

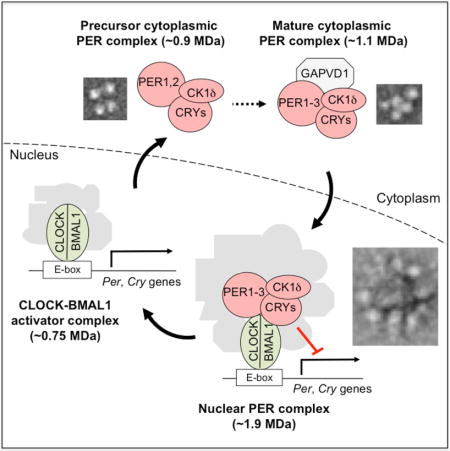
Abstract
The mammalian circadian clock is built on a feedback loop in which PER and CRY proteins repress their own transcription. We found that in mouse liver nuclei all three PERs, both CRYs, and Casein Kinase-1δ (CK1δ) are present together in a ~1.9-MDa repressor assembly that quantitatively incorporates its CLOCK-BMAL1 transcription factor target. Prior to incorporation, CLOCK-BMAL1 exists in a 750-kDa complex. Single-particle electron microscopy (EM) revealed nuclear PER complexes purified from mouse liver to be quasi-spherical ~40-nm structures. In the cytoplasm, PERs, CRYs, and CK1δ were distributed into several complexes of ~0.9–1.1 MDa that appear to constitute an assembly pathway regulated by GAPVD1, a cytoplasmic trafficking factor. Single-particle EM of two purified cytoplasmic PER complexes revealed ~20-nm and ~25-nm structures, respectively, characterized by flexibly-tethered globular domains. Our results define the macromolecular assemblies comprising the circadian feedback loop and provide an initial structural view of endogenous eukaryotic clock machinery.
TOC image
Aryal et al. report that core circadian clock proteins in the nucleus are present together in a ~1.9-MDa, quasi-spherical, 40-nm complex. In the cytoplasm, clock proteins are incorporated into several complexes of ~0.9–1.1 MDa, representing a likely assembly pathway. Two are flexible, multi-globular structures of ~20 and ~25 nm, respectively.
INTRODUCTION
Circadan clocks are molecular oscillators with ~24-hour periods that drive daily biological rhythms. Such clocks are found in animals, plants, fungi, and cyanobacteria (Hurley et al., 2016). They likely represent ancient timekeeping mechanisms important for predicting daily environmental cycles on a rotating planet. In mammals, circadian clocks are present in most if not all cells (Balsalobre et al., 1998; Dibner et al., 2010). These distributed clocks control a myriad of processes, in aggregate creating coherent 24-hour programs of physiology, metabolism, and behavior (Durgan et al., 2006; Lamia et al., 2009; Marcheva et al., 2010).
The mammalian clock is built on a conserved transcriptional feedback loop in which the PERIOD (PER) and CRYPTOCHROME (CRY) proteins inhibit their own expression (Partch et al., 2014). The three PER and two CRY proteins accumulate in the cytoplasm and interact with Casein Kinase-1 (CK1), a protein kinase important for clock function that is thought to regulate PER and CRY stability and nuclear entry (Cheong and Virshup, 2011). Accumulating evidence indicates that CK1δ is the isoform important for circadian rhythms (Xu et al., 2005; Etchegaray et al., 2009; Walton et al., 2014; Kim et al., 2014). Little is known about the organization of clock proteins in the cytoplasm or the factors regulating their cytoplasmic accumulation, interactions, or nuclear entry.
The PERs, CRYs, and CK1δ enter the nucleus, where they form at least one assembly, the nuclear PER complex, of >1 MDa in mass (Brown et al., 2005; Kim et al., 2014). The nuclear PER complex incorporates several independent, pre-existing transcriptional repressor complexes (including chromatin modifiers and remodelers) and is comprised of about thirty proteins (Kim et al., 2014). PER and CRY proteins within the complex interact with DNA-bound CLOCK-BMAL1, the transcription factor activating Per and Cry gene expression (Partch et al., 2014), thereby delivering the cargo of repressors to CLOCK-BMAL1 and adjacent chromatin (Duong et al., 2011; Padmanabhan et al., 2012; Duong and Weitz, 2014; Kim et al., 2014). Although CLOCK-BMAL1 is customarily described as a heterodimer, whether it is present as such in vivo is unknown. Additional findings suggest that PERs and CRYs promote dissociation of CLOCK-BMAL1 from DNA (Chiou et al., 2016) and that CRY1 might act independently of PERs in negative feedback (Koike et al., 2012; Michael et al., 2017). Various single Per and Cry null mutations lead to distinct circadian phenotypes (Liu et al., 2007), sometimes interpreted as indicating that different PERs and CRYs function as physically independent repressors. Thus the number and nature of circadian clock protein complexes is unclear.
Factors outside the core feedback loop are also important for circadian rhythms. A coupled transcriptional loop involving REV-ERBα and ROR regulates circadian amplitude (Zhao et al., 2016). Additional CLOCK-BMAL1 inhibitors (Zhao et al., 2006; Duffield et al., 2009; Robles et al., 2010; Anafi et al., 2014) and post-transcriptional processes (Menet et al., 2012) also influence the circadian cycle. Physiological inputs to the transcriptional cycle include NAD sensing (Asher and Schibler, 2011) and heme metabolism (Klemz et al., 2016).
Although incompletely characterized, the nuclear PER complex is clearly elaborate, dynamic, and responsive to multiple signals, attributes of a macromolecular protein machine (Alberts, 1998). In contrast to other macromolecular machines like the ribosome (Yusupova and Yusupov, 2014) or the proteasome (Kish-Trier and Hill, 2013), virtually nothing is known about its integrated function. Crystal structures of several core clock proteins in monomeric or heterodimeric form (Huang et al., 2012; Czarna et al., 2013; Nangle et al., 2014; Schmalen et al., 2014) represent important early steps toward this goal, but at present there is not even a rudimentary understanding of the structural organization of the PER complex, the factors governing its assembly, or how its many components work together.
With the long-term goal of understanding the circadian clock in terms of the integrated functions of its macromolecular machines, we developed assays to monitor native circadian clock protein complexes across the circadian cycle, and we optimized the affinity purification of endogenous PER complexes from mouse tissues for in vitro biochemistry and single-particle analysis by electron microscopy (EM). The results define the organization of circadian clock proteins in the cell, provide a new view of the nuclear PER complex as an integrated whole, and reveal a likely assembly pathway for the PER complex in the cytoplasm.
RESULTS
Mass and circadian profile of the nuclear PER complex
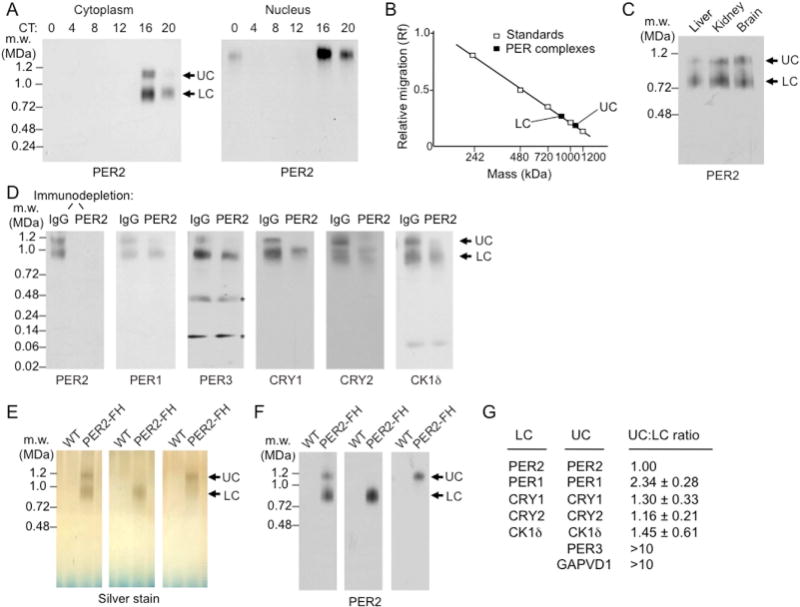
Our previous work indicated that the nuclear PER complex is >1.2 MDa in mass (Kim et al., 2014). To obtain a more precise estimate, we developed a modified electrophoretic assay, blue native-agarose polyacrylamide gel electrophoresis (BN-APAGE), which allows complexes of up to 6 MDa or more to be resolved. We prepared nuclear extracts from mouse liver at circadian time 18 hours (CT18), the approximate peak of PER-CRY negative feedback. The extraction efficiently removes PER complexes from chromatin, and the solubilized complexes retain the incorporated CLOCK-BMAL1 target (Kim et al., 2014). The nuclear extracts were resolved by BN-APAGE, and the resulting immunoblots were probed for PER2, which showed an apparent single band migrating at 1.9 ± 0.1 MDa (SEM; n = 4) (Figure 1A,B). Nuclear extracts from mouse liver, brain, and kidney exhibited an apparent single band of virtually identical migration (Figure 1C), indicating that an exclusive PER2-containing complex of this mass is a common or universal feature of the clock.
Figure 1. Nuclear PER complex: mass, circadian profile, and clock proteins.
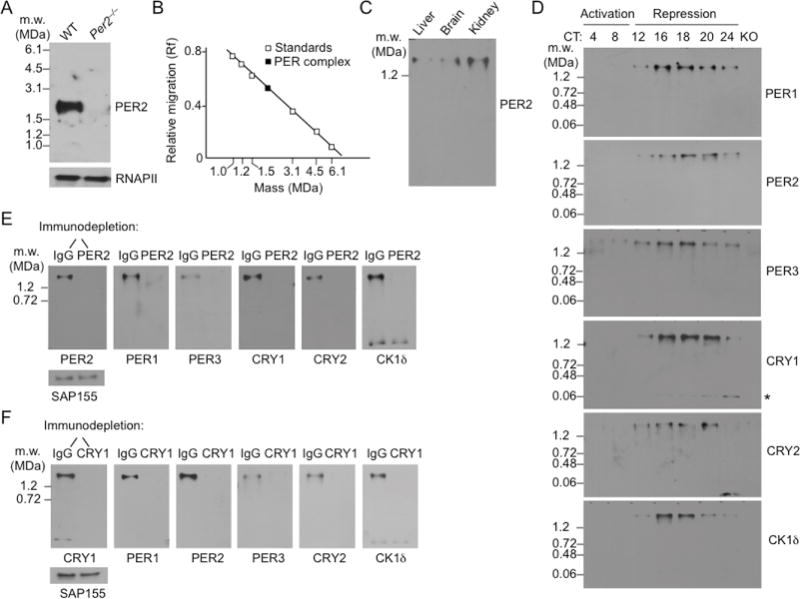
(A) Mass estimate of nuclear PER complex from mouse liver (CT18). Top, Immunoblot of a BN-APAGE gel probed for PER2. Genotypes labeled at top. Molecular weight markers, left. MDa, megadaltons. Bottom, SDS-PAGE immunoblot loading control, RNA Polymerase II large subunit (RNAPII).
