

Abstract
Introduction
Sin3B serves as a scaffold for chromatin-modifying complexes that repress gene transcription to regulate distinct biological processes. Sin3B-containing complexes are critical for cell cycle withdrawal, and abrogation of Sin3B-dependent cell cycle exit impacts tumor progression.
Areas covered
In this review, we discuss the biochemical characteristics of Sin3B-containing complexes and explore how these complexes regulate gene transcription. We focus on how Sin3B-containing complexes, through the association of the Rb family of proteins, repress the expression of E2F target genes during quiescence, differentiation, and senescence. Finally, we speculate on the potential benefits of the inhibition of Sin3B-containing complexes for the treatment of cancer.
Expert opinion
Further identification and characterization of specific Sin3B-containing complexes provide a unique opportunity to prevent the pro-tumorigenic effects of the senescence-associated secretory phenotype, and to abrogate cancer stem cell quiescence and the associated resistance to therapy.
Keywords: Cancer stem cell, cellular senescence, chromatin modifiers, quiescence, senescence associated secretory phenotype, Sin3B
1. Introduction
In the USA, cancer is the second-leading cause of death with approximately 600,000 deaths in 2016 [1]. The progress made in basic, translational, and clinical research in the past half-century have significantly improved the prognosis for certain cancer types including chronic myeloid leukemia, colon cancer, and prostate cancer. However, the prognosis for other cancer types such as pancreatic ductal adenocarcinoma (PDAC) and lung cancer remains abysmal [1]. While targeted therapies have been added to the arsenal of cancer treatments, resistance almost invariably emerges. As such, there continues to be a pressing need to identify new cellular targets that can specifically affect tumor cells and/or modulate the tumor microenvironment.
The past decade has highlighted the importance of epigenetic processes in the initiation and the progression of almost all types of cancers. Mutations in numerous chromatin modifiers have been identified in cancers and are thought to be early events that promote cell transformation [2]. It is generally thought that these mutations alter the transcriptional status of the cells who harbor them, leading to the inappropriate expression of specific genes. These defects result in the impairment of normal differentiation and/or promote aberrant cellular division and self-renewal. Because histone modifications and the modulation of chromatin architecture are reversible processes, targeting the specific factors responsible for abnormal chromatin status in cancer cells represents a promising therapeutic strategy. In fact, numerous drugs that inhibit chromatin-modifying enzymatic activities are currently being used clinically or are being tested in clinical trials [2].
In this review, we focus on the chromatin modifier Sin3B and its associated complexes. We discuss the mechanisms underlying the alteration of chromatin structure by Sin3B-containing complexes and the relevance of these mechanisms in normal biological processes. We further explore the role of Sin3B in cancer and highlight the potential therapeutic benefits of inhibiting Sin3B-containing complexes in cancer.
2. The Sin3 complex
2.1. The discovery of Sin3
Sin3 (switch independent 3) was initially discovered in a genetic screen investigating factors important for mating-type switching in S. cerevisiae [3,4]. Subsequently, Sin3 was identified through mutations associated with inappropriate gene derepression [5]. From these studies and further biochemical purifications, it quickly became apparent that Sin3 functions in a transcriptional repressive complex that contains numerous core factors and at least one class I histone deaceytlase (HDAC). In mammals, two isoforms of Sin3, Sin3A, and Sin3B, were isolated in studies investigating novel proteins associated with either Max interacting protein 1 (Mxi1) or mitotic arrest deficient gene 1 (Mad1), both transcription factors that associate with the Myc partner Max (Myc-associated factor X) to repress transcription of its target genes [6,7]. Similar to Sin3’s function in yeast, both Sin3A and Sin3B are required for the repressive functions of Mxi1 and Mad1 proteins [6,7]. While Sin3 is known to interact with chromatin to repress gene transcription, it lacks any intrinsic enzymatic activity and is devoid of known DNA-binding sequences suggesting that interaction with transcription factors and/or chromatin-binding partners is required for its localization on chromatin.
2.2. Structural insight into Sin3 function
Sin3’s structure provided important insight into its function. In yeast, Sin3 is a 175-kDa protein that contains six evolutionarily conserved regions, including four paired amphipathic helices (PAH 1–4), one HDAC interaction domain (HID), and one highly conserved region (HCR) (Figure 1) [8]. Structural studies revealed that the PAH domains of Sin3 form helix-loop-helix folds similar to those of the Myc family of transcription factors suggesting that PAH domains may serve as protein-binding interfaces. Indeed, through these domains, Sin3 interacts with a plethora of proteins and serves as a scaffold to tether different enzymatic activities to sequence-specific transcription factors (Figure 1). Biochemical studies have identified 8 core components of the Sin3 complex: Sin3, HDAC1/2, RBBP4/7 (Retinoblastoma-binding protein), SAP18 (Sin3-associated protein), SAP30, and SDS3 [8]. Additional Sin3-interacting factors including SAP130, SAP180, BRMS1 (breast cancer metastasis suppressor 1), ING1/2 (inhibitor of growth protein), RBBP1, the plant homeodomain (PHD) protein PF1, and the chromodomain protein MRG15 have subsequently been identified, and are believed to stabilize Sin3 interactions with various components of the complex and/or chromatin [9–12]. Sin3 has also been reported to associate with chromatin-modifying enzymes and transcription factors such as the histone demethylase KDM5A, Rb (Retinoblastoma), p130, p53, the histone methylase ESET, NCoR (nuclear receptor corepressor), the transcription factor E2F4, and the ATP-dependent helicase BRG1, which tether Sin3 complexes to genomic loci and facilitate the modification of chromatin structure [10,13–19].
Figure 1.
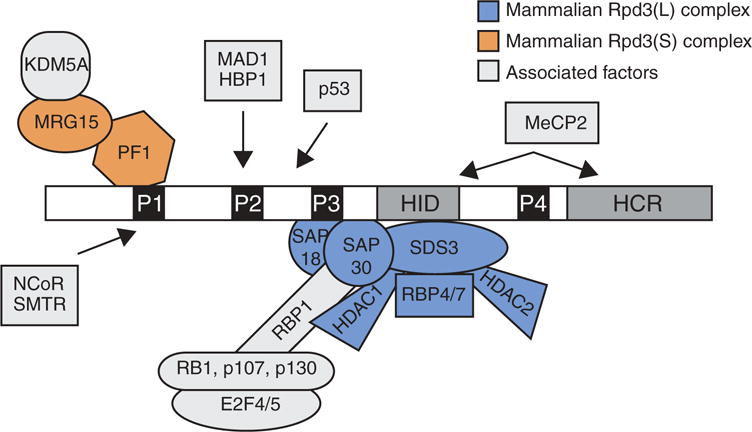
Sin3 and its associated proteins.
Sin3 has 6 evolutionary conserved domains: 4 paired amphipathic domains (PAH, P1–4 in figure), 1 HDAC interacting domain (HID), and 1 highly conserved region (HCR). The core complex assembles around the PAH3 and HID domain whereas transcription factors are thought to associate with the PAH1 and PAH2 domains.
Exactly how Sin3 can interact with so many different enzymes and/or transcription factors in a cellular context-specific manner remains unclear. One hypothesis is that the PAH1 and PAH2 domains of Sin3 are flexible and can fold into numerous confirmations generating a diverse repertoire of specific protein-protein interfaces [20–22]. Whether extrinsic forces influence Sin3’s protein structure to favor certain interactions over others, however, is not known. Additionally, Sin3 is known to interact with adaptor proteins like SAP30 to bridge other factors such as RBBP1 and the Rb family of proteins to Sin3 complexes [10]. Phosphorylation of RBBP1 and Rb by cyclin-dependent kinase (CDK)-Cyclin complexes during cell cycle blocks their association and prevents Sin3-HDAC and E2F4 repressive complexes from repressing E2F target genes [23]. It is likely that other protein modifications occur either on Sin3, core factors of the Sin3 complex, or associated factors that modulate their interactions [24].
In mammals, Sin3A and Sin3B have highly similar sequences and structures when associated with known interactors [25]. This explains how both isoforms associate with similar factors such as Mad1. Yet, other factors such as Foxk1/2 (forkhead box protein) and the methyl CpG-binding protein MeCP2 or PF1 and E2F4 preferentially interact with Sin3A or Sin3B, respectively, despite the almost ubiquitous expression of both Sin3A and Sin3B proteins [17,26–28]. One possible explanation for this specificity may be the differences in structure observed for the apo-form of the PAH2 domain between Sin3A and Sin3B [29]. Additionally, the differences in amino acid sequences between Sin3A and Sin3B outside the six evolutionarily conserved domains may be sufficient to generate unique protein-interaction sites. It will therefore be important to perform biochemical and proteomic studies on Sin3A and Sin3B individually to identify novel protein interactions instead of relying on one isoform to infer interactions with the other.
