, Figure 3).
Figure 2.
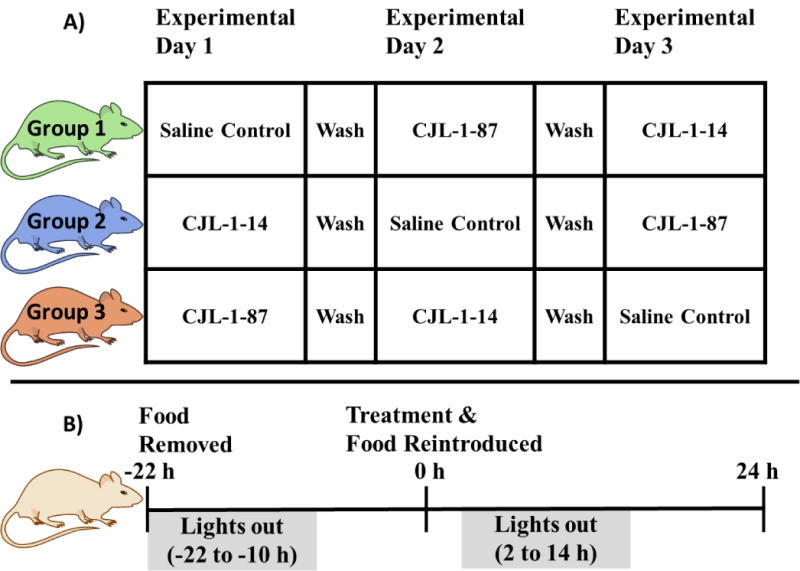
Experimental paradigms used to study energy homeostasis. A) Head to head crossover design of experimental groups. B) Fasting-refeeding experimental paradigm in which significant results were observed. Saline is the vehicle control and ligand treatment consisted of a 5 nmol dose.
Figure 3.
Investigation of 5 nmol bivalent ligand CJL-1-87 (
 ) compared to 5 nmol monovalent ligand CJL-1-14 (
) compared to 5 nmol monovalent ligand CJL-1-14 (
 ) (A) and saline (
) (A) and saline (
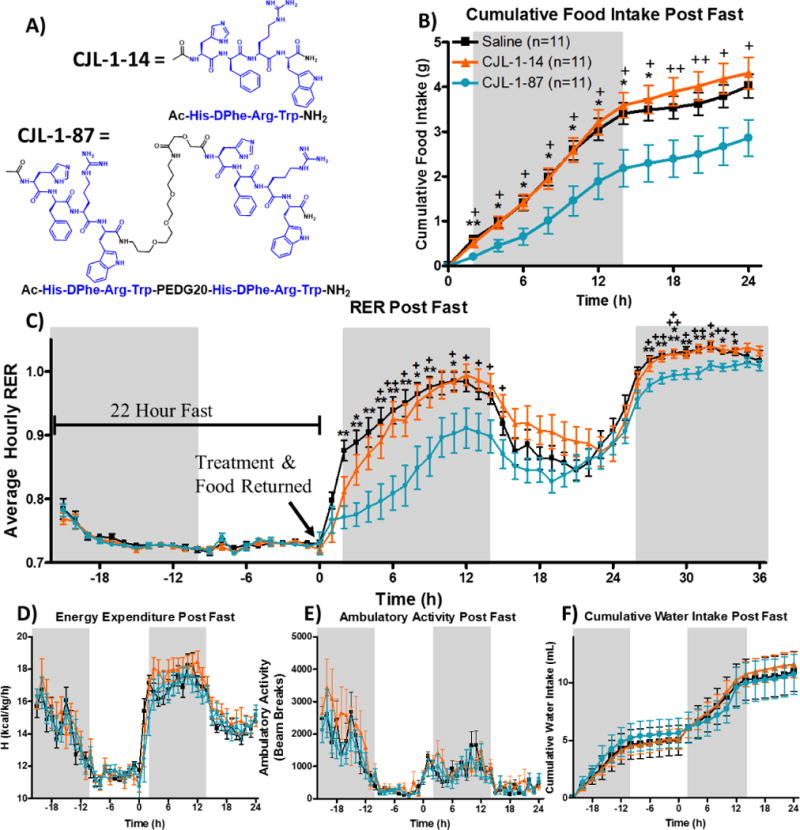
 ) on energy homeostasis in TSE metabolic cages following a cross-over paradigm (Figure 2A). Male mice were fasted starting at the previous light cycle (t=−22 h) until 2 h prior to the light cycle (t=0 h). Treatment was given ICV at time 0 h and food was returned immediately thereafter (Figure 2B). Cumulative food intake (B) was significantly reduced by CJL-1-87 compared to saline and CJL-1-14. The RER (C) was significantly reduced after CJL-1-87 treatment compared to saline and CJL-1-14. No significant effects were observed between saline, CJL-1-14, and CJL-1-87 on energy expenditure (D), ambulatory activity (E), or water intake (F). No significant effects were observed on any parameters past 24 h other than RER and, therefore, data is not shown. Grey boxes represent lights off. *p<0.05; **p<0.01, ***p<0.001 for CJL-1-87 compared to saline. +p<0.05; ++p<0.01, +++p<0.001 CJL-1-87 for compared to CJL-1-14.
) on energy homeostasis in TSE metabolic cages following a cross-over paradigm (Figure 2A). Male mice were fasted starting at the previous light cycle (t=−22 h) until 2 h prior to the light cycle (t=0 h). Treatment was given ICV at time 0 h and food was returned immediately thereafter (Figure 2B). Cumulative food intake (B) was significantly reduced by CJL-1-87 compared to saline and CJL-1-14. The RER (C) was significantly reduced after CJL-1-87 treatment compared to saline and CJL-1-14. No significant effects were observed between saline, CJL-1-14, and CJL-1-87 on energy expenditure (D), ambulatory activity (E), or water intake (F). No significant effects were observed on any parameters past 24 h other than RER and, therefore, data is not shown. Grey boxes represent lights off. *p<0.05; **p<0.01, ***p<0.001 for CJL-1-87 compared to saline. +p<0.05; ++p<0.01, +++p<0.001 CJL-1-87 for compared to CJL-1-14.
In the fasting-refeeding paradigm, food was removed from mice immediately before the lights out cycle on the previous day. Treatment occurred two hours prior to lights out and food was reintroduced (Figure 2B). Due to the fast, a hyperphagic response occurs during refeeding and this robust response can aid in the detection of effects. However, fasting can also mask subtle effects due to the strong desire to eat.66 During fasting many hormone levels change to promote feeding. Of important relevance to the melanocortin system, the endogenous MC3R/MC4R antagonist agouti-related peptide (AGRP) is upregulated in the hypothalamic regions that are also innervated by proopiomelanocortin (POMC) neurons.67–70 AGRP blocks the agonism of endogenous melanocortin agonist peptides (e.g. α-MSH, β-MSH, γ-MSH, ACTH) and is also known to function as an inverse agonist to increase feeding and lower energy expenditure.57, 68, 71 In addition, fasting upregulates several other hormones including ghrelin, corticosterone, and neuropeptide Y (NPY) while several hormones are also downregulated including insulin, leptin, and resistin (for a review see Jensen, 2013.72) These changes in hormonal expression levels are hypothesized to drive the hyperphagic response. Although the maximal signal and, therefore, signal to noise is increased in a fasting-refeeding paradigm compared to a nocturnal feeding paradigm due to the increased baseline or control food intake, achieving a measurable decrease in food intake can sometimes be challenging. This is because the experimental compounds need to overcome these robust hormone changes to have observable activity.
In the current fasting-refeeding experiments, food intake after 5 nmol CJL-1-87 compared to saline administration was significantly reduced between 2 to 16 hours after ICV administration as expected for a melanocortin receptor agonist (Figure 3B). Approximately 50% less food was consumed compared to saline and 5 nmol CJL-1-14 at the 2-8 hour time points. Notably, there was no significant difference in food intake after fasting between CJL-1-14 and saline groups. Compared to CJL-1-14 food intake after CJL-1-87 treatment was significantly reduced at every time point from 2 to 24 hours after compound administration (Figure 3B). It is worth noting that the current experiments were performed in a crossover design paradigm, such that all 11 age matched male mice included in the study were administered saline, 5 nmol CJL-1-14 and 5 nmol CJL-1-87 on different days with a washout period of one week in between administrations and no mice were excluded (Figure 2A).
The RER was determined by dividing the volume of CO2 produced by the volume of O2 consumed by an animal.56, 73, 74 Because the oxidation of carbohydrates or fatty acids produces different amounts of CO2 for each O2 molecule utilized, the RER indicates whether mice are using carbohydrates or fats as the primary fuel source. RER values of about 1.0 indicate that carbohydrates are the primary fuel source being utilized, whereas values of approximately 0.7 indicate that fats are primarily being utilized. 56, 73, 74 The RER in the current experiment was indirectly calculated by measuring the amount of CO2 and O2 entering and exiting the sealed metabolic cages as previously described.56, 73, 74
During the fast, RER values for all groups dropped to slightly above 0.7 indicating that fats are primarily being used as a source of energy (Figure 3C). This is expected due to the lack of carbohydrates available from food during the fast that results in a reliance of fat storage for energy, an effect previously described.75, 76 At the 0 h time point, the compound or saline is administered and food is reintroduced to the cage. A rapid increase in the RER is observed in the saline treatment group until the fuel source is primarily carbohydrates during the initial dark cycle as anticipated. Treatment with both CJL-1-87 and CJL-1-14 resulted in a more gradual increase in RER (Figure 3C). Significant reduction of RER values were observed 2–9 and 11 hours after CJL-1-87 treatment compared to after saline treatment during the first dark cycle. Significant reduction in RER values were observed 5–9 and 11–15 hours after CJL-1-87 treatment compared to CJL-1-14 treatment. The compound specific reduction in RER suggests that the mice are using more fats after CJL-1-87 administration compared to either saline or CJL-1-14. No significant differences in energy expenditure (kcal/kg/h) was observed between any treatment groups (Figure 3D). The energy expenditure taken together with the RER data indicates that the mice are burning approximately the same amount of calories upon different treatments, however the CJL-1-87 treated mice appear to be utilizing more fats for their energy in lieu of carbohydrates (Figure 3C–D).
During the following light cycle (t = 14–26 h), the RER decreased for all groups most likely due to decreased feeding and decreased activity that is expected with nocturnal feeders like mice. The RER increases again preceding and during the second dark cycle (t = 26–38 h). Although no clear trend was observed in food intake beyond 24 hours, the RER was still significantly lower after CJL-1-87 treatment compared to saline or CJL-1-14 at time points 27–34 h (Figure 3C). This second time period of significantly decreased RER in the absence of decreased food intake suggests that compound CJL-1-87 has long-lasting effects on energy homeostasis that do not appear to directly correlate to food intake. This is not observed with the monovalent ligand CJL-1-14.
The length of time that CJL-1-87 affected energy homeostasis was unexpected since previous nocturnal paradigm studies showed no significant effects on food intake past 8 hours with this compound,21 and less than 3% of the CJL-1-87 remained intact after 24 hours in the in vitro serum stability assays (Figure 1). However, the serum stability studies give only an indication of the metabolic stability of CJL-1-87 in the brain as the proteases present in the cerebrospinal fluid (CSF) may be different than those in the serum. Another factor in the interpretation of the long lasting effects on RER of CJL-1-87 is the rate of clearance from the ventricular system of the brain into the peripheral. Although the rate of clearance is currently unknown for CJL-1-87 and CJL-1-14, it has previously been reported that CSF empting time and compound clearance from the CSF is rapid after ICV administration. 77–79 These data suggest the long-lasting response was the result of a pharmacological effect due to receptor binding, and not due to the drug being present in the cerebrospinal fluid (CSF) for an extended period of time. It could be that once CJL-1-87 is bound to a receptor, it avoids degradation and clearance from the brain while promoting long lasting effects, or that the binding of CJL-1-87 to melanocortin receptors may cause unique downstream signaling cascades not observed with its monovalent counterpart. Either way, the increased duration of action on RER with increased utilization of fat stores for energy is a distinct characteristic of the bivalent ligand that is not observed with the monovalent ligand. Further studies into the CSF clearance rate of CJL-1-87 and CJL-1-14 as well as studies thoroughly characterizing the absorption, distribution, metabolism, and excretion of these ligands will be necessary to draw clear conclusions about the long-lasting (>24 h) effects.
Whenever an experiment’s measurements are based on loss of function, it can raise questions about assay artifacts. In this particular assay paradigm, it may be questioned whether the reduction in food intake and RER are a consequence of an adverse reaction or toxicity to CJL-1-87. In order to address this concern, mice were monitored visually for at least 2 hours post treatment and no adverse reactions in any of the three treatment groups were observed. Additionally, no significant effect was observed between saline, CJL-1-87 or CJL-1-14 on locomotor activity (beam breaks), water intake (mL), or energy expenditure (kcal/h/kg) (Figure 3D–F). If sick-like behavior or toxicity were suspect for the observed decrease in food intake and RER, it would be expected that these other parameters would be lowered as well. In particular, locomotor activity, as quantified by infrared beam breaks along the side of the cages (X-axis), would be negatively affected because a sick mouse wound typically be hunched and inactive in the corner of the cage (Figure 3E). The lack of effect observed on activity between all treatment groups supports that visceral illness or other adverse reactions were not likely to be contributing factors to the significant effects observed.