(B) Nuclear PER complex: relative migration in BN-APAGE vs. log molecular mass.
(C) BN-APAGE immunoblot of nuclear extracts from mouse liver, brain, and kidney (CT18) probed for PER2.
(D) Circadian profiles of nuclear complexes containing the core negative feedback proteins. Nuclear extracts from mouse livers collected at the indicated circadian times (CT) were analyzed by BN-APAGE, and immunoblots were probed for the proteins indicated at the right of each panel. KO, control sample (CT18) from a null mutant lacking the particular protein probed on each panel, except for CK1δ, which was from Per2−/−. Asterisk, CRY1 monomer. See Figure S1.
(E) Mouse liver nuclear extracts (CT18) were immunodepleted with control IgG or anti-PER2 antibody, and the depleted (unbound) fraction was analyzed by BN-APAGE. Immunoblots were probed for the proteins indicated at the bottom of each panel. Nuclear protein SAP155 (SDS-PAGE) shows input.
(F) As in (E), except extracts were immunodepleted with control IgG or anti-CRY1 antibody.
To identify native nuclear complexes into which the core clock proteins are assembled, we prepared nuclear extracts from mouse livers obtained at time points spanning the circadian cycle, resolved complexes by BN-APAGE, prepared immunoblots, and probed the blots for PER1, PER2, PER3, CRY1, CRY2, and CK1δ. We aimed to determine whether the various clock proteins were distributed into different protein complexes that might function independently in negative feedback, as is often assumed. The results showed that all six of the core clock proteins migrated at the same ~1.9-MDa mass from the earliest point in the cycle that they were detectable in the nucleus (the onset of negative feedback) until they turned over (the termination of negative feedback), and we detected no complexes migrating at other masses for any of the six proteins (Figure 1D). We thus found no positive evidence for the co-existence of different circadian clock repressor complexes, although it is possible that such complexes exist but are labile in our extraction conditions. This assessment was supported by long exposures of the immunoblots, which showed exclusive incorporation of all six clock proteins into one or more complexes migrating at ~1.9 MDa, with the slight exceptions of CRY1 and CK1δ, which showed in addition a single minor band migrating at the size of the CRY1 or CK1δ monomer, respectively (Figure S1A). There was no evidence for nuclear PER-CRY or PER-PER dimers, PER or CRY ternary complexes, or other small assemblies that are commonly invoked. The results do not exclude the possibility that such assemblies are present at low abundance or appear transiently during the cycle.
The core clock proteins are present together in the nuclear PER complex
The analysis presented in Figure 1 does not exclude the possibility that clock proteins are distributed into independent repressor complexes if the hypothetical complexes have masses too similar to resolve by BN-APAGE. To determine which specific clock proteins are together in complexes, we performed immunodepletions from mouse liver nuclear extracts (CT18) and analyzed the samples by BN-APAGE immunoblots. Quantitative immunodepletion of complexes containing PER2 resulted in the specific, quantitative co-depletion of PER1, PER3, CRY1, CRY2, and CK1 δ (except for the small amount of CK1δ monomer, as would be expected) (Figure 1E), indicating that each of these proteins is present exclusively in one or more complexes that includes PER2. Similarly, quantitative immunodepletion of complexes containing CRY1 resulted in the specific, quantitative co-depletion of PER1, PER2, PER3, CRY2, and CK1δ (Figure 1F), indicating that each of these proteins is present exclusively in one or more complexes that includes CRY1. Because all of the CRY1 (other than the monomer) is associated with PER2, all of the PER2 is associated with CRY1, and both PER2 and CRY1 are quantitatively associated with each of the remaining proteins, we can conclude that all six of the core clock proteins are exclusively present together in individual complexes.
Nuclear PER complexes quantitatively incorporate CLOCK-BMAL1
We next used BN-APAGE immunoblot analysis of liver nuclear extracts to monitor CLOCK-BMAL1 over the circadian cycle. During the circadian transcriptional activation phase, when little or no PER complex is present in the nucleus, BMAL1 and CLOCK were predominantly or exclusively present in a ~750-kDa complex; no signals for either protein were detected at the mass corresponding to the free heterodimer, ~185 kDa, or at the masses of the respective monomers, ~75 kDa and ~110 kDa (Figure 2A). As the PER complex accumulated in the nucleus from CT12–18 (Figure 1D), the signals for BMAL1 and CLOCK at ~750 kDa progressively diminished, while at the same time new signals for BMAL1 and CLOCK appeared and gradually intensified at the migration position of the ~1.9-MDa PER complex (Figure 2A); this large CLOCK-BMAL1-containing complex followed the abundance cycle expected for the PER complex (Figure 1D). Experiments with one-hour time resolution (CT16–20) indicated that CLOCK-BMAL1 became quantitatively incorporated into this high-molecular mass complex at about CT19, with only an occasional trace of BMAL1 or CLOCK signals remaining at the ~750-kDa position (examples in Figure 2B).
Figure 2. CLOCK-BMAL1 becomes fully incorporated into the PER complex.
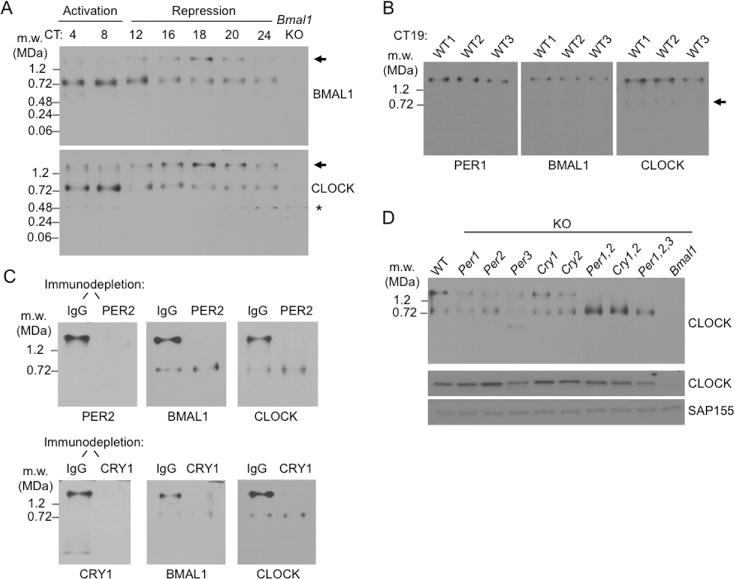
(A) BN-APAGE immunoblots showing circadian profiles of liver nuclear protein complexes containing BMAL1 (top) or CLOCK (bottom) at the indicated circadian times. Arrow, position of PER complex. Asterisk, non-specific. Loss of CLOCK signal in Bmal1−/− (Bmal1 KO) suggests instability of CLOCK monomer. See Figure S2.
(B) Quantitative incorporation of CLOCK-BMAL1 at the peak of the repression phase. BN-APAGE immunoblots of nuclear extracts from livers collected at CT19 from three wildtype mice, probed for the proteins indicated at bottom. Arrow, trace ~750-kDa complex containing CLOCK-BMAL1. Three biological replicates shown.
(C) High molecular-weight complex containing CLOCK-BMAL1 is the PER complex. Mouse liver nuclear extracts (CT19) were immunodepleted with control IgG or anti-PER2 antibody (top), or IgG and anti-CRY1 antibody (bottom), and the depleted (unbound) fraction was analyzed by BN-APAGE. Immunoblots of the gels were probed for proteins indicated at bottom.
(D) Genetic analysis of incorporation of CLOCK-BMAL1 into the PER complex. Top, BN-APAGE immunoblot of liver nuclear extracts (CT18) from wildtype (WT) or mutants null for the circadian clock genes indicated at the top (KO) probed for CLOCK. Middle and bottom, SDS-PAGE immunoblots showing CLOCK expression and SAP155 loading control, respectively.
To confirm that this result reflected incorporation into the PER complex, we immunodepleted complexes containing PER2 or CRY1 from liver nuclear extracts (CT19) and analyzed the samples by BN-APAGE immunoblots. In both PER2 and CRY1 immunodepletions, we observed essentially complete co-depletion of BMAL1 and CLOCK from the high-molecular-mass complex but no depletion from the residual ~750-kDa complex (Figure 2C). In addition, both the 750-kDa complex and the high-molecular-mass complex were observed in wildtype mice and in single clock gene mutants that have at least some clock function, whereas only the 750-kDa complex was detected in double Cry1−/−; Cry2−/− (van der Horst et al., 1999) or triple Per1−/−; Per2−/−; Per3−/− (Dallman and Weaver, 2010) mutants that lack negative feedback and clock function (Figure 2D). Together the results indicate that CLOCK-BMAL1 becomes quantitatively incorporated into the PER complex.
To investigate the ~750-kDa CLOCK-BMAL1 complex, we enriched the complexes from liver nuclear extract (CT4) with an anti-CLOCK antibody, resolved the eluted, partially-purified complexes by BN-APAGE, prepared immunoblots, and probed the blots for six factors known to interact with CLOCK-BMAL1. Notably, the 750-kDa CLOCK-BMAL1 complex was not detectably labeled by antibodies against any of the six proteins, namely MLL1 (Katada and Sassone-Corsi, 2010), JARID1a (DiTacchio et al., 2011), TRAP150 (Lande-Diner et al., 2013), CHD4 or MTA2 (Kim et al., 2014), or DDB1 (Tamayo et al., 2015) (Figure S2A,B), even though each was present in the extract, specifically co-immunoprecipitated with CLOCK (Figure S2C), and was detectable in one or more complexes in BN-APAGE analysis of the crude extract (Figure S2B). Thus it appears that none of the six CLOCK-BMAL1-associated proteins is a stoichiometric component of the ~750-kDa CLOCK-BMAL1 complex, although it is formally possible that one or more is present in the complex but undetectable because of a sterically masked epitope. Instead, each appears to be associated with only a small fraction of CLOCK-BMAL1.
Nuclear PER complexes are quasi-spherical 40-nm particles
Nothing is known about the structural organization of endogenous circadian clock machinery from any eukaryote. To investigate the basic structure of the nuclear PER complex, we affinity purified PER complexes (Figure S3) from liver nuclei (CT18) of PER2-FH mice (Figures 3A,B), a line in which the PER2 protein was replaced by a PER2 fusion protein tandem-tagged at the C terminus with FLAG and Hemagglutinin epitopes (Duong et al., 2011). Control samples, mock-purified from wildtype mice (i.e., mice with untagged PER2), or samples of affinity-purified nuclear PER complexes from PER2-FH mice were negatively stained with uranyl formate and analyzed by single-particle EM. Control samples showed few if any particles of characteristic appearance (Figure 3C), whereas purified PER complexes appeared as beaded particles of about 40-nm diameter (Figure 3D,E), a size compatible with a ~1.9-MDa mass. To obtain improved structural detail, we generated two-dimensional class averages, which revealed a quasi-spherical structure in which a large central core is studded with smaller globular domains (Figures 3F,G and S4). The images presented a consistent picture of the general architecture of the complex, but the variability in size and fine details suggests that the class averages represent not only different angles of view of the complex but also complexes with compositional and conformational heterogeneity.
