

Abstract
INTRODUCTION
Design and construction of an extensively modified yeast genome is a direct means to interrogate the integrity, comprehensiveness, and accuracy of the knowledge amassed by the yeast community to date. The international synthetic yeast genome project (Sc2.0) aims to build an entirely designer, synthetic Saccharomyces cerevisiae genome. The synthetic genome is designed to increase genome stability and genetic flexibility while maintaining cell fitness near that of the wild type. A major challenge for a genome synthesis lies in identifying and eliminating fitness-reducing sequence variants referred to as “bugs.”
RATIONALE
Debugging is imperative for successfully building a fit strain encoding a synthetic genome. However, it is time-consuming and laborious to replace wild-type genes and measure strain fitness systematically. The Sc2.0 PCRTag system, which specifies recoded sequences within open reading frames (ORFs), is designed to distinguish synthetic from wild-type DNA in a simple polymerase chain reaction (PCR) assay. This system provides an opportunity to efficiently map bugs to the related genes by using a pooling strategy and subsequently correct them. Further, as we identify bugs in designer sequences, we will identify gaps in our knowledge and gain a deeper understanding of genome biology, allowing refinement of future design strategies.
RESULTS
We chemically synthesized yeast chromosome X, synX, designed to be 707,459 base pairs. A high-throughput mapping strategy called pooled PCRTag mapping (PoPM) was developed to identify unexpected bugs during chromosome assembly. With this method, the genotypes of pools of colonies with normal or defective fitness are assessed by PCRTag analysis. The PoPM method exploits the patchwork structure of synthetic and wild-type sequences observed in the majority of putative synthetic DNA integrants or meiotic progeny derived from synthetic/wild-type strain backcross. PCRTag analysis with both synthetic and wild-type specific primers, carried out with genomic DNA extracted from the two pools of clones (normal fitness versus a specific growth defect), can be used to identify regions of synthetic DNA missing from the normal fitness pool and, analogously, sections of wild-type DNA absent from the specific growth-defect pool. In this way, the defect can be efficiently mapped to a very small overlapping region, and subsequent systematic analysis of designed changes in that region can be used to identify the bug. Several bugs were identified and corrected, including a growth defect mapping to a specific synonymously recoded PCRTag sequence in the essential FIP1 ORF and the effect of introducing a loxPsym site that unexpectedly altered the the promoter function of a nearby gene, ATP2. In addition, meiotic crossover was employed to repair the massive duplications and rearrangements in the synthetic chromosome. The debugged synX strain exhibited high fitness under a variety of conditions tested and in competitive growth with the wild-type strain.
CONCLUSION
Synthetic yeast chromosome X was chemically synthesized from scratch, a rigorous, incremental step toward complete synthesis of the whole yeast genome. Thousands of designer modifications in synX revealed extensive flexibility of the yeast genome. We developed an efficient mapping method, PoPM, to identify bugs during genome synthesis, generalizable to any watermarked synthetic chromosome, and several details of yeast biology were uncovered by debugging. Considering the numerous gene-associated PCRTags available in the synthetic chromosomes, PoPM may represent a powerful tool to map interesting phenotypes of mutated synthetic strains or even mutated wild-type strains to the relevant genes. It may also be useful to study yeast genetic interactions when an unexpected phenotype is generated by alterations in two or more genes, substantially expanding understanding of yeast genomic and cellular functions. The PoPM method is also likely to be useful for mapping phenotype(s) resulting from the genome SCRaMbLE system.
Graphical Abstract
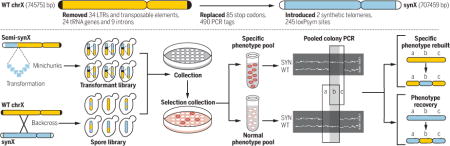
Characterization of synX and debugging by pooled PCRTag mapping. (Top)
Design overview of synthetic chromosome X. (Bottom) Flow diagram of pooled PCRTag mapping (PoPM).
Saccharomyces cerevisiae was the first eukaryote to have its genome fully sequenced (1). Our understanding of yeast physiology, metabolism, and network biology is extensive (2–5). Design and construction of an extensively modified yeast genome, Sc2.0, is a direct means to interrogate the integrity, comprehensiveness, and accuracy of knowledge amassed by the yeast community to date. However, a major challenge for a successful genome synthesis lies in identifying and eliminating fitness-reducing variants, sequences we refer to as “bugs” (6). This process can be time-consuming and laborious. Further, as we identify bugs in designer sequences, we will identify gaps in our knowledge and gain a deeper understanding of genome biology, allowing refinement of future design strategies.
The international Synthetic Yeast Genome Project (Sc2.0) aims to build an entirely synthetic, designer S. cerevisiae genome. The synthetic genome is designed to increase genome stability and genetic flexibility while maintaining cell fitness near that of the wild type. Here, we describe chemical synthesis of yeast chromosome X, synX, designed according to Sc2.0 principles (7, 8). We have developed an efficient strategy to map bugs, pooled PCRTag mapping, PoPM, which enabled us to identify fitness-reducing sequence alterations and subsequently revert them to the wild-type sequence. The debugged synX strain has high fitness under all conditions tested and in competitive growth with wild-type cells. Our detailed analysis of unexpected phenotypes has led to new understanding of genome structure and regulation.