Along similar lines, it is now appreciated that Sin3 is a scaffold for several complexes that function in unique cellular processes. In both yeast and mammalian cells, a large (Rpd3L) and a small (Rpd3S) Sin3-containing complex have been described [28,30,31]. In yeast, these complexes share the core components Sin3, Rpd3, and Ume1 but also contain unique components such as Sds3 for Rpd3(L) and the PHD-containing Rco1 and the chromodomain-containing Eaf3 for Rpd3(S). Moreover, the molecular mechanism engaged by each complex to repress transcription is distinct. The Rpd3(L) complex, via its interaction with sequence-specific transcription factors, is tethered to the promoters of discrete genes and deacetylates histones to prevent access for the transcription machinery. On the other hand, the Rpd3(S) complex binds to gene bodies of actively transcribed genes through the direct recognition of H3K36me3 by Eaf3. There, Rpd3(S) deacetylates histones in the wake of PolII progression to prevent intragenic cryptic transcription [32]. In mammals, a homologous complex to the Rpd3(S) complex has been described, and it similarly associates within actively transcribed gene bodies [28]. Intriguingly, this complex contains Sin3B but not Sin3A, demonstrating isoform specificity within Sin3 complexes in mammals. In support of this, a recent study described a Sin3A-specific complex that associates with Foxk1 at the promoters of autophagy genes in muscle cells to repress their expression during normal growth conditions [26].
2.3. Nonredundant functions of Sin3A and Sin3B
While the sequences of Sin3A and Sin3B are highly similar, mouse Sin3A is more closely related to human Sin3A than mouse Sin3B, suggesting that each isoform evolved to perform specialized functions. This is supported by biochemical data showing that Sin3A and Sin3B can exist in distinct complexes, as presented above, and by studies investigating the role of both Sin3A and Sin3B in development. In Drosophila, Sin3 null mutants die during larval development. However, each of the three Sin3 isoforms in Drosophila (Sin3 187, Sin3 190, Sin3 220) are expressed temporally and in a tissue-specific manner during development, suggesting that the different Sin3 isoforms regulate distinct genes and pathways [33,34]. Similarly, genetic deletion of either Sin3A or Sin3B prevents embryonic development in the mouse [35–37]. Yet, while Sin3A−/− embryos did not survive past embryonic day 6.5 (E6.5), Sin3B−/− embryos were detected as late as E18.5, indicating nonredundant cellular functions for Sin3A and Sin3B [35,36]. The defects of Sin3A−/− embryos were due to a requirement for Sin3A for cell survival whereas the failure for Sin3B−/− embryos to develop correlated with the failure of certain cellular lineages including erythrocytes and chondrocytes to properly differentiate [35–37].
These differences were also apparent in somatic cells deleted for either Sin3A or Sin3B. Sin3A−/− mouse embryonic fibroblasts (MEFs) lose proliferative capacity and rapidly die via apoptosis [35]. Similar observations were described for thymocytes and mouse germ cells acutely deleted for Sin3A [37,38]. In contrast, Sin3B is not required for cell viability in normal growth conditions, but instead is required for cell cycle withdrawal such as cellular quiescence, senescence, and differentiation following the appropriate cellular signals [36,39,40]. While the precise mechanisms Sin3A engages to promote viability in normal and neoplastic cells remain unclear, a growing literature supports a model where Sin3B functions with the Rb family of proteins (Rb, p107, and p130) and E2F4 to repress the transcription of E2F target genes during cell cycle withdrawal [10,13,14,17,36].
2.4 Sin3B, E2F4, and the Rb family of proteins: partners in gene repression to promote cell cycle exit
The regulation of cell cycle gene expression is a highly coordinated process and ensures that the appropriate genes are expressed according to the stage of cell cycle. The restriction point, the time in cell cycle where a cell commits to either fully complete a cell cycle and divide or instead to exit cell cycle, is regulated by the coordinated action of the Rb family of proteins and CDKs [41]. Hypophosphorylated Rb associates with and sequesters activating E2F transcription factors (E2F1, E2F2, and E2F3), thus preventing their ability to bind their target genes and to promote cell cycle progression [41,42]. Additionally, recent work has identified a complex in mammalian cells named DREAM, containing p130 (and p107 in instances when p130 is absent), E2F4, the transcription factor DP1, and the MuvB (multi-vulval class B) core, that is essential for the repression of pro-proliferative cell cycle genes in quiescence [43–45]. When a cell commits to entering cell cycle, p130 dissociates from DREAM at least in part due to changes in its phosphorylation status [43,46]. In tandem, CDK4/6-Cyclin-D complexes phosphorylate Rb and prevent its binding to activating E2F complexes. These actions simultaneously relieve the transcriptional repression of cell cycle genes and free activating E2F complexes to associate at the promoters of their target genes. Accordingly, the cell upregulates pro-proliferative cell cycle genes and initiates the replication of its DNA [42,47].
The initial hints of functional interplay between Sin3 and the Rb family came through the observations that the Rb family of proteins requires class I HDACs for part of their repressive activity [48–50]. Additionally, Sin3B and HDAC1 associate with the DREAM complex supporting the earlier observations that, in quiescent cells, p130 and E2F4 association at specific promoters correlates with histone hypoacetylation [17,51]. RBBP1, a bridging factor that binds to Rb family members, associates with SAP30 and recruits Sin3-HDAC to Rb family complexes (Figure 1). This recruitment of Sin3-HDAC to Rb accounted for all of Rb’s HDAC repressive activity demonstrating a functional link between Sin3 and Rb [10]. Moreover, in quiescent cells, E2F4, p107, p130, HDAC1, and Sin3B colocalize at genes, such as cyclin A and E2F1, whose expression is initially repressed but increases as a cell progresses through G1 [17]. Consistently, the recruitment of Sin3B and HDAC1 to the promoter of these genes required p107, p130, and an intact E2F4-binding site [17].
The generation and characterization of Sin3B−/− embryos and cells provided genetic evidence for a functional association between Sin3B, the Rb family of proteins, and E2F4. In Sin3B−/− embryos, chondrocytes and erythrocytes failed to terminally differentiate similar to observations made for p130−/− p107−/−, E2F4−/−, and Rb1−/− embryos [52–54]. In vitro, Sin3B−/− MEFs failed to exit cell cycle following the overexpression of the tumor suppressor p16Ink4a again reminiscent of p130−/− p107−/−, E2F4−/−, and Rb1−/− MEFs [52,54]. In hepatocytes, which are a largely quiescent, Sin3B localized to the promoters of E2F targets cyclin E, E2F1, and b-myb and was required for their repression [36]. These studies support a model where Sin3B along with HDAC1 is recruited to the promoters of E2F target genes by the Rb family of proteins and E2F4 to deacetyate their histones and repress their transcription (Figure 2).
Figure 2.
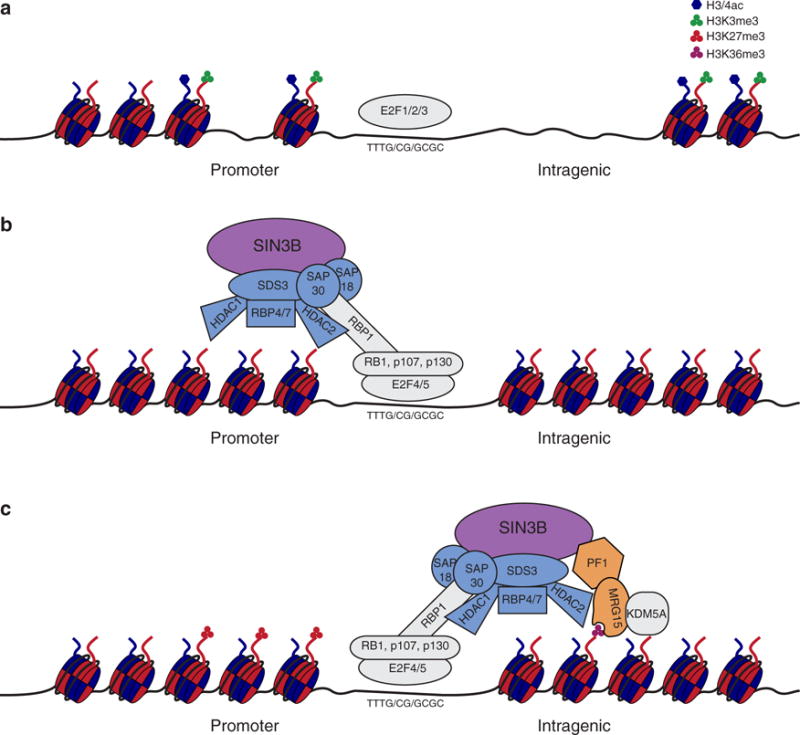
Proposed model of Sin3B mediated gene repression during cell cycle exit.