Figure 3. Purification and single-particle EM analysis of mouse liver nuclear PER complex (CT18).

(A) Silver-stained BN-PAGE gel showing wildtype (WT) control (untagged PER2) sample and purified nuclear PER complex from PER2-FH mice. Arrow, purified complex. See Figure S3.
(B) PER2 immunoblot of gel in (A).
(C–F) Single-particle EM analysis of purified negatively stained PER complexes.
(C) Negative control, representative EM image of negatively stained sample from mock-purification of extract from wildtype mice. Scale bar as indicated.
(D) Representative EM image of negatively stained PER complexes from PER2-FH mice.
(E) Gallery of negatively stained PER complexes obtained in independent experiments. Boxes, 100 nm.
(F) Class averages of negatively stained nuclear PER complexes obtained from classifying 10,866 particles into 100 classes. Boxes, 78.3 nm. Asterisks, class average images enlarged in (G). See Figure S4.
(G) Enlarged views of the class averages marked in (F).
Activity of CK1δ in purified PER complexes
In pilot experiments, we found affinity-purified nuclear PER complexes to be stable in vitro for at least a few hours at room temperature, enabling us to perform kinase assays to examine the actions of CK1δ within its native environment, the endogenous PER complex. After incubation with γ-32P-ATP at 25°C for 1 h, conventional blue native gel (BN-PAGE) autoradiography showed that intact PER complexes were radioactively labeled (Figure 4A), indicating that the purified complex both has intrinsic protein kinase activity and includes one or more substrates. Most of the labeling occurred on only two distinguishable protein bands, one corresponding to PERs and the other to CLOCK (Figures 4B and S5A). BMAL1, CRY1, and CK1δ were minor phosphorylation targets (Figure S5A); CK1 is known to autophosphorylate (Cheong and Virshup, 2011). In the presence of the CK1δ/ε inhibitor PF-670462 (slightly selective for CK1δ: IC50 values—13 and 80 nM for CK1δ and CK1ε, respectively; Walton et al., 2009), phosphorylation in the complex was nearly abolished in a dose-dependent manner, whereas the CK1ε-selective inhibitor PF-4800567 (IC50 values—711 and 32 nM for CK1δ and CK1ε, respectively; Walton et al., 2009) had no discernible effect (Figure 4C), indicating that virtually all of the detected activity can be ascribed to CK1 δ.
Figure 4. Action of CK1δ within the purified nuclear PER complex.
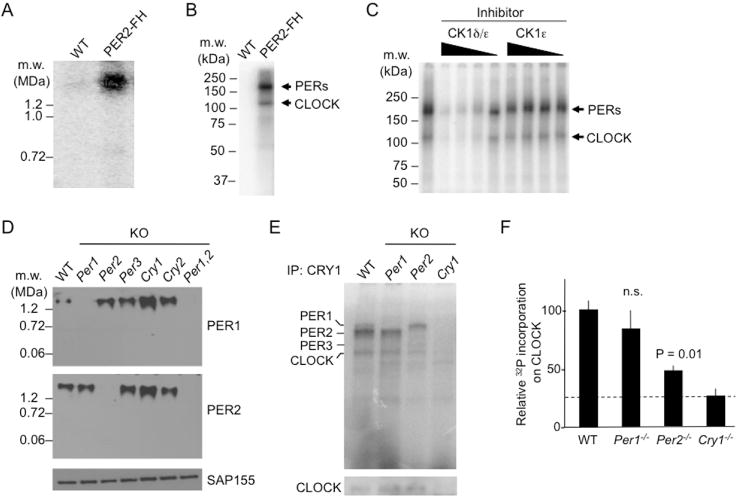
(A) Protein kinase activity of nuclear PER complex. BN-PAGE autoradiogram showing control sample (WT) or affinity-purified nuclear PER complex (PER2-FH) after incubation with γ-32P-ATP (25°C, 1 h).
(B) SDS-PAGE autoradiogram of material analyzed in (A). See Figure S5.
(C) CK1δ in purified PER complex phosphorylates PERs and CLOCK. SDS-PAGE autoradiogram as in (B). Leftmost lane, no inhibitor. Remaining lanes, decreasing concentrations (625, 125, 25, or 5 nM, represented by wedges) of PF-670462, a CK1δ/ε inhibitor (CK1δ/ε), or PF-4800567, a CK1ε-selective inhibitor (CK1ε).
(D) Top and Middle, BN-APAGE immunoblots of mouse liver nuclear extracts (CT18) from wildtype (WT) or single mutants null for the circadian clock genes indicated at the top (KO) probed for proteins indicated at right. Bottom, SDS-PAGE SAP155 immunoblot loading control.
(E) Top, SDS-PAGE autoradiogram of liver nuclear extracts (CT18) from genotypes indicated at top after immunoprecipitation with anti-CRY1 antibody and incubation with γ-32P-ATP (25°C, 10 min; linear range for wildtype). Bottom, SDS-PAGE immunoblot for CLOCK served as loading control for PER complexes.
(F) Phosphor-imaging quantification of labeled CLOCK bands from (E) after normalization to CLOCK immunoblot signals from (E). Shown are mean ± SEM; t-test (two-tailed). Dashed line, background.
The finding that CLOCK is a major target of CK1δ in the endogenous complex suggests that delivery of CK1δ to CLOCK-BMAL1 might represent an unrecognized negative feedback action of the PER complex. We found that nuclear PER complexes (which include CLOCK-BMAL1) bind specifically to the CLOCK-BMAL1 E-box DNA binding site in vitro (Figure S5B). Pre-treatment of the complexes with a non-specific phosphatase resulted in a substantial increase in the binding of the complex to the E-box, whereas subsequent treatment with active CK1δ reduced the E-box binding to its original level (Figure S5C,D). The results suggest that CK1δ phosphorylation of CLOCK-BMAL1 or other proteins in the complex might promote the dissociation of CLOCK-BMAL1 from DNA.
To investigate CK1δ action in more detail, we analyzed PER complexes from clock mutant mice. First we found that the absence of a single PER or CRY does not dramatically affect the assembly or accumulation of nuclear PER complexes—PER complexes from mice singly lacking PER1, PER2, PER3, CRY1, or CRY2 migrated as a single band at a mass similar to wildtype (Figure 4D). Next we compared the intrinsic kinase activity of native PER complexes from wildtype, Per1−/−, and Per2−/− mice. We isolated the PER complexes from liver nuclear extracts (CT18) by immunoprecipitaton with an anti-CRY1 antibody, incubated the isolated complexes with γ-32P-ATP, and resolved labeled proteins by SDS-PAGE autoradiography. In wildtype complexes, PER1, PER2, and CLOCK were the major phosphorylation substrates; no labeled band was detected at the expected migration of PER3 (Figure 4E), and the pattern was essentially identical to that of affinity-purified PER complexes from PER2-FH mice. In complexes lacking PER1, the band for PER1 was absent, as expected, and the labeling of PER2 and CLOCK were similar to that of wildtype (Figure 4E). In contrast, in complexes lacking PER2, the band for PER2 was absent, as expected, but the labeled band for PER1 showed substantially increased intensity, a new labeled band appeared at the expected migration of PER3, and CLOCK was significantly hypophosphorylated (Figure 4E,F). Thus loss of PER1 or PER2 differentially affects the actions of CK1δ within the nuclear PER complex.
Identification of cytoplasmic PER complexes
To examine the possible macromolecular organization of clock proteins in the cytoplasm, we prepared cytoplasmic and nuclear extracts from livers obtained at time points spanning the circadian cycle, resolved complexes by BN-PAGE, prepared immunoblots, and probed the blots for PER2. The results revealed two PER2-containing cytoplasmic complexes that exhibited circadian abundance cycles paralleling the nuclear PER complex (Figure 5A). The “upper complex” (UC) and the “lower complex” (LC) migrated as assemblies of 1.1 ± 0.03 MDa and 0.9 ± 0.03 MDa, respectively (SEM, n = 4) (Figure 5B). Cytoplasmic extracts of liver, kidney, and brain exhibited essentially identical results (Figure 5C), indicating that the two PER2-containing complexes are a common or universal feature of the clock. Both complexes are within the size range for nuclear import (Pante and Kann; 2002), so in principle either one could serve as a precursor of the much larger nuclear PER complex.
Figure 5. Identification and characterization of cytoplasmic PER complexes.
(A) BN-PAGE immunoblots for PER2 showing circadian cycle of PER complexes (arrows) in mouse liver cytoplasm (left) and nucleus (right). UC, upper cytoplasmic complex; LC, lower cytoplasmic complex.
(B) Cytoplasmic PER complexes: relative migration in BN-PAGE vs. log molecular mass.
(C) BN-PAGE immunoblot of cytoplasmic extracts from mouse liver, brain, and kidney (CT18) probed for PER2.
(D) Mouse liver cytoplasmic extracts (CT18) were immunodepleted with control IgG or anti-PER2 antibody, and the depleted (unbound) fraction was analyzed by BN-PAGE. Immunoblots were probed for proteins indicated at the bottom of each panel. Asterisks, non-specific.
(E) Silver-stained BN-PAGE gel showing wildtype (WT) control sample and purified UC and LC without (left) and with (middle and right) glycerol-gradient separation of the two.
(F) PER2 immnuoblot of gels in (E). See Figure S6.
(G) Proteins of LC and UC and their relative representation in the two complexes determined by quantitative TMT mass spectrometry normalized to PER2 (mean ± SEM; n= 3).
To examine the distribution of clock proteins in the complexes, we immunodepleted PER2-containing complexes from liver cytoplasmic extracts (CT18) and analyzed the samples by BN-PAGE immunoblots. In mock-depleted control extracts, all of the clock proteins were present in complexes migrating at ~0.9–1.1 MDa (Figure 5D, IgG lanes). In addition to complexes at the UC and LC positions, CRY2 and possibly CRY1 and CK1δ appeared to be present in one or more complexes of ~1.0 MDa, intermediate to UC and LC (Figure 5D, IgG lanes). There was no evidence for monomers, dimers, or small assemblies of any of the clock proteins, with the exception of CK1δ, for which we observed the monomer (Figure 5D, IgG lanes), as in the nucleus. Quantitative immunodepletion of PER2-containing complexes resulted in complete or nearly complete co-depletion of PER1, PER3, CRY1, CRY2, and CK1δ at the UC migration position (Figure 5D), indicating that all of the proteins are in a ~1.1-MDa complex with PER2. In contrast, a large fraction of each of the clock proteins remained at the LC migration position (Figure 5D), indicating the presence of additional clock complexes with a mass similar to LC (or intermediate to LC and UC) that do not include PER2 (or in which the PER2 epitope is sterically masked).