Design and assembly of synX
The sequence of synX was created in silico with the genome-editing suite BioStudio, starting from the native sequence of chromosome X (8–10) (fig. S1). Modifications in synX (Fig. 1A) include deletion of retrotransposons, subtelomeric repeats, and introns. In addition, 24 tRNA genes were removed and a single-copy tRNA gene, tR(CCU)J (11), was relocated to the HO locus (table S1). All TAG stop codons were replaced by TAA. A total of 490 pairs of synonymous sequences in open reading frames (ORFs), or PCRTags, served as a DNA watermarking system. We inserted 245 loxPsym sites in the 3′ untranslated region (UTR) of nonessential genes and at the locations of most deleted features to enable the inducible evolution system SCRaMbLE (synthetic chromosome rearrangement and modification by loxP-mediated evolution) (8, 12, 13). SynX was split into 18 “megachunks” (A to R) of 30 to 60 kb DNA fragments and further divided into 171 “minichunks” of ~5-kb DNA fragments. All of the minichunks were synthesized by DNA synthesis providers and used for stepwise incorporation to replace native chromosome X (fig. S2 and table S2) (10, 14). PCRTag analysis verified the presence of all synthetic amplicons and the corresponding absence of wild-type amplicons, consistent with complete incorporation (Fig. 1B and fig. S3).
Fig. 1. Characterization and fitness testing of synX strain.
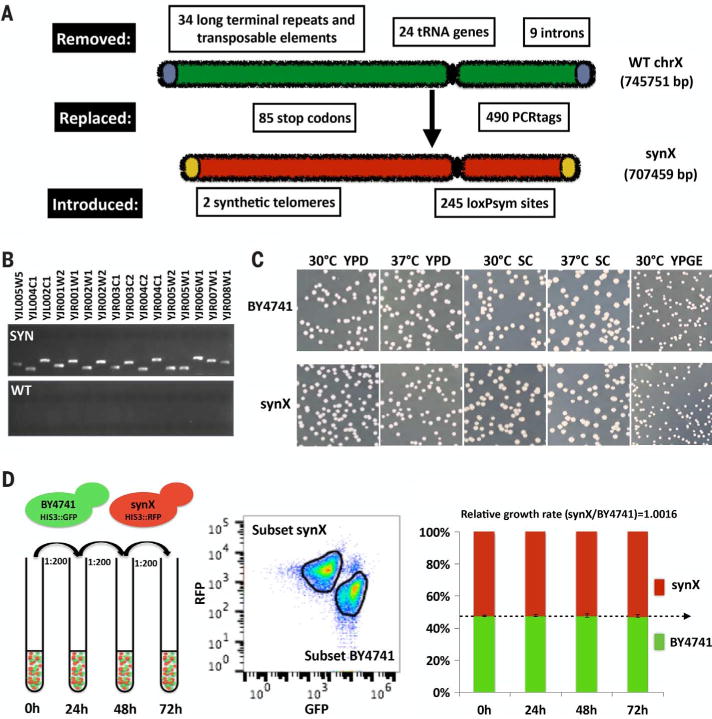
(A) Design overview of synthetic chromosome X. (B) PCRTag analysis of synX (a pericentromeric region). Analysis of the complete set of PCRTags is shown in fig. S3. (C) Growth fitness of synX strain yYW0115 on various types of media. (D) Competitive growth assay of synX strain yYW0115 and native strain BY4741. Samples were analyzed by flow cytometry to quantify the ratio of RFP-positive to GFP-positive cells. Relative growth rate was calculated based on the ratio of synX to BY4741 at each time point.
Characterization of synX
The initial designed sequence of synX (yeast_chr10_3_37) included three fitness-reducing bugs that we mapped to designer changes. The synX designed sequence was modified to reflect that the three designer changes were reverted to the wild-type sequence (table S3). Sequencing of synX strain (yYW0077) identified 11 sequence variants (table S4) and three residual wild-type regions compared to the designed chromosome (yeast_chr10_3_40); the latter three were corrected to the designed sequence in the physical strain (fig. S4). Two unexpected massive structural variations were also identified unexpectedly, and subsequently restored to the desired structure by an intercross. We observed that the native 2-micron plasmids were completely lost in synX strain yYW0077, as revealed by whole-genome sequencing and RNA sequencing (figs. S5 and S6). There was no apparent growth defect associated with loss of the 2-micron plasmid (fig. S7), and the 2-micron plasmid was reintroduced to the synX strain yYW0115 as a consequence of the intercross to remove the massive duplication. The 2-micron plasmid was previously reported to have no impact on yeast life span, and it is hypothesized that this element may be a form of parasitic DNA (15, 16). In the intercrossed synX strain, 2-micron levels were normal (fig. S5), suggesting there are no negative effects associated with 2-micron maintenance. It is possible that a stochastic 2-micron loss occurred in one or more of the many single-colony purification steps performed during the construction of synX.
The debugged synX strain yYW0115 exhibited phenotypes very similar to those of the wild type (BY4741) under a variety of culture conditions (Fig. 1C). Notably, synX strain yYW0115 exhibited an elongated cell morphology. However, after an endoreduplication backcross to wild type, the morphology of synX cells became similar to that of wild type (fig. S8). To detect more subtle differences in growth properties, we implemented a competitive growth assay to characterize fitness of synX with high sensitivity (Fig. 1D) (17). Cells encoding synX (yYW0115) and native chrX (BY4741) were tracked by red fluorescent protein (RFP) and green fluorescent protein (GFP) expression, respectively. After inoculation at a 1:1 cell ratio, the two strains maintained a steady population ratio over a 72-hour coculture period (Fig. 1D), suggesting identical growth properties under the competitive growth conditions tested.