Depicted is a proposed model for Sin3B mediated repression of E2F target genes during cell cycle withdrawal. (a) Depicted is a gene locus targeted by activating E2F transcription factors during normal cell growth where the promoter has activating histone marks. The chromatin is accessible for binding of the transcription machinery. (b) During cell quiescence, Sin3B-HDAC complexes associate at the promoters of E2F target genes through the recruitment of E2F4 and the Rb family of proteins. There, Sin3B-HDAC complexes deacetylate histones to impair to accessibility of the locus. (c) During differentiation, a Sin3B-HDAC-KDM5A complex bind to intragenic regions of E2F target genes to deaceylate and demethylate histones in order to compact chromatin and repress transcription. Whether MRG15 contributes to the binding to H3K36me3 is not known.
However, it is clear that Sin3B is essential for cellular differentiation both in vitro and in vivo [13,14,36], yet acetylation/deacetylation of histones is not believed to be sufficient for the stable silencing of genes. In this regard, it was shown that Rb is required for H3K27me3, a repressive histone mark, to be deposited and maintained on cell cycle genes for their permanent silencing during the differentiation of myoblasts into myotubes in vitro [55]. Similarly, an analysis of the genomic occupancy of Sin3A/B, E2F4, and KDM5A, a H3K4 demethylase, demonstrated that all four proteins associate at a subset of cell cycle gene promoters in order to modify their histones, compact the surrounding chromatin, and permanently turn off their expression as myoblasts differentiate into myotubes (Figure 2) [14]. Accordingly, mice with muscle-specific deletion of Sin3A and/or Sin3B revealed a coordinated role for these proteins in the repression of cell cycle genes and maintenance of differentiated muscle cells in vivo [13]. Surprisingly, KDM5A was shown to require MRG15 to associate specifically with Sin3B within the mammalian homolog of the Rpd3(S) complex [14,28,56,57]. How KDM5A associates at similar genomic loci as Sin3A in differentiating muscle cells remains unclear, but one possibility is that Sin3B-containing complexes associate with Sin3A-containing complexes, consistent with immunoprecipitation studies that have demonstrated that Sin3A and Sin3B interact at sub-stoichiometric levels [14,26]. A recent report demonstrated coordinated repression of cell cycle targets by p130, E2F4, and KDM5A as embryonic stem cells differentiate [58]. Whether the mammalian Rpd3(S) complex is required for the recruitment of KDM5A to these loci and more broadly whether Sin3B participates in gene silencing as stem cells differentiate is not known.
3. Sin3B expression in cancer
The analysis of numerous public databases documenting genomic alterations failed to identify recurring mutations within the Sin3B coding regions in any cancer type. However, interrogation of Sin3B expression in different tumors revealed that in pancreatic cancer, Sin3B protein levels are inversely correlated with the disease stage [39,40]. Similarly, analysis of publicly available expression databases revealed that Sin3B is significantly downregulated in human prostate adenocarcinoma compared to normal prostate tissue [59]. These results were confirmed by the observation that the transcriptional program engaged by the Sin3B-HDAC complex is altered in human prostate adenocarcinoma, even in cases where Sin3B expression was not altered [59]. These observations suggest that a better readout for Sin3B alterations in cancer may lie in the assessment of its target genes’ expression, rather than Sin3B expression itself. Such an approach would be consistent with the notion that in addition to Sin3B, alterations of other components of its associated complex(es) would result to a similar functional outcome. In that aspect, it is important to note that the expression levels of several components of the Sin3B small complex are altered in solid tumors. These include EMSY, which was first identified as amplified in breast cancer and in higher-grade ovarian cancer [60] as well as PF1 and KDM5A, which are all significantly overexpressed in breast cancers [60,61]. Additionally, while Sin3B expression levels are not significantly different between healthy individuals and patients affected by blood malignancies, increased Sin3B levels are predictive of a poor prognosis in acute myeloid leukemia (AML; discussed below) [62]. The bases for such effects are currently unknown, but may be related to Sin3B’s ability to regulate HSC differentiation and quiescence [63]. The fact that the expression levels of Sin3B or its associated partners may be either increased or decreased depending on malignancy type likely reflects the wide range of Sin3B targets and its organ-specific biological functions.
These data coupled with the fact that Sin3B-containing complexes are important for the transcriptional repression of cell cycle genes during differentiation and quiescence support further investigation of how Sin3B-mediated cell cycle exit affects cancer development and progression. Cellular senescence, a permanent form of cell cycle withdrawal, has recently emerged as a central regulator of cancer initiation and progression, warranting the investigation of the role of the Sin3B-containing complexes in this biological process.
4. Sin3B mediated cellular senescence
4.1. The dual roles of cellular senescence
Cellular senescence is a form of cell cycle exit distinct from quiescence and terminal differentiation that has been implicated in numerous disease processes such as aging, wound healing, and cancer [64]. Numerous cellular stressors including genotoxic agents, oncogene activation, loss of tumor suppressors, exposure to reactive oxygen species, and aging can trigger the loss of proliferative capacity and promote entry into cellular senescence. The mechanisms that promote entry into senescence remain incompletely understood, but are known to include the activation of the DNA damage response and the engagement of cellular effectors such as p53, p16Ink4a, and Rb that enforce the molecular programs required for cell cycle exit [64,65]. Additionally, chromatin-modifying complexes are essential for the reorganization of chromatin structure in senescent cells and the maintenance of the senescent state [66]. In senescent cells, the promoters of E2F target genes are enriched for Rb, histone methyltransferases (i.e. Suv39H1), HDACs, and heterochromatic protein 1 (HP1) and are embedded in senescence-associated heterochromatic foci (SAHF), resulting in the seemingly irreversible silencing of transcription at E2F loci [67]. While SAHF are a characteristic of senescent cells, they alone are not sufficient to identify senescent cells. Instead, a set of senescence hallmarks should be used to identify senescent cells such as senescence-associated beta-galactosidase (SA-β-gal) positivity, the upregulation of p16Ink4a, the generation and secretion of the senescence-associated secretory phenotype (SASP), and the presence of SAHF [68,69].
In the context of cancer, senescence was initially hypothesized to serve as a barrier to tumor progression. This hypothesis stemmed from several observations. First, ectopic expression of oncogenes caused primary cells to prematurely senesce in vitro in a process termed oncogene-induced senescence (OIS) [70]. Second, senescent cells are detected in preneoplastic lesions, but not in the corresponding cancerous tissues, in both mice and humans [71]. And third, genetic ablation of the effector programs of senescence (i.e. p53, p16Ink4a, and Rb) in mouse models of cancer leads to accelerated disease progression and decreased disease-free survival [72–74]. Together, these observations support a model where oncogene activation drives a cell into cellular senescence, ultimately preventing the proliferation of a potentially deleterious pre-cancerous cell.
In addition to exiting the cell cycle, senescent cells secrete a distinct set of proinflammatory cytokines, growth factors, and matrix metalloproteases collectively termed the SASP [75,76]. The SASP is thought to signal in both an autocrine and paracrine manner to reinforce cellular senescence and promote senescence in neighboring cells [75,77,78]. Furthermore, in early preneoplastic lesions, the SASP recruits immune cells that are responsible for clearing potentially deleterious senescent cells. Consistently, abrogation of the SASP or immune cell depletion prevents senescent cell clearance and accelerates tumor progression in various mouse models of cancer, supporting an antitumorigenic role for the SASP [79–81]. Conversely, studies have also demonstrated that conditioned medium from senescent fibroblasts promotes the growth and invasion of cancer cells in vitro [76,82,83]. Additionally, coculture with senescent fibroblasts accelerates the growth of cancer cells in mouse xenografts [76,82,83]. In specific mouse models of cancer, the SASP can also promote an inflammatory microenvironment that accelerates cancer progression [40,84,85]. A recent study using a mouse model of hepatocellular carcinoma (HCC) reported that the SASP played distinct roles depending on the stage of tumorigenesis [85]. As discussed above, in precancerous lesions, the SASP recruited immune cells that eliminated preneoplastic senescent cells and prevented tumor progression. However, in the presence of fully transformed cells, the inflammatory microenvironment generated by the SASP fueled their growth, resulting in accelerated tumor progression [85]. These studies support a model where the SASP initially restricts the progression of preneoplastic lesions (Figure 3). However, as cells bypass or escape senescence and become fully transformed cancer cells, the SASP promotes their growth and invasive potential (Figure 3). Therefore, targeting senescent cells and/or the SASP at specific stages of cancer progression may represent a novel strategy for the treatment of various cancers [86].