We affinity purified (Figure S6) LC and UC from liver cytoplasm of PER2-FH mice (Figure 5E,F), analyzed their compositions by mass spectrometry, and quantitatively compared their compositions by tandem mass tag (TMT) mass spectrometry (Online Methods). The results (Figure 5G) indicate that LC consists of only five polypeptides: PER1, PER2, CRY1, CRY2, and CK1δ (no PER3 was detected). If the analysis provided the complete composition, then some proteins must be present in greater than one copy to account for the mass of the complex. UC consists of seven polypeptides: the same five found in LC plus PER3 and GAPVD1 (also called RAP6 and GAPEX5), a guanine nucleotide exchange factor for Rab GTPases involved in receptor-mediated endocytosis and other cytoplasmic trafficking pathways (Hunker et al., 2006; Lodhi et al., 2007; Chua et al., 2014). TMT analysis suggested that UC has an additional copy of PER1 compared to LC (Figure 5G). SDS-PAGE immunoblots of the purified cytoplasmic complexes confirmed the mass spectrometry assignments (Figure 6A); the trace detection of PER3 in the LC sample was likely derived from a trace of contaminating UC. BN-PAGE immunoblots further confirmed that GAPVD1 is present in UC but not detectable in LC (Figure 6B), and PER2 co-immunoprecipitations showed that GAPVD1 is associated with PER2 in the cytoplasm, as expected, but not detectably associated with PER2 in the nucleus (Figure 6C). Thus unlike the other proteins in UC, GAPVD1 does not become part of the nuclear PER complex, consistent with its reported function as a cytoplasmic trafficking factor.
Figure 6. PER3 and GAPVD1 are detected in UC but not LC.
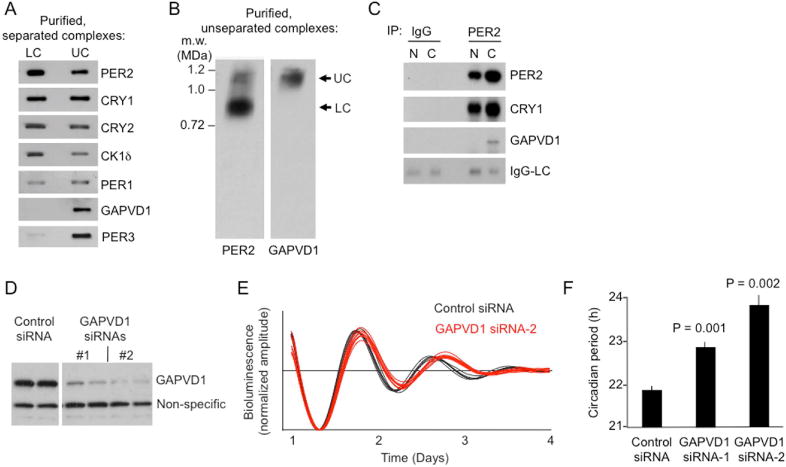
(A) SDS-PAGE immunoblots of purified LC and UC, probed for proteins indicated at right.
(B) BN-PAGE immunoblot of purified cytoplasmic PER complexes, without prior glycerol gradient sedimentation to separate UC from LC, probed for PER2 or GAPVD1, as indicated.
(C) Co-immunoprecipitation (IP) with control IgG or anti-PER2 antibody, as indicated, from liver nuclear (N) or cytoplasmic (C) extracts (CT18), probed for proteins indicated at right. IgG-LC, IgG light chain immunoprecipitation positive control.
(D) Immunoblots showing depletion of GAPVD1 from bioluminescent circadian reporter cells by two non-overlapping GAPVD1 siRNAs in comparison with control siRNA (duplicate samples).
(E) Examples of circadian oscillations of bioluminescence in synchronized reporter fibroblasts after delivery of control siRNA (black) or GAPVD1 siRNA-2 (red). Four independent cultures shown for each.
(F) Circadian period lengths of bioluminescent circadian reporter cells after delivery of control siRNA, GAPVD1 siRNA-1, or GAPVD1-siRNA-2, as indicated (mean ± SEM; n = 4; two-tailed, paired t-test).
To assess the importance of GAPVD1 for clock function, we introduced small interfering RNAs (siRNAs) into Bli circadian reporter cells (Robles et al., 2010) to deplete GAPVD1, and we monitored real-time circadian oscillations of bioluminescence. Depletion of GAPVD1 by either of two non-overlapping siRNAs produced a significant lengthening of circadian period compared to controls (Figure 6D–F). Thus GAPVD1 is a cytoplasmic factor important for clock function.
Single-particle EM analysis of cytoplasmic PER complexes
To investigate the structural organization of cytoplasmic PER complexes, we affinity-purified LC and UC from liver cytoplasm of PER2-FH mice (Figure S6) and analyzed the negatively stained complexes by single-particle EM. The control sample, mock-purified from wildtype mice, showed few particles, whereas the LC sample showed abundant particles of a characteristic appearance (Figure 7A). The LC particles were ~20 nm in diameter, and many appeared to have four globular densities that could be variably arranged (Figure 7B), an impression supported by class averages (Figures 7C and S7A). A similar analysis for UC revealed the complexes to be slightly larger, ~25 nm in diameter (Figure 7D). Class averages showed that UC is similar to LC in structural organization, except that it appears to have at least one additional globular domain (Figure 7E–G, Figure S7B), consistent with its greater mass and additional components.
Figure 7. Single-particle EM analysis of mouse liver cytoplasmic PER complexes (CT18).
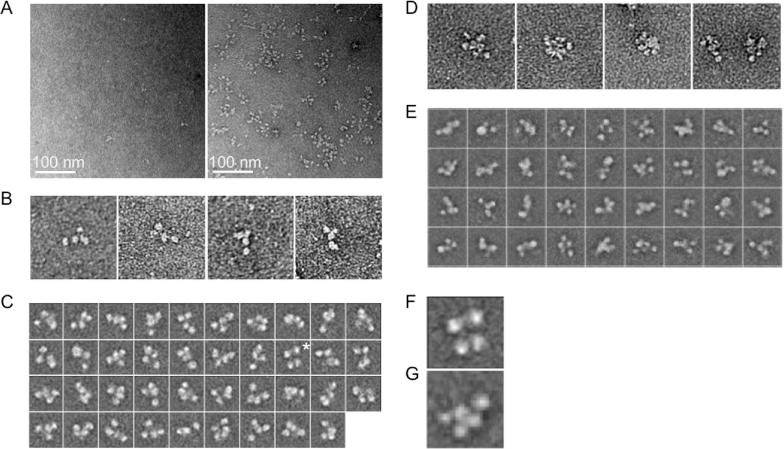
(A) Left, negative control: representative EM image of negatively stained sample from LC purification procedure performed on extract from wildtype mice. Scale bar as indicated. Right, representative image of negatively stained LC from PER2-FH mice.
(B) Examples of negatively stained LC particles, showing typical four-domain structure. Boxes, 100 nm.
(C) The 39 class averages of negatively stained LC obtained by using the iterative stable alignment and classification (ISAC) procedure and stringent classification parameters (100 images per group, pixel error threshold of 0.7). Boxes, 33.4 nm. Asterisk, class average enlarged in (F). See Figure S7A.
(D) Gallery of negatively stained UC particles showing typical multi-lobed appearance. Boxes, 100 nm.
(E) Some class averages of negatively stained UC generated during analysis with ISAC. Boxes, 52.2 nm. See Figure S7B.
(F) Enlarged view of a single LC class average showing four-domain structure.
(G) Enlarged view of a single UC class average showing five-domain structure.
DISCUSSION
The core proteins of the mammalian circadian clock have been known for nearly two decades (Young and Kay, 2001), yet little is known about how they are organized in the cell. The work reported here is directed towards characterizing the identities, properties, behavior, and structures of the macromolecular machines governing the circadian cycle. Our results show that the PERs, CRYs, and CK1δ are exclusively assembled into a small number of protein complexes that together comprise the nuclear and cytoplasmic segments of the circadian feedback loop.
In the nucleus, we found evidence for two circadian clock complexes, a ~1.9-MDa PER negative feedback complex, which includes the CLOCK-BMAL1 target and virtually all of the PER1, PER2, PER3, CRY1, CRY2, and CK1δ, with the exception of a small amount of CRY1 and CK1δ monomer, and a ~750-kDa CLOCK-BMAL1 activation complex, which includes all or nearly all of the CLOCK and BMAL1 present in the activation phase (when little or no PER complex is present). Apart from CLOCK and BMAL1, the components of the ~750-kDa complex remain to be determined.
The nuclear PER complex appears fully formed, as it were, at the earliest time PERs and CRYs are detected in the nucleus. Thus once a presumptive precursor PER-CRY-CK1δ structure enters the nucleus, recruitment of the transcriptional repressor cargo and incorporation of CLOCK-BMAL1 from the ~750-kDa activation complex must be rapid, at least on a circadian timescale. The eventual complete incorporation of CLOCK-BMAL1 implies that little if any of the CLOCK-BMAL1 distributed on hundreds or thousands of genes across the genome (Rey et al., 2011; Koike et al., 2012) would escape negative feedback repression.
In addition to bringing the cargo of repressors to chromatin at CLOCK-BMAL1 DNA binding sites, this process brings CK1δ to CLOCK-BMAL1. Our data, which provide an assessment of the role of CK1δ in its native structural environment, suggest that actions of CK1δ within the complex reduce the binding affinity of CLOCK-BMAL1 to DNA, perhaps contributing to the dissociation of CLOCK-BMAL1 from DNA late in the repressive phase. This phenomenon could be related to the PER-dependent release from DNA and sequestration of the homologous CLOCK-CYC in Drosophila (Menet et al., 2010). CK1δ might thus play a direct role in circadian negative feedback.
Rather than being distributed into physically independent repressors, all of the PERs and CRYs are virtually exclusively present together in the nuclear PER complex. Thus the distinct circadian phenotypes observed in different Per or Cry null mutations (Liu et al., 2007) result not because of different actions of independent PER and CRY repressors but rather because the loss of particular PERs or CRYs differentially perturbs the function or dynamics of the ~1.9-MDa PER complex. This view is further supported by the differences we found in CK1δ action in isolated PER complexes lacking PER1 as compared with those lacking PER2.
Chromatin immunoprecipitation data suggest that CRY1 could act independently of PERs to repress CLOCK-BMAL1, particularly in the late repressive phase (Koike et al., 2012). We found no positive evidence of a CRY1 complex corresponding to such an action. The only nuclear CRY1 complex that we detected at any point in the cycle is the ~1.9-MDa PER complex. However, we invariably detected a small amount of CRY1 monomer (but not CRY2 or PER protein monomers), raising the possibility that it could participate in transient interactions or arise from a labile complex lacking PER proteins. While our results provide no support for the proposal, they do not exclude it.
The single-particle EM analysis of purified nuclear PER complexes, which reveals a quasi-spherical ~40-nm assembly, provides an initial view of the structural organization of endogenous eukaryotic circadian clock machinery. We hypothesize that the inner core of the particle is comprised of a PER-CRY-CK1δ scaffold and the outer globular domains correspond to transcriptional regulatory cargo. Because the purified complex can bind specifically to the E-box DNA sequence, CLOCK-BMAL1 (or at least its DNA-binding domain) must remain on the surface after solubilization.
The variability of structural details and the presence of apparent substoichiometric proteins in the purified nuclear PER complex (Kim et al., 2014) suggest that it might be a collection of closely-related compositional variants rather than a single entity with a uniquely defined structure. We envision that an invariant PER-CRY-CK1δ core is festooned with a somewhat variable set of transcriptional effectors, either because the recruitment and assembly process is inherently variable or because there are time-dependent changes in PER complex composition (Duong and Weitz, 2014) that produce a spectrum of complexes as newly-assembled nuclear PER complexes are added to pre-existing ones.