Mapping bugs by using pooled PCRTag mapping
We have now encountered sparsely distributed bugs in most assembled Sc2.0 chromosomes (14, 18–20). One efficient strategy to debug synthetic chromosomes is to correlate genotype and phenotype of many putative integrants after integration of each megachunk (typically 30 to 60 kb of synthetic DNA). In this way, slow growth defects may be identified immediately and assigned to a specific segment of synthetic DNA. We have developed a high-throughput bug mapping strategy called pooled PCRTag mapping (PoPM) (Fig. 2A). Here, the genotype of pools of colonies derived from integration experiments is assessed by using PCRTag analysis, a simple polymerase chain reaction (PCR)–based assay that distinguishes synthetic DNA from wild-type DNA. The PoPM method exploits the patchwork structure of synthetic and wild-type sequences observed in target regions of the majority of putative synthetic DNA integrants. After first phenotyping all integrants under selective conditions (e.g., high temperature and/or growth on a nonfermentable carbon source), each clone is then binned into one of two pools—normal fitness or a specific growth defect. PCRTag analysis with both synthetic and wild-type primer pairs, carried out with genomic DNA extracted from the two pools of clones, can be used to identify regions of synthetic DNA missing from the normal fitness pool and, analogously, sections of wild-type DNA absent from the specific growth defect pool. In this way, the defect can be efficiently mapped to a very small region, and subsequent systematic analysis of designed changes in that region can be used to identify the bug. We can then update the design and correct the physical sequence. This strategy is generic and may be applied to the construction and debugging of any watermarked chromosome or even multisynthetic watermarked chromosome strains. Further, PoPM can be applied to meiotic progeny derived from synthetic/wild-type strain backcross experiments, which can markedly reduce the effort associated with PCRTagging of many tetrads (14). PoPM can also be used to map synthetic sick or lethal interactions indicated by the absence of two or more wild-type DNA fragments from the growth defect pool without changes in the normal fitness pool (fig. S9A). Similarly, PoPM can be used to map multiple bugs simultaneously, indicated by the absence of multiply synthetic DNA fragments from the normal fitness pool without changes in the defect pool (fig. S9B). In addition, quantitative PCR (qPCR) can improve the PoPM strategy to avoid noisy amplification due to a very few maverick clones.
Fig. 2. Debugging by pooled PCRTag mapping.
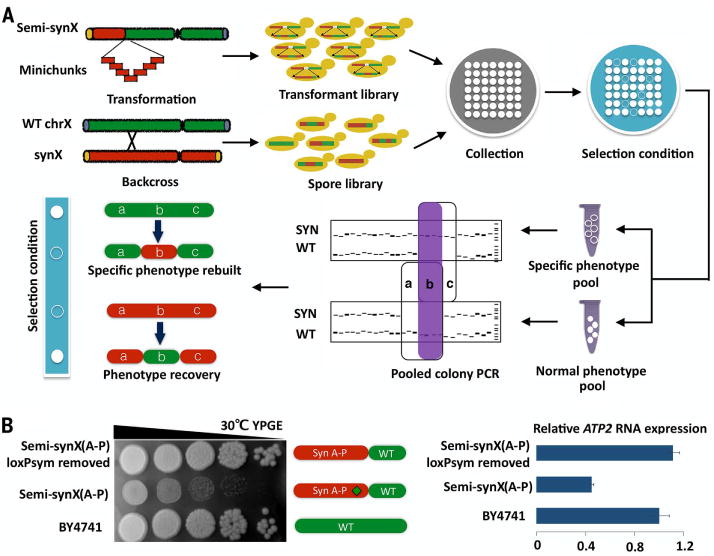
(A) Flow diagram of pooled PCRTag mapping (PoPM). Chromosome X strains with patchworks of synthetic and native sequences, generated by incorporation of synthetic DNA minichunks (transformant library) or by backcross with wild-type (WT) cells (spore library), are subjected to phenotype testing under a selective condition (filled circle, high fitness; open circle, low fitness). PCRTagging is carried out on pools of high-fitness colonies as well as pools of low-fitness colonies to enable mapping of the defect to a small segment of synthetic DNA. The purple-shaded region “b” in the PCR indicates the region containing the “bug.” (B) The insertion of a loxPsym site in the 3′ UTR of YJR120W disrupts expression of neighboring gene ATP2, leading to a growth defect on the nonfermentable carbon source glycerol/ethanol (YPGE).
We used PoPM to debug synX in two separate instances during assembly. First, by applying PoPM to the transformants derived from incorporation of megachunk P, we identified the loxPsym site in the 3′ UTR of YJR120W as being responsible for a growth defect of semisynthetic synX strain (A-P) (yYW0062) on YPGE (Fig. 2B). Because of the short gap [131 base pairs (bp)] between the terminator of YJR120W and the neighboring coding region of ATP2 (9), we hypothesized that insertion of the loxPsym site in the 3′ UTR of YJR120W, annotated as a “protein of unknown function” that codes for an ORF of 116 codons, disrupts the promoter region of ATP2, which encodes a subunit of the mitochondrial F1F0 ATP synthase (21). ATP2 mRNA expression in semi-synX(A-P) was decreased to about 40% of that observed in the wild-type strain; deletion of the loxPsym site in the semi-synX(A-P) strain restored expression of ATP2 and repaired the growth defect on YPGE (Fig. 2B). We conclude that YJR120W is not a functional gene at all, but rather a “dubious ORF,” and that its deletion produces a transcriptional hypomorph of ATP2, a situation variously referred to as an “off by one error” (22) or “neighboring gene effect” (23).