Figure 3.
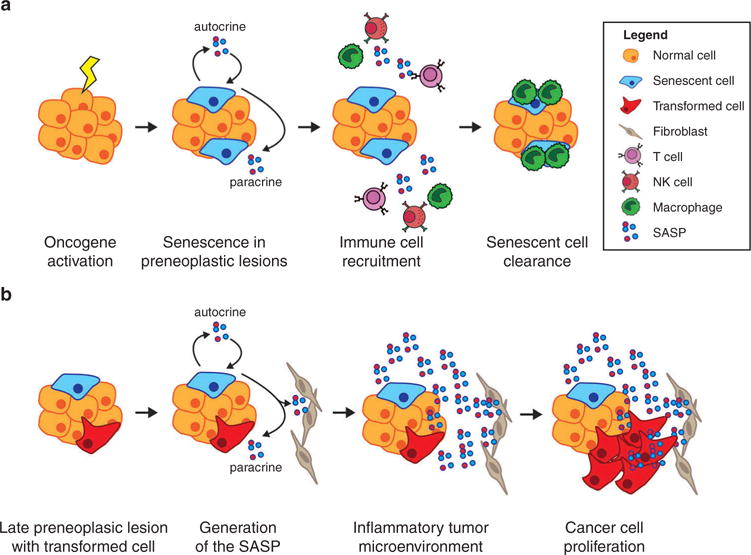
The dual role of the SASP in tumor progression.
Depicted is a model for the role of the SASP at distinct stages of cancer development. (a) In early preneoplastic lesions, cells that acquire oncogenes become senescent. These cells exit cell cycle and generate the SASP, which functions in an autocrine manner to enforce senescence and in a paracrine manner to promote neighboring cells to become senescent. The SASP further recruits various types of immune cells, which recognize and clear senescent cells. (b) In more developed neoplastic lesions that contain fully transformed cells, the SASP generates a pro-inflammatory environment. Fibroblasts are also involved in generating the proinflammatory SASP. This in turn increases the proliferative and invasive capacity of transformed cells.
4.2. Targeting Sin3B-mediated cellular senescence
We previously demonstrated that Sin3B is required for both replicative senescence and OIS in primary fibroblasts, due to its ability to silence the expression of pro-proliferative E2F target genes [39]. As such, targeting Sin3B may serve as an effective method to prevent the detrimental consequences of SASP production in cancer. In support of this, we found that pancreatic-specific genetic inactivation of Sin3B impaired PDAC progression in mice [40]. In Sin3B-deleted pancreata, pancreatic intraepithelial neoplasms (PanIN), the preneoplastic lesion in PDAC, were devoid of senescent cells and did not express SASP factors such as IL-1α and IL-6. Accordingly, the recruitment of immune cells and the activation of inflammatory pathways were significantly decreased in Sin3B-deleted pancreata [40]. Other observations similarly point to the possibility that targeting SASP factors improves PDAC prognosis. IL-1α expression, which has been proposed to be an initiating event for the generation of the SASP [87], predicts poor prognosis in PDAC patients, and the pharmacological inhibition of the IL-1 receptor (IL-1R) reduces tumor burden in mice [88,89]. Moreover, IL-6 was required for PanIN progression and its deletion prevented PDAC formation [90]. Additionally, targeting of the NF-κβ pathway, which functions downstream of IL-1R activation, abolishes the SASP, prevents the generation of an inflammatory microenvironment, and greatly improves overall survival in a mouse model of PDAC [88,91]. Together these studies demonstrate that targeting both upstream mediators of the SASP, such as Sin3B, and the effector arms of the SASP, such as IL-1α and IL-6, efficiently prevents PDAC progression. It will therefore be important to assess whether inhibition of Sin3B similarly mitigates the negative impact of the SASP in other types of cancer, such as HCC.
However, because loss of Sin3B also prevents the cell cycle withdrawal of cells expressing oncogenes [39], inhibiting Sin3B may affect different cancer types in distinct ways. One study reported that decreased Sin3B levels correlated with worse survival and increased metastasis in breast cancer [92]. Additionally, in lymphoma cells derived from Eμ-tTA/tet-O-MYC mice, c-myc-induced miR-17–92 targeted Sin3B mRNA transcripts for degradation. This miR-17–92-dependent decrease of Sin3B levels, along with other miR-17–92 targets, prevented the premature entry into senescence of lymphoma cells [93]. Recently, we assessed the role of Sin3B in prostate cancer progression using PTEN deletion to model prostate adenocarcinoma (PCa) in mice [59]. Similar to our observations in PDAC, Sin3B was required for senescence of prostatic intraepithelial neoplasms, the preneoplastic lesions in PCa. However, unlike what was reported in PDAC, Sin3B deletion promoted PCa progression and PCa cell invasion [59].
The differences in the impact of Sin3B inactivation in PDAC and PCa progression likely highlight the different contributions of senescence and SASP in these cancers. PDAC is a highly inflammatory disease and increased inflammation is associated with worse prognosis in patients. Moreover, as discussed above, studies that target the effector arms of the SASP have proven effective at improving disease outcome [88–90]. In contrast, enhancing senescence and the SASP in PCa sensitizes prostate cancer cells to cytotoxic therapies and improves survival in mice [94–96]. Moving forward, it will be important to determine the role of the SASP in specific subtypes of cancers in order to determine whether pro-senescent or anti-senescent therapies are appropriate. Additionally, studies should attempt to perturb the SASP at different stages of cancer development using either chemical compounds or temporal ablation of SASP effector pathways, instead of using mouse genetic models where specific genes are deleted prior to cancer development. These studies could then better reflect the treatment of cancer patients in the clinic and provide important insight into the therapeutic potential of SASP inhibition at different stages of cancer progression.
5. Targeting Sin3B-mediated quiescence to eliminate cancer stem cells
5.1. Targeting cancer stem cell quiescence sensitizes cells to cytotoxic therapy
Studies over the past decades have identified that cancer cell populations are heterogeneous and hierarchically organized. In leukemia as well as in some solid tumors, it is thought that a subpopulation of self-renewing cancer stem cells (CSCs) sustains the long-term growth of the cancer. Critically, in both mouse models of cancer and in clinical studies, CSCs have been found to be resistant to commonly employed therapies and are often responsible for disease relapse [97]. Therefore, in order to provide long-term disease remission to patients, new therapeutic strategies are needed that target and eliminate CSCs.
A common theme across numerous cancerous types such as AML, melanoma, breast cancer, and colon cancer is the observation that CSCs are a slow-cycling population of cells [98–100]. In these cancers, treatment with traditional cytotoxic therapies that target actively cycling cells fails to eradicate quiescent CSCs. These quiescent CSCs then drive disease relapse following treatment [101]. One strategy that has been utilized to sensitize CSCs to therapy is through the ablation of cellular pathways required for the maintenance of quiescence. For example, in chronic myelogenous leukemia (CML), which is known to contain quiescent CSCs that are resistant to imatinib, targeting FBXW7 prevented CSC quiescence and synergized with imatinib to eradicate CSCs and improve disease outcome in mice [102,103]. Similarly, forcing AML CSC reentry into cell cycle through the administration of G-CSF sensitized these cells to cytotoxic therapy and improved survival in mice [104]. Finally, inhibiting CSC quiescence in breast cancer also sensitized cells to therapy, suggesting that this strategy has efficacy across multiple cancer types [105]. Therefore, the identification of novel strategies to prevent quiescence in CSCs will likely be useful for the treatment of cancer patients.