The Effect of ICV Administration of CJL-1-14 and CJL-1-87 on Body Composition
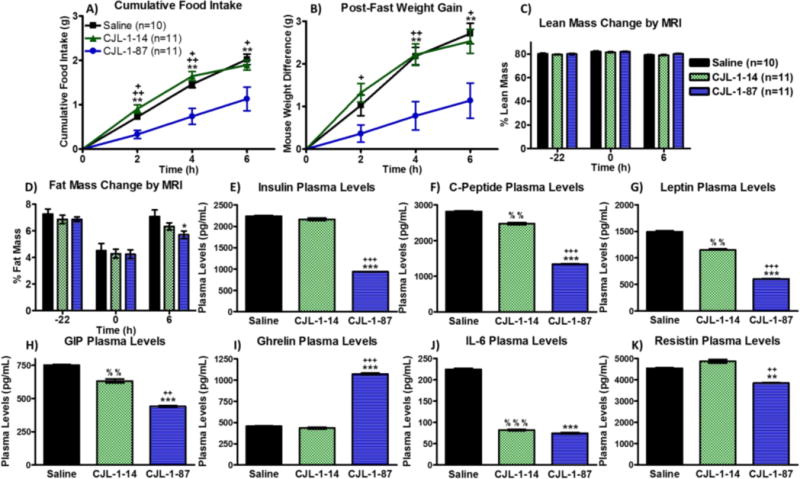
To study the effect of CJL-1-87 and CJL-1-14 on body composition and metabolically active hormone levels in the blood plasma of mice, a new cohort of 32 male age matched mice (saline, n=10; CJL-1-14, n=11; CJL-1-87, n=11) underwent cannulation surgery and placement validation as previously discussed. The animals were administered a single treatment of saline vehicle control, 5 nmol CJL-1-14, or 5 nmol CJL-1-87 and sacrificed 6 h post-treatment in the fasting-refeeding paradigm described above but in conventional cages (Figure 2B). Once again, CJL-1-87 treatment decreased food intake as compared to CJL-1-14 and saline (Figure 3B, Figure 4A). As expected the decreased food intake was accompanied by a significant reduction in the amount of weight gained after the food was reintroduced with CJL-1-87 treatment compared to CJL-1-14 for the first 6 h post-treatment (Figure 4B). Mice treated with CJL-1-87 gained back 55% less weight than saline and CJL-1-14 treated mice 6 h post fast. The CJL-1-87 treated mice also gained significantly less weight than saline treated mice 4 and 6 h post-fast (Figure 4B).
Figure 4.
A new cohort of male mice received a single treatment of saline vehicle control (
 , n=10), 5 nmol CJL-1-14 (
, n=10), 5 nmol CJL-1-14 (
 , n=11), or 5 nmol CJL-1-87 (
, n=11), or 5 nmol CJL-1-87 (
 , n=11) in the fasting paradigm used above (Figure 2B). CJL-1-87 treatment resulted in lowered refeeding food intake (A) corresponding to slower regain of body weight (B) measured manually. Measurements using an ECHOMRI-100H system showed no change in lean body mass percentage (C), but a significant decrease in body fat mass percentage was observed 6 h after treatment with CJL-1-87. (D). Mice were sacrificed 6 h post treatment and their trunk blood was analyzed using an Luminex Milliplex systems to examine insulin (E), C-peptide (F), leptin (G), GIP (H), ghrelin (I), IL-6 (J), and resistin (K). Hormone and cytokine levels are reported as pg per mL of plasma. Time 0 h was defined as the time of treatment. *p<0.05; **p<0.01; ***p<0.001 for CJL-1-87 compared to saline. +p<0.05; ++ p<0.01; +++p<0.001 for CJL-1-87 compared to CJL-1-14. % p<0.05; %% p<0.01; %%% p<0.001 for CJL-1-14 compared to saline.
, n=11) in the fasting paradigm used above (Figure 2B). CJL-1-87 treatment resulted in lowered refeeding food intake (A) corresponding to slower regain of body weight (B) measured manually. Measurements using an ECHOMRI-100H system showed no change in lean body mass percentage (C), but a significant decrease in body fat mass percentage was observed 6 h after treatment with CJL-1-87. (D). Mice were sacrificed 6 h post treatment and their trunk blood was analyzed using an Luminex Milliplex systems to examine insulin (E), C-peptide (F), leptin (G), GIP (H), ghrelin (I), IL-6 (J), and resistin (K). Hormone and cytokine levels are reported as pg per mL of plasma. Time 0 h was defined as the time of treatment. *p<0.05; **p<0.01; ***p<0.001 for CJL-1-87 compared to saline. +p<0.05; ++ p<0.01; +++p<0.001 for CJL-1-87 compared to CJL-1-14. % p<0.05; %% p<0.01; %%% p<0.001 for CJL-1-14 compared to saline.
The amount of lean mass and fat mass was measured pre-fast, immediately before compound administration, and 6 h post-administration just before sacrificing the animals using an EchoMRI-100H system. There were no significant differences between the saline, CJL-1-14, and CJL-1-87 treatment in their effect on body percentage of lean mass before the fast, at compound administration, or 6 h after compound administration (Figure 4C). There were no significant differences in body fat mass percentage before the fast, or immediately before treatment between saline, CJL-1-14, and CJL-1-87 as expected. The body fat percentage was significantly lower in mice that received CJL-1-87 treatment compared to saline treatment 6 h post-administration (Figure 4D). Mice treated with CJL-1-87 gained back 45% less fat mass 6 h after the fast compared to saline treated mice. Mice treated with CJL-1-14 gained back 15% less fat mass compared to saline treated mice, however there were no significant differences in body fat percentage between CJL-1-14 compared to saline or CJL-1-87. The decreased body fat percentage after CJL-1-87 treatment is consistent with the lowered RER values in the metabolic cage studies supporting the hypothesis that fat, and not carbohydrates, is the primary fuel source after CJL-1-87 treatment (Figure 3C).
The Effect of ICV Administration of CJL-1-14 and CJL-1-87 on Hormone Levels
The same 32 mice from the body composition studies were sacrificed and trunk blood was collected 6 h post ICV treatment with saline, 5 nmol CJL-1-14, or 5 nmol CJL-1-87. The plasma was analyzed using a multiplex Luminex Milliplex system to assess six hormones and one cytokine: insulin, C-peptide, leptin, ghrelin, glucose-dependent insulinotropic polypeptide (GIP), resistin, and interleukin 6 (IL-6) (Figure 4 E–K). Mice treated with CJL-1-87 had significantly lower levels of insulin, C-peptide, leptin, GIP and resistin compared to mice receiving either saline or CJL-1-14 (Figure 4E–H and K). Also CJL-1-87 treated mice had significantly lower levels of IL-6 compared to saline, but there was no significant difference between CJL-1-14 and CJL-1-87 treatment (Figure 4J). Mice receiving CJL-1-87 had significantly increased ghrelin levels compared to mice receiving saline or CJL-1-14 (Figure 4I). Mice receiving CJL-1-14 had significantly reduced C-peptide, leptin, GIP, and IL-6 compared to mice receiving saline (Figure 4F–H and J).
The differences in metabolic hormone levels observed between all three treatment groups demonstrate unique effects of the melanocortin bivalent agonist ligand design. The unique effects were evident in the significant differences in the levels of insulin, C-peptide, leptin, GIP, ghrelin, and resistin between CJL-1-14 treatment and CJL-1-87 treatment (Figure 4E–I and K). It should be noted that although the same molar amount of peptide (5 nmols) was administered, the bivalent ligand CJL-1-87 contains twice as many pharmacophores. It could, therefore, be hypothesized that differences between CJL-1-14 and CJL-1-87 on C-peptide, leptin, and GIP were due to a “dose-dependent” effect of the pharmacophores. (Figure 4F, H, and I). However, this is an intricate argument, since a similar “dose-dependent” effect was not observed with insulin, ghrelin, IL-6, or resistin. Doubling of the pharmacophores could be a contributing factor, but the complexity of the in vivo pharmacology suggests that the effects are due to characteristics of CJL-1-87 that are distinct from CJL-1-14. The unique characteristics may originate from the bivalent design strategy, differences in receptor selective (e.g.MC3R vs MC4R), different receptors being responsible for the different responses (e.g. hormone release vs food intake), or the different distribution of CJL-1-87 and CJL-1-14 after ICV delivery. Further experimentation will be necessary to determine the exact neuronal mechanism of action of the observed in vivo response differences. However, it is clear that in the current fasting-refeeding in vivo experimental paradigm that CJL-1-87 behaviors differently than its monovalent counterpart. Possible in vitro molecular mechanisms focused on how the bivalent ligand CJL-1-87 may be interacting with melanocortin dimers will be discussed below.
The effects of the melanocortin ligands on the hormone plasma levels are consistent with previous literature reports (Table 2). 80–92 For example, it has previously been reported that melanocortin agonists decreased insulin serum and plasma levels, regardless of food intake.80–82 This is consistent with the decreased insulin levels observed herein with CJL-1-87 treatment, albeit no effect was observed with CJL-1-14 treatment (Figure 4E). As would be anticipated, C-peptide was also decreased by melanocortin agonism (Figure 4F), because insulin and C-peptide are produced in equimolar amounts from the cleavage of proinsulin.83, 84 Similarly, decreased leptin levels in the serum or plasma have been reported following administration of melanocortin agonists as observed herein (Figure 4G).81, 82 There is also evidence that melanocortin agonist administration decreases GIP, similar to the current results (Figure 4H).85 At least two studies have reported that the melanocortin agonist MTII has no effect on resistin serum levels although one study found that resistin mRNA expression was upregulated after MTII treatment. 86, 87 Also, the MC3R and MC4R antagonist SHU9119 was reported to increase resistin serum levels making interpretation of the data inconclusive.86, 87 The significant decrease in resistin levels after CJL-1-87 treatment appears to be a unique effect of the bivalent ligand compared to the monovalent ligand CJL-1-14 and the previous studies (Figure 4K).86, 87 Previous reports of melanocortin agonists decreasing IL-6 gene expression and secretion after administration in mice which is also consistent with the current results (Figure 4J).88–90 However, this effect is confounded by conflicting data indicating that α-MSH increases IL-6 expression.91 Although ghrelin is usually thought of as an upstream regulator of the melanocortin pathway, there is some evidence that α-MSH can directly affect the release of ghrelin (Figure 4I).92 It is therefore unclear if the current increase in ghrelin after CJL-1-87 treatment is a direct pharmacological effect or an effect of the decreased food intake.
Table 2.
Summary of the effects of fasting and melanocortin (MC) receptor agonism on hormone and cytokine levels from the literature (Lit.) and the current study. Results from fasting and melanocortin (MC) agonism are from the literature with citations referenced directly to their right. A ? mark indicates conflicting results. Results from 5 nmol CJL-1-14 and CJL-1-87 treatments are summarized from the current study. In the current studies, the significance of treatment compared to saline treatment is indicated such that one arrow is p<0.05, two arrows is p<0.01, and three arrows is p<0.001.
| Hormone | Fasting from Lit. | Fasting Citations | MC Agonism from Lit. | MC Agonism Citations | CJL-1-14 (Current Study) | CJL-1-87 (Current Study) |
|---|---|---|---|---|---|---|
| Insulin | ↓ | 72, 94 | ↓ | 80–82 | ↔ | ↓↓↓ |
| C-Peptide | ↓ | 72, 83, 84, 94 | ↓ | 80–84 | ↓↓ | ↓↓↓ |
| Leptin | ↓ | 72, 94 | ↓ | 81, 82 | ↓↓ | ↓↓↓ |
| GIP | ↓ | 85, 95 | ↓ | 85 | ↓↓ | ↓↓↓ |
| Ghrelin | ↑ | 72, 96 | ? ↑ | 92 | ↔ | ↑↑↑ |
| IL-6 | ↑ | 97 | ? ↓ | 88–91 | ↑↑↑ | ↑↑↑ |
| Resistin | ↓ | 72, 93 | ? | 86, 87 | ↔ | ↓↓ |
Although the above literature provides evidence that some of the changes in hormone levels may be from activation of melanocortin receptors, it is possible that some of the changes in hormones are due to lowered refeeding of CJL-1-87 treated mice. During the fasting state prior to compound administration, the lack of food intake is likely to cause a decrease in the serum concentration of insulin, C-peptide, leptin, resistin, and GIP. 72, 85, 93–95 Inversely, ghrelin is normally elevated during fasting and decreased after food intake (Table 2).72, 96 IL-6 is also thought to be increased during fasting.97 While it is possible that direct pharmacological agonism causes the changes in the plasma hormone levels after compound administration, the decrease in food intake resulting from CJL-1-87 administration may slow the rate of change in hormones levels from the fasting state. However, such slowing cannot explain the significantly reduced plasma levels of C-peptide, leptin, GIP and IL-6 observed with CJL-1-14 treated mice compared to saline treated mice that had the same food intake. This indicates that these hormone changes are a direct effect of agonizing the melanocortin receptors with CJL-1-14. The effects of CJL-1-87 on hormone levels may be attributed to pharmacological agonism of the melanocortin receptors, decreased food intake, or some combination of both. Regardless, it appears that CJL-1-87 has physiological effects that significantly reduce food intake after fasting in spite of the increased levels of the orexigenic peptide ghrelin and the decreased levels of the anorexigenic peptides leptin and insulin.