A growth defect on YPD at 30°C was noted in strain semi-synX(A-O) (yYW0055) (Fig. 3A). Applying PoPM to the transformants derived from incorporation of megachunk O, we mapped this to the reverse PCRTag within the synthetic FIP1 allele, a set of 10 synonymously recoded codons. By comparing wild type with synthetic sequences, we identified a putative binding site for transcription factor Rap1p in the synthetic sequence (Fig. 3B) (24–28). Rap1p binding within transcription units is known to mediate steric down-regulation of gene expression and was shown to lead to RNA polymerase stalling and a reduction in full-length transcript level similar to that observed in our RNA analysis of FIP1 (27). Replacement with individual wild-type codons across the entire PCRTag showed that codons disrupting the Rap1p binding site repaired the growth defect and restored FIP1 mRNA level (Fig. 3C). PCRTag swaps in synVI and synXII are also reported to lead to growth defects (14, 18).
Fig. 3. PCRTag in FIP1 causes a growth defect.
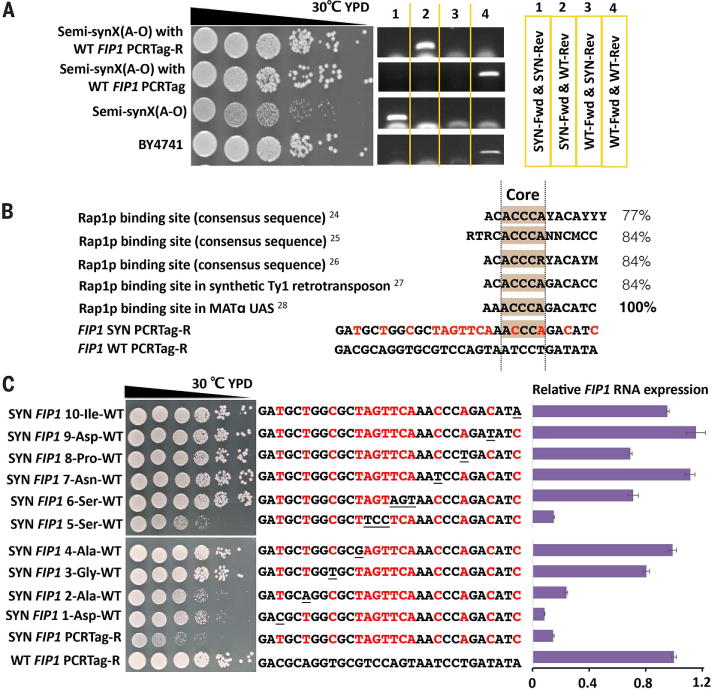
(A) The reverse (R) synthetic PCRTag recoded within YJR093C (FIP1) causes a growth defect. (B) FIP1 SYN PCRTag-R introduces a Rap1p recognition site. Percentages indicate similarity of FIP1 SYN PCRTag-R to known Rap1p binding sites. Red letters show differences between synthetic and native PCRTag-R sequences. (C) Growth assay and relative FIP1 RNA expression of codon-by-codon swap strains in FIP1 SYN PCRTag-R. Red letters represent SYN-specific bases; underlined letters represent restored WT codons. RNA level is quantified relative to WT FIP1.
Massive duplications and rearrangements
Electrophoretic karyotype analysis of the synthetic chromosome X strain synX(A-R) (yYW0077) by pulsed-field gel electrophoresis revealed a considerably slower-migrating species than expected for synX as compared to native chromosome X (Fig. 4A). Genome sequencing, structural variation analysis, and plots of read depth revealed multiple regions on synX with amplifications of complex structure (Fig. 4B). qPCR to evaluate the copy number of genes located in each of the triplicated regions suggested that they arose during transformation with megachunk C (Fig. 4C). Analysis of junction sequences suggested a tandem duplication for the structure of the triplicated regions (Fig. 4B and fig. S10A). The aberrant junctions underlying the amplifications and their position relative to the incoming fragments during integrative transformation suggested that homologous recombination occurred at near-terminal loxPsym sites in one case and nonhomologous recombination occurred at the Not I sites at the minichunk termini in the other case. Other amplification structures in megachunk D and E were also analyzed, and qPCR indicated that the duplications occurred during megachunk E transformation (fig. S10, B and C). Structural rearrangements mediated by loxPsym sites and cohesive end termini were also seen in synII and synV (19, 20).
Fig. 4. Large duplications and rearrangements and restoration to desired structure.
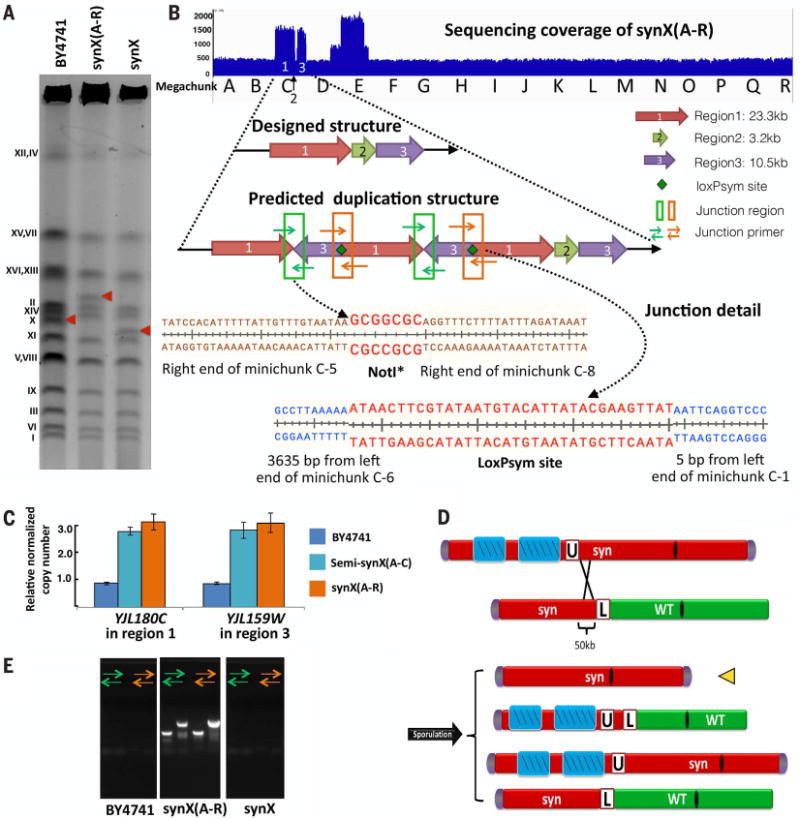
(A) Karyotypic analysis of synX(A-R), yYW0077 and corrected synX strain, yYW0115 by pulsed-field gel electrophoresis. (B) Two large duplications and rearrangements occurred in the synX(A-R) strain. Sequencing coverage of synX(A-R) strain revealed two large duplications of synthetic fragments. The NotI* site and loxPsym site mediated junction of duplicated segments. (C) Massive duplications and rearrangements occurred during integrative transformation. Intermediate assembly strain semi-synX(A-C) (yYW0007) and synX(A-R) have identical copy numbers in the duplicated region. (D) Meiotic crossover to generate a synX strain lacking amplified segments. (E) Verification of the absence of duplication regions in synX strain yYW0115 by means of junction primers.