5.2. Sin3B as a potential mediator of CSC quiescence
Recently, we identified a critical role for Sin3B in maintaining quiescence of hematopoietic stem cells (HSCs) [63]. Similar to observations made in somatic cells, Sin3B loss resulted in the upregulation of E2F target genes in HSCs. Moreover, we found that Sin3B associated with E2F4, KDM5A, and HDAC1 in the HSC-like HPC-7 cell line suggesting that Sin3B complexes that regulate cell cycle exit in somatic cells likely mediate similar effects in HSCs [63]. Factors that have been reported to be required for HSC quiescence are often required for leukemic stem cell (LSC) quiescence, and approaches that target LSC quiescence have been successful at eliminating LSCs in mouse models of AML and have been used in patients [99,106]. In this regard, targeting Sin3B-containing complexes may be an attractive approach to perturb LSC quiescence and sensitize LSCs to cytotoxic therapies. Indeed, we identified a subset of AML patients with elevated Sin3B expression levels that had worse overall survival than patients with no alteration in Sin3B expression levels (Figure 4) [62]. It will be important for future studies to determine whether Sin3B levels are causative or correlative in AML prognosis, and whether targeting Sin3B may sensitize LSCs to cytotoxic therapies due to their increased propensity to reenter cell cycle.
Figure 4.
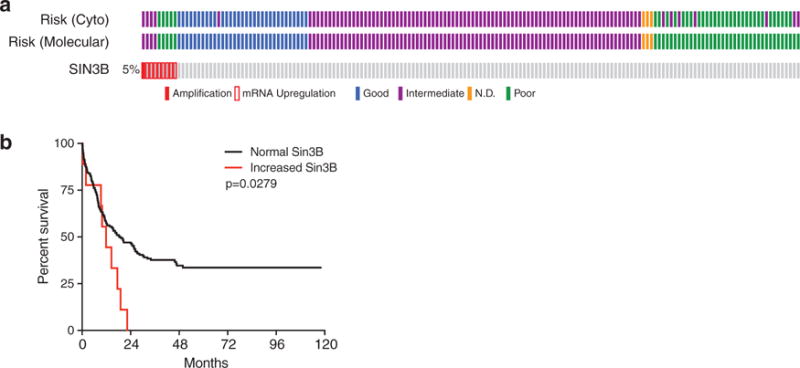
Increased Sin3B expression correlates with poor prognosis in acute myeloid leukemia.
Data was acquired and visualized through cbioportal from the study done by The Cancer Genome Atlas Research Network [62]. (a) Oncomine plot for Sin3B status (both exome-sequencing and RNA-sequencing) showing that Sin3B is upregulated or amplified in a small subset of patients. High Sin3B levels correlates with intermediate or poor risk disease. (b) Worse overall survival for patients with increased Sin3B expression. Curves were compared using the log-rank test.
The risk of targeting factors important for normal HSC function is poor tolerance due to unwanted side effects. For example, while broad HDAC inhibition is effective at eliminating LSCs in a mouse model of chronic myeloid leukemia, it similarly impacts normal HSCs, limiting its efficacy [107,108]. We found that Sin3B loss only mildly perturbs steady-state hematopoiesis [63]. This suggests that a therapeutic window may exist for Sin3B inhibition in AML patients. Since Sin3B likely exists in a variety of complexes that engage different cellular pathways, strategies should attempt to identify the specific complex that is biologically relevant to the regulation of quiescence and disrupt its assembly and/or function [109]. One potential Sin3B containing complex that could be targeted is DREAM, which has previously been shown to promote quiescence in gastrointestinal stromal tumors (GIST). Inhibition of DREAM assembly synergized with imatinib therapy and increased GIST apoptosis [110].
6. The use of decoy molecules to inhibit specific Sin3B complexes
As discussed above, Sin3B provides a scaffold for the assembly of several chromatin-modifying complexes that regulate gene transcription in distinct biological processes [8]. One potential strategy to inhibit these complexes is the use of chemical compounds that inhibit the specific enzymatic activity of these complexes such as HDAC inhibitors or histone demethylase inhibitors. However, because these enzymes are often found in numerous complexes, their inhibition will likely lead to unwanted side effects. A more promising approach would be to develop inhibitors that prevent the assembly of specific Sin3B complexes. An intriguing approach is the use of decoy molecules that mimic the structure of Sin3B domains and competitively inhibit Sin3B associated factors from binding to Sin3B complexes. Sin3-interacting domain (SID) decoys, which resemble the PAH2 domain of Sin3A and Sin3B, have been successfully used to inhibit breast cancer growth both in vitro and in vivo [111,112]. These studies demonstrate that decoy molecules effectively disrupt Sin3A and Sin3B complex assembly and inhibit their associated activities to target cancer cells. It will therefore be important to continue to identify novel Sin3B-associated factors that mediate specific Sin3B complex functions. By subsequently identifying the critical interacting domain for such interactions coupled with structural studies of the conserved domains of Sin3B, decoy molecules can be designed that prevent the assembly of Sin3B complexes and subsequently inhibit their function.
7. Expert opinion
The past couple decades of research has identified essential functions for Sin3B complexes in numerous physiological and pathological processes [8]. While it is appreciated that Sin3B functions as a scaffold for multiple chromatin-modifying complexes, the identity of these complexes remains elusive [8,28,56]. Much of the work identifying Sin3B interactions relied on targeted immunoprecipitation experiments based on previous work performed on yeast Sin3 or the closely related Sin3A. Since it is clear that Sin3A and Sin3B mediate nonredundant cellular functions in mammalian cells, delineating differences in complex composition between the two paralogues will likely inform the specific cellular pathways regulated by each Sin3 protein. Unbiased approaches should be utilized to identify specific Sin3B complexes in both primary and cancer cells.
Along similar lines, many of the studies into Sin3B complex function have yet to identify the specific interactions that mediate its functions. While there is clear evidence that the Rb family of proteins recruit and associate with Sin3B complexes at E2F target genes to repress their expression during cellular quiescence, differentiation, and senescence [17,36], little is known about the exact domains within Sin3B that are responsible for this function. Moreover, studies have suggested that distinct Sin3B complexes regulate different forms of cell cycle withdrawal through unique mechanisms. For example, Sin3B complexes are often associated at the promoters of E2F loci in quiescent cells whereas they are found in intragenic regions of E2F loci in differentiating muscle cells [14,17,36]. Furthermore, KDM5A interacts with Sin3B and associates at similar E2F loci as muscle cells differentiate. This suggests that the mammalian Rpd3(S) complex containing Sin3B likely contributes to cell cycle exit during myoblast differentiation. Whether this complex is similarly important for quiescence and senescence or whether the mammalian Rpd3(L) complex containing Sin3B mediates these functions remains unknown. While interaction studies shed light into the mechanisms used by Sin3B to mediate these forms of cell cycle exit, future experiments should attempt to identify the critical Sin3B interactions and Sin3B domains for quiescence, differentiation, and senescence through the use of Sin3B mutants that prevent specific associations. This would aide in the development of decoy molecules that prevent specific Sin3B-containing complex formation and functions in cells.
In this review, we discuss how eliminating the SASP prevents PDAC progression and inhibits the growth and invasion potential of cancer cells [40,76,88,90]. Sin3B inhibition likely prevents the SASP by abrogating the senescence process entirely. As such, its inhibition in cancer may pose a risk by increasing the proliferative capacity of cancer cells as has been observed in lymphoma and PCa cells [59,93]. An alternative approach could be to identify the mechanisms downstream of Sin3B that specifically upregulate the SASP. We observed that Sin3B modulated IL-1α production in PDAC both in vitro and in vivo [40]. It has been proposed that activation of IL-1α is an initiating event for the SASP [83,87], yet the mechanisms that activate IL-1α are not known. A better understanding of the mechanisms both upstream and downstream of IL-1α activation will greatly improve our understanding of how the SASP is initiated and maintained and provide numerous molecules that can be targeted to inhibit the SASP.
In conclusion, we believe that an improved understanding of the composition of specific Sin3B containing complexes will enable the generation of novel therapeutic strategies for the treatment of cancer. Specifically, we propose that Sin3B inhibition may be useful for targeting the CSC population and for dampening the proinflammatory, pro-tumorigenic aspects of the SASP. Moving forward, studies should not merely investigate the functions of Sin3B, but instead delineate the specific Sin3B interactions that mediate specific physiological and pathological processes.
Article highlights.
Sin3B is a scaffold for the assembly of various chromatin-modifying complexes.
Sin3B-containing complexes, through their association with the Rb family of proteins, E2F4, and KDM5A are critical for the repression of E2F pro-proliferative target genes during cellular quiescence, differentiation, and senescence.