Little is known about cytoplasmic events in the circadian cycle. Our results reveal that in the cytoplasm the core clock proteins are fully incorporated into at least four complexes of ~0.9–1.1 MDa, two of which, LC and UC, include PER2, and at least two of which likely do not. These latter complexes must include some combination of PER1, PER3, CRY1, CRY2, or CK1δ. UC resembles the nuclear PER complex in that it includes all of the core clock proteins together, as indicated by immunodepletion and mass spectrometry. In comparison, the other complexes appear “incomplete” in that they are missing PER3 (LC) or PER2 and possibly other clock proteins.
We cannot exclude a complicated dynamical relationship between the nuclear PER complex and the cytoplasmic PER complexes mediated, for example, by bidirectional nuclear transport of clock proteins. But a simpler hypothesis (and our working model) is that the smaller “incomplete” cytoplasmic PER complexes represent intermediates in a temporally-controlled assembly pathway that generates UC, a “complete” PER-CRY-CK1δ scaffold structure, which, apart from its regulatory subunit GAPVD1, enters the nucleus and forms the core of the nuclear PER complex. The period-length phenotype caused by depletion of GAPVD1 indicates that its presence in UC is important for the dynamics of the circadian transcriptional cycle, suggesting that it plays a role in regulating the rate of assembly, accumulation, trafficking, or nuclear entry of a mature precursor complex. It remains to be determined if the assembly or trafficking of cytoplasmic PER complexes somehow involves vesicles or other membrane-delimited compartments, like other processes in which GAPVD1 has been implicated.
On this model, the accumulation of PER complexes in the cytoplasm reflects bottlenecks in assembly, maturation, and nuclear entry. Such a process would lead to a gradual accumulation of nuclear PER complexes during the repression phase, as we observed (Figure 1D); a similar gradual circadian accumulation of nuclear PER2 has been measured in living cells (Smyllie et al., 2016). Although not a binary gate for nuclear entry, like that described for PER and TIM in Drosophila (Curtin et al., 1995; Meyer et al., 2006), such bottlenecks could nonetheless generate the delay in negative feedback required for oscillations (Friesen et al., 1993). Our results show that from the onset of negative feedback, about seven hours are required for the nuclear PER complex to accumulate to a concentration sufficient to saturate CLOCK-BMAL1.
The flexible four- and five-domain structures of LC and UC are mysterious. We do not know how these globular structural features relate to the protein compositions or functions of the complexes. It is tempting to speculate that they reflect modular aspects of the assembly process.
STAR * METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Charles J. Weitz (cweitz@hms.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice and Tissue Collection
Adult mice (10–32 weeks of age, males and females were used) were entrained to a 12:12-hour light-dark cycle for at least 12 days and then placed in constant darkness for 1–2 days for circadian tissue collection. All genotypes were in a mixed C57Bl/6;129 background. Mice were euthanized in the dark, and tissues were dissected under room light and processed immediately for preparation of nuclear or cytoplasmic extracts. Studies were performed in accordance with the protocol approved by the Harvard Medical School Standing Committee on Animals.
METHOD DETAILS
Antibodies
Antibodies for western blots (WB) and co-immunoprecipitations (IP) were used at the following dilutions: PER2— IP, WB 1:2000, PER3— WB 1:1000, cytosolic blue native 1:500; CRY2— WB 1:8000, cytoplasmic blue native 1:2000; CRY1—IP, WB 1:2000, blue native 1:5000; JARID1a— WB 1:2000; PER1 (Abcam)— WB 1:100; CK1™-WB 1:2000; CLOCK-WB 1:5000; PER1 (Thermo) — WB 1:10,000 blue native; TRAP150— WB 1:10,000; DDB1— WB 1:5000; RNA-Pol II-CTD-phospho-S2— WB 1:10,000; CHD4— WB 1:5000; MTA2— WB 1:2000; BMAL1— WB 1:10,000; MLL1— WB 1:1000; SAP155— WB 1:5000; GAPVD1/GAPEX5— WB 1:2000.
Preparation of Extracts
Nuclear extracts
Mice were entrained to a 12:12 h light-dark cycle for at least 14 days, transferred to constant darkness, and euthanized under infrared light. Livers (or other tissues) were immediately harvested under room light and each placed in 15 ml of ice cold PBS. After washing (20 ml of ice-cold PBS) and slicing the tissue into small pieces (a few mm in each dimension), the preparation was washed twice with ice cold PBS (20 ml each) and pelleted (500 × g, 2 min). Nuclear homogenization buffer (10 mM HEPES pH 7.6, 1.5 mM MgCl2, 100 mM KCl, 0.1 mM EGTA [pH 8.0], 0.1 mM EDTA [pH 8.0], 10% glycerol, protease inhibitors [Roche] and phosphatase inhibitors 2 and 3 [Sigma]), 1 mM DTT, 0.15 mM spermine, and 0.5 mM spermidine) was added (4 ml/g tissue), and the tissue was homogenized using a chilled 7-ml Douncer (Kontes Glass) with Pestle-A and Pestle-B 20 times each. Homogenates were mixed with 20 ml of iced-cold buffer (nuclear homogenization buffer containing 1.96 M sucrose and without DTT). The sample (25 ml) was carefully layered on a 10 ml chilled sucrose cushion (10 mM HEPES, pH 7.6, 15 mM KCl, 2 mM EDTA [pH 8.0], 2mM EGTA [pH 8.0], and 2.05 M sucrose) in an ultracentrifugation tube (polypropylene, 25 × 89 mm). Nuclei were pelleted (25,000 rpm, 70 min, 4°C; Rotor: Beckman Coulter SW32Ti). Nuclei were rinsed with and resuspended in 500 μl of ice-cold nuclear pellet washing buffer (10 mM HEPES, pH 7.6, 100 mM KCl, 1.5 mM MgCl2, 0.1 mM EGTA [pH 8.0], 0.1 mM EDTA [pH 8.0], 10% glycerol, and protease and phosphatase inhibitors as above) and transferred to chilled 1.5 ml Eppendorf tube. Nuclei were pelleted (benchtop Eppendorf centrifuge, 4000 rpm, 5 min, 4°C) and incubated for 30 min (with vortexing every 5 min) with ice-cold nuclear lysis buffer (10 mM HEPES pH 7.9, 100 mM KCl, 1.5 mM MgCl2, 0.1 mM EDTA [pH 8.0], 0.1 mM EGTA [pH 8.0], 1 mM DTT, 20% glycerol, 0.5% Triton X-100, protease and phosphatase inhibitors as above; 1.5 ml of nuclear lysis buffer /1 mg nuclear pellet). KCl (3.3 M) was added drop-wise to a final concentration of 400 mM KCl, and the sample was incubated for 20 min (shaking at 20 rpm, 4°C). Insoluble materials were removed by centrifugation (21,000 × g, 20 min, 4°C). The nuclear extract (supernatant) was diluted to 150 mM KCl (final) by addition of dilution buffer (10 mM HEPES pH 7.9, 1.5 mM MgCl2, 0.1 mM EDTA, [pH 8.0], 0.1 mM EGTA [pH 8.0], 20% glycerol, and protease and phosphatase inhibitors as above). The nuclear extract was aliquoted, snap-frozen, and stored at −80°C.
Cytoplasmic extracts
Harvested livers (or other tissues) were placed immediately in ice cold PBS, pH 7.4, sliced into small pieces (~a few mm in each dimension), and washed three times with ice cold PBS (15 ml each). Homogenization buffer (10mM HEPES pH 7.6, 250 mM sucrose, protease inhibitors [Roche] and phosphatase inhibitors 2 and 3 [Sigma]) (4 ml/g tissue) was added, and tissue was homogenized with a 7-ml Douncer (Kontes Glass) with Pestle-A 8 times. Nuclei and debris were cleared by multiple centrifugation steps (4°C) as follows: 1) 1,000 × g, 10 min; 2) 2,000 × g, 15 min; 3) 8,000 × g, 5 min; 4) 21,000 × g, 30 min. Each time, supernatant was transferred to a fresh chilled tube. Cytoplasmic extracts (supernatants) were snap-frozen and stored at −80°C.
Blue Native Gel Analysis
Blue native-agarose polyacrylamide gel (BN-APAGE)
We prepared an 18 × 8-cm glass plate by lightly rubbing the top 2–3 cm with sandpaper (the sanded surface faces the gel). Combs were made by combining a 1.5-mm 10-well comb (Hoefer), a central spacer made from a piece of film, and an additional piece of film cut in the same shape as the first comb. The comb was placed against the scratched portion of the plate. The temperature of all the reagents and apparatus used for casting was kept at room temperature.
BN-APAGE gels (2 mm thick, with a superimposed decreasing gradient of agarose and an increasing gradient of acrylamide) were prepared at room temperature with agarose (Sigma, Type IX-A, Ultra-low Gelling Temperature) and acrylamide, each in a gel buffer containing 166 mM ε-aminocaproic acid, 50 mM Bis-Tris, pH 7.5. Acrylamide solution (room temperature) was placed in the outlet chamber, and agarose solution (prepared by boiling for ~1 min and cooling to room temperature) was placed in the other chamber of a gradient mixture, and the gel was cast as in a conventional gradient acrylamide gel. Once the gel was poured, the comb was placed immediately (wells, 5–8 mm deep). First, polymerization of the acrylamide was allowed to proceed at room temperature for at least 30 min, followed by polymerization of agarose by transferring the gel to 4°C for at least 1 h. To facilitate removing the comb, a layer of cathode buffer was placed at the interface of comb and gel for at least 5 min. Residual gel in the wells was removed by pipetting.
The temperature of all the reagents and the apparatus (Hoefer SE 600 Series Vertical Electrophoresis Systems) used for BN-APAGE were adjusted to 4°C before running the samples. Unstained Native Markers (Invitrogen), Mega HA ladder (Hyalose), and samples were prepared in the sample buffer (0.5% Coomassie blue G-250 and 50 mM ε-aminocaproic acid in 10 mM Bis-Tris, pH 7.5, final). Electrophoresis was performed using BN-APAGE (0.8–0% agarose; 0–12% or 0–8% polyacrylamide) in 1× anode buffer (50 mM Bis-Tris HCl, pH 7.0) and 1× cathode buffer (50 mM Tricine, 15 mM Bis-Tris, 0.0015% G-250, pH 7.0) for at least 18 h using relatively low voltage and low current (e.g., 30 V, 3 mA, for four gels running ~18 h). For PER complex size estimation, Mega HA ladders (Hyalose) were calibrated with Unstained Native Markers (Invitrogen), and the Mega HA ladder was stained as described (Cowman et al, 2011). Blue native polyacrylamide agarose gel (BN-PAGE) For conventional blue native polyacrylamide gel analysis of PER complexes (Kim et al., 2014), all reagents and equipment were kept at 4°C. Samples or NativeMark Unstained Protein Standard (Invitrogen) were mixed with loading dye to final concentrations of 0.5% Coomassie blue G and 50 mM ε-aminocaproic acid in 10 mM Bis-Tris, pH 7.0. Samples were separated on 10-well NativePAGE 3–12% Bis-Tris gels (Invitrogen) using anode buffer (50 mM Bis-Tris HCl, pH 7.0) and blue cathode buffer (50 mM Tricine, 15 mM Bis-Tris, 0.004% G-250, pH 7.0; compatible with silver staining). PER complexes were separated by overnight electrophoresis at 4°C using a voltage limit of 180 V and a current limit of 10 mA. For immunoblot analysis, proteins in BN-PAGE gels were transferred onto PVDF membrane (Millipore) by wet-transfer system (Bio-RAD) in transfer buffer (20 mM Tris, pH 7.0, 150 mM Glycine, 0.02% SDS, and 20% methanol).