125I-AGRP Competitive Binding Studies
Although the bivalent ligand CJL-1-87 is advantageous in reducing food intake, body weight, and fat mass gained when utilizing a fasting-refeeding paradigm compared to CJL-1-14, the exact molecular mechanism is unclear. Two major hypotheses can be envisioned based on the bivalent design concept: 1) The c.a 20-fold increased binding affinity of CJL-1-87 compared CJL-1-14 helps it compete more effectively with endogenous AGRP that is upregulated in the fasting state. 2) The bivalent ligands are interacting with melanocortin homodimers or heterodimers in a unique fashion that is different than the monovalent counterpart. In order to investigate how effectively CJL-1-87 can displace AGRP, competitive radioligand binding assays using 125I-AGRP87–132 were performed (Table 3). Previously reported results of CJL-1-14 and CJL-1-87 competition against 125I-NDP-MSH and results from antagonist monovalent tetrapeptide Ac-His-DNal(2′)-Arg-Trp-NH2 and antagonist homobivalent ligand CJL-1-140, Ac-His-DNal(2′)-Arg-Trp-(PEDG20)-His-DNal(2′)-Arg-Trp-NH2, are provided for comparison (Table 3).21
Table 3.
The experimental compounds were used to displace either 125I-NDP-MSH or 125I-AGRP in a dose-response manner to calculate the IC50 values. The % represents the amount of 125I-NDP-MSH signal reduction at 100 µM. The reported errors are the standard error of the mean (SEM) determined from at least two independent experiments. aThe IC50 values obtained by competition with 125I-NDP-MSH were reported previously, but are included herein for direct comparison.21
| 125I-NDP-MSH IC50 (nM)a | 125I-AGRP IC50 (nM) | ||||
|---|---|---|---|---|---|
| Compound | Structure | mMC3R | mMC4R | mMC3R | mMC4R |
| Mean±SEM | Mean±SEM | Mean±SEM | Mean±SEM | ||
| NDP-MSH | 4.2±0.6 | 1.1±0.1 | 3.3±0.08 | 2.6±0.09 | |
| CJL1-14 | Ac- -NH2 | 60% @100 µM | 210±60 | 6200±1700 | 130±20 |
| CJL1-87 | Ac- -(PEDG20)- -NH2 | 3500±500 | 9.9±3 | 330±40 | 7.5±2 |
| CJL1-80 | Ac- -NH2 | 1400±200 | 26±3 | 660±20 | 12±4 |
| CJL1-140 | Ac- -(PEDG20)- -NH2 | 350±80 | 10±0.03 | 120±10 | 11±2 |
At the mMC4R, the IC50 values obtained by competing experimental ligands against 125I-AGRP(87-132) are within experimental error (less than 3-fold change) of those obtained by competing against 125I-NDP-MSH.21 Bivalent ligand CJL-1-87 had a 17-fold higher binding affinity when competing against 125I-AGRP(87-132) compared to its monovalent counterpart CJL-1-14 at the mMC4R. At the mMC3R, the experimental agonist ligands tended to be better at displacing 125I-AGRP(87-132) compared to 125I-NDP-MSH. In fact, compound CJL-1-14 and CJL-1-87 had approximately a 10-fold shift in the IC50 values obtained when displacing 125I-AGRP(87-132) compared to 125I-NDP-MSH at the mMC3R. Unlabeled NDP-MSH and antagonist ligand Ac-His-DNal(2′)-Arg-Trp-NH2 had less than 3-fold changes in IC50 values to compete 125I-NDP-MSH and 125I-AGRP(87-132). Homobivalent antagonist CJL-1-140 had nearly a 3-fold shift in its IC50 value when competing against 125I-AGRP(87-132) compared to the value obtained when competing against 125I-NDP-MSH. The homobivalent agonist ligand CJL-1-87 had an 18-fold higher binding affinity compared to its monovalent counterpart CJL-1-14 when both are competing against 125I-AGRP(87-132) at the mMC3R (Table 3). This increase in binding affinity of the agonist ligands when competing against 125I-AGRP(87-132) compared to 125I-NDP-MSH at the mMC3R and not the mMC4R may implicate that the mMC3R has increased regulation in reducing refeeding from the fasting state. This observation is consistent with a previous report that MC3R knockout mice have lowered food intake after fasting compared to wild type mice.69
During fasting, the expression of endogenous MC3R and MC4R antagonist/inverse agonist AGRP is upregulated and promotes a state of hunger that drives refeeding. 67–70 This may allow melanocortin agonists to have two functional effects by both activating the melanocortin receptors, and functionally antagonizing AGRP’s orexigenic effects by blocking the endogenous AGRP from binding the melanocortin receptors. This dual action may be able to explain a mechanism of how bivalent ligand CJL-1-87 has more significant effects in the fasting-refeeding paradigm than the monovalent ligand CJL-1-14, but not in the nocturnal paradigm. Although CJL-1-87 was previously reported to possess only about 3- to 5-fold increased agonist potency to stimulate cAMP signaling compared to CJL-1-14,21 the increased in vitro binding affinity of 17- to 18-fold may increase CJL-1-87’s ability to compete effectively with AGRP in vivo. In the nocturnal paradigm in which there would be lower amounts of AGRP present, the increase in binding affinity would be less of a driving force in decreasing feeding and, therefore, CJL-1-14 and CJL-1-87 would have more similar in vivo effects based on the closer functional cAMP potencies.21, 57 However, in the fasting state there would be a higher concentration of AGRP and the ability of CJL-1-87 to compete with and antagonize AGRP’s orexigenic effects would become increasingly important. Monovalent ligand CJL-1-14 may not have a strong enough binding affinity to effectively compete with AGRP and antagonize its orexgenic effects, whereas CJL-1-87 with a 17- to 21-fold increased binding affinity at both the mMC3R and mMC4R would block the orexigenic effects of AGRP in vivo resulting in significantly decreased refeeding.
It should be noted that although the hypothesized binding affinity mechanism may explain the current observation of bivalent ligand CJL-1-87’s advantages in the fasting paradigm over the monovalent ligand CJL-1-14, this proposed mechanism does not exclude the possibility that melanocortin receptor dimerization is also responsible. In fact, the increased binding affinity of CJL-1-87 is hypothesized to be a result of synergistic bivalent binding at melanocortin dimers.21 It is possible that the dimerization of melanocortin receptors may play a functional role beyond synergistic bivalent binding in the pharmacology of CJL-1-87 which will be discussed further below. Regardless of the exact mechanism, it does appear that the bivalent design strategy is responsible either by synergistic binding, or by targeting melanocortin dimers for the ability of the bivalent ligand, and not the monovalent ligand, to decrease feeding in the fasting-refeeding paradigm.
Bioluminescence Resonance Energy Transfer (BRET) Supports mMC3R-mMC4R Heterodimerization
The second hypothesis for the significant differences between CJL-1-14 and CJL-1-87 in the fasting-refeeding paradigm could be the interaction of the compounds with melanocortin receptor dimers. Portoghese and coworkers have demonstrated that bivalent ligands can be designed to selectively target opioid heterodimers.1–7 It has previously been supported that the melanocortin receptors can form homodimers, heterodimers, or higher-order oligomers utilizing various techniques to demonstrate proximity and association.98–108 Furthermore, it has recently been demonstrated that coexpression of the MC1R and MC5R can create ligand-dependent signal modulation providing evidence that melanocortin heterodimerization can have functional consequences.108 Considering that the bivalent ligands were designed to interact with melanocortin receptor dimers, it is possible that the unique in vivo effects of CJL-1-87 versus CJL-1-14 could be from bivalent binding specifically to homodimers or heterodimers. In order to explore whether these effects could be mediated through a MC3R-MC4R heterodimer, bioluminescence resonance energy transfer (BRET) studies were undertaken to show association of the two receptors. Additionally, cAMP-based AlphaScreen experiments were performed on cells coexpressing both the mMC3R and mMC4R to begin elucidating the possible functional significance of a mMC3R-mMC4R heterodimer.
BRET is a biophysical technique that can be used to demonstrate the association of two proteins. It has previously been utilized to show that the mMC3R and mMC4R are in close proximity suggesting that they form homodimers or higher-order oligomers.98–100 It has also been shown that the hMC1R closely associates with the hMC3R by BRET suggesting heterodimerization.98 To our knowledge, the MC3R and MC4R have never been studied by BRET for heterodimerization. In rats, mRNA of both the MC3R and MC4R have been detected in select regions of the brain such as the anteroventral periventricular (AVPV), ventral premammillary (PMV), and posterior hypothalamic nuclei suggesting possible in vivo coexpression.109–111 Also in vitro coexpression of the MC3R and MC4R in neuronal cells colocalize on the cell membrane suggesting heterodimerization is possible.110 This hypothesized heterodimer may explain some of the ambiguous pharmacology of the melanocortin system such as the mechanism of the synergistic effects observed in the MC3R-MC4R double knockout mice.112, 113 Furthermore, due to the differential expression profile of the mMC3R and mMC4R, ligands that would target a mMC3R-mMC4R heterodimer could be developed to be tissue selective acting only in the distinct regions within the brain that coexpress the receptors. Further studies will be needed to demonstrate whether individual neurons do in fact coexpress the MC3R and MC4R.
BRET studies were performed on HEK293 cells expressing only the mMC3R, only the mMC4R, or coexpressing the mMC3R and mMC4R (Figure 5). The Promega NanoBRET™ Protein:Protein interaction system was utilized with limited modifications to develop mMC3R and mMC4R fused to both the NanoLuc® fusion protein and the HaloTag® fusion protein. HEK293 cells expressing the mMC3R-NanoLuc® and the mMC3R-HaloTag® resulted in a BRET ratio of 90 ± 5 mBU, supporting mMC3R homodimerization. This data is similar to a previous result observed with a slightly different BRET system that showed a BRET ratio of 350 mBU with the hMC3R. The decrease in signal currently observed might be explained by differences in assay paradigms including using Cos-7 cells, the human receptors, or DeepBlueC substrate.98 HEK293 cells expressing the mMC4R-NanoLuc® and the mMC4R-HaloTag® resulted in a high BRET ratio of 100 ± 10 mBU. This is in good agreement with previous results reported by two independent laboratories using different BRET conditions (e.g. using Cos-7 cells versus HEK293 cells) that reported BRET ratios of 75 mBU and 110 mBU with the hMC4R.99, 100
Figure 5.
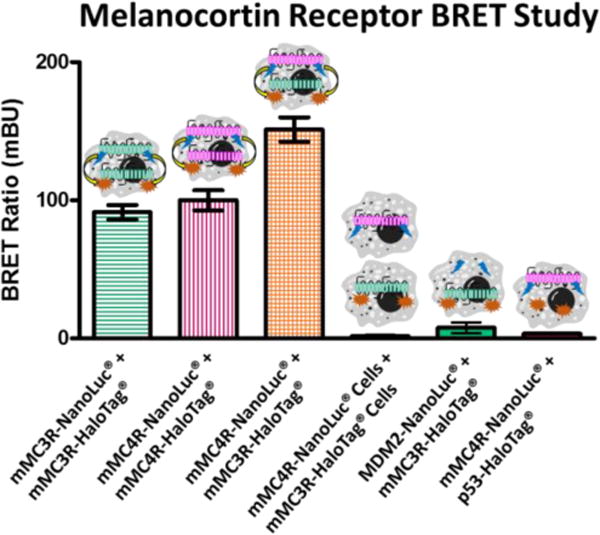
Bioluminescence resonance energy transfer (BRET) of the mMC3R and mMC4R. As previously shown in similar systems, coexpression of the mMC3R-NanoLuc® and the mMC3R-HaloTag® result in high BRET signal supporting homodimerization. Similar results were achieved when the mMC4R-NanoLuc® and the mMC4R-HaloTag® were coexpressed. When the mMC4R-NanoLuc® and the mMC3R-HaloTag® are coexpressed an even higher BRET signal is observed suggesting heterodimerization. Cells expressing mMC4R-NanoLuc® were mixed with cells expressing the mMC3R-HaloTag® in equal amounts and produced minimal signal. As a negative control, unrelated mouse double minute 2 (MDM2)-NanoLuc® and the mMC3R-HaloTag® are coexpressed as well as mMC4R-NanoLuc® and the p53-HaloTag®. Both resulted in minimal signal. Receptors were expressed at a 1:4 donor NanoLuc® plasmid to acceptor HaloTag® plasmid. Data are the mean ± standard error of the mean (SEM) determined from three independent experiments, except for the mMC4R-NanoLuc® and the mMC3R-HaloTag® coexpression which was performed in six independent experiments.
HEK293 cells that were transfected to coexpress mMC4R-NanoLuc® and the mMC3R-HaloTag® gave a high BRET ratio of 150 ± 10 mBU (Figure 5). As a negative control, a plate of cells was transiently transfected with the mMC4R-NanoLuc® and a separate plate was transfected with mMC3R-HaloTag®. These cells were mixed in equal amounts and assayed together which resulted in minimal BRET signal (1.6 ± 0.5 mBU). This suggested that BRET signal was indeed from specific mMC3R-mMC4R interactions on the same cell membrane, and not from non-specific interactions of the receptors being assayed at the same time. As a further negative control, unrelated mouse double minute 2 (MDM2)-NanoLuc® and the mMC3R-HaloTag® were coexpressed as well as mMC4R-NanoLuc® and the p53-HaloTag® that resulted in minimal signal (7.7 ± 4.0 mBU and 3.4 ± 0.3 mBU, respectively).
The increased signal from mMC4R-NanoLuc® and mMC3R-HaloTag® coexpression compared to singly expressed mMC3R or mMC4R suggests that the mMC3R and the mMC4R form heterodimers in addition to homodimers. The ratio of homodimers to heterodimers is yet to be determined. It also should be noted that the relative BRET signal could be affected by the expression levels of the transiently transfected receptors which was not quantified in this study. It is also possible that higher-order oligomers are responsible for the high BRET signal of both homodimers and heterodimers as it is possible that the receptors could form tetramers (or even higher-order oligomers) that are “heterodimers” of the homodimer species. This would cause strong BRET signal of both homo- and heterodimers as observed herein.