Targeting Sin3B ablates the senescence-associated secretory phenotype (SASP) and prevents pancreatic ductal adenocarcinoma (PDAC) progression in mice.
Sin3B is required for hematopoietic stem cell (HSC) quiescence and its inhibition could force quiescent cancer stem cells to cycle and sensitize them to cytotoxic therapies.
Decoy molecules effectively prevent the assembly of Sin3 complexes and represent a unique strategy for the inhibition of specific Sin3B-containing complexes.
This box summarizes key points contained in the article.
Acknowledgments
Funding
This work was funded by the National Institute of Health (R01CA148639, R21CA155736 and R21 CA206013.
Footnotes
Declaration of interest
G. David has received support from The Samuel Waxman Cancer Research Foundation snd a Feinberg NYU individual grant. D. J. Cantor was supported by a predoctoral NIH/NCI training grant (T32CA009161) and a predoctoral NIH/NCI NRSA (F30CA203047). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Feinberg AP, Koldobskiy MA, Gondor A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17:284–299. doi: 10.1038/nrg.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sternberg PW, Stern MJ, Clark I, et al. Activation of the yeast HO gene by release from multiple negative controls. Cell. 1987;48:567–577. doi: 10.1016/0092-8674(87)90235-2. [DOI] [PubMed] [Google Scholar]
- 4.Nasmyth K, Stillman D, Kipling D. Both positive and negative regulators of HO transcription are required for mother-cell-specific mating-type switching in yeast. Cell. 1987;48:579–587. doi: 10.1016/0092-8674(87)90236-4. [DOI] [PubMed] [Google Scholar]
- 5.Silverstein RA, Ekwall K. Sin3: a flexible regulator of global gene expression and genome stability. Curr Genet. 2005;47:1–17. doi: 10.1007/s00294-004-0541-5. [DOI] [PubMed] [Google Scholar]
- 6.Ayer DE, Lawrence QA, Eisenman RN. Mad-Max transcriptional repression is mediated by ternary complex formation with mammalian homologs of yeast repressor Sin3. Cell. 1995;80:767–776. doi: 10.1016/0092-8674(95)90355-0. [DOI] [PubMed] [Google Scholar]
- 7.Schreiber-Agus N, Chin L, Chen K, et al. An amino-terminal domain of Mxi1 mediates anti-Myc oncogenic activity and interacts with a homolog of the yeast transcriptional repressor SIN3. Cell. 1995;80:777–786. doi: 10.1016/0092-8674(95)90356-9. [DOI] [PubMed] [Google Scholar]
- 8.Grzenda A, Lomberk G, Zhang JS, et al. Sin3: master scaffold and transcriptional corepressor. Biochim Biophys Acta. 2009;1789:443–450. doi: 10.1016/j.bbagrm.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fleischer TC, Yun UJ, Ayer DE. Identification and characterization of three new components of the mSin3A corepressor complex. Mol Cell Biol. 2003;23:3456–3467. doi: 10.1128/MCB.23.10.3456-3467.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10•.Lai A, Kennedy BK, Barbie DA, et al. RBP1 recruits the mSIN3-histone deacetylase complex to the pocket of retinoblastoma tumor suppressor family proteins found in limited discrete regions of the nucleus at growth arrest. Mol Cell Biol. 2001;21:2918–2932. doi: 10.1128/MCB.21.8.2918-2932.2001. This study describes the association of a Sin3-HDAC complex with the pocket proteins through RBP1, and suggests that pocket protein recruitment of Sin3 complexes is required for E2F gene repression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meehan WJ, Samant RS, Hopper JE, et al. Breast cancer metastasis suppressor 1 (BRMS1) forms complexes with retinoblastoma-binding protein 1 (RBP1) and the mSin3 histone deacetylase complex and represses transcription. J Biol Chem. 2004;279:1562–1569. doi: 10.1074/jbc.M307969200. [DOI] [PubMed] [Google Scholar]
- 12.Kuzmichev A, Zhang Y, Erdjument-Bromage H, et al. Role of the Sin3-histone deacetylase complex in growth regulation by the candidate tumor suppressor p33(ING1) Mol Cell Biol. 2002;22:835–848. doi: 10.1128/MCB.22.3.835-848.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Oevelen C, Bowman C, Pellegrino J, et al. The mammalian Sin3 proteins are required for muscle development and sarcomere specification. Mol Cell Biol. 2010;30:5686–5697. doi: 10.1128/MCB.00975-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14•.van Oevelen C, Wang J, Asp P, et al. A role for mammalian Sin3 in permanent gene silencing. Mol Cell. 2008;32:359–370. doi: 10.1016/j.molcel.2008.10.015. This study identifies a complex containing Sin3B, E2F4, and KDM5A that associates at and represses pro-proliferative E2F target genes during cellular differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murphy M, Ahn J, Walker KK, et al. Transcriptional repression by wild-type p53 utilizes histone deacetylases, mediated by interaction with mSin3a. Genes Dev. 1999;13:2490–2501. doi: 10.1101/gad.13.19.2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balciunaite E, Spektor A, Lents NH, et al. Pocket protein complexes are recruited to distinct targets in quiescent and proliferating cells. Mol Cell Biol. 2005;25:8166–8178. doi: 10.1128/MCB.25.18.8166-8178.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17••.Rayman JB, Takahashi Y, Indjeian VB, et al. E2F mediates cell cycle-dependent transcriptional repression in vivo by recruitment of an HDAC1/mSin3B corepressor complex. Genes Dev. 2002;16:933–947. doi: 10.1101/gad.969202. The authors demonstrate that Sin3B and HDAC1 are recruited to the promoters of E2F pro-proliferative genes by the pocket proteins and E2F4 in quiescent fibroblasts, and that this recruitment is required for gene repression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang L, Mei Q, Zielinska-Kwiatkowska A, et al. An ERG (ets-related gene)-associated histone methyltransferase interacts with histone deacetylases 1/2 and transcription co-repressors mSin3A/B. Biochem J. 2003;369:651–657. doi: 10.1042/BJ20020854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De La Serna IL, Ohkawa Y, Imbalzano AN. Chromatin remodelling in mammalian differentiation: lessons from ATP-dependent remodellers. Nat Rev Genet. 2006;7:461–473. doi: 10.1038/nrg1882. [DOI] [PubMed] [Google Scholar]
- 20.Brubaker K, Cowley SM, Huang K, et al. Solution structure of the interacting domains of the Mad-Sin3 complex: implications for recruitment of a chromatin-modifying complex. Cell. 2000;103:655–665. doi: 10.1016/s0092-8674(00)00168-9. [DOI] [PubMed] [Google Scholar]
- 21.Swanson KA, Knoepfler PS, Huang K, et al. HBP1 and Mad1 repressors bind the Sin3 corepressor PAH2 domain with opposite helical orientations. Nat Struct Mol Biol. 2004;11:738–746. doi: 10.1038/nsmb798. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Zhang Z, Demeler B, et al. Coupled unfolding and dimerization by the PAH2 domain of the mammalian Sin3A corepressor. J Mol Biol. 2006;360:7–14. doi: 10.1016/j.jmb.2006.04.069. [DOI] [PubMed] [Google Scholar]
- 23.Suryadinata R, Sadowski M, Steel R, et al. Cyclin-dependent kinase-mediated phosphorylation of RBP1 and pRb promotes their dissociation to mediate release of the SAP30.mSin3.HDAC transcriptional repressor complex. J Biol Chem. 2011;286:5108–5118. doi: 10.1074/jbc.M110.198473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ellenrieder V, Zhang JS, Kaczynski J, et al. Signaling disrupts mSin3A binding to the Mad1-like Sin3-interacting domain of TIEG2, an Sp1-like repressor. Embo J. 2002;21:2451–2460. doi: 10.1093/emboj/21.10.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sahu SC, Swanson KA, Kang RS, et al. Conserved themes in target recognition by the PAH1 and PAH2 domains of the Sin3 transcriptional corepressor. J Mol Biol. 2008;375:1444–1456. doi: 10.1016/j.jmb.2007.11.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bowman CJ, Ayer DE, Dynlacht BD. Foxk proteins repress the initiation of starvation-induced atrophy and autophagy programs. Nat Cell Biol. 2014;16:1202–1214. doi: 10.1038/ncb3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nan X, Ng HH, Johnson CA, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 28.Jelinic P, Pellegrino J, David G. A novel mammalian complex containing Sin3B mitigates histone acetylation and RNA polymerase II progression within transcribed loci. Mol Cell Biol. 2011;31:54–62. doi: 10.1128/MCB.00840-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He Y, Radhakrishnan I. Solution NMR studies of apo-mSin3A and -mSin3B reveal that the PAH1 and PAH2 domains are structurally independent. Protein Sci. 2008;17:171–175. doi: 10.1110/ps.073097308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carrozza MJ, Li B, Florens L, et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–592. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 31.Keogh MC, Kurdistani SK, Morris SA, et al. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell. 2005;123:593–605. doi: 10.1016/j.cell.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 32.Li B, Gogol M, Carey M, et al. Combined action of PHD and chromo domains directs the Rpd3S HDAC to transcribed chromatin. Science. 2007;316:1050–1054. doi: 10.1126/science.1139004. [DOI] [PubMed] [Google Scholar]