For silver staining, the gel was first incubated in fixative solution (40% ethanol, 10% acetic acid) for 1 h with shaking, then incubated with fresh fixative solution for another 8–16 h. After fixation, the gel was processed according the instructions provided with the silver staining kit (SilverQuest, Invitrogen).
Immunoblot Analysis
Samples run on blue native or SDS-PAGE gels were transferred onto PVDF membrane by wet-transfer system in transfer buffer. Membranes from blue native gels run with Coomassie blue were quickly de-stained (100% methanol). Membranes were blocked with 5% non-fat milk in 1× TBST (50 mM Tris, pH 7.4, 150 mM NaCl, 0.1% Tween-20) for 1 h at room temperature and incubated with diluted primary antibody (overnight) followed by incubation with HRP-conjugated secondary antibody (1 h). Signals were detected by ECL Prime Western Blotting Detection Reagent (GE Healthcare).
Co-immunoprecipitation of native PER or CLOCK Complexes
Nuclear extract from wildtype mice prepared at CT4 and CT19 were incubated with appropriate antibody or control IgG bound to Dynabeads protein G (Invitrogen) approximately for 3 hours at 4°C. Beads were preci pitated and washed 3 × by IP wash buffer (50 mM Tris-HCl, pH 7.4, 250 mM NaCl, 1.5mM MgCl2 and 0.2% Triton X-100) and bound proteins were eluted by 1 × SDS sample buffer.
Immunodepletion
Nuclear or cytoplasmic extract (25–30 μg) was incubated with 1 μg of antibody or control IgG bound to 10 μl (net) Dynabeads (Invitrogen) at least for 2.5 h. Unbound fractions were directly analyzed by BN-APAGE (nuclear extracts) or BN-APAGE (cytoplasmic extracts).
Enrichment of CLOCK-BMAL1 complex
Nuclear extract (140 μg) was incubated for 2 hours with 1 μg of control IgG or CLOCK antibody loaded onto 25 μl (net) Protein A/G Plus Ultralink Resin. Beads were precipitated and washed 3× by IP wash buffer. Bound materials were eluted by 0.25 μg/μl CLOCK peptide prepared in elution buffer (10 mM HEPES pH 7.9, 100 mM KCL, 0.1 mM EDTA, pH 8.0, 0.1 mM EGTA, pH 8.0, 1.5 mM MgCl2, and 20% glycerol). Eluted materials were analyzed by BN-APAGE.
Nuclear PER complex purification for EM
Nuclear PER complexes were purified for EM by FLAG-HA dual affinity purification and density gradient centrifugation. First, anti-FLAG M2 affinity agarose resin (Sigma) was washed three times with ice-cold 150 buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 3 mM MgCl2, 1 mM EDTA, 0.2% Triton X-100). Nuclear lysate prepared from sixteen Per2-FH mice and wildtype (control) mice was thawed on ice, followed by incubation with 1 ml of washed anti-FLAG M2 resin at 4°C for 1 h in a head-over-head rotator. Unbound proteins were washed away using three washes of five volumes of 150 buffer, two washes of 375 buffer (50 mM Tris-Cl pH 7.4, 375 mM NaCl, 3 mM MgCl2, 1 mM EDTA, 0.2% Triton X-100), and two more washes using 150 buffer. Bound proteins were eluted in 5 ml of 150 buffer containing 100 μg/ml FLAG peptide (Sigma) for 30 min at 4°C. Eluted proteins were concentrated to a volume of 200 μl using 100 kDa MWCO centrifugal concentrators (Vivaspin). The concentrate was layered on a 1.8-ml glycerol gradient (15–55% glycerol, 50 mM HEPES-NaOH pH 7.6, 3 mM MgCl2, 1 mm EDTA, 150 mM NaCl) that had been cooled at 4°C. Density gradient centrifugation was performed using a TLS55 rotor spun at 55,000 rpm for 3 h at 4°C. The peak fraction containing nuclear PER complex was incubated with 20 μl of anti-HA resin (Santa Cruz Biotechnology) and placed in a thermomixer (Eppendorf) for overnight capture at 4°C, 1,200 rpm. Unbound proteins were removed by washing four times with 150 buffer, and once with EM elution buffer (50 mM Tris-Cl pH 7.4, 150 mM NaCl, 3 mM MgCl2, 1 mM EDTA, 0.005% Triton X-100). Bound proteins were eluted for 30 min at 4°C using 20 μl of EM elution buffer containing 500 μg/ml HA peptide (ThermoFisher). After elution, supernatants were spun through low-binding filter columns (Ultrafree-MC HV Centrifugal Filter; EMD Millipore) to eliminate any residual resin. Nuclear PER complexes were prepared for EM within 30 min after elution, as described (Ohi et al., 2004). Briefly, 3 μl of sample was adsorbed to a glow-discharged carbon-coated copper grid, washed with 2 drops of deionized water, and stained with two drops of 0.75% uranyl formate.
Cytoplasmic PER complexes were affinity-purified with the protocol described above for nuclear complexes, but included the following adaptations: 80 ml of cytoplasmic liver extract of from Per2-FH mice and wildtype (control) mice were used for parallel FLAG affinity purifications. Density gradient centrifugation was performed using a TLS55 rotor spun at 55,000 rpm at 4°C for 4 h to maximize separation between the lower and upper cytoplasm complexes. BN-PAGE was used to identify fractions enriched for either lower or upper cytoplasmic PER complexes prior to anti-HA resin capture.
Mass Spectrometry
Following purification, lower and upper cytoplasmic PER complexes were run on BN-PAGE and localized by Coomassie staining or silver staining (SilverQuest, Invitrogen) and excised from the gel. The complexes were denatured and digested, labeled with isobaric mass tags (TMT, Thermo Scientific) as previously reported (Ting et al., 2011), pooled, and subjected to liquid chromatography tandem mass spectrometry analysis (LC-MS/MS, Taplin Biological Mass Spectrometry Facility; n = 3). In two of the three experiments, the heavy and light isotope tags were switched between the LC and UC, respectively. In the third experiment, quantification was performed using label-independent LC-MS/MS quantification (peak intensities over three LC-MS/MS runs of the same sample). Peptides of proteins found in all three experiments in the PER2-FH sample but not the controls were included in the analysis.
EM and single-particle analysis
Purified samples were negatively stained with 0.75% (w/v) uranyl formate as described (Ohi et al., 2004). Low-dose images were recorded using a Tecnai T12 electron microscope (FEI) equipped with an Ultrascan 895 4K × 4K CCD camera (Gatan) and operated at an acceleration voltage of 120 kV. The calibrated magnification was 57,555 x, corresponding to a pixel size of 2.61 Å at the specimen level, and the defocus was set to −1.5 μm.
For the nuclear PER complex, 10,866 particles were interactively selected from 559 images using BOXER (Ludtke et al., 1999) and windowed into 300 × 300-pixel images using SPIDER (Frank et al., 1996). After image normalization and particle centering, the particle images were classified into 100, 200, and 500 groups using k-means classification procedures implemented in SPIDER (Figures 3F and S4). For the lower cytoplasmic PER complex, 15,591 particles were selected from 626 images using BOXER and windowed into 128 × 128-pixel images. For UC (upper cytoplasmic PER complex), 22,598 particles were selected from 1,172 images and windowed into 200 × 200-pixel images. The particle images were reduced to 64 × 64 pixels for classification with the iterative stable alignment and clustering (ISAC) procedure (Yang et al., 2012) implemented in SPARX (Hohn et al., 2007). For LC (lower cytoplasmic PER complex), specifying 100 images per group and a pixel error threshold of 0.7, eight generations of ISAC yielded 39 classes (Figure 7C), accounting for 678 particles (4.35% of the entire data set). Specifying 100 images per group and a pixel error threshold of 2.0, nine generations of ISAC yielded 108 classes (Figure S7A), accounting for 1,980 particles (12.7% of the entire data set). For UC, ISAC did not yield any stable class averages, so the 256 candidate class averages are shown that resulted from specifying 50 images per group and a pixel error threshold of 0.7 (Figures 7E and S7B).
Kinase assays
For purified PER2-FH nuclear PER complexes, anti-FLAG M2 Magnetic Beads (Sigma M8823) were used to capture complexes for the kinase assays. For nuclear PER complexes from other mouse lines, affinity beads for kinase assays were prepared as follows: Magnetic Protein G beads (Life technologies) were washed three times with 150 buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 3 mM MgCl2, 1 mM EDTA, 0.2% Triton X-100). A sample 10 μl of washed beads was incubated with 2 μg of anti-CRY1 antibody (Bethyl) in a volume of 100 μl 150 buffer for 30 min.
After washing three times with 150 buffer to remove unbound material, the antibody-bead mixture was incubated with 20 μl of nuclear extract for 1 h at 4°C to capture target protein complexes. Unbound proteins from the lysate were removed by three washes with 150 buffer. Beads were resuspended in 10 μl of reaction buffer (50 mM Tris-Cl, pH 7.4, 150 mM NaCl, 3 mM MgCl2, 1 mM EDTA, 0.005% Triton X-100) containing 200 nM γ-32P-ATP. Kinase assays were performed (25°C 1 h for PER2-FH complexes or 10 min with 100 nM γ-32P-ATP [in the linear range] for complexes isolated from other mouse lines) in a thermomixer (Eppendorf) at maximum speed. Reducing SDS sample buffer was added to each sample followed by heating for 10 min at 100°C Samples were separated on a NuPAGE Novex 4–12% Bis-Tris Protein Gel (Life Technologies) or a 4–15% Mini-Protean TGX (BIO-RAD) gel along with pre-stained protein standards. The gel was run until the migration front was almost at the bottom of the gel and the front was then excised to remove free radioactive ATP. The gel was dried, exposed to an imaging plate, and read the next day using a phosphorimager (Storm, Molecular Dynamics).