Coexpression of mMC3R and mMC4R effects on Functional Potency
Based upon the BRET data indicating mMC3R-mMC4R heterodimerization, cAMP AlphaScreen assays were performed on cells coexpressing both the mMC3R and mMC4R to elucidate functional significance. In these experiments, HEK293 cells stably expressing one receptor subtype were transiently transfected with the second receptor subtype to achieve dual expressing cells (Figure 6). To determine if the effects were due to the receptors being expressed on the same cell membrane (Figure 6 C, and D) versus the receptors being assayed together; an equal mixture of both individual stable cells lines was assayed as a control (Figure 6 E). Five cell categories (i.e. stable mMC3R cells, stable mMC4R cells, stable mMC3R cells transiently expressing the mMC4R, stable mMC4R cells transiently expressing the mMC3R, and an equal mixture of stable mMC3R cells and stable mMC4R cells) were screened in parallel at the same time to control for inherent day to day assay variability (Figure 6, Table 4). Only cells that coexpressed the mMC3R and mMC4R could possibly contain mMC3R-mMC4R heterodimers (Figure 6 C and D), whereas the mixture of mMC3R stable cells with mMC4R stable cells would contain no heterodimers even though both cell types are assayed together (Figure 6 E). At least three independent experiments were performed on separate days. The EC50 values for ligands NDP-MSH, α-MSH, MTII, CJL-1-87, and CJL-1-14 at the singly expressed mMC3R and mMC4R were consistent with the field.21, 114, 115
Figure 6.
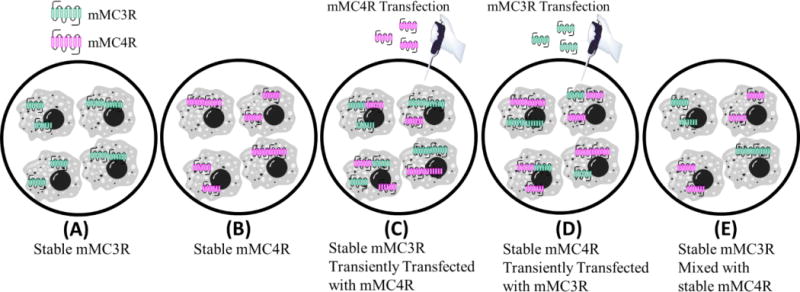
Cell categories for cAMP AlphaScreen® functional assays during coexpression experiments (Table 4). (A) Cells stably expressing mMC3R. (B) Cells stably expressing the mMC4R. (C) Cells stably expressing the mMC3R that were then transiently transfected with mMC4R plasmid. (D) Cells stably expressing the mMC4R that were then transiently transfected with mMC3R plasmid. (E) Cells stably expressing the mMC3R were mixed 1:1 with cells stably expressing the mMC4R. This mixture cannot contain MC3R-MC4R heterodimers.
Table 4.
Functional cAMP AlphaScreen® assays were performed to determine in vitro potency of compounds to induce cAMP signaling in five cell categories (Figure 6): (A) stable mMC3R cells, (B) stable mMC4R cells, (C) stable mMC3R cells transiently transfected with mMC4R, (D) stable mMC4R cells transiently transfected with mMC3R, and (E) an equal mixture of stable mMC3R cells and mMC4R cells. All assays and cell categories were run in parallel. The reported standard error of the mean (SEM) was determined from at least three independent experiments.
| Agonist cAMP Based Alpha Screen Results EC50 (nM) | |||||
|---|---|---|---|---|---|
| Compound | mMC3R | mMC4R | mMC3R Trans mMC4R | mMC4R Trans mMC3R | mMC3R Mixed with mMC4R |
| Mean±SEM | Mean±SEM | Mean±SEM | Mean±SEM | Mean±SEM | |
| NDP-MSH | 0.13±0.02 | 0.46±0.07 | 0.19±0.03 | 0.12±0.01 | 0.13±0.02 |
| α-MSH | 0.21±0.02 | 3.1±0.7 | 0.27±0.05 | 0.20±0.03 | 0.19±0.03 |
| MTII | 0.10±0.02 | 0.11±0.03 | 0.09±0.02 | 0.06±0.01 | 0.08±0.02 |
| CJL1-14 | 23±3 | 8.5±1 | 7.7±1 | 4.0±0.5 | 9.9±2 |
| CJL1-87 | 2.6±0.4 | 1.2±0.3 | 1.4±0.3 | 0.63±0.1 | 2.0±0.4 |
The EC50 values for the four monovalent ligands (i.e. NDP-MSH, α-MSH, MTII, and CJL-1-14) were within the inherent experimental error (<3-fold difference) when comparing the coexpressing cells (with possible heterodimers) to the mixed stable cell category (with no possible heterodimers) (Table 4). The values obtained for coexpressed cell lines corresponded well with the most potent EC50 at the individually expressed cells lines (i.e. stable mMC3R or stable mMC4R cells) as expected based on the principle of the harmonic mean.116 This indicates that there is no observable effect on the potency of these monovalent compounds (in our hands) of the mMC3R and mMC4R being coexpressed in the same cells compared to on different cells. Although it should be noted that the monovalent ligand CJL-1-14 was observed to trend towards increased potency (2.5 fold increase) when the mMC4R stable cells were transiently transfected with the mMC3R plasmid, but this is within the inherent 3-fold experimental error associated with these assays in our laboratory.
Bivalent ligand CJL-1-87 possessed subnanomolar potency that was over 3-fold more potent compared to results achieved with the stable cell mixture when the mMC3R was transiently transfected into the stable mMC4R cell line (Table 4). These results may suggest that the bivalent ligand induced a synergistic effect during coexpression of the mMC3R and mMC4R presumably through a mMC3R-mMC4R heterodimer. However, the increase observed was slight (3-fold) especially considering the monovalent CJL-1-14 resulted in a trending increase as well (2.5-fold). Also the opposite transfection order in which stable mMC3R cells were transiently transfected with the mMC4R did not result in a difference (see Supplemental Materials for a further discussion). Therefore, these results are currently inconclusive, but warrant further studies to establish the functional effects of melanocortin coexpression, and whether the mMC3R-mMC4R heterodimer may be a future neuronal molecular drug target.
Conclusions
The current work demonstrates that the homobivalent ligand CJL-1-87 has a distinct pharmacological in vivo profile compared to the monovalent control ligand CJL-1-14. The findings reported herein, provide new knowledge and molecular probes for future anti-obesity drug design and in vivo mechanism of action studies. The bivalent ligand CJL-1-87 had increased ability to reduce food intake, promote fat utilization, modulate metabolic hormones, and decrease percent body fat when utilized in a mouse fasting-refeeding experimental paradigm as compared to the monovalent control ligand CJL-1-14. This extends previous reports that CJL-1-87 has c.a. 20-fold increased binding affinity and moderately increased functional activity in vitro.21 However, in this current report, in vivo paradigm specific effects of CJL-1-87 could not have been predicted by in vitro assays. Compound CJL-1-87 appears to be effective at reducing food intake in spite of peripheral hormone signals that stimulate hunger and refeeding including elevated ghrelin levels and depressed insulin and leptin levels.
It is postulated that the current in vivo differences reported herein, between CJL-1-87 and CJL-1-14, are due to the bivalent ligand design strategy. Two possible mechanisms to explain the distinct pharmacology of CJL-1-87 compared to CJL-1-14 based on the homobivalent ligand design have been presented. In the first proposed mechanism, the lowered entropy of the bivalent ligand binding allows CJL-1-87 to compete more effectively at displacing the endogenous AGRP MC3R/MC4R antagonist (that is upregulated in the fasting state) resulting in both agonism of the melanocortin receptors and antagonism of the orexigenic effects of AGRP. The monovalent ligand CJL-1-14 has a lower binding affinity is speculated to not compete as effectively with endogenous AGRP. In the second proposed mechanism, it is postulated that CJL-1-87 interacts with melanocortin receptor homo- and/or heterodimers in a distinct molecular mechanism as compared to the monovalent counterpart CJL-1-14. The unique pharmacology observed in vivo may be a result of interacting with MC3R homodimers, MC4R homodimers, or MC3R-MC4R heterodimers that were all identified to form during in vitro BRET studies. It is possible that both of these postulated mechanisms play a synergistic role in the present in vivo observations. However, additional experimental studies are necessary to validate the postulated physiological significance of melanocortin dimers in vivo.
The result presented herein provides “proof-of-concept” and a foundation that homobivalent ligands can achieve unique in vivo effects that are discrete from than their monovalent counterparts. Thus, validating melanocortin homobivalent ligands as a design strategy for therapeutically relevant compounds towards the treatment of energy homeostasis disorders such as obesity or cachexia. The advantage of CJL-1-87 compared to CJL-1-14 is increased in vitro binding affinity, increased in vivo functional potency, increased the duration of action, decreased food intake after fasting, decreased body fat percentage, and differentially altered metabolic hormone levels.
Methods
Compound Preparation
All compounds were prepared as reported previously following standard solid phase peptide synthesis and Fmoc methodology.21 All compounds were purified to greater than 95% purity by RP-HPLC and the mass was confirmed using ESI-MS (University of Minnesota Department of Chemistry Mass Spectrometry Laboratory). The compounds used for animal studies were dissolved in sterile saline (0.9%; Hospira, Lake Forest, IL) to a stock solution of 10−2 M. The compounds used for in vitro serum stability studies, competitive binding studies, and functional cAMP AlphaScreen assays were first dissolved to a 10−2 M stock in DMSO. The stock solution was stored at −20 °C. The day of the assays the 10−2 M stock was diluted to the appropriate concentration in the proper assay buffer (e.g. saline for animal studies.)
Serum Stability Studies
The experimental ligands at an initial concentration of 10 µM in mouse serum (Cat # M5905; Sigma-Aldrich) were incubated at 37 °C on an orbital shaker. At the time points 0, 0.5, 1.5, 3, 6, 8, 24, 48, and 72 h time points, a 50 µL aliquot was taken and the reaction was quenched with 150 µL of cold (4 °C) 66% aqueous acetonitrile. Samples were incubated in a bucket of wet ice for 10–15 min then centrifuged at 12900 rpm at 4 °C. The supernatant was collected into pre-labeled tubes and stored at −80 °C until liquid chromatography-positive electrospray ionization-tandem mass spectrometry (LC-ESI+-MS/MS) analysis was performed. Liquid chromatography was carried out with a reverse-phase linear gradient and a flow rate of 15 µL/min on a 0.5 × 150 mm Zorbax SB-C18 5 µm column (Agilent, Santa Clara, CA). The gradient was from 95% aqueous TFA (0.1%) and 5% acetonitrile to 35% aqueous TFA (0.1%) and 65% acetonitrile in 15 min. This was followed by a washout and re-equilibration period at initial conditions.
Mass spectrometry was performed on a Finnigan TSQ Quantum Discovery MAX triple quadrupole mass analyzer (Thermo Scientific, Waltham, MA). Selected reaction monitoring (SRM) mass transitions were optimized for each compound from the MS/MS product ion spectra of the initial control sample. Collision energy was 35 eV and scan width was 1.0 amu. The following SRM transitions were monitored for each compound: NDP-MSH, m/z 824.2 → 136.1; α-MSH, m/z 555.9 → 136.1 and 833.3 → 136.1; CJL 1-14, m/z 343.9 → 110.1 and 686.7 → 180.1; CJL 1-87, m/z 544.9 → 110.2 and 816.3 → 110.0; CJL 5-35-4, m/z 503.1 → 110.1; CJL 1-116, m/z 482.1 → 156.1 and 321.9 → 156.1; CJL 1-31, m/z 746.8 → 179.9; CJL 5-35-1, m/z 806.4 → 172.0 and 806.4 → 152.0; and CJL 1-41, m/z 785.6 → 445.1 and 524.2 → 195.1. All samples were run in two technical replicates. The signal intensity at time point 0 h was arbitrarily set as 100% and the % intact peptide at each time point was calculated relative to this signal. Data was graphed and half-lives were calculated using PRISM software (v 4.0; GraphPad Inc.).
Animals
All studies were performed in accordance with the Institutional Animal Care and Use Committee (IACUC) of the University of Minnesota. The mice used were all male wild type mice with a mixed genetic background from the C57BL/6J and 129/Sv inbred strains as previously used.21, 56, 57, 60 All mice were maintained on a 12 h light/dark cycle (Lights out was at 11:00 AM) in a temperature controlled room (23–25 °C) with free access to tap water. Excluding the fast prior to injection, mice had free access to a normal chow (Harlan Teklad 2018 Diet: 18.6% crude protein, 6.2% crude fat, 3.5% crude fiber, with energy density of 3.1 kcal/g). The hPYY validation experiments (see below) and the Luminex Multiplex hormone panel experiments took place in standard mouse polycarbonate conventional cages provided by University of Minnesota’s Research Animal Resources (RAR). All cages were changed weekly by lab research staff.
Cannulation Surgery and Placement Validation
Cannulation surgeries were performed to place a cannula into the lateral cerebral ventricle as previously reported.21, 56, 57 All mice were age matched to have surgeries at 8 weeks old. Briefly, mice were anesthetized with a mixture of xylazine (5 mg/kg) and ketamine (100 mg/kg) administered intraperitoneal (IP). Mice were placed in a stereotaxic apparatus (David Kopf Instruments) that was used to guide the cannula placement. A 26-guage cannula (Cat # 8IC315GS4SPC; PlasticsOne, Roanoke, VA) was placed into the lateral cerebral ventricle at the coordinates 1.0 mm lateral and 0.46 mm posterior to bregma and 2.3 mm ventral to the skull.117 Dental cement (C&B-Metabond Adhesive Cement Kit # S380) followed by Lang’s Jet™ Denture Repair Kit (Jet Denture Repair Powder, Ref #1220; Jet Liquid, Ref # 1403) was used to secure the cannula. Flunixin meglumine (FluMegluine, Clipper Distribution Company) and 0.5 mL of 0.9% saline (Hospira, Lake Forrest, IL) was administered subcutaneously after surgery to aid in recovery. Mice were given at least seven days to recover before cannula validation. Mice were housed individually after surgery and for the remainder of the experiments.