- 33.Pile LA, Spellman PT, Katzenberger RJ, et al. The SIN3 deacetylase complex represses genes encoding mitochondrial proteins: implications for the regulation of energy metabolism. J Biol Chem. 2003;278:37840–37848. doi: 10.1074/jbc.M305996200. [DOI] [PubMed] [Google Scholar]
- 34.Sharma V, Swaminathan A, Bao R, et al. Drosophila SIN3 is required at multiple stages of development. Dev Dyn. 2008;237:3040–3050. doi: 10.1002/dvdy.21706. [DOI] [PubMed] [Google Scholar]
- 35.Dannenberg JH, David G, Zhong S, et al. mSin3A corepressor regulates diverse transcriptional networks governing normal and neoplastic growth and survival. Genes Dev. 2005;19:1581–1595. doi: 10.1101/gad.1286905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36••.David G, Grandinetti KB, Finnerty PM, et al. Specific requirement of the chromatin modifier mSin3B in cell cycle exit and cellular differentiation. Proc Natl Acad Sci U S A. 2008;105:4168–4172. doi: 10.1073/pnas.0710285105. The initial characterization of Sin3B−/− embryos and mammalian cells demonstrating an essential role for Sin3B in cell cycle exit. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cowley SM, Iritani BM, Mendrysa SM, et al. The mSin3A chromatin-modifying complex is essential for embryogenesis and T-cell development. Mol Cell Biol. 2005;25:6990–7004. doi: 10.1128/MCB.25.16.6990-7004.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pellegrino J, Castrillon DH, David G. Chromatin associated Sin3A is essential for male germ cell lineage in the mouse. Dev Biol. 2012;369:349–355. doi: 10.1016/j.ydbio.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39•.Grandinetti KB, Jelinic P, DiMauro T, et al. Sin3B expression is required for cellular senescence and is up-regulated upon oncogenic stress. Cancer Res. 2009;69:6430–6437. doi: 10.1158/0008-5472.CAN-09-0537. The authors demonstrate that Sin3B is required for OIS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40•.Rielland M, Cantor DJ, Graveline R, et al. Senescence-associated SIN3B promotes inflammation and pancreatic cancer progression. J Clin Invest. 2014;124:2125–2135. doi: 10.1172/JCI72619. The authors demonstrate that Sin3B promotes PDAC progression and propose a model where the SASP generates a proinflammatory and protumorigenic microenvironment in PDAC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bertoli C, Skotheim JM, De Bruin RA. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol. 2013;14:518–528. doi: 10.1038/nrm3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol. 2013;14:297–306. doi: 10.1038/nrm3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Litovchick L, Florens LA, Swanson SK, et al. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev. 2011;25:801–813. doi: 10.1101/gad.2034211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44•.Litovchick L, Sadasivam S, Florens L, et al. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol Cell. 2007;26:539–551. doi: 10.1016/j.molcel.2007.04.015. The initial characterization of the DREAM complex in mammalian cells. [DOI] [PubMed] [Google Scholar]
- 45.Sadasivam S, DeCaprio JA. The DREAM complex: master coordinator of cell cycle-dependent gene expression. Nat Rev Cancer. 2013;13:585–595. doi: 10.1038/nrc3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tedesco D, Lukas J, Reed SI. The pRb-related protein p130 is regulated by phosphorylation-dependent proteolysis via the protein-ubiquitin ligase SCF(Skp2) Genes Dev. 2002;16:2946–2957. doi: 10.1101/gad.1011202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henley SA, Dick FA. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div. 2012;7:10. doi: 10.1186/1747-1028-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48•.Luo RX, Postigo AA, Dean DC. Rb interacts with histone deacetylase to repress transcription. Cell. 1998;92:463–473. doi: 10.1016/s0092-8674(00)80940-x. This study along with ref. 49 and ref. 50 identify a biochemical interaction between the pocket proteins and HDAC1/2 and further demonstrate that HDAC1/2 is required for the transcriptional repression of pocket protein target genes. [DOI] [PubMed] [Google Scholar]
- 49•.Brehm A, Miska EA, McCance DJ, et al. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601. doi: 10.1038/35404. See comment for ref. 48. [DOI] [PubMed] [Google Scholar]
- 50•.Ferreira R, Magnaghi-Jaulin L, Robin P, et al. The three members of the pocket proteins family share the ability to repress E2F activity through recruitment of a histone deacetylase. Proc Natl Acad Sci U S A. 1998;95:10493–10498. doi: 10.1073/pnas.95.18.10493. See comment for ref. 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sandoval R, Pilkinton M, Colamonici OR. Deletion of the p107/p130-binding domain of Mip130/LIN-9 bypasses the requirement for CDK4 activity for the dissociation of Mip130/LIN-9 from p107/p130-E2F4 complex. Exp Cell Res. 2009;315:2914–2920. doi: 10.1016/j.yexcr.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gaubatz S, Lindeman GJ, Ishida S, et al. E2F4 and E2F5 play an essential role in pocket protein-mediated G1 control. Mol Cell. 2000;6:729–735. doi: 10.1016/s1097-2765(00)00071-x. [DOI] [PubMed] [Google Scholar]
- 53.Humbert PO, Rogers C, Ganiatsas S, et al. E2F4 is essential for normal erythrocyte maturation and neonatal viability. Mol Cell. 2000;6:281–291. doi: 10.1016/s1097-2765(00)00029-0. [DOI] [PubMed] [Google Scholar]
- 54.Cobrinik D, Lee MH, Hannon G, et al. Shared role of the pRB-related p130 and p107 proteins in limb development. Genes Dev. 1996;10:1633–1644. doi: 10.1101/gad.10.13.1633. [DOI] [PubMed] [Google Scholar]
- 55.Blais A, Van Oevelen CJ, Margueron R, et al. Retinoblastoma tumor suppressor protein-dependent methylation of histone H3 lysine 27 is associated with irreversible cell cycle exit. J Cell Biol. 2007;179:1399–1412. doi: 10.1083/jcb.200705051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Malovannaya A, Lanz RB, Jung SY, et al. Analysis of the human endogenous coregulator complexome. Cell. 2011;145:787–799. doi: 10.1016/j.cell.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hayakawa T, Ohtani Y, Hayakawa N, et al. RBP2 is an MRG15 complex component and down-regulates intragenic histone H3 lysine 4 methylation. Genes Cells. 2007;12:811–826. doi: 10.1111/j.1365-2443.2007.01089.x. [DOI] [PubMed] [Google Scholar]
- 58.Beshiri ML, Holmes KB, Richter WF, et al. Coordinated repression of cell cycle genes by KDM5A and E2F4 during differentiation. Proc Natl Acad Sci U S A. 2012;109:18499–18504. doi: 10.1073/pnas.1216724109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bainor AJ, Deng FM, Wang Y, et al. Chromatin-associated protein SIN3B prevents prostate cancer progression by inducing senescence. Cancer Res. 2017 doi: 10.1158/0008-5472.CAN-16-3410. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hughes-Davies L, Huntsman D, Ruas M, et al. EMSY links the BRCA2 pathway to sporadic breast and ovarian cancer. Cell. 2003;115:523–535. doi: 10.1016/s0092-8674(03)00930-9. [DOI] [PubMed] [Google Scholar]
- 61.Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ley TJ, Miller C, Ding L, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63••.Cantor DJ, David G. The chromatin-associated Sin3B protein is required for hematopoietic stem cell functions in mice. Blood. 2017;129:60–70. doi: 10.1182/blood-2016-06-721746. The authors report a critical role for Sin3B for the maintenance of HSC quiescence and the ability of HSCs to properly differentiate following transplantation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482–496. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- 65.Micco R, Fumagalli M, Cicalese A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nat Cell Biol. 2006;444:638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 66.David G. Regulation of oncogene-induced cell cycle exit and senescence by chromatin modifiers. Cancer Biol Ther. 2012;13:992–1000. doi: 10.4161/cbt.21116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Narita M, Nunez S, Heard E, et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- 68.Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15:397–408. doi: 10.1038/nrc3960. [DOI] [PubMed] [Google Scholar]
- 69.Lau L, David G. Senescence phenotypes induced by Ras in primary cells. Methods Mol Biol. 2017;1534:17–30. doi: 10.1007/978-1-4939-6670-7_2. [DOI] [PubMed] [Google Scholar]