RNA silencing and real-time monitoring of circadian oscillations
A mouse fibroblast reporter cell line (Bli, Bmal1-Luc reporter) was cultured as described (Padmanabhan et al., 2012), with some amendments. Cells were grown in DMEM (Gibco, 1 g/L glucose, (+) L-glutamine, 110 mg/l sodium pyruvate) supplemented with 10% heat-inactivated FBS (Atlas), penicillin/streptomycin (Corning) and MEM nonessential amino acids (Cellgro) at 37°C. Cells were passaged using Trypsin/EDTA (Corning). In 300 μl Opti-MEM Medium (Life Technologies), 30 pmol of siRNA were incubated with 9 μl Lipofectamine RNAiMAX (Life Technologies) and incubated for 5 min at room temperature. Cells were grown to 80–90% confluence and then passaged onto 10 mm × 35 mm dishes at a density of 250,000 cells/dish in a final volume of 2 ml of medium. To each dish was added 250 μl of siRNA/Lipofectamine RNAiMAX mixture, and incubated for 18 h at 37°C Medium was then replaced with LumiCycle Medium, composed of 1 × DMEM, without dye (Sigma), 10 mM HEPES, ph. 7.4, 0.35% D-glucose, 350 mg/l bicarbonate, 10% FBS, 350 mg/l bicarbonate, penicillin/streptomycin, MEM nonessential amino acids and 100 μM d-Luciferin. Dishes were sealed with high vacuum grease (Dow Corning) and microscope cover glass (Fisherbrand), and bioluminescence was continuously recorded in a LumiCycle (Actimetrics) placed within an incubator (37°C, 5% CO2). Period lengths of bioluminescence rhythms were determined using LumiCycle Analysis software (Actimetrics). siRNAs target GPVD1 (si83714, si83716, Life Technologies). siControl is Silencer Select Negative Control No. 1 siRNA (Life Technologies).
DNA Binding of purified nuclear PER complexes
DNA binding of native nuclear PER complexes from nuclear extracts was performed as previously described (Tamayo et al., 2015) with some modifications. Briefly, sense and anti-sense strands of ssDNA Binding/Quantitation Oligonucleotides were combined (1 μM final) and heated to 94°C for 10 min in High Salt Annealing Buffer (10 mM Tris-HCl, ph. 7.0, 300 mM NaCl, 2.5 mM MgCl2, 0.05% Tween-20), then allowed to cool for 1 h at 25°C to form dsDNA. 150 μl of dsDNA (83 bp) was incubated with 50 μl Dynabeads M-270 Streptavidin (Life Technologies) for 30 min at room temperature. Unbound DNA was washed away with Nuclear Lysis Buffer. 50 μl of immobilized DNA was incubated with 150 μl of nuclear extract for 1 h at 4°C. Beads were then washed 3 times with Nuclear Lysis Buffer prior to release of DNA by incubation with 50 μl Restriction Enzyme Digestion Buffer (NEB buffer 4, 1 μl XhoI, 1 μl SmaI) for 1 h at 25°C.
Electrophoretic Mobility Shift Assay (EMSA)
A radiolabeled probe was generated from a known CLOCK-BMAL1 binding site from the Cry1 locus containing an E-box sequence (CACGTG) or control DNA containing a scrambled E-box (CGATCG) using a standard procedure. Briefly, in 100 μl of T4 Polynucleotide Kinase Reaction Buffer, 250 nM ssDNA EMSA Oligonucleotide was incubated with 5 μl of T4 polynucleotide kinase (NEB) and 18 μl of 3.3 μM 3000 Ci/mmol ATP, [γ-32P] (Perkin-Elmer) for 1 h at 37°C Free ATP was removed using Illustra MicroSpin G-25 columns (GE). ssDNA was phenol-chloroform extracted. Glycoblue (Life Technologies) was used a co-precipitant during ethanol precipitation. To form the dsDNA EMSA probe, ssDNA was resuspended in annealing buffer (10 mM Tris-HCl, pH 7.5, 50 mM NaCl, 1 mM MgCl2, 0.1 mM EDTA) to a final concentration of 500 nM, then heated to 95°C for 5 min, and slowly cooled to room temperature. Nuclear PER complex, purified as previously described (Kim et al, 2014), was incubated with 15 nM EMSA probe and a range of 50 nM to 0 nM unlabeled EMSA probe in a final volume of 10 μl for 1 hr at 30°C. Samples were then run on a Native PAGE 3–12% Bis-Tris gel (Life Technologies). Gels were dried using a gel dryer (Bio-Rad) and visualized using a phosphor imager (Molecular Dynamics).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as mean ± SEM. Sample number (n) indicates the number of independent biological samples in each experiment. Data were analyzed by Student’s t-test (two-tailed), and differences were considered statistically significant at p <0.05. The analysis of data was not blinded to condition.
DATA AND SOFTWARE AVAILABILITY
This work has not generated data suitable for public repositories, nor did it involve custom software. The raw EM images can be obtained by contacting the authors.
Supplementary Material
Highlights.
Macromolecular organization of the core circadian clock proteins in the cell
Evidence for a cytoplasmic assembly pathway for circadian clock PERIOD complexes
Electron microscopy images of nuclear and cytoplasmic PERIOD complexes
Acknowledgments
This work was supported by the G. Harold and Leila Y. Mathers Charitable Foundation (C.J.W.), the U.S. National Institutes of Health (NS095977 to C.J.W.), the U.S. National Institutes of Health Training Grant in Fundamental Neurobiology (T32NS007484 to R.P.A.), a Mahoney Postdoctoral Fellowship (to R.P.A.), an Alice and Joseph Brooks Fund Postdoctoral Fellowship (to P.B.K.), the U.S. National Institutes of Health Training Grant in Sleep, Circadian, and Respiratory Neurobiology (T32HL07901 to A.G.T), and a research fellowship from the Deutsche Forschungsgemeinschaft (GE 2557/1-1 to M.G.). We thank David Ginty for helpful comments, Mariska Twaalfhoven for superb technical assistance and Ming Liu for expert mouse colony management.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
C.J.W. conceived and oversaw the project, analyzed data, and wrote the paper; T.W. oversaw the electron microscopy, analyzed data, and contributed to the writing of the paper; R.P.A., P.B.K., A.G.T., and P.-L.C. performed experiments, analyzed data, contributed to the planning, design, and interpretation of experiments and to the writing of the paper; M.G. performed preliminary experiments for the project.
SUPPLEMENTAL INFORMATION
References
- Alberts B. The cell as a collection of protein machines: preparing the next generation of molecular biologists. Cell. 1998;92:291–294. doi: 10.1016/s0092-8674(00)80922-8. [DOI] [PubMed] [Google Scholar]
- Anafi RC, Lee Y, Sato TK, Venkataraman A, Ramanathan C, Kavakli IH, Hughes ME, Baggs JE, Growe J, Liu AC, Kim J, Hogenesch JB. Machine learning helps identify CHRONO as a circadian clock component. PLoS Biol. 2014;15:e1001840. doi: 10.1371/journal.pbio.1001840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher G, Schibler U. Crosstalk between components of circadian and metabolic cycles in mammals. Cell Metab. 2011;13:125–137. doi: 10.1016/j.cmet.2011.01.006. [DOI] [PubMed] [Google Scholar]
- Balslobre A, Damiola Fl, Schibler U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell. 1998;93:929–937. doi: 10.1016/s0092-8674(00)81199-x. [DOI] [PubMed] [Google Scholar]
- Brown SA, Ripperger J, Kadener S, Fleury-Olela F, Vilbois F, Rosbash M, Schibler U. PERIOD1-associated proteins modulate the negative limb of the mammalian circadian oscillator. Science. 2005;308:693–696. doi: 10.1126/science.1107373. [DOI] [PubMed] [Google Scholar]
- Cheong JK, Virshup DM. Casein kinase 1: Complexity in the family. Int J Biochem Cell Biol. 2011;43:465–469. doi: 10.1016/j.biocel.2010.12.004. [DOI] [PubMed] [Google Scholar]
- Chiou YY, Yang Y, Rashid N, Ye R, Selby CP, Sancar A. Mammalian Period represses and de-represses transcription by displacing CLOCK-BMAL1 from promoters in a Cryptochrome-dependent manner. Proc Natl Acad Sci USA. 2016;113:E6072–E6079. doi: 10.1073/pnas.1612917113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua CE, Tang BL. Engagement of the small GTPase Rab31 protein and its effector, early endosome antigen 1, is important for trafficking of the ligand-bound epidermal growth factor receptor from the early to the late endosome. J Biol Chem. 2014;289:12375–12389. doi: 10.1074/jbc.M114.548321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowman MK, Chen CC, Pandya M, Han Yuan H, Ramkishun D, LoBello J, Bhilocha S, Russell-Puleri S, Skendaj E, Mijovic J, Jing W. Improved agarose gel electrophoresis method and molecular mass calculation for high molecular mass hyaluronan. Anal Biochem. 2011;417:50–56. doi: 10.1016/j.ab.2011.05.023. [DOI] [PubMed] [Google Scholar]
- Curtin KD, Huang ZJ, Rosbash M. Temporally regulated nuclear entry of the Drosophila period protein contributes to the circadian clock. Neuron. 1995;14:365–372. doi: 10.1016/0896-6273(95)90292-9. [DOI] [PubMed] [Google Scholar]
- Czarna A, Berndt A, Singh HR, Grudziecki A, Ladurner AG, Timinszky G, Kramer A, Wolf E. Structures of Drosophila cryptochrome and mouse cryptochrome1 provide insight into circadian function. Cell. 2013;153:1494–1405. doi: 10.1016/j.cell.2013.05.011. [DOI] [PubMed] [Google Scholar]
- Dallman R, Weaver DR. Altered body mass regulation in male mPeriod mutant mice on high-fat diet. Chronobiol Int. 2010;27:1317–1328. doi: 10.3109/07420528.2010.489166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibner C, Schibler U, Albrecht U. The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu Rev Physiol. 2010;72:517–549. doi: 10.1146/annurev-physiol-021909-135821. [DOI] [PubMed] [Google Scholar]
- DiTacchio L, Le HD, Vollmers C, Hatori M, Witcher M, Secombe J, Panda S. Histone lysine demethylase JARID1a activates CLOCK-BMAL1 and influences the circadian clock. Science. 2011;333:1881–1885. doi: 10.1126/science.1206022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffield GE, Watson NP, Mantani A, Peirson SN, Robles-Murguia M, Loros JJ, Israel MA, Dunlap JC. A role for Id2 in regulating photic entrainment of the mammalian circadian system. Curr Biol. 2009;19:297–304. doi: 10.1016/j.cub.2008.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong HA, Robles MS, Knutti D, Weitz CJ. A molecular mechanism for circadian clock negative feedback. Science. 2011;332:1436–1439. doi: 10.1126/science.1196766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong HA, Weitz CJ. Temporal orchestration of repressive chromatin modifiers by circadian clock Period complexes. Nat Struct Mol Biol. 2014;21:126–132. doi: 10.1038/nsmb.2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durgan DJ, Trexler NA, Egbejimi O, McElfresh TA, Suk HY, Petterson LE, Shaw CA, Hardin PE, Bray MS, Chandler MP, Chow CW, Young ME. The circadian clock within the cardiomyocyte is essential for responsiveness of the heart to fatty acids. J Biol Chem. 2006;281:24254–24269. doi: 10.1074/jbc.M601704200. [DOI] [PubMed] [Google Scholar]
