

Abstract
Many cellular processes are modulated by cyclic AMP and nucleotide phosphodiesterases (PDEs) regulate this second messenger by catalyzing its breakdown. The major unique function of testicular Leydig cells is to produce testosterone in response to luteinizing hormone (LH). Treatment of Leydig cells with PDE inhibitors increase cAMP levels and the activity of its downstream effector, cAMP-dependent protein kinase (PKA), leading to a series of kinase-dependent signaling and transcription events that ultimately increase testosterone release. We have recently shown that PDE4B, PDE8A and PDE8B are highly expressed in rodent Leydig and adrenocortical cells and that combined inhibition of PDE4 and PDE8 lead to dramatically increased steroid biosynthesis. Here we investigated and the effect of PDE4 and PDE8 inhibitions on the molecular mechanisms of cAMP in a mouse MA10 Leydig cell line model with SILAC mass spectrometry-based phosphoproteomics. We treated MA10 cells either with PDE family specific PDE4 inhibitor (Rolipram) and PDE8 family specific inhibitor (PF-04957325) alone or in combination and quantified the resulting phosphorylation changes at five different time points between 0 and 180 minutes. We identified 28,336 phosphosites from 4837 proteins and observed significant regulation of 749 sites in response to PDE4 and PDE8 inhibitor treatment. Of these, 132 phosphosites were consensus PKA sites. Our data strongly suggest that PDE4 and PDE8 inhibitors synergistically regulate phosphorylation of proteins required for many different cellular processes, including cell cycle progression, lipid and glucose metabolism, transcription, endocytosis and vesicle transport. Our data suggests cAMP, PDE4 and PDE8 coordinate regulation of steroidogenesis by acting on not one rate-limiting step but rather multiple pathways. Moreover, the pools of cAMP controlled by these PDEs also coordinate many other metabolic processes that must be regulated to assure timely and sufficient testosterone secretion in response to LH.
Keywords: Proteomics, Phosphorylation, Phosphodiesterase, SILAC, Steroidogenesis, Leydig cells
Graphical Abstract
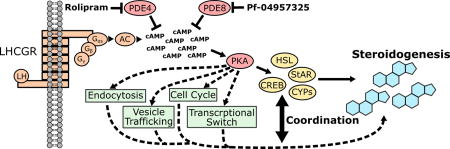
Image size: Please provide an image with a minimum of 531 × 1328 pixels (h × w) or proportionally more. The image should be readable at a size of 5 × 13 cm using a regular screen resolution of 96 dpi.
1 Introduction
Many regulatory enzymes that control cellular signaling cascades exist as multiple isoforms with similar functions. These isoforms are typically encoded by closely related families of genes, each often having alternative splice variants. This is true of enzymes regulating the cellular flux of the ubiquitous second messenger 3´,5´-cyclic adenosine monophosphate (cAMP) and its downstream effectors such as protein kinase A (PKA). The cellular concentration of cAMP is controlled in a spatiotemporal manner by the relative activities of adenylyl cyclases (ACs) and cyclic nucleotide phosphodiesterases (PDEs). In mammals there are ten different AC genes (ADCY1-10) and 21 genes encoding different isoforms of PDEs that are classified into 11 different gene families (PDEs 1-11) [1–4]. Most of the PDE genes are known to have multiple mRNA variants formed by alternative splicing or alternate transcriptional start sites.
PDEs can localize to specific subcellular compartments (microdomains), and selective ablation or deactivation of PDE isoforms can dysregulate specific cellular functions [3]. The specificity of PDE isozyme function is facilitated by interaction of PDEs with an array of scaffolding proteins, most prominently A-kinase anchoring proteins (AKAPs), that are localized to discrete cellular compartments [5, 6]. The degree of functional overlap or “cooperation” in cAMP signaling between different PDE family members is not very well understood. It is important to know how each PDE contributes to the overall functional roles of cAMP not only to understand cellular signaling mechanisms in general, but also to rationally design effective PDE inhibitors. For instance, if multiple PDEs cooperatively regulate a cellular signaling pathway, then an isoform selective inhibitor to a single PDE may not have a major effect on this pathway. Thus, to rationally design drugs with desired actions on cellular signaling networks that govern disease we first need to acquire detailed knowledge about the spatio-temporal wiring of cAMP microdomains regulated by the various PDE species [7].