Cannula placement was validated by the increase in feeding after the administration of 2.5 µg of human (h)PYY3–36 (Cat # H8585; Bachem) as described previously.21, 56, 57, 60 The mice each received a saline treatment and a hPYY treatment on different days separated by a washout period of at least 3 days in a cross-over design nocturnal feeding paradigm. In the nocturnal feeding paradigm, mice have free access to food and water throughout the experiment. Compound or saline is administered two hours prior to lights out (t = 0 h) and body weight and food intake are measured. A mouse with a validated properly placed cannula consumed at least 0.8 g more food after hPYY administration compared to saline administration at the 4 h time point. On average mice ate 0.4 ± 0.1 g at the 4 h time point after saline administration compared to 1.6 ± 0.1 g after hPYY administration (Supplemental Figure 1).
In Vivo Energy Metabolism Studies
The first cohort of mice with validated cannula placement were transitioned into sealed metabolic home cages and allowed one week to acclimate to the cages. The TSE PhenoMaster metabolic cage system (TSE Systems, Berlin Germany) was configured to measure food intake, water intake, oxygen uptake, carbon dioxide production, and locomotor activity in 15 minute bins. After the acclimation period, a three group crossover design paradigm was utilized to assess the differences between CJL-1-14, CJL-1-87, and saline (Figure 2A). In the fasting-refeeding paradigm, food was removed from the mice at the start of the lights out on the previous day (t = −22 h) for a 22 hour fast. Two hours before lights out on the day of the assay (t = 0 h) mice received the indicated dose (5 nmol compound or saline vehicle control) and food was returned to the cage (Figure 2B). Compound was administered through an internal cannula (Cat# 8IC315IS4SPC; PlasticsOne, Roanoke, VA) in 3 µL of saline.
The cumulative food and water intakes were reported in two hour increments. The energy expenditure and RER were calculated from the oxygen uptake and carbon dioxide production. The oxygen uptake, carbon dioxide production, and locomotor activity were recorded in 15 minute bins. The RER of four 15 minute recordings were averaged for each reported hourly RER. Similarly, the 15 minute energy expenditure readings (kcal/h) were normalized to the animals’ pre-treatment weight (kcal/kg/h) and were then averaged into one hour bins. Activity measurements were the ambulatory activity (XA) defined as consecutive beam breaks of two different X-axis beams. Activity readings of the four 15 minute intervals were summed for each one hour bin. Figures were made using PRISM software (v 4.0; GraphPad Inc.). Statistical analysis was performed using SPSS V23 software (IBM) utilizing a multivariate general linear model followed by a Bonferroni post hoc test. Statistical significance was defined as p < 0.05.
Body Composition Studies
A second cohort of 32 male age matched littermate mice (saline, n=10; 5 nmol CJL-1-14, n=11; 5 nmol CJL-1-87, n=11) remained in standard mouse polycarbonate conventional cages. The second cohort was used for both body composition analysis and Luminex Milliplex assays in a block design. Food intake and body weight were manually measured using a standard top-loading laboratory balance at time points −22, 0, 2, 4, and 6 hours. The amount of lean body mass and fat mass were measured using an EchoMRI-100H™ (Echo Medical Systems LLC, Houston TX, USA) at time points −22, 0, and 6 hours. The lean body mass percentage and body fat mass percentage were calculated based on the amount of lean mass or fat mass measured divided by the manually recorded weight immediately prior to the MRI measurements. Results are presented as the Mean ± SEM.
Luminex Milliplex Hormone Panel Studies
Trunk blood of the second cohort was collected six hours after treatment using 1.5 mL EDTA-K2 coated tubes (Milian, USA) and placed on ice. To prevent hormone degradation, a series of inhibitors were added to each EDTA-K2 coated tube. DPPIV (Cat. No. DPP4, EMD Millipore Corporation, Billerica, MA) was added at a concentration of 10 µL/mL of whole blood. Pefabloc/AEBSF (Product No. 11873601001, Roche, Indianapolis, IN) was added at 1 mg/mL of whole blood. Protease Inhibitor Cocktail (Part No. P8340, Sigma-Aldrich, St. Louis, MO) was added at 10 µL/mL of whole blood. Aprotinin (Part No. A6279, Sigma-Aldrich, Indianapolis, IN) was added at 500 KIU/mL of whole blood. The total whole blood collected per tube was about 500 µL. Whole blood samples were spun at 10,000 rpm for 10 minutes at 4 °C. Plasma was collected from the supernatant and aliquoted to avoid multiple freeze/thaw cycles. The samples were then frozen at −20 °C until they were needed. Plasma hormone levels were measured in duplicate from a 10 µL sample using the Mouse Metabolic Hormone Magnetic Bead Panel Milliplex Kit (Cat. No. MMHMAG-44K, EMD Millipore Corporation, Billerica, MA), which is commercially available. Hormone levels were read and acquired using Magpix instrument and Luminex xPonent 4.2 software (Cat. No. 40-072, EMD Millipore Corporation, Billerica, MA). Data was analyzed using GraphPad Prism.
Cell Culture
Female HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 1% penicillin/streptomycin and 10% newborn calf serum (NCS) at 37 °C in humidified atmosphere of 5% CO2 and 95% air. Stable cell lines were generated using wildtype mMC4R and mMC3R-Flag DNA in a pCDNA3 expression vector (20 µg) utilizing the calcium phosphate transfection method.118 The stable cell populations were selected for using G418 (0.7–1.0 mg/mL). The ligands were all assayed as TFA salts.
Competitive Radioligand Binding Affinity Studies
The human AGRP(87-132) or NDP-MSH peptides were radioiodinated using Na125I following the chloramine T procedure.119 Monoradioiodinated peptide was isolated from uniodinated or diradioiodinated peptide by HPLC using a C18 column. It was eluted isocratically in a mobile phase of 24% acetonitrile and 76% triethylamine phosphate (pH 3.0).
Binding experiments were performed on HEK293 cells stably expressing the mMC3R and mMC4R. Cells were plated in 12-well tissue culture plates (Cat. # 353043, Corning Life Sciences) 1–2 days preceding the competition experiment. Cells were 90–100% confluency on the day of the assay. Media was gently aspirated and cells were treated with a freshly diluted aliquot of experimental non-labeled ligand at the appropriate concentrations (10−12 to 10−4 M) in assay buffer (DMEM and 0.1% bovine serum albumin (BSA)) and a constant amount of 125I-NDP-MSH or 125I-AGRP (100,000 cpm/well). After a one hour incubation at 37 °C, media was gently aspirated and cells were washed once with assay buffer. The assay buffer was gently removed and cells were lysed with NaOH (500 µL; 0.1 M) and Triton X-100 (500 µL; 1%) for a minimum of 10 minutes. The cell lysate was transferred to 12×75 mm polystyrene tubes and radioactivity was quantified on a WIZARD2 Automatic Gamma Counter (PerkinElmer). All experiments were performed with duplicate data points and in at least two independent experiments. The non-specific values were defined as signal from 10−6 M unlabeled NDP-MSH or AGRP(87-132), corresponding to the respective 125I-labeled peptide. Data was analyzed by a nonlinear regression method utilizing the PRISM program (v4.0; GraphPad Inc.) to generate and calculate concentration-effect curves and IC50 values. The standard error of the mean (SEM) was derived from the IC50 values from at least two independent experiments.
Bioluminescence Resonance Energy Transfer (BRET) Studies
In order to examine the association and proximity of the melanocortin receptors, the NanoBRET™ Protein:Protein Interaction System was utilized according to manufacturer’s instructions. Plasmids were constructed to incorporate the NanoLuc® fusion protein and the HaloTag® fusion protein onto the C-terminus of both the mMC3R and mMC4R of the plasmids described above. Competitive binding assays with 125I-NDP-MSH were utilized to support proper cell membrane expression and ligand binding (Supplemental Figure 2, Supplemental Table 2). On the first day, cells were plated in the morning into 6 well plates. In the afternoon of the same day, cells were transiently transfected with melanocortin receptor by adding FuGene6 Transfection (8 µL/well, Promega), DNA (2 µg/well) in OptiMem medium (Invitrogen) at a total volume of 100 µL/well. The ratio of donor NanoLuc® to acceptor HaloTag® DNA was optimized in preliminary experiments and a ratio of 1 Receptor-NanoLuc® plasmid: 4 Receptor-HaloTag® plasmid was utilized for all current experiments. Cells were incubated with transfection reagent overnight at 37° C at 5 % CO2. One day after the transfection, cells were re-plated into 96-well black clear bottom plate (Cat # 3603, Corning Life Sciences) at 30,000 cells in 100 µL of assay buffer (4% FBS in OptiMem). As a control, a mixture of mMC4R-NanoLuc® cells and mMC3R-HaloTag® cells was made by mixing 15,000 cells of each cell type together. In these experimental conditions, the mMC4R-NanoLuc® and mMC3R-HaloTag® should not associate since they are on separate cell membranes.
To each well, 1 µL of 0.1 mM HaloTag® NanoBRET™ 618 ligand was added and incubated 18–24 h at 37° C at 5 % CO2. As a negative control, each assay also included; no acceptor controls in which 1 µL of DMSO was added instead of 618 ligand rendering the BRET relay system incomplete providing the background signal. This background signal was subtracted from the final experimental signal. Plates were then developed 48 to 60 hours after transfection. To develop plates, 25 µL of 5× solution of NanoBRET™ Nano-Glo® Substrate in Opti-MEM® was added to each well. Plates were then read within 10 min on a FlexStation® 3 plate reader (Molecular Devices) at the donor emission wavelength (460 nm) and acceptor emission wavelength (618 nm). The milli BRET Units (mBUs) were calculated by dividing the acceptor emission of 618 nm by the donor emission at 460 nm and multiplying it by 1000. All assays were performed in at least three independent experiments.
AlphaScreen cAMP Functional Bioassay Studies
The AlphaScreen® technology (Cat # 6760625M, PerkinElmer Life Sciences) that was utilized to study the cAMP signaling was performed as described by the manufacturer and described previously by our lab.21, 120 Briefly on the day of the assay, cells that were 70–90% confluency were removed from 10 cm plates using Gibco® Versene solution. Cells were pelleted by centrifugation (Sorvall Super T21 high speed centrifuge, swinging bucket rotor) at 800 rpm for five minutes. The media was gently removed, cells were resuspended in in Dulbecco’s phosphate buffered saline solution (DPBS 1X [−] without calcium and magnesium chloride, Gibco ® Cat # 14190-144), and an aliquot was removed for manual cell counting. Cells were re-pelleted by centrifugation and DPBS was removed. The cells were resuspended in stimulation buffer (Hank’s Balanced Salt Solution [HBSS 10X [−] sodium bicarbonate] and [−] phenol red, Gibco®], 0.5 mM isobutylmethylxanthine [IBMX], 5 mM HEPES buffer solution [1M, Gibco®], 0.1% bovine serum albumin [BSA] in Milli-Q water, pH=7.4) and anti-cAMP acceptor beads (1.0 unit per well, AlphaScreen®). This resuspended cell/acceptor bead solution was added manually to a 384 well microplate (OptiPlate-384; PerkinElmer). The final concentration was 10,000 cells/well and 1.0 Unit of anti-cAMP beads/well. The equal mixture of stable mMC3R cells and stable mMC4R cells was 5,000 cells/well of each cell type for a total of 10,000 cells/well. Cells were stimulated for two hours in a dark laboratory drawer with ligand diluted in stimulation buffer from 10−13 to 10−4 M.
During incubation, a three component biotinylated cAMP/streptavidin donor bead working solution was made with streptavidin donor beads (1 Unit/well, AlphaScreen®), biotinylated cAMP (1 Unit/well, AlphaScreen®), and lysis buffer (10% Tween-20, 5 mM HEPES buffer solution [1M, Gibco®], 0.1% bovine serum albumin [BSA] in Milli-Q water, pH=7.4). After the incubation, the biotinylated cAMP/streptavidin donor bead working solution was added and mixed into each well in the 384 well plate under green light. Cells were incubated for another two hours in the dark. The plate was read using a pre-normalized assay protocol set by the manufacturer on an EnSpire™ Alpha plate reader. All assays were performed in at least three independent experiments with duplicate data points. The potency EC50 values were calculated utilizing nonlinear regression methods using the PRISM software (v4.0; GraphPad Inc.).
Supplementary Material
Acknowledgments
We would like to thank Katlyn Fleming, Mike Powers, and Dr. Mark Ericson for their insightful dialogue, comments, and revisions while preparing this manuscript.
Funding: This work has been supported by NIH Grant R01DK091906 (C.H.-L.). C.J.L. and A.T.Z. were provided support from the University of Minnesota Doctoral Dissertation Fellowship. C.J.L. was provided additional support by the University of Minnesota College of Pharmacy Olsteins Graduate Fellowship. Radioligands were provided through the Georgetown-Howard University Peptide Radioiodination Shared Resource.