Meiotic crossover was employed to repair the amplifications of synthetic DNA in synX(A-R) (Fig. 4D). The selectable marker URA3 was inserted adjacent to the amplified region in synX, while a second semisynthetic chrX strain (yYW0098), which had a single copy of the amplified regions that extended from the left arm up to megachunk F and LEU2 as a selectable marker inserted between the synthetic and wild-type regions, was constructed. Synthetic megachunk F (~50 kb) served as the homologous region for crossover of the two chromosome X. After mating the two strains and sporulating, spores unable to grow on medium lacking uracil and medium lacking leucine were selected from tetrads (fig. S10D). One out of four tetrads showed crossover events in the region of interest. The deletion of the amplified region was verified by using junction primers and pulsed-field gel electrophoresis analysis (Fig. 4, A and E, and fig. S10E). Finally, one out of six of the Ura–Leu– spores had all the synthetic PCRTags, and genome sequencing verified the presence of single-copy DNA across all previously amplified regions.
Synthetic yeast chromosome X was chemically synthesized from scratch, which is a rigorous, incremental step toward the complete synthesis of the whole yeast genome. Thousands of designer modifications in synX, along with the massive duplication and the absence of native 2-micron plasmids in the draft synX strain, revealed the extensive flexibility of the yeast genome. However, one of the most challenging and time-consuming processes in a genome-scale DNA synthesis is debugging, considering that none of the modifications are permitted to substantially decrease the cell growth and fitness. We developed an efficient mapping method called PoPM to identify unexpected bugs during genome synthesis. By means of the debugging process, several details of yeast biology were uncovered, including the effects of unwittingly introducing a loxPsym site in the 3′ UTR of YJR120W affecting promoter function of the nearby gene ATP2, and the identification of a putative binding site for transcription factor Rap1p in the recoded essential gene FIP1. Use of a competitive growth assay established that the debugged synX strain exhibits high fitness.
The yeast 2.0 genome is being designed and built by chemical synthesis, but many phenotypes are not readily mapped to the underlying gene(s), especially complex phenotypes related to multiple genes. Considering that numerous gene-associated PCRTags are present in synthetic chromosomes, PoPM may represent a powerful tool for mapping interesting phenotypes of mutated synthetic strains or even mutated wild-type strains to the relevant genes. It may also be useful for studying yeast genetic interactions when an unexpected phenotype is generated by alterations in two or more genes. Thus, PoPM can substantially expand the understanding of yeast genomic and cellular functions. In addition, the PoPM method is also likely to be very useful for mapping phenotype(s) resulting from the genome SCRaMbLE system.
Materials and methods
SynX design
SynX was produced in silico with the genome-editing suite BioStudio (fig. S1)_(10). Three design principles were defined as follows: first, a synthetic chromosome should result in a (near) wild-type phenotype and fitness; second, a synthetic chromosome should lack destabilizing elements such as tRNA genes or transposons; and third, a synthetic chromosome should have genetic flexibility to facilitate future studies (7). We updated three versions of synX in silico after the debugging process from yeast_chr10_3_37 to yeast_chr10_3_40 (table S9). The length of synX (version: yeast_chr10_3_40) is 707,459 bp, which is 38,292 bp shorter than wild-type chromosome X.
SynX synthesis and assembly
The designed synX sequence was divided into 18 megachunks (30 to 60 kb of synthetic DNA) and then further divided into 171 minichunks of ~5 kb (table S1). All minichunks were synthesized by providers (Genewiz and Life Technologies). Adjacent minichunks overlapped by 400 bp, and the last minichunks of each megachunk contain alternating selectable marker (URA3 or LEU2), except the last megachunk R. With an average of 10 minichunks and alternating selectable marker in each incorporation, the native chromosome X was systematically replaced by its synthetic counterpart in 18 successive rounds of transformation (fig. S2).
Yeast colony PCR for rapid screening of WT and SYN PCRTags
Yeast cells from single colonies were resuspended in 50 μl of 20 mM NaOH followed by three cycles (99°C for 60 s, 4°C for 60 s) in a thermocycler; 1 μl of this mix was used as a template with regular PCR mix and for performing PCR cycles.