We have shown previously that the cAMP-specific PDEs, 4B, 8A and 8B are highly expressed in the steroidogenic Y1 and MA10 cell lines derived from rodent adrenal and Leydig cells. Using PDE4 and PDE8 selective small molecule (SM) inhibitors and gene knockout models, we found that these PDEs regulate steroid synthesis under both basal and luteinizing hormone (LH) stimulated conditions [8]. Notably, dual inhibition of PDEs 4 and 8 caused a very large synergistic increase in steroidogenesis in the Leydig cell line that exceeds the effect of maximal LH stimulation alone. These findings lead to the hypotheses that i) signaling microdomains regulated by these PDEs cooperate to modulate steroidogenesis and ii) that dual inhibition of PDEs 4 and 8 might be a suitable means to treat pathologies caused by defects in steroidogenesis. Additionally, these findings also raise the question how many other cAMP dependent processes in the cell might be regulated in a similar manner.
The LH/choriogonadotropin (CG) receptor (LHCGR) and adrenocorticotropic hormone (ACTH) receptor are thought to couple predominantly to Gαs and thus affect cAMP/PKA signaling [9, 10]. PKA is known to directly phosphorylate and activate several transcription factors, most prominently cyclic AMP-responsive element-binding protein (Creb) [11], leading to induction of key steroidogenic enzymes, e.g. steroidogenic acute regulatory protein (StAR) and several cytochrome P450 (CYP) family members necessary for steroid hormone biosynthesis. PKA also phosphorylates and likely activates StAR and stimulates cholesterol ester hydrolysis by phosphorylation of hormone sensitive lipase (HSL) [12, 13].
Despite an emerging recognition that multiple pathways cooperate to regulate LH/ACTH dependent steroidogenesis [12, 14], the molecular mechanisms underlying the synergy between PDE4 and PDE8 inhibitors as activators of steroidogenesis remain unknown. Because we were particularly interested in how PDE4 and PDE8 inhibition synergizes to facilitate efficient steroid biosynthesis, we used PDE-isoform selective drugs to knock-down PDE4 (Rolipram) and PDE8 (PF-04957325) activity in the MA10 mouse Leydig cell model [15]. We profiled global phosphorylation changes in response to PDE inhibition with unbiased phosphoproteomics methods [16, 17]. Triple encoded SILAC labeling [18] of MA10 cell populations allowed us to accurately quantify 16,732 phosphorylation sites and of these, 749 were significantly regulated by PDE inhibition. These data shed light on the molecular pathways likely to be affected by the dramatic synergism between PDE4 and PDE8 in steroidogenesis and confirm the importance of PDE8 isoforms in the basal, non LH-stimulated state. More importantly, the approach strongly suggests cAMP/PDE regulation of a wide array of signaling pathways not traditionally associated with gonadotropin/cAMP signaling in steroidogenic cells. Tantalizingly, PDEs 4B, 8A, and 8B appear to cooperate in different functional microdomains to regulate multiple pathways that ultimately facilitate steroidogenesis and coordinate steroid production with other cAMP dependent processes in the cells.
2 Materials and Methods
2.1 Materials
Mouse MA10 Leydig tumor cells were a kind gift of Dr. Mario Ascoli (University of Iowa). Rolipram was purchased from Tocris Bioscience (Bristol, UK). PF-04957325 was a kind gift of Pfizer Inc. (Groton, CT). Isotope labeled amino acids for SILAC were obtained from Cambridge Isotope Labs (Andover, MA) or Sigma-Isotec (St Louis, MO). Custom RPMI medium (−Lys/−Arg) was obtained from Caisson Labs (North Logan, UT). Dialyzed FBS and PHOS-Select Iron affinity gel were from Sigma (St. Louis, MO). C18 reverse phase material for nano-LC columns (3 µm Reprosil-C18.aq) was obtained from Dr. Maisch (Ammerbuch, DE). Protease and phosphatase inhibitor cocktails as well as the Pierce 660 nm protein assay were purchased from Thermo Scientific (Rockford, IL). Sequencing grade endoproteinase LysC and Trypsin were obtained from Promega (Madison, WI). Oasis C18 cartridges for peptide extraction were from Waters (Milford, MA).