Abbreviations
- GPCR
G protein-coupled receptor
- MC3R
melanocortin 3 receptor
- MC4R
melanocortin 4 receptor
- MSH
melanocyte-stimulating hormone
- NDP-MSH
[Nle4,DPhe7]-α-MSH
- PEDG20
19-amino-5-oxo-3,10,13,−16 tetraoxa-6-azanonadecan-1-oic acid
- cAMP
cyclic adenosine monophosphate
- ICV
intracerebroventricular
- RER
respiratory exchange ratio
- AGRP
agouti-related peptide
- POMC
proopiomelanocortin
- ACTH
adrenocorticotropic hormone
- MRI
magnetic resonance imagining
- GIP
gastric inhibitory polypeptide or glucose-dependent insulinotropic peptide
- IL-6
interleukin-6
- MTII
melanotan II
- DNal(2′)
D-(2-naphthyl)alanine
- BRET
bioluminescence resonance energy transfer
- HEK293
human embryonic kidney 293
- RP-HPLC
reverse-phase high-pressure liquid chromatography
- ESI-MS
electro-spray ionization mass spectrometry
- DMSO
dimethyl sulfoxide
- TFA
trifluoroacetic acid
- EDTA
ethylenediaminetetraacetic acid
- DPPIV
dipeptidyl peptidase-4
Footnotes
Associated Content
Supporting Information: Table of amount intact peptide from serum stability assays (S1). Table of IC50 values from binding assays on the BRET receptor constructs (S2) hPYY validation data (S3) Figure of competitive radioligand binding assays on the BRET receptor constructs (S4) Discussion of expression levels during coexpression cAMP studies (S5)
Author Contributions: All animal studies were performed by C.J.L., D.N.A. and S.L.W. Competitive binding assays were performed by C.J.L., K.T.F. and S.M.S. BRET assays were performed by K.T.F. AlphaScreen assays were performed by C.J.L. Radiolabeled compounds were prepared by R.C.S. All experimental compounds were synthesized and prepared by C.J.L. Serum stability studies were performed by C.J.L. and A.T.Z. The manuscript was written by C.J.L. with contributions from all authors.
Conflict of Interest: The authors declare no competing financial interests.
References
- 1.Bhushan RG, Sharma SK, Xie ZH, Daniels DJ, Portoghese PS. A bivalent ligand (KDN-21) reveals spinal delta and kappa opioid receptors are organized as heterodimers that give rise to delta(1) and kappa(2) phenotypes. Selective targeting of delta-kappa heterodimers. J Med Chem. 2004;47:2969–2972. doi: 10.1021/jm0342358. [DOI] [PubMed] [Google Scholar]
- 2.Xu LP, Josan JS, Vagner J, Caplan MR, Hruby VJ, Mash EA, Lynch RM, Morse DL, Gillies RJ. Heterobivalent ligands target cell-surface receptor combinations in vivo. Proc Natl Acad Sci U S A. 2012;109:21295–21300. doi: 10.1073/pnas.1211762109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Portoghese PS, Ronsisvalle G, Larson DL, Yim CB, Sayre LM, Takemori AE. Opioid agonist and antagonist bivalent ligands as receptor probes. Life Sci. 1982;31:1283–1286. doi: 10.1016/0024-3205(82)90362-9. [DOI] [PubMed] [Google Scholar]
- 4.Portoghese PS, Larson DL, Sayre LM, Yim CB, Ronsisvalle G, Tam SW, Takemori AE. Opioid agonist and antagonist bivalent ligands: The relationship between spacer length and selectivity at multiple opioid receptors. J Med Chem. 1986;29:1855–1861. doi: 10.1021/jm00160a010. [DOI] [PubMed] [Google Scholar]
- 5.Portoghese PS, Nagase H, Lipkowski AW, Larson DL, Takemori AE. Binaltorphimine-related bivalent ligands and their kappa-opioid receptor antagonist selectivity. J Med Chem. 1988;31:836–841. doi: 10.1021/jm00399a026. [DOI] [PubMed] [Google Scholar]
- 6.Akgün E, Javed MI, Lunzer MM, Powers MD, Sham YY, Watanabe Y, Portoghese PS. Inhibition of inflammatory and neuropathic pain by targeting a mu opioid receptor/chemokine receptor5 heteromer (MOR-CCR5) J Med Chem. 2015;58:8647–8657. doi: 10.1021/acs.jmedchem.5b01245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daniels DJ, Lenard NR, Etienne CL, Law PY, Roerig SC, Portoghese PS. Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc Natl Acad Sci U S A. 2005;102:19208–19213. doi: 10.1073/pnas.0506627102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Erez M, Takemori AE, Portoghese PS. Narcotic antagonistic potency of bivalent ligands which contain beta-naltrexamine. Evidence for bridging between proximal recognition sites. J Med Chem. 1982;25:847–849. doi: 10.1021/jm00349a016. [DOI] [PubMed] [Google Scholar]
- 9.Conn PM, Rogers DC, Stewart JM, Niedel J, Sheffield T. Conversion of a gonadotropin-releasing hormone antagonist to an agonist. Nature. 1982;296:653–655. doi: 10.1038/296653a0. [DOI] [PubMed] [Google Scholar]
- 10.Blum JJ, Conn PM. Gonadotropin-releasing hormone stimulation of luteinizing hormone release: A ligand-receptor-effector model. Proc Natl Acad Sci U S A. 1982;79:7307–7311. doi: 10.1073/pnas.79.23.7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karellas P, McNaughton M, Baker SP, Scammells PJ. Synthesis of bivalent beta(2)-adrenergic and adenosine A(1) receptor ligands. J Med Chem. 2008;51:6128–6137. doi: 10.1021/jm800613s. [DOI] [PubMed] [Google Scholar]
- 12.Huang G, Pemp D, Stadtmuller P, Nimczick M, Heilmann J, Decker M. Design, synthesis and in vitro evaluation of novel uni- and bivalent ligands for the cannabinoid receptor type 1 with variation of spacer length and structure. Bioorg Med Chem Lett. 2014;24:4209–4214. doi: 10.1016/j.bmcl.2014.07.038. [DOI] [PubMed] [Google Scholar]
- 13.Nimczick M, Pemp D, Darras FH, Chen X, Heilmann J, Decker M. Synthesis and biological evaluation of bivalent cannabinoid receptor ligands based on hCB(2)R selective benzimidazoles reveal unexpected intrinsic properties. Bioorg Med Chem. 2014;22:3938–3946. doi: 10.1016/j.bmc.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 14.Russo O, Berthouze M, Giner M, Soulier JL, Rivail L, Sicsic S, Lezoualc’h F, Jockers R, Berque-Bestel I. Synthesis of specific bivalent probes that functionally interact with 5-HT(4) receptor dimers. J Med Chem. 2007;50:4482–4492. doi: 10.1021/jm070552t. [DOI] [PubMed] [Google Scholar]
- 15.Singh N, Hazari PP, Prakash S, Chuttani K, Khurana H, Chandra H, Mishra AK. A homodimeric bivalent radioligand derived from 1-(2-methoxyphenyl) piperazine with high affinity for in vivo 5-HT1A receptor imaging. Medchemcomm. 2012;3:814–823. [Google Scholar]
- 16.Soulier JL, Russo O, Giner M, Rivail L, Berthouze M, Ongeri S, Maigret B, Fischmeister R, Lezoualc’h F, Sicsic S, Berque-Bestel I. Design and synthesis of specific probes for human 5-HT4 receptor dimerization studies. J Med Chem. 2005;48:6220–6228. doi: 10.1021/jm050234z. [DOI] [PubMed] [Google Scholar]
- 17.Kuhhorn J, Hubner H, Gmeiner P. Bivalent dopamine D2 receptor ligands: Synthesis and binding properties. J Med Chem. 2011;54:4896–4903. doi: 10.1021/jm2004859. [DOI] [PubMed] [Google Scholar]
- 18.Kuhhorn J, Gotz A, Hubner H, Thompson D, Whistler J, Gmeiner P. Development of a Bivalent Dopamine D-2 Receptor Agonist. J Med Chem. 2011;54:7911–7919. doi: 10.1021/jm2009919. [DOI] [PubMed] [Google Scholar]
- 19.Xu Y, Duggineni S, Espitia S, Richman DD, An J, Huang Z. A synthetic bivalent ligand of CXCR4 inhibits HIV infection. Biochem Biophys Res Commun. 2013;435:646–650. doi: 10.1016/j.bbrc.2013.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Busnelli M, Kleinau G, Muttenthaler M, Stoev S, Manning M, Bibic L, Howell LA, McCormick PJ, Di Lascio S, Braida D, Sala M, Rovati GE, Bellini T, Chini B. Design and Characterization of Superpotent Bivalent Ligands Targeting Oxytocin Receptor Dimers via a Channel-Like Structure. J Med Chem. 2016;59:7152–7166. doi: 10.1021/acs.jmedchem.6b00564. [DOI] [PubMed] [Google Scholar]
- 21.Lensing CJ, Freeman KT, Schnell SM, Adank DN, Speth RC, Haskell-Luevano C. An in Vitro and in Vivo Investigation of Bivalent Ligands That Display Preferential Binding and Functional Activity for Different Melanocortin Receptor Homodimers. J Med Chem. 2016;59:3112–3128. doi: 10.1021/acs.jmedchem.5b01894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carrithers MD, Lerner MR. Synthesis and characterization of bivalent peptide ligands targeted to G-protein-coupled receptors. Chem Biol. 1996;3:537–542. doi: 10.1016/s1074-5521(96)90144-1. [DOI] [PubMed] [Google Scholar]
- 23.Alleti R, Vagner J, Dehigaspitiya DC, Moberg VE, Elshan NG, Tafreshi NK, Brabez N, Weber CS, Lynch RM, Hruby VJ, Gillies RJ, Morse DL, Mash EA. Synthesis and characterization of time-resolved fluorescence probes for evaluation of competitive binding to melanocortin receptors. Bioorg Med Chem. 2013;21:5029–5038. doi: 10.1016/j.bmc.2013.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brabez N, Lynch RM, Xu LP, Gillies RJ, Chassaing G, Lavielle S, Hruby VJ. Design, synthesis, and biological studies of efficient multivalent melanotropin ligands: Tools toward melanoma diagnosis and treatment. J Med Chem. 2011;54:7375–7384. doi: 10.1021/jm2009937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elshan NGRD, Jayasundera T, Anglin BL, Weber CS, Lynch RM, Mash EA. Trigonal scaffolds for multivalent targeting of melanocortin receptors. Org Biomol Chem. 2015;13:1778–1791. doi: 10.1039/c4ob02094d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fernandes SM, Lee YS, Gillies RJ, Hruby VJ. Synthesis and evaluation of bivalent ligands for binding to the human melanocortin-4 receptor. Bioorg Med Chem. 2014;22:6360–6365. doi: 10.1016/j.bmc.2014.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Handl HL, Sankaranarayanan R, Josan JS, Vagner J, Mash EA, Gillies RJ, Hruby VJ. Synthesis and evaluation of bivalent NDP-alpha-MSH(7) peptide ligands for binding to the human melanocortin receptor 4 (hMC4R) Bioconjugate Chem. 2007;18:1101–1109. doi: 10.1021/bc0603642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vagner J, Handl HL, Monguchi Y, Jana U, Begay LJ, Mash EA, Hruby VJ, Gillies RJ. Rigid linkers for bioactive peptides. Bioconjugate Chem. 2006;17:1545–1550. doi: 10.1021/bc060154p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vagner J, Handl HL, Gillies RJ, Hruby VJ. Novel targeting strategy based on multimeric ligands for drug delivery and molecular imaging: Homooligomers of alpha-MSH. Bioorg Med Chem Lett. 2004;14:211–215. doi: 10.1016/j.bmcl.2003.09.079. [DOI] [PubMed] [Google Scholar]
- 30.Bowen ME, Monguchi Y, Sankaranarayanan R, Vagner J, Begay LJ, Xu L, Jagadish B, Hruby VJ, Gillies RJ, Mash EA. Design, synthesis, and validation of a branched flexible linker for bioactive peptides. J Org Chem. 2007;72:1675–1680. doi: 10.1021/jo062276g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jagadish B, Sankaranarayanan R, Xu L, Richards R, Vagner J, Hruby VJ, Gillies RJ, Mash EA. Squalene-derived flexible linkers for bioactive peptides. Bioorg Med Chem Lett. 2007;17:3310–3313. doi: 10.1016/j.bmcl.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dehigaspitiya DC, Navath S, Weber CS, Lynch RM, Mash EA. Synthesis and bioactivity of MSH4 oligomers prepared by an A + B strategy. Tetrahedron Lett. 2015;56:3060–3065. doi: 10.1016/j.tetlet.2014.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dehigaspitiya DC, Anglin BL, Smith KR, Weber CS, Lynch RM, Mash EA. Linear scaffolds for multivalent targeting of melanocortin receptors. Organic & biomolecular chemistry. 2015;13:11507–11517. doi: 10.1039/c5ob01779c. [DOI] [PubMed] [Google Scholar]
- 34.Bapst JP, Froidevaux S, Calame M, Tanner H, Eberle AN. Dimeric DOTA-alpha-melanocyte-stimulating hormone analogs: synthesis and in vivo characteristics of radiopeptides with high in vitro activity. J Recept Signal Transduction Res. 2007;27:383–409. doi: 10.1080/10799890701723528. [DOI] [PubMed] [Google Scholar]
- 35.Bagutti C, Stolz B, Albert R, Bruns C, Pless J, Eberle AN. [111In]-DTPA-labeled analogues of alpha-melanocyte-stimulating hormone for melanoma targeting: receptor binding in vitro and in vivo. Int J Cancer. 1994;58:749–755. doi: 10.1002/ijc.2910580521. [DOI] [PubMed] [Google Scholar]