Yeast genomic DNA preparation for PCRTag analysis
Yeast cells from single colonies were resuspended in 100 μl of 200 mM LiOAc–1% SDS and incubated at 70°C for 15 min. A volume of 300 μl of 100% ethanol was added, and the suspension was vortexed for 30 s. DNA was collected at 15,000 rpm for 3 min and then dissolved in 100 μl of Tris-HCl-EDTA buffer. Cell debris was centrifuged at 15,000 rpm for 1 min, and 1 μl of supernatant was used for regular PCR or 10 nl for 384-well PCR.
Complete set of PCRTags analyses of synX
At each of the intermediate incorporation steps, as well as on completion of synX, PCRTag analysis (table S3) revealed the presence of synthetic PCRTags and absence of native PCRTags (fig. S3). Amplification of PCRTags was performed with GoTaq Hot Start Polymerase Master Mixes (Promega, Madison, WI) with 400 nM each of forward and reverse primers, and synX genomic DNA in a 2.5-μl final reaction volume. All PCR reagents were delivered in 384-well PCR plates with an automated dispenser (GoTaq Master Mixes were dispensed by a Cobra liquid handling system, Art Robbins Instruments; primers and genomic DNA were dispensed by an Echo 550 Liquid Handler, Labcyte). Touchdown PCR was performed as follows: 95°C for 3 min; 20 cycles of (95°C for 30 s, 64°C for 20 s, –0.3°C per cycle, 72°C for 30 s), followed by 15 cycles of 95°C for 30 s, 58°C for 20 s, 72°C for 30 s; and a final extension of 72°C for 5 min.
Yeast genomic DNA preparation for DNA sequencing; RNA isolation from yeast for RNA sequencing; nucleotide sequence analysis of synX; RNA-Seq analysis of synX
These experiments were performed as described in a previous study (8).
Dilution assay on various types of media
Yeast strains were grown for 24 hours in YPD at 30°C with rotation. Saturated cultures were diluted 10,000-fold in water, and 12 μl of diluted cultures were plated onto each type of medium (YPD, SC, and YPGE). Plates were incubated at 30°C or 37°C for 2 days (YPD and SC) or 4 days (YPGE). YPGE was prepared with 3% (v/v) glycerol and 3% ethanol. YPD, yeast extract peptone dextrose; SC, synthetic complete medium; YPGE, yeast extract peptone glycerol ethanol.
Serial dilution assay on various types of media
Yeast strains were grown overnight in YPD at 30°C with rotation. Cultures were serially diluted in 10-fold increments in water and plated onto each type of medium (YPD, SC, and YPGE). Plates were incubated at 30°C for 2 days (YPD) or 4 days (YPGE).
Cell morphology
Cells were grown to mid-log phase in YPD medium at 30°C. Bright-field images were collected with an Olympus CX41 microscope.
Pulsed-field gel electrophoresis
Full-length yeast chromosomes were prepared in agarose plugs as described previously (29). The karyotype of wild-type strain BY4741 was used as the control. A 1% gel was prepared with low–melting point agarose (Lonza, 50100). Chromosomes were separated by clamped homogeneous electric field (CHEF) gel electrophoresis by using the CHEF-DR III Pulsed Field Electrophoresis Systems (Bio-Rad) with the following settings: 6 V/cm, switch time 60 to 120 s over 24 hours, 14°C, 0.5× Tris-borate–EDTA buffer. Gels were stained with 5 μg of ethidium bromide per milliliter of buffer solution and washed three times before being imaged.
Copy-number analysis in duplication regions
Semi-synX(A-C), semi-synX(A-E), synX(A-R), and control (BY4741) strain were grown overnight at 30°C in 5 ml of YPD. Genomic DNA was prepared following the protocol “Yeast genomic DNA preparation for DNA sequencing.” DNA was eluted in 50 μl of water. Primers were designed by using the PrimeQuest Design tool on the IDT website and are listed in table S6. Primers were selected to anneal outside PCRTag sequences and, thus, amplify both synthetic and native DNA. qPCR reactions were carried out in a final volume of 4 μl by using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad; 172-5272). Each reaction included 75 nl of cDNA and 50 nl of forward/reverse primers mix (50 μM each) introduced into each well of a Hard-Shell Thin-Wall 384-Well Skirted PCR Plate (Bio-Rad; HSP- 3905) by using the Echo (LabCyte). Gene expression analysis was performed by using the CFX Software Manager (Bio-Rad). qPCR was performed on technical triplicates and data were analyzed by using two reference genes (TAF10 and UBC6).
mRNA expression analysis of ATP2 and FIP1
mRNA expression analysis was performed as described in a previous report (30). Strains were grown overnight at 30°C in 5 ml of YPD. RNA was prepared from 250 μl of culture by using the RNeasy Minikit (Qiagen; catalog no. 74106) per the manufacturer’s instructions. In brief, cells were lysed enzymatically, and eluted RNA was treated with deoxyribonuclease (DNase) (Qiagen; catalog no. 79254) in solution before passage over a second column and eluted in 30 μl of water. cDNA was prepared from ∼5 μg of RNA by using SuperScript III Reverse Transcriptase (Invitrogen; catalog no. 18080044) per the manufacturer’s instructions. Complete digestion of gDNA was confirmed in a control qPCR experiment lacking reverse transcriptase by using 100 ng of prepared RNA and two different primer pairs corresponding to the reference genes (UBC6 and TAF10). Primers were designed with the Prime-Quest Design and are listed in table S6. qPCR reactions were carried out as described above. Each reaction included 75 nl of cDNA and 50 nl of forward/reverse primers mix (50 μM each) introduced into each well of a Hard-Shell Thin-Wall 384-Well Skirted PCR Plate (Bio-Rad; HSP-3905) by using the Echo (LabCyte). Gene expression analysis was performed by using the CFX Software Manager (Bio-Rad). For the ATP2 experiment, qPCR was performed on technical triplicates, and data were analyzed by using a single reference gene (TAF10). For the FIP1 experiment, qPCR was performed on technical triplicates, and data were analyzed by using two reference genes (TAF10 and UBC6).