2.2 Cell culture, harvest and lysate preparation
For SILAC-labeling MA10 cells were cultured in custom RPMI medium supplemented with 15% dialyzed fetal bovine serum (dFBS), 100 units/ml Penicillin, 100 µg/ml Streptomycin, 292 µg/ml glutamine, 20 µg/ml proline and the corresponding isotope labelled Lys (0.11 mM) and Arg (0.57 mM) (light label = Lys0/Arg0, medium label = Lys4/Arg6, heavy label = Lys8/Arg10). Cells were split every two to three days and grown for seven cell doublings on 15 cm cell culture dishes at 37°C and 5% CO2 to achieve complete incorporation of isotope-labeled Lys/Arg. Before drug treatment, the growth medium was aspirated, cells washed twice with warm, sterile PBS (25 ml), and serum-free SILAC growth medium was added (same as SILAC growth medium but without dFBS, 25 ml). After a 3 h starvation period, cells were treated with either inhibitor or DMSO control (0.1 % DMSO in cell culture medium final) and incubated for the time indicated. The medium was then aspirated and the cells washed twice quickly with 25 ml of ice cold PBS. 750 ul of freshly prepared Tris-buffered 8 M urea, pH 7.8, containing protease and phosphatase inhibitor cocktail was added to each 15 cm culture dish of MA10 cells while being kept on ice. Cells were harvested with a cell scraper and sonicated for 20 × 30 sec. (90 sec intermediate pause, 40 min. total) at 75 W in an ice-water bath and then clarified by centrifugation at 4°C and 21,000 rcf for 20 min. The protein content of each sample was determined with the Pierce 660 nm protein quantification assay.
2.3 Preparation of peptide samples
Individual SILAC-labelled MA10 lysate samples (light, medium or heavy) were combined according to the experimental design with a protein ratio of 1:1:1. For reduction of proteins and capping of cysteines, the lysate was treated sequentially with tris(2-carboxyethyl)phosphine (TCEP, 1 mM final), chloroacetamide (CAM, 2 mM final) and again TCEP (1 mM final) each for 20 min at 37°C with gentle agitation. The lysate was then diluted 2× with 100 mM aq. tetraethylammonium bicarbonate solution (TEAB; urea concentration ca. 4 M final) and the pH was adjusted to 8.5 with 1 N aq. NaOH solution. Then endoproteinase LysC was added (1:50 enzyme:substrate calculated on protein content) and the mixture incubated at 37°C with gentle agitation for 2h. The mixture was then further diluted 2× with 100 mM TEAB (urea concentration ca. 2 M final) and trypsin (1:50) was added. The mixture was then incubated for 16 h at 37°C with gentle agitation. The crude peptide mixture was diluted 2× with 5% aq. acetonitrile (ACN) containing 0.1% trifluoroacetic acid (TFA) and the pH adjusted to 2 by adding formic acid (FA, 1% final). The mixture was then clarified by centrifugation at 10,000 rcf for 10 min. and peptides were extracted from the supernatant using Oasis C18 cartridges. Bound peptides were washed with Solvent A (0.1% TFA in water), eluted using 80% aq. ACN containing 0.1 % TFA (Solvent B) and dried in a vacuum concentrator. For strong cation exchange (SCX) chromatography, peptides were re-suspended in SCX buffer A and fractionated on a Polysulfoethyl A column (200 × 9.4 mm, 5 uM, 200 Å) from PolyLC (Columbia, MD) using a Dionex UltiMate 3000 analytical UHPLC system (Sunnyvale, CA) according to the previously published protocol[19]. 24 fractions were collected, concentrated in vacuum to remove excess ACN and combined into 12 fractions according to the published protocol. 2% of each fraction was then sampled for desalting on StageTip [20] and background proteome analysis and the remaining 98% subjected to peptide extraction using Oasis C18 cartridges. Peptides were then eluted with Solvent B, dried in vacuum and phosphopeptides were enriched using the published Fe-IMAC in solution protocol [19]. using the PHOS-Select iron affinity gel. Recovered phosphopeptides were desalted on StageTip and stored at −20°C until analyzed.