- 36.Morais M, Raposinho PD, Oliveira MC, Correia JDG, Santos I. Evaluation of novel Tc-99m(I)-labeled homobivalent alpha-melanocyte-stimulating hormone analogs for melanocortin-1 receptor targeting. J Biol Inorg Chem. 2012;17:491–505. doi: 10.1007/s00775-011-0871-y. [DOI] [PubMed] [Google Scholar]
- 37.Smeester BA, Lunzer MM, Akgun E, Beitz AJ, Portoghese PS. Targeting putative mu opioid/metabotropic glutamate receptor-5 heteromers produces potent antinociception in a chronic murine bone cancer model. Eur J Pharmacol. 2014;743:48–52. doi: 10.1016/j.ejphar.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Akgun E, Javed MI, Lunzer MM, Smeester BA, Beitz AJ, Portoghese PS. Ligands that interact with putative MOR-mGluR5 heteromer in mice with inflammatory pain produce potent antinociception. Proc Natl Acad Sci U S A. 2013;110:11595–11599. doi: 10.1073/pnas.1305461110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perez M, Pauwels PJ, Fourrier C, Chopin P, Valentin JP, John GW, Marien M, Halazy S. Dimerization of sumatriptan as an efficient way to design a potent, centrally and orally active 5-HT1B agonist. Bioorg Med Chem Lett. 1998;8:675–680. doi: 10.1016/s0960-894x(98)00090-0. [DOI] [PubMed] [Google Scholar]
- 40.Zhang YN, Gilliam A, Maitra R, Damaj MI, Tajuba JM, Seltzman HH, Thomas BF. Synthesis and Biological Evaluation of Bivalent Ligands for the Cannabinoid 1 Receptor. J Med Chem. 2010;53:7048–7060. doi: 10.1021/jm1006676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harikumar KG, Akgun E, Portoghese PS, Miller LJ. Modulation of cell surface expression of nonactivated cholecystokinin receptors using bivalent ligand-induced internalization. J Med Chem. 2010;53:2836–2842. doi: 10.1021/jm100135g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yekkirala AS, Kalyuzhny AE, Portoghese PS. An immunocytochemical-derived correlate for evaluating the bridging of heteromeric mu-delta opioid protomers by bivalent ligands. ACS Chem Biol. 2013;8:1412–1416. doi: 10.1021/cb400113d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Le Naour M, Akgun E, Yekkirala A, Lunzer MM, Powers MD, Kalyuzhny AE, Portoghese PS. Bivalent ligands that target mu opioid (MOP) and cannabinoid1 (CB1) receptors are potent analgesics devoid of tolerance. J Med Chem. 2013;56:5505–5513. doi: 10.1021/jm4005219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Giuliani D, Galantucci M, Neri L, Canalini F, Calevro A, Bitto A, Ottani A, Vandini E, Sena P, Sandrini M, Squadrito F, Zaffe D, Guarini S. Melanocortins protect against brain damage and counteract cognitive decline in a transgenic mouse model of moderate Alzheimers disease. Eur J Pharmacol. 2014;740:144–150. doi: 10.1016/j.ejphar.2014.06.063. [DOI] [PubMed] [Google Scholar]
- 45.Giuliani D, Bitto A, Galantucci M, Zaffe D, Ottani A, Irrera N, Neri L, Cavallini GM, Altavilla D, Botticelli AR, Squadrito F, Guarini S. Melanocortins protect against progression of Alzheimer’s disease in triple-transgenic mice by targeting multiple pathophysiological pathways. Neurobiol Aging. 2014;35:537–547. doi: 10.1016/j.neurobiolaging.2013.08.030. [DOI] [PubMed] [Google Scholar]
- 46.Giuliani D, Neri L, Canalini F, Calevro A, Ottani A, Vandini E, Sena P, Zaffe D, Guarini S. NDP-alpha-MSH induces intense neurogenesis and cognitive recovery in Alzheimer transgenic mice through activation of melanocortin MC4 receptors. Mol Cell Neurosci. 2015;67:13–21. doi: 10.1016/j.mcn.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 47.Uckert S, Bannowsky A, Albrecht K, Kuczyk MA. Melanocortin receptor agonists in the treatment of male and female sexual dysfunctions: results from basic research and clinical studies. Expert Opin Invest Drugs. 2014;23:1477–1483. doi: 10.1517/13543784.2014.934805. [DOI] [PubMed] [Google Scholar]
- 48.Van der Ploeg LH, Martin WJ, Howard AD, Nargund RP, Austin CP, Guan X, Drisko J, Cashen D, Sebhat I, Patchett AA, Figueroa DJ, DiLella AG, Connolly BM, Weinberg DH, Tan CP, Palyha OC, Pong SS, MacNeil T, Rosenblum C, Vongs A, Tang R, Yu H, Sailer AW, Fong TM, Huang C, Tota MR, Chang RS, Stearns R, Tamvakopoulos C, Christ G, Drazen DL, Spar BD, Nelson RJ, MacIntyre DE. A role for the melanocortin 4 receptor in sexual function. Proc Natl Acad Sci U S A. 2002;99:11381–11386. doi: 10.1073/pnas.172378699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Penagarikano O, Lazaro MT, Lu XH, Gordon A, Dong HM, Lam HA, Peles E, Maidment NT, Murphy NP, Yang XW, Golshani P, Geschwind DH. Exogenous and evoked oxytocin restores social behavior in the Cntnap2 mouse model of autism. Sci Transl Med. 2015;7 doi: 10.1126/scitranslmed.3010257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barrett CE, Modi ME, Zhang BC, Walum H, Inoue K, Young LJ. Neonatal melanocortin receptor agonist treatment reduces play fighting and promotes adult attachment in prairie voles in a sex-dependent manner. Neuropharmacology. 2014;85:357–366. doi: 10.1016/j.neuropharm.2014.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Joppa MA, Ling N, Chen C, Gogas KR, Foster AC, Markison S. Central administration of peptide and small molecule MC4 receptor antagonists induce hyperphagia in mice and attenuate cytokine-induced anorexia. Peptides. 2005;26:2294–2301. doi: 10.1016/j.peptides.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 52.Deboer MD, Marks DL. Cachexia: lessons from melanocortin antagonism. Trends Endocrinol Metab. 2006;17:199–204. doi: 10.1016/j.tem.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 53.DeBoer MD, Marks DL. Therapy insight: Use of melanocortin antagonists in the treatment of cachexia in chronic disease. Nat Clin Pract Endocrinol Metab. 2006;2:459–466. doi: 10.1038/ncpendmet0221. [DOI] [PubMed] [Google Scholar]
- 54.Doering SR, Todorovic A, Haskell-Luevano C. Melanocortin antagonist tetrapeptides with minimal agonist activity at the mouse melanocortin-3 receptor. ACS Med Chem Lett. 2015;6:123–127. doi: 10.1021/ml500340z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ericson MD, Wilczynski A, Sorensen NB, Xiang Z, Haskell-Luevano C. Discovery of a beta-Hairpin Octapeptide, c[Pro-Arg-Phe-Phe-Dap-Ala-Phe-DPro], Mimetic of Agouti-Related Protein(87-132) [AGRP(87-132)] with Equipotent Mouse Melanocortin-4 Receptor (mMC4R) Antagonist Pharmacology. J Med Chem. 2015;58:4638–4647. doi: 10.1021/acs.jmedchem.5b00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lensing CJ, Adank DN, Doering SR, Wilber SL, Andreasen A, Schaub JW, Xiang Z, Haskell-Luevano C. Ac-Trp-DPhe(p-I)-Arg-Trp-NH2, a 250-Fold Selective Melanocortin-4 Receptor (MC4R) Antagonist over the Melanocortin-3 Receptor (MC3R), Affects Energy Homeostasis in Male and Female Mice Differently. ACS Chem Neurosci. 2016;7:1283–1291. doi: 10.1021/acschemneuro.6b00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Irani BG, Xiang Z, Yarandi HN, Holder JR, Moore MC, Bauzo RM, Proneth B, Shaw AM, Millard WJ, Chambers JB, Benoit SC, Clegg DJ, Haskell-Luevano C. Implication of the melanocortin-3 receptor in the regulation of food intake. Eur J Pharmacol. 2011;660:80–87. doi: 10.1016/j.ejphar.2010.10.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 59.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 60.Marsh DJ, Hollopeter G, Huszar D, Laufer R, Yagaloff KA, Fisher SL, Burn P, Palmiter RD. Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nat Genet. 1999;21:119–122. doi: 10.1038/5070. [DOI] [PubMed] [Google Scholar]
- 61.Clemmensen C, Finan B, Fischer K, Tom RZ, Legutko B, Sehrer L, Heine D, Grassl N, Meyer CW, Henderson B, Hofmann SM, Tschop MH, Van der Ploeg LH, Muller TD. Dual melanocortin-4 receptor and GLP-1 receptor agonism amplifies metabolic benefits in diet-induced obese mice. EMBO Mol Med. 2015;7:288–298. doi: 10.15252/emmm.201404508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Singh A, Dirain ML, Wilczynski A, Chen C, Gosnell BA, Levine AS, Edison AS, Haskell-Luevano C. Synthesis, biophysical, and pharmacological evaluation of the melanocortin agonist AST3-88: modifications of peptide backbone at Trp 7 position lead to a potent, selective, and stable ligand of the melanocortin 4 receptor (MC4R) ACS Chem Neurosci. 2014;5:1020–1031. doi: 10.1021/cn5000953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, Hadley ME. 4-Norleucine, 7-D-Phenylalanine-Alpha-Melanocyte-Stimulating Hormone - a Highly Potent Alpha-Melanotropin with Ultralong Biological-Activity. P Natl Acad Sci-Biol. 1980;77:5754–5758. doi: 10.1073/pnas.77.10.5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Castrucci AMD, Hadley ME, Sawyer TK, Hruby VJ. Enzymological Studies of Melanotropins. Comp Biochem Phys B. 1984;78:519–524. doi: 10.1016/0305-0491(84)90090-7. [DOI] [PubMed] [Google Scholar]
- 65.Langendonk JG, Balwani M, Anderson KE, Bonkovsky HL, Anstey AV, Bissell DM, Bloomer J, Edwards C, Neumann NJ, Parker C, Phillips JD, Lim HW, Hamzavi I, Deybach JC, Kauppinen R, Rhodes LE, Frank J, Murphy GM, Karstens FPJ, Sijbrands EJG, de Rooij FWM, Lebwohl M, Naik H, Goding CR, Wilson JHP, Desnick RJ. Afamelanotide for Erythropoietic Protoporphyria. N Engl J Med. 2015;373:48–59. doi: 10.1056/NEJMoa1411481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ellacott KL, Morton GJ, Woods SC, Tso P, Schwartz MW. Assessment of feeding behavior in laboratory mice. Cell Metab. 2010;12:10–17. doi: 10.1016/j.cmet.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haskell-Luevano C, Chen PL, Li C, Chang K, Smith MS, Cameron JL, Cone RD. Characterization of the neuroanatomical distribution of agouti-related protein immunoreactivity in the rhesus monkey and the rat. Endocrinology. 1999;140:1408–1415. doi: 10.1210/endo.140.3.6544. [DOI] [PubMed] [Google Scholar]
- 68.Marsh DJ, Miura GI, Yagaloff KA, Schwartz MW, Barsh GS, Palmiter RD. Effects of neuropeptide Y deficiency on hypothalamic agouti-related protein expression and responsiveness to melanocortin analogues. Brain Res. 1999;848:66–77. doi: 10.1016/s0006-8993(99)01962-9. [DOI] [PubMed] [Google Scholar]
- 69.Renquist BJ, Murphy JG, Larson EA, Olsen D, Klein RF, Ellacott KL, Cone RD. Melanocortin-3 receptor regulates the normal fasting response. Proc Natl Acad Sci U S A. 2012;109:E1489–1498. doi: 10.1073/pnas.1201994109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hahn TM, Breininger JF, Baskin DG, Schwartz MW. Coexpression of Agrp and NPY in fasting-activated hypothalamic neurons. Nat Neurosci. 1998;1:271–272. doi: 10.1038/1082. [DOI] [PubMed] [Google Scholar]
- 71.Goodin SZ, Keichler AR, Smith M, Wendt D, Strader AD. Effect of gonadectomy on AgRP-induced weight gain in rats. Am J Physiol-Reg I. 2008;295:R1747–R1753. doi: 10.1152/ajpregu.90345.2008. [DOI] [PubMed] [Google Scholar]
- 72.Jensen TL, Kiersgaard MK, Sorensen DB, Mikkelsen LF. Fasting of mice: a review. Lab Anim. 2013;47:225–240. doi: 10.1177/0023677213501659. [DOI] [PubMed] [Google Scholar]
- 73.Speakman JR. Measuring energy metabolism in the mouse - theoretical, practical, and analytical considerations. Front Physiol. 2013;4:34. doi: 10.3389/fphys.2013.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Meyer CW, Reitmeir P, Tschop MH. Exploration of Energy Metabolism in the Mouse Using Indirect Calorimetry: Measurement of Daily Energy Expenditure (DEE) and Basal Metabolic Rate (BMR) Curr Protoc Mouse Biol. 2015;5:205–222. doi: 10.1002/9780470942390.mo140216. [DOI] [PubMed] [Google Scholar]
- 75.Tanner JM, Kearns DT, Kim BJ, Sloan C, Jia Z, Yang T, Abel ED, Symons JD. Fasting-induced reductions in cardiovascular and metabolic variables occur sooner in obese versus lean mice. Exp Biol Med (Maywood) 2010;235:1489–1497. doi: 10.1258/ebm.2010.010171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marvyn PM, Bradley RM, Mardian EB, Marks KA, Duncan RE. Data on oxygen consumption rate, respiratory exchange ratio, and movement in C57BL/6J female mice on the third day of consuming a high-fat diet. Data in brief. 2016;7:472–475. doi: 10.1016/j.dib.2016.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pardridge WM. Drug delivery to the brain. J Cereb Blood Flow Metab. 1997;17:713–731. doi: 10.1097/00004647-199707000-00001. [DOI] [PubMed] [Google Scholar]