Integration of tR(CCU)J into HO locus
The wild-type tR(CCU)J tRNA gene, with 380 bp of upstream sequence and 100 bp of downstream sequence, was amplified with primers tR(CCU)J-HO-Bsa I (table S6). The PCR product was assembled into HO integration plasmid pSIB843 (from the Boeke lab) by using yeast Golden Gate (31). The tR(CCU)J-URA3 fragment flanked by 500 bp of homologous HO genomic sequence was integrated into the HO locus to generate synX strains from yYW0113 to yYW0115.
Replacement of residual wild-type sequence
Three residual wild-type regions were observed after genome sequencing of strain yYW0077. We subsequently replaced the wild-type sequence with their synthetic counterparts. First, we deleted the wild-type sequence by integrating URA3; second, we removed URA3 by incorporating the corresponding synthetic minichunk, using medium with 5-fluoroorotic acid (5-FOA) to screen for the loss of URA3. Three wild-type regions were replaced successively (fig. S7A).
Incorporation of the synthetic YJR092W4 into synX by cotransformation of minichunk O-5 with integration of tR(CCU)J-URA3 into HO locus
PCRTag analysis revealed that the synthetic YJR092W4 was not originally incorporated into the synX strain yYW0113, which was confirmed by whole-genome sequencing. However, we were able to replace the wild-type YJR092W4 fragment by cotransforming minichunk O-5 with tR(CCU)J-URA3 fragment, which allowed two necessary integrations with one selective marker. tR(CCU)J-URA3 with HO homology fragment, along with a 10-fold molar excess of minichunk O-5 fragment, was transformed into strain yYW0113 and plated on SC–Ura medium. By using yeast colony PCR, transformants were screened to confirm both integrations (fig. S7B).
Mating-type change
Yeast strain yYW0094, semi-237 kb-synX(A-F), was transformed with HO gene plasmid pJDE152 (from the Boeke lab) and selected on SC–Ura medium. Single colonies were restreaked on a SC–Ura plate and incubated for 2 days at 30°C. Single colonies from restreaked plates were cultured in YPD liquid medium for 24 hours at 30°C and spread on 5-FOA to select strains that had lost the plasmid. Single colonies were restreaked on YPD plates and incubated for 2 days at 30°C.The mating type of different colonies were tested by using mating-type tester yeast strains or mating-type test primers (table S6). Diploid strains that were unable to mate with either MATa Tester or MATα Tester strains were sporulated to generate four spores with 2:2 MATa to MATα.
Competitive growth assay of synX with native strain BY4741
GFP-labeled BY4741 and RFP-labeled synX strain were cocultured in 5 ml of YPD at 30°C with overnight cultures diluted to an initial absorbance at 600 nm (A600) of 0.05. Competitive growth mixtures were diluted by 1:200 in 5 ml of fresh YPD every 24 hours for 72 hours. Competitive growth assay was performed on technical triplicates. At each 24-hour time point, samples were analyzed by flow cytometry (BD Accuri C6) to count for RFP-positive and GFP-positive cells. A total of 30,000 events were counted in each sample. The gates for GFP and RFP populations were set by FlowJo (Single cell Analysis Software) auto-gate. Relative growth rate was calculated from the ratio of synX to BY4741 at each time point.
Supplementary Material
Acknowledgments
Work in Tianjin University was funded by “863” program: 2012AA02A708, “973” program: 2014CB745100, and international cooperative project: 2015DFA00960 from the Ministry of Science and Technology, and China National Natural Science Foundation of China: 21390203 and 21621004. U.S. research was funded by U.S. NSF grants MCB-1026068, MCB-1158201 to J.D.B. and MCB-1445545 to J.S.B., and U.S. Department of Energy DE-FG02097ER25308 to S.M.R.. J.D.B. and J.S.B. are founders and directors of Neochromosome Inc. J.D.B. serves as a scientific advisor to Recombinetics Inc. and Sample6, Inc. These arrangements are reviewed and managed by the committees on Conflict of interest at NYU Langone Medical Center (J.D.B.) and Johns Hopkins University (J.S.B.). U.K. research was funded by a Chancellor’s Fellowship from the University of Edinburgh, a start-up fund from Scottish Universities Life Sciences Alliance (SULSA), and Biotechnology and Biological Sciences Research Counci grants BB/M005690/1, BB/M025640/1, and BB/M00029X/1 to Y.C. We thank A. Heguy and the staff at NYU’s Genome Technology Center for outstanding deep-sequencing services. Additional information (synX design diagram, PCRTag sequences, Feature summary table (wild-type X, designed synX, physical strain yYW0115; yeast_chr10_9_01), Variants in physical strain (yeast_chr10_9_01), Minichunk plasmids, PCR primers) related to synX can be accessed on the Sc2.0 website http://syntheticyeast.org, under main menu Sc2.0→Data.
All genomic data for this paper are available under the Sc2.0 umbrella BioProject accession number PRJNA351844.