2.4 LC-MS/MS and data analysis
Peptides were separated on a Thermo-Dionex RSLCNano UHPLC instrument (Sunnyvale, CA) with 10 cm long fused silica capillary columns made in-house with a laser puller (Sutter, Novato CA) and packed with 3 micron reversed phase C18 beads. The LC gradient was 90 min long with 3–35% B at 200 nL/min. LC solvent A was 0.1% acetic acid and LC solvent B was 0.1% acetic acid, 99.9% acetonitrile. MS data was collected with a Thermo Orbitrap Elite. Data-dependent analysis was applied using Top15 selection with CID fragmentation. Raw files were analyzed by MaxQuant version 1.5.2.8 using protein, peptide and site FDRs of 0.01 and a score minimum of 40 for modified peptides, 0 for unmodified peptides; delta score minimum of 17 for modified peptides, 0 for unmodified peptides. MS/MS spectra were searched against the UniProt mouse database (updated July 22nd, 2015). MaxQuant search parameters: Variable modifications included Oxidation (M), phosphorylation (STY). Carbamidomethyl (C) was a fixed modification. Max. labeled amino acids was 3, max. missed cleavages was 2, enzyme was Trypsin/P, max charge was 7, multiplicity was 3, SILAC labels were Arg0/Lys0 (light), Arg6/Lys4 (medium), Arg10/Lys8 (heavy). The initial search tolerance for FTMS scans was 20 ppm and 0.5 Da for ITMS MS/MS scans. Data was further processed using the Perseus software package (version 1.5.2.6), the R environment, Origin Pro 8.0 and Microsoft Excel.
Time series data was clustered using the Mfuzz package for R. Missing values allowed were 0, minimum standard deviation 0.2 (corresponding to an abs. log2 SILAC ration of ≥0.2), number of clusters was restricted to 6 and the fuzzifier factor, m, was set to 2.559. Upstream kinase candidates responsible for phosphorylation of a given phosphosite and protein-protein interactions were predicted with the NetPhorest web application (http://www.netphorest.info/) using mouse FASTA sequences and the cognate human kinase substrate consensus site information used by NetPhorest [21]. A minimum NetPhorest Score of 0.1 was required for consideration as a putative upstream kinase/interaction. Sequence logos for phosphosites regulated in response to stimulation were produced using IceLogo (version 1.2) [22]. GO term enrichment analysis was performed using the GOrilla web application (http://cbl-gorilla.cs.technion.ac.il/) [23]. Further, all regulated phosphosites were searched against the Phosphosite Plus database (http://www.phosphosite.org/) and all phosphosites reported in the following are annotated according to the major mouse protein isoform listed in the Phosphosite Plus database except in the case where a phosphorylation site is isoform specific.
3 Results and discussion
3.1 Experimental setup and workflow
In this study, we examined changes in the global phosphoproteome in response to PDE inhibition in the steroidogenic mouse Leydig MA10 cell model. We wanted to investigate i) how inhibition of PDE4 and PDE8 enzymes affects cellular cAMP signaling compartments and ii) how combined PDE4 and 8 inhibition can lead to the previously observed, synergistic effects on cellular signaling and steroidogenesis [8]. We expected that treatment of MA10 cells with PDE inhibitors would lead to a significant activation of PKA and downstream signaling events. We applied stable isotope labeling by amino acids in cell culture (SILAC)-based quantitative MS [24] and phosphoproteomics to quantify changes in signaling events downstream of PDE inhibition. PDE4 and 8 inhibition is thought to mimic the cAMP/PKA component of LH signaling. To ensure that PDE4 and 8 inhibition does not lead to a “flooding” of the cells with cAMP and a non-physiological hyper activation of cellular signaling we measured cAMP levels in MA10 cells following inhibitor treatment. We found a 1.8-fold increase of cellular cAMP after 10 min of combined PDE4 and 8 inhibitor treatment (1.8 ng/mg protein). This was lower than cAMP levels typically achieved using 10 ng/ml LH as the stimulus, leading to the conclusion that hyper-activation of the cells did not occur (SI Figure 1).