- 78.Maness LM, Kastin AJ, Farrell CL, Banks WA. Fate of leptin after intracerebroventricular injection into the mouse brain. Endocrinology. 1998;139:4556–4562. doi: 10.1210/endo.139.11.6319. [DOI] [PubMed] [Google Scholar]
- 79.Pardridge WM. Drug transport in brain via the cerebrospinal fluid. Fluids and barriers of the CNS. 2011;8:7. doi: 10.1186/2045-8118-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fan W, Dinulescu DM, Butler AA, Zhou J, Marks DL, Cone RD. The central melanocortin system can directly regulate serum insulin levels. Endocrinology. 2000;141:3072–3079. doi: 10.1210/endo.141.9.7665. [DOI] [PubMed] [Google Scholar]
- 81.Obici S, Feng ZH, Tan JZ, Liu LS, Karkanias G, Rossetti L. Central melanocortin receptors regulate insulin action. J Clin Invest. 2001;108:1079–1085. doi: 10.1172/JCI12954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pierroz DD, Ziotopoulou M, Ungsunan L, Moschos S, Flier JS, Mantzoros CS. Effects of acute and chronic administration of the melanocortin agonist MTII in mice with diet-induced obesity. Diabetes. 2002;51:1337–1345. doi: 10.2337/diabetes.51.5.1337. [DOI] [PubMed] [Google Scholar]
- 83.Steiner DF, Cunningham D, Spigelman L, Aten B. Insulin biosynthesis: evidence for a precursor. Science. 1967;157:697–700. doi: 10.1126/science.157.3789.697. [DOI] [PubMed] [Google Scholar]
- 84.Rubenstein AH, Clark JL, Melani F, Steiner DF. Secretion of Proinsulin C-Peptide by Pancreatic [beta] Cells and its Circulation in Blood. Nature. 1969;224:697–699. [Google Scholar]
- 85.Panaro BL, Tough IR, Engelstoft MS, Matthews RT, Digby GJ, Moller CL, Svendsen B, Gribble F, Reimann F, Holst JJ, Holst B, Schwartz TW, Cox HM, Cone RD. The Melanocortin-4 Receptor Is Expressed in Enteroendocrine L Cells and Regulates the Release of Peptide YY and Glucagon-like Peptide 1 In Vivo. Cell metabolism. 2014;20:1018–1029. doi: 10.1016/j.cmet.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee JH, Bullen JW, Jr, Stoyneva VL, Mantzoros CS. Circulating resistin in lean, obese, and insulin-resistant mouse models: lack of association with insulinemia and glycemia. Am J Physiol Endocrinol Metab. 2005;288:E625–632. doi: 10.1152/ajpendo.00184.2004. [DOI] [PubMed] [Google Scholar]
- 87.Lin J, Choi YH, Hartzell DL, Li CL, Della-Fera MA, Baile CA. CNS melanocortin and leptin effects on stearoyl-CoA desaturase-1 and resistin expression. Biochem Biophys Res Commun. 2003;311:324–328. doi: 10.1016/j.bbrc.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 88.Huang QH, Hruby VJ, Tatro JB. Systemic alpha-MSH suppresses LPS fever via central melanocortin receptors independently of its suppression of corticosterone and IL-6 release. Am J Physiol-Reg I. 1998;275:R524–R530. doi: 10.1152/ajpregu.1998.275.2.R524. [DOI] [PubMed] [Google Scholar]
- 89.Ottani A, Neri L, Canalini F, Calevro A, Rossi R, Cappelli G, Ballestri M, Giuliani D, Guarini S. Protective effects of the melanocortin analog NDP-alpha-MSH in rats undergoing cardiac arrest. Eur J Pharmacol. 2014;745:108–116. doi: 10.1016/j.ejphar.2014.10.022. [DOI] [PubMed] [Google Scholar]
- 90.Bohm M, Apel M, Sugawara K, Brehler R, Jurk K, Luger TA, Haas H, Paus R, Eiz-Vesper B, Walls AF, Ponimaskin E, Gehring M, Kapp A, Raap U. Modulation of basophil activity: A novel function of the neuropeptide alpha-melanocyte-stimulating hormone. J Allergy Clin Immunol. 2012;129:1085–1093. doi: 10.1016/j.jaci.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 91.Jun DJ, Na KY, Kim W, Kwak D, Kwon EJ, Yoon JH, Yea K, Lee H, Kim J, Suh PG, Ryu SH, Kim KT. Melanocortins induce interleukin 6 gene expression and secretion through melanocortin receptors 2 and 5 in 3T3-L1 adipocytes. J Mol Endocrinol. 2010;44:225–236. doi: 10.1677/JME-09-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Engelstoft MS, Park WM, Sakata I, Kristensen LV, Husted AS, Osborne-Lawrence S, Piper PK, Walker AK, Pedersen MH, Nohr MK, Pan J, Sinz CJ, Carrington PE, Akiyama TE, Jones RM, Tang C, Ahmed K, Offermanns S, Egerod KL, Zigman JM, Schwartz TW. Seven transmembrane G protein-coupled receptor repertoire of gastric ghrelin cells. Molecular metabolism. 2013;2:376–392. doi: 10.1016/j.molmet.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA. The hormone resistin links obesity to diabetes. Nature. 2001;409:307–312. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- 94.Champy MF, Selloum M, Piard L, Zeitler V, Caradec C, Chambon P, Auwerx J. Mouse functional genomics requires standardization of mouse handling and housing conditions. Mamm Genome. 2004;15:768–783. doi: 10.1007/s00335-004-2393-1. [DOI] [PubMed] [Google Scholar]
- 95.Elliott RM, Morgan LM, Tredger JA, Deacon S, Wright J, Marks V. Glucagon-like peptide-1(7–36)amide and glucose-dependent insulinotropic polypeptide secretion in response to nutrient ingestion in man: acute post-prandial and 24-h secretion patterns. J Endocrinol. 1993;138:159–166. doi: 10.1677/joe.0.1380159. [DOI] [PubMed] [Google Scholar]
- 96.Asakawa A, Inui A, Kaga T, Yuzuriha H, Nagata T, Ueno N, Makino S, Fujimiya M, Niijima A, Fujino MA, Kasuga M. Ghrelin is an appetite-stimulatory signal from stomach with structural resemblance to motilin. Gastroenterology. 2001;120:337–345. doi: 10.1053/gast.2001.22158. [DOI] [PubMed] [Google Scholar]
- 97.Lavin DN, Joesting JJ, Chiu GS, Moon ML, Meng J, Dilger RN, Freund GG. Fasting induces an anti-inflammatory effect on the neuroimmune system which a high-fat diet prevents. Obesity. 2011;19:1586–1594. doi: 10.1038/oby.2011.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mandrika I, Petrovska R, Wikberg J. Melanocortin receptors form constitutive homo- and heterodimers. Biochem Biophys Res Commun. 2005;326:349–354. doi: 10.1016/j.bbrc.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 99.Nickolls SA, Maki RA. Dimerization of the melanocortin 4 receptor: A study using bioluminescence resonance energy transfer. Peptides. 2006;27:380–387. doi: 10.1016/j.peptides.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 100.Kopanchuk S, Veiksina S, Mutulis F, Mutule I, Yahorava S, Mandrika I, Petrovska R, Rinken A, Wikberg JE. Kinetic evidence for tandemly arranged ligand binding sites in melanocortin 4 receptor complexes. Neurochem Int. 2006;49:533–542. doi: 10.1016/j.neuint.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 101.Zanna PT, Sanchez-Laorden BL, Perez-Oliva AB, Turpin MC, Herraiz C, Jimenez-Cervantes C, Garcia-Borron JC. Mechanism of dimerization of the human melanocortin 1 receptor. Biochem Biophys Res Commun. 2008;368:211–216. doi: 10.1016/j.bbrc.2008.01.060. [DOI] [PubMed] [Google Scholar]
- 102.Sebag JA, Hinkle PM. Opposite effects of the melanocortin-2 (MC2) receptor accessory protein MRAP on MC2 and MC5 receptor dimerization and trafficking. J Biol Chem. 2009;284:22641–22648. doi: 10.1074/jbc.M109.022400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Piechowski CL, Rediger A, Lagemann C, Muhlhaus J, Muller A, Pratzka J, Tarnow P, Gruters A, Krude H, Kleinau G, Biebermann H. Inhibition of melanocortin-4 receptor dimerization by substitutions in intracellular loop 2. J Mol Endocrinol. 2013;51:109–118. doi: 10.1530/JME-13-0061. [DOI] [PubMed] [Google Scholar]
- 104.Rediger A, Piechowski CL, Habegger K, Gruters A, Krude H, Tschop MH, Kleinau G, Biebermann H. MC4R dimerization in the paraventricular nucleus and GHSR/MC3R heterodimerization in the arcuate nucleus: Is there relevance for body weight regulation? Neuroendocrinology. 2012;95:277–288. doi: 10.1159/000334903. [DOI] [PubMed] [Google Scholar]
- 105.Biebermann H, Krude H, Elsner A, Chubanov V, Gudermann T, Gruters A. Autosomal-dominant mode of inheritance of a melanocortin-4 receptor mutation in a patient with severe early-onset obesity is due to a dominant-negative effect caused by receptor dimerization. Diabetes. 2003;52:2984–2988. doi: 10.2337/diabetes.52.12.2984. [DOI] [PubMed] [Google Scholar]
- 106.Chapman KL, Findlay JB. The melanocortin 4 receptor: oligomer formation, interaction sites and functional significance. Biochim Biophys Acta. 2013;1828:535–542. doi: 10.1016/j.bbamem.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 107.Elsner A, Tarnow P, Schaefer M, Ambrugger P, Krude H, Gruters A, Biebermann H. MC4R oligomerizes independently of extracellular cysteine residues. Peptides. 2006;27:372–379. doi: 10.1016/j.peptides.2005.02.027. [DOI] [PubMed] [Google Scholar]
- 108.Kobayashi Y, Hamamoto A, Takahashi A, Saito Y. Dimerization of melanocortin receptor 1 (MC1R) and MC5R creates a ligand-dependent signal modulation: Potential participation in physiological color change in the flounder. Gen Comp Endocrinol. 2016;230–231:103–109. doi: 10.1016/j.ygcen.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 109.Mountjoy KG, Mortrud MT, Low MJ, Simerly RB, Cone RD. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol Endocrinol. 1994;8:1298–1308. doi: 10.1210/mend.8.10.7854347. [DOI] [PubMed] [Google Scholar]
- 110.Wachira SJ, Hughes-Darden CA, Nicholas HB, Jr, Taylor CV, Robinson TJ. Neural melanocortin receptors are differentially expressed and regulated by stress in rat hypothalamic-pituitary-adrenal axis. Cell Mol Biol (Noisy-le-grand) 2004;50:703–713. [PubMed] [Google Scholar]
- 111.Mountjoy KG. Distribution and Function of Melanocortin Receptors within the Brain. Melanocortins: Multiple Actions and Therapeutic Potential. 2010;681:29–48. doi: 10.1007/978-1-4419-6354-3_3. [DOI] [PubMed] [Google Scholar]
- 112.Atalayer D, Robertson KL, Haskell-Luevano C, Andreasen A, Rowland NE. Food demand and meal size in mice with single or combined disruption of melanocortin type 3 and 4 receptors. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1667–1674. doi: 10.1152/ajpregu.00562.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao L, Metzger JM, Strack AM, Camacho RE, Mellin TN, Nunes CN, Min W, Fisher J, Gopal-Truter S, MacIntyre DE, Chen HY, Van der Ploeg LH. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat Genet. 2000;26:97–102. doi: 10.1038/79254. [DOI] [PubMed] [Google Scholar]
- 114.Ericson MD, Schnell SM, Freeman KT, Haskell-Luevano C. A fragment of the Escherichia coli ClpB heat-shock protein is a micromolar melanocortin 1 receptor agonist. Bioorg Med Chem Lett. 2015;25:5306–5308. doi: 10.1016/j.bmcl.2015.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tala SR, Schnell SM, Haskell-Luevano C. Microwave-assisted solid-phase synthesis of side-chain to side-chain lactam-bridge cyclic peptides. Bioorg Med Chem Lett. 2015;25:5708–5711. doi: 10.1016/j.bmcl.2015.10.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Santos RG, Giulianotti MA, Dooley CT, Pinilla C, Appel JR, Houghten RA. Use and Implications of the Harmonic Mean Model on Mixtures for Basic Research and Drug Discovery. Acs Comb Sci. 2011;13:337–344. doi: 10.1021/co100065a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. Academic Press; San Diego: 1997. [Google Scholar]
- 118.Chen CA, Okayama H. Calcium phosphate-mediated gene transfer: a highly efficient transfection system for stably transforming cells with plasmid DNA. Biotechniques. 1988;6:632–638. [PubMed] [Google Scholar]
- 119.Hunter WM, Greenwood FC. Preparation of iodine-131 labelled human growth hormone of high specific activity. Nature. 1962;194:495–496. doi: 10.1038/194495a0. [DOI] [PubMed] [Google Scholar]
- 120.Singh A, Tala SR, Flores V, Freeman K, Haskell-Luevano C. Synthesis and Pharmacology of alpha/beta(3)-Peptides Based on the Melanocortin Agonist Ac-His-dPhe-Arg-Trp-NH2 Sequence. ACS Med Chem Lett. 2015;6:568–572. doi: 10.1021/acsmedchemlett.5b00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.