- 70•.Serrano M, Lin AW, McCurrach ME, et al. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. A important discovery demonstrating that oncogenes drive fibroblasts into premature cellular senescence. [DOI] [PubMed] [Google Scholar]
- 71•.Collado M, Gil J, Efeyan A, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. A seminal study helping to formulate the hypothesis that cellular senescence is a tumor-suppressive mechanism in vivo. [DOI] [PubMed] [Google Scholar]
- 72.Chen Z, Trotman LC, Shaffer D, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Braig M, Lee S, Loddenkemper C, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–665. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- 74.Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 75.Kuilman T, Michaloglou C, Vredeveld LC, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 76.Coppe JP, Patil CK, Rodier F, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Acosta JC, O’Loghlen A, Banito A, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 78.Acosta JC, Banito A, Wuestefeld T, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978–990. doi: 10.1038/ncb2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Krizhanovsky V, Yon M, Dickins RA, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–667. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lujambio A, Akkari L, Simon J, et al. Non-cell-autonomous tumor suppression by p53. Cell. 2013;153:449–460. doi: 10.1016/j.cell.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kang TW, Yevsa T, Woller N, et al. Senescence surveillance of premalignant hepatocytes limits liver cancer development. Nature. 2011;479:547–551. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- 82•.Davalos AR, Coppe JP, Campisi J, et al. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010;29:273–283. doi: 10.1007/s10555-010-9220-9. An excellent review highlighting the proinflammatory and protumorigenic aspects of the SASP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Laberge RM, Sun Y, Orjalo AV, et al. MTOR regulates the protumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015;17:1049–1061. doi: 10.1038/ncb3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yoshimoto S, Loo TM, Atarashi K, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- 85.Eggert T, Wolter K, Ji J, et al. Distinct functions of senescence-associated immune responses in liver tumor surveillance and tumor progression. Cancer Cell. 2016;30:533–547. doi: 10.1016/j.ccell.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tchkonia T, Zhu Y, van Deursen J, et al. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123:966–972. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Orjalo AV, Bhaumik D, Gengler BK, et al. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci U S A. 2009;106:17031–17036. doi: 10.1073/pnas.0905299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ling J, Kang Y, Zhao R, et al. KrasG12D-induced IKK2/β/NF-κB activation by IL-1α and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:105–120. doi: 10.1016/j.ccr.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhuang Z, Ju HQ, Aguilar M, et al. IL1 receptor antagonist inhibits pancreatic cancer growth by abrogating NF-kappaB activation. Clin Cancer Res. 2016;22:1432–1444. doi: 10.1158/1078-0432.CCR-14-3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang Y, Yan W, Collins MA, et al. Interleukin-6 is required for pancreatic cancer progression by promoting MAPK signaling activation and oxidative stress resistance. Cancer Res. 2013;73:6359–6374. doi: 10.1158/0008-5472.CAN-13-1558-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Daniluk J, Liu Y, Deng D, et al. An NF-kappaB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J Clin Invest. 2012;122:1519–1528. doi: 10.1172/JCI59743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pablo G-S, Quintanilla A, Lafita MC, et al. Sin3b interacts with Myc and decreases Myc levels. J Biol Chem. 2014;289:22221–22236. doi: 10.1074/jbc.M113.538744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li YL, Choi PS, Casey SC, et al. MYC through miR-17–92 suppresses specific target genes to maintain survival, autonomous proliferation, and a neoplastic state. Cancer Cell. 2014;26:262–272. doi: 10.1016/j.ccr.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Di Mitri D, Toso A, Chen JJ, et al. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature. 2014;515:134–137. doi: 10.1038/nature13638. [DOI] [PubMed] [Google Scholar]
- 95.Kalathur M, Toso A, Chen JJ, et al. A chemogenomic screening identifies CK2 as a target for pro-senescence therapy in PTEN-deficient tumours. Nat Commun. 2015;6:7227. doi: 10.1038/ncomms8227. [DOI] [PubMed] [Google Scholar]
- 96.Revandkar A, Perciato ML, Toso A, et al. Inhibition of Notch pathway arrests PTEN-deficient advanced prostate cancer by triggering p27-driven cellular senescence. Nat Commun. 2016;7:13719. doi: 10.1038/ncomms13719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275–291. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 98.Roesch A, Fukunaga-Kalabis M, Schmidt EC, et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141:583–594. doi: 10.1016/j.cell.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Thomas D, Majeti R. Biology and relevance of human acute myeloid leukemia stem cells. Blood. 2017;129:1577–1585. doi: 10.1182/blood-2016-10-696054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kreso A, O’Brien CA, van Galen P, et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science. 2013;339:543–548. doi: 10.1126/science.1227670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen W, Dong J, Haiech J, et al. Cancer stem cell quiescence and plasticity as major challenges in cancer therapy. Stem Cells Int. 2016;2016:1–16. doi: 10.1155/2016/1740936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Reavie L, Buckley SM, Loizou E, et al. Regulation of c-Myc ubiquitination controls chronic myelogenous leukemia initiation and progression. Cancer Cell. 2013;23:362–375. doi: 10.1016/j.ccr.2013.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Takeishi S, Matsumoto A, Onoyama I, et al. Ablation of Fbxw7 eliminates leukemia-initiating cells by preventing quiescence. Cancer Cell. 2013;23:347–361. doi: 10.1016/j.ccr.2013.01.026. [DOI] [PubMed] [Google Scholar]
- 104.Saito Y, Uchida N, Tanaka S, et al. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat Biotechnol. 2010;28:275–280. doi: 10.1038/nbt.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lu D, Chen S, Tan X, et al. Fra-1 promotes breast cancer chemo-sensitivity by driving cancer stem cells from dormancy. Cancer Res. 2012;72:3451–3456. doi: 10.1158/0008-5472.CAN-11-2536. [DOI] [PubMed] [Google Scholar]
- 106.Essers MA, Trumpp A. Targeting leukemic stem cells by breaking their dormancy. Mol Oncol. 2010;4:443–450. doi: 10.1016/j.molonc.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wilting RH, Yanover E, Heideman MR, et al. Overlapping functions of Hdac1 and Hdac2 in cell cycle regulation and haematopoiesis. Embo J. 2010;29:2586–2597. doi: 10.1038/emboj.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang B, Strauss AC, Chu S, et al. Effective targeting of quiescent chronic myelogenous leukemia stem cells by histone deacetylase inhibitors in combination with imatinib mesylate. Cancer Cell. 2010;17:427–442. doi: 10.1016/j.ccr.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kadamb R, Mittal S, Bansal N, et al. Sin3: insight into its transcription regulatory functions. Eur J Cell Biol. 2013;92:237–246. doi: 10.1016/j.ejcb.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 110.Boichuk S, Parry JA, Makielski KR, et al. The DREAM complex mediates GIST cell quiescence and is a novel therapeutic target to enhance imatinib-induced apoptosis. Cancer Res. 2013;73:5120–5129. doi: 10.1158/0008-5472.CAN-13-0579. [DOI] [PubMed] [Google Scholar]
- 111•.Farias EF, Petrie K, Leibovitch B, et al. Interference with Sin3 function induces epigenetic reprogramming and differentiation in breast cancer cells. Proc Natl Acad Sci U S A. 2010;107:11811–11816. doi: 10.1073/pnas.1006737107. The authors demonstrate the utilization of decoy molecules to prevent the assembly of Sin3 complexes and target breast cancer growth. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bansal N, Petrie K, Christova R, et al. Targeting the SIN3A-PF1 interaction inhibits epithelial to mesenchymal transition and maintenance of a stem cell phenotype in triple negative breast cancer. Oncotarget. 2015;6:34087–34105. doi: 10.18632/oncotarget.6048. [DOI] [PMC free article] [PubMed] [Google Scholar]