Footnotes
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Johnston M. Genome sequencing: The complete code for a eukaryotic cell. Curr Biol. 1996;6:500–503. doi: 10.1016/S0960-98220200526-2. [DOI] [PubMed] [Google Scholar]
- 2.Tong AH, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- 3.Breitkreutz A, et al. A global protein kinase and phosphatase interaction network in yeast. Science. 2010;328:1043–1046. doi: 10.1126/science.1176495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Picotti P, et al. A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis. Nature. 2013;494:266–270. doi: 10.1038/nature11835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghaemmaghami S, et al. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 6.Baker M. Synthetic genomes: The next step for the synthetic genome. Nature. 2011;473:403–408. 405–408. doi: 10.1038/473403a. [DOI] [PubMed] [Google Scholar]
- 7.Dymond JS, et al. Synthetic chromosome arms function in yeast and generate phenotypic diversity by design. Nature. 2011;477:471–476. doi: 10.1038/nature10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Annaluru N, et al. Total synthesis of a functional designer eukaryotic chromosome. Science. 2014;344:55–58. doi: 10.1126/science.1249252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galibert F, et al. Complete nucleotide sequence of Saccharomyces cerevisiae chromosome X. EMBO J. 1996;15:2031–2049. [PMC free article] [PubMed] [Google Scholar]
- 10.Richardson SM, et al. Design of a synthetic yeast genome. Science. 2017;355:1040–1044. doi: 10.1126/science.aaf4557. [DOI] [PubMed] [Google Scholar]
- 11.Yona AH, et al. tRNA genes rapidly change in evolution to meet novel translational demands. eLife. 2013;2:e01339. doi: 10.7554/eLife.01339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dymond J, Boeke J. The Saccharomyces cerevisiae SCRaMbLE system and genome minimization. Bioeng Bugs. 2012;3:168–171. doi: 10.4161/bbug.19543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen Y, et al. SCRaMbLE generates designed combinatorial stochastic diversity in synthetic chromosomes. Genome Res. 2016;26:36–49. doi: 10.1101/gr.193433.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitchell LA, et al. Synthesis, debugging, and effects of synthetic chromosome consolidation: synVI and beyond. Science. 2017;355:eaaf4831. doi: 10.1126/science.aaf4831. [DOI] [PubMed] [Google Scholar]
- 15.Falcon AA, Rios N, Aris JP. 2-micron circle plasmids do not reduce yeast life span. FEMS Microbiol Lett. 2005;250:245–251. doi: 10.1016/j.femsle.2005.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Futcher B, Reid E, Hickey DA. Maintenance of the 2 micron circle plasmid of Saccharomyces cerevisiae by sexual transmission: An example of a selfish DNA. Genetics. 1988;118:411–415. doi: 10.1093/genetics/118.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Breslow DK, et al. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat Methods. 2008;5:711–718. doi: 10.1038/nmeth.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang W, et al. Engineering the ribosomal DNA in a megabase synthetic chromosome. Science. 2017;355:eaaf3981. doi: 10.1126/science.aaf3981. [DOI] [PubMed] [Google Scholar]
- 19.Xie ZX, et al. “Perfect” designer chromosome V and behavior of a ring derivative. Science. 2017;355:eaaf4704. doi: 10.1126/science.aaf4704. [DOI] [PubMed] [Google Scholar]
- 20.Shen Y, et al. Deep functional analysis of synII, a 770-kilobase synthetic yeast chromosome. Science. 2017;355:eaaf4791. doi: 10.1126/science.aaf4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saltzgaber-Muller J, Kunapuli SP, Douglas MG. Nuclear genes coding the yeast mitochondrial adenosine triphosphatase complex. Isolation of ATP2 coding the F1-ATPase beta subunit. J Biol Chem. 1983;258:11465–11470. [PubMed] [Google Scholar]
- 22.Pan X, et al. A DNA integrity network in the yeast Saccharomyces cerevisiae. Cell. 2006;124:1069–1081. doi: 10.1016/j.cell.2005.12.036. [DOI] [PubMed] [Google Scholar]
- 23.Ben-Shitrit T, et al. Systematic identification of gene annotation errors in the widely used yeast mutation collections. Nat Methods. 2012;9:373–378. doi: 10.1038/nmeth.1890. [DOI] [PubMed] [Google Scholar]
- 24.Idrissi FZ, Piña B. Functional divergence between the half-sites of the DNA-binding sequence for the yeast transcriptional regulator Rap1p. Biochem J. 1999;341:477–482. doi: 10.1042/bj3410477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graham IR, Chambers A. Use of a selection technique to identify the diversity of binding sites for the yeast RAP1 transcription factor. Nucleic Acids Res. 1994;22:124–130. doi: 10.1093/nar/22.2.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lieb JD, Liu X, Botstein D, Brown PO. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat Genet. 2001;28:327–334. doi: 10.1038/ng569. [DOI] [PubMed] [Google Scholar]
- 27.Yarrington RM, Richardson SM, Lisa Huang CR, Boeke JD. Novel transcript truncating function of Rap1p revealed by synthetic codon-optimized Ty1 retrotransposon. Genetics. 2012;190:523–535. doi: 10.1534/genetics.111.136648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shore D, Nasmyth K. Purification and cloning of a DNA binding protein from yeast that binds to both silencer and activator elements. Cell. 1987;51:721–732. doi: 10.1016/0092-86748790095-X. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz DC, Cantor CR. Separation of yeast chromosome-sized DNAs by pulsed field gradient gel electrophoresis. Cell. 1984;37:67–75. doi: 10.1016/0092-86748490301-5. [DOI] [PubMed] [Google Scholar]
- 30.Mitchell LA, Boeke JD. Circular permutation of a synthetic eukaryotic chromosome with the telomerator. Proc Natl Acad Sci USA. 2014;111:17003–17010. doi: 10.1073/pnas.1414399111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agmon N, et al. Yeast Golden Gate (yGG) for the Efficient Assembly of S. cerevisiae Transcription Units. ACS Synth Biol. 2015;4:853–859. doi: 10.1021/sb500372z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.