- Etchegaray JP, Machida KK, Noton E, Constance CM, Dallmann R, Di Napoli MN, DeBruyne JP, Lambert CM, Yu EA, Reppert SM, Weaver DR. Casein kinase 1 delta regulates the pace of the mammalian circadian clock. Mol Cell Biol. 2009;29:3853–3866. doi: 10.1128/MCB.00338-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Friesen WO, Block GD, Hocker CG. Formal approaches to understanding biological oscillators. Annu Rev Physiol. 1993;55:661–681. doi: 10.1146/annurev.ph.55.030193.003305. [DOI] [PubMed] [Google Scholar]
- Hohn M, Tang G, Baldwin PR, Huang Z, Penczek PA, Yang C, Glaeser RM, Adams PD, Ludtke SL. SPARX, a new environment for cryo-EM image processing. J Struct Biol. 2007;157:47–55. doi: 10.1016/j.jsb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Huang N, Chelliah Y, Shan Y, Taylor CA, Yoo SH, Partch C, Green CB, Zhang H, Takahashi JS. Crystal structure of the heterdimeric CLOCK:BMAL1 transcriptional activator complex. Science. 2012;337:189–194. doi: 10.1126/science.1222804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunker CM, Galvis A, Kruk I, Giambini H, Veisaga ML, Barbieri ML. Rab5-activating protein 6, a novel endosomal protein with a role in endocytosis. Biochem Biophys Res Commun. 2006;340:967–975. doi: 10.1016/j.bbrc.2005.12.099. [DOI] [PubMed] [Google Scholar]
- Hurley JM, Loros JJ, Dunlap JC. Circadian oscillators: around the transcription-translation feedback loop and on to output. Trends Biochem Sci. 2016;41:834–846. doi: 10.1016/j.tibs.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katada S, Sassone-Corsi P. The histone methyltransferase MLL1 permits the oscillation of circadian gene expression. Nat Struct Mol Biol. 2010;17:1414–1421. doi: 10.1038/nsmb.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Kwak PB, Weitz CJ. Specificity in circadian clock feedback from targeted reconstitution of the NuRD co-repressor. Mol Cell. 2014;56:738–748. doi: 10.1016/j.molcel.2014.10.017. [DOI] [PubMed] [Google Scholar]
- Kim JY, Kwak PB, Gebert M, Duong HA, Weitz CJ. Purification and analysis of PERIOD protein complexes of the mammalian circadian clock. Meth Enzymol. 2015;551:197–210. doi: 10.1016/bs.mie.2014.10.013. [DOI] [PubMed] [Google Scholar]
- Kish-Trier E, Hill CP. Structural biology of the proteasome. Annu Rev Biophys. 2013;42:29–49. doi: 10.1146/annurev-biophys-083012-130417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemz R, Reischl S, Wallach T, Witte N, Jürchott K, Klemz S, Lang V, Lorenzen S, Knauer M, Heidenreich S, Xu M, Ripperger JA, Schupp M, Stanewsky R, Kramer A. Reciprocal regulation of carbon monoxide metabolism and the circadian clock. Nat Struct Mol Biol. 2017;24:15–22. doi: 10.1038/nsmb.3331. [DOI] [PubMed] [Google Scholar]
- Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, Takahashi JS. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science. 2012;338:349–354. doi: 10.1126/science.1226339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamia KA, Storch KF, Weitz CJ. Physiological significance of a peripheral tissue circadian clock. Proc Natl Acad Sci USA. 2008;105:15172–15177. doi: 10.1073/pnas.0806717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lande-Diner L, Boyault C, Kim JY, Weitz CJ. A positive feedback loop links circadian clock factor CLOCK-BMAL1 to the basic transcriptional machinery. Proc Natl Acad Sci USA. 2013;110:16021–16026. doi: 10.1073/pnas.1305980110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu AC, Welsh DK, Ko CH, Tran HG, Zhang EE, Priest AA, Buhr ED, Singer O, Meeker K, Verma IM, Doyle FJ, 3rd, Takahashi JS, Kay SA. Intercellular coupling confers robustness against mutations in the SCN circadian clock network. Cell. 2007;129:605–616. doi: 10.1016/j.cell.2007.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodhi IJ, Chiang S, Chang L, Vollenweider D, Watson RT, Inoue M, Pessin JE, Saltiel AR. Gapex-5, a Rab31 guanine nucleotide exchange factor that regulates Glut4 trafficking in adipocytes. Cell Metab. 2007;5:59–72. doi: 10.1016/j.cmet.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- Marcheva B, Ramsey KM, Buhr ED, Kobayashi Y, Su H, Ko CH, Ivanova G, Omura C, Mo S, Vitaterna MH, Lopez JP, Philipson LH, Bradfield CA, Crosby SD, JeBailey L, Wang X, Takahashi JS, Bass J. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. 2010;466:627–631. doi: 10.1038/nature09253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menet JS, Abruzzi KC, Desrochers J, Rodriguez J, Rosbash M. Dynamic PER repression mechanisms in the Drosophila circadian clock: from on-DNA to off-DNA. Genes Dev. 2010;24:358–367. doi: 10.1101/gad.1883910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menet JS, Rodriguez J, Abruzzi KC, Rosbash M. Nascent-Seq reveals novel features of mouse circadian transcriptional regulation. Elife. 2012;1:e00011. doi: 10.7554/eLife.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer P, Saez L, Young MW. PER-TIM interactions in living Drosophila cells: an interval timer for the circadian clock. Science. 2006;311:226–229. doi: 10.1126/science.1118126. [DOI] [PubMed] [Google Scholar]
- Michael AK, Fribourgh JL, Chelliah Y, Sandate CR, Hura GL, Schneidman-Duhovny D, Tripathi SM, Takahashi JS, Partch CL. Formation of a repressive complex in the mammalian circadian clock is mediated by the secondary pocket of CRY1. Proc Natl Acad Sci USA. 2017;114:1560–1565. doi: 10.1073/pnas.1615310114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nangle SN, Rosensweig C, Koike N, Tei H, Takahashi JS, Green CB, Zheng N. Molecular assembly of the period-cryptochrome circadian transcriptional repressor complex. Elife. 2014;3:e03674. doi: 10.7554/eLife.03674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohi M, Li Y, Cheng Y, Walz T. Negative staining and image classification-powerful tools in modern electron microscopy. Biol Proced Online. 2004;6:23–34. doi: 10.1251/bpo70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan K, Robles MS, Westerling T, Weitz CJ. Feedback regulation of transcriptional termination by the mammalian circadian clock PERIOD complex. Science. 2012;337:599–602. doi: 10.1126/science.1221592. [DOI] [PubMed] [Google Scholar]
- Pante N, Kann M. Nuclear Pore Complex Is Able to Transport Macromolecules with Diameters of ~39 nm. Mol Biol Cell. 2002;13:425–434. doi: 10.1091/mbc.01-06-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partch CL, Green CB, Takahashi JS. Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 2014;24:90–99. doi: 10.1016/j.tcb.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey G, Cesbron F, Rougemont J, Reinke H, Brunner M, Naef F. Genome-wide and phase-specific DNA-binding rhythms of BMAL1 control circadian output functions in mouse liver. PLoS Biol. 2011;9:e1000595. doi: 10.1371/journal.pbio.1000595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles MS, Boyault C, Knutti D, Padmanabhan K, Weitz CJ. Identification of RACK1 and protein kinase C-alpha as integral components of the mammalian circadian clock. Science. 2010;327:463–466. doi: 10.1126/science.1180067. [DOI] [PubMed] [Google Scholar]
- Schmalen I, Reischl S, Wallach T, Klemz R, Grudziecki A, Prabu JR, Benda C, Kramer A, Wolf E. Interaction of circadian clock proteins CRY1 and PER2 is modulated by zinc binding and disulfide bond formation. Cell. 2014;157:1203–1215. doi: 10.1016/j.cell.2014.03.057. [DOI] [PubMed] [Google Scholar]
- Smyllie NJ, Pilorz V, Boyd J, Meng QJ, Saer B, Chesham JE, Maywood ES, Krogager TP, Spiller DG, Boot-Handford R, White MR, Hastings MH, Loudon AS. Visualizing and quantifying intracellular behavior and abundance of the core circadian clock protein PERIOD2. Curr Biol. 2016;26:1880–1886. doi: 10.1016/j.cub.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamayo AG, Duong HA, Robles MS, Mann M, Weitz CJ. Histone mono-ubiquitination by a Clock-Bmal1 complex marks Per genes for circadian negative feedback. Nat Struct Mol Biol. 2015;22:759–766. doi: 10.1038/nsmb.3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting L, Rad R, Gygi SP, Haas W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nature Methods. 2011;8:937–940. doi: 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Horst GT, Muijtjens M, Kobayashi K, Takano R, Kanno S, Takao M, de Wit J, Verkerk A, Eker AP, van Leenen D, Buijs R, Bootsma D, Hoeijmakers JH, Yasui A. Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms. Nature. 1999;398:627–630. doi: 10.1038/19323. [DOI] [PubMed] [Google Scholar]
- Walton KM, Fisher K, Rubitski D, Marconi M, Meng QJ, Sládek M, Adams J, Bass M, Chandrasekaran R, Butler T, Griffor M, Rajamohan F, Serpa M, Chen Y, Claffey M, Hastings M, Loudon A, Maywood E, Ohren J, Doran A, Wager TT. Selective inhibition of casein kinase 1 epsilon minimally alters circadian clock period. J Pharmacol Exp Ther. 2009;330:430–439. doi: 10.1124/jpet.109.151415. [DOI] [PubMed] [Google Scholar]
- Xu Y, Padiath QS, Shapiro RE, Jones CR, Wu SC, Saigoh N, Saigoh K, Ptácek LJ, Fu YH. Functional consequences of a CKIdelta mutation causing familial advanced sleep phase syndrome. Nature. 2005;434:640–644. doi: 10.1038/nature03453. [DOI] [PubMed] [Google Scholar]
- Yang Z, Eng J, Chittuluru J, Asturias FJ, Penczek PA. Iterative stable alignment and clustering of 2D transmission electron microscope images. Structure. 2012;20:237–247. doi: 10.1016/j.str.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MW, Kay SA. Time zones: a comparative genetics of circadian clocks. Nat Rev Genet. 2001;2:2–15. doi: 10.1038/35088576. [DOI] [PubMed] [Google Scholar]
- Yusupova G, Yusupov M. High-resolution structure of the eukaryotic 80S ribosome. Annu Rev Biochem. 2014;83:467–486. doi: 10.1146/annurev-biochem-060713-035445. [DOI] [PubMed] [Google Scholar]
- Zhao X, Hirota T, Han X, Cho H, Chong LW, Lamia K, Liu S, Atkins AR, Banayo E, Liddle C, Yu RT, Yates JR, 3rd, Kay SA, Downes M, Evans RM. Circadian amplitude regulation via FBXW7-targeted REV-ERBα degradation. Cell. 2016;165:1644–1657. doi: 10.1016/j.cell.2016.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao WN, Malinin N, Yang FC, Staknis D, Gekakis N, Maier B, Reischl S, Kramer A, Weitz CJ. CIPC is a mammalian circadian clock protein without invertebrate homologues. Nat Cell Biol. 2006;9:268–275. doi: 10.1038/ncb1539. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.