We used two experimental sets of triple SILAC labeled MA10 cells (Figure 1A) to compare firstly, DMSO control, PDE4 inhibition (10 mM Rolipram), and combined (10 mM Rolipram/200 nM PF-04957325) treatment, and secondly, a DMSO control, PDE8 inhibition (200 nM PF-04957325), and combined PDE inhibitor treatment for 1 h. The overlapping experimental sets allowed us to directly interrogate and quantify differences in combination treatment vs. individual phosphodiesterase family inhibition, and compare the effects of PDE4 vs. PDE8 inhibition.
Figure 1.

A) Experimental setup for the triple SILAC labeling experiments in MA10 cells comparing single PDE4 (Rolipram, Rol) or PDE8 (PF-04957325, PF) inhibitor treatment with PDE4 and 8 inhibitor combination treatment for 1 h and the setup of the time series experiment with PDE4 and 8 inhibitor treatment for 0, 1, 15, 60 and 180 minutes. B) Phosphoproteomics workflow according to which MA10 cell samples were processed and analyzed.
Another major objective of this study was to examine if MA10 cell treatment with a PDE4 and 8 inhibitor combination mimicked the signaling dynamics of tropic hormone signaling (i.e. LH in MA10 cells). In order to determine the dynamic changes in phosphorylation in response to PDE inhibition, we applied time-series analyses to compare the status of the phosphoproteome at 0, 1, 15, 60, and 180 minutes after addition of PDE inhibitors. To achieve robust quantification across all five time points of the experiment we joined two triple label SILAC experiments by means of a common control time point. We chose the 15 min. time point as the control time point and set up paired experiments comparing control (ctrl, 15 min), 1 min., 180 min., and ctrl (15 min), 0 min., 60 min (Figure 1A). Figure 1B illustrates the workflow of sample processing (see also 2.3 and 2.4).
3.2 Summary of the dataset
We analyzed a total of eight triple SILAC PDE4 and PDE8 inhibition experiments; 12 sample fractions were collected from each experiment and analyzed in single nanoLC-MS/MS runs resulting in a total of 96 analytical runs for phosphopeptide–enriched samples and 72 runs for proteome background samples.
Analysis of background samples identified 8234 proteins expressed in MA10 cells and 5269 of these proteins could be quantified in at least one experiment. For further analysis of the MA10 proteome we ranked identified proteins according to the MaxQuant intensity calculated for each protein (as an approximate measure for cellular abundance) and then analyzed each intensity quartile for enrichment of Gene Ontology (GO) terms (biological process (BP), molecular function (MF) and cellular component (CC). This GO-term analysis revealed an enrichment of terms related to lipid metabolism, oxidoreductase activity as well as vesicular and mitochondrial membrane proteins in the top intensity quartile (Figure 2a). This is consistent with a cell line functionally specialized to produce steroid hormones. On the lower end of protein abundance we find an enrichment of proteins that regulate transcription as well as integral membrane proteins/receptors. A complete list of proteins identified in the MA10 cell proteome is given in SI-Excel file 1. Notably, no proteins were observed to change in abundance beyond log2 SILAC ratio of 0.5 in any of our experiments; we conclude, therefore, that de-novo protein synthesis and degradation play at most only a minor role in our experiments. Background analysis with 2% of the peptide did not achieve comparable quantification for proteins at every given time point of the experiment as compared to our phosphoproteomic analysis (peptide level). Thus, we found that normalization of phosphopeptide ratios to the corresponding protein ratio in the time series experiment led to a large number of missing values. Since we observed minimal changes in protein abundance (< 1.4 fold) in the proteome, we used non-normalized phosphopeptide ratios in our analyses.
Figure 2.

A) Plot of the log10 protein intensity and the protein intensity rank (n = 8234). Abundance of a protein was estimated to increase in parallel with its intensity. The proteins were divided into quartiles of intensity and a GO term enrichment was conducted for each of the quartiles using the GOrilla web application. Shown are examples for enriched terms for GOBP, GOMF and GOCC. B) Venn diagram comparing the proteins identified in the background proteome and the phosphoproteome. C) Venn diagram comparing the proteins quantified in the background proteome and the phosphoproteome. D) Comparison of the number of identified phosphosites, quantified phosphosites and regulated phosphosites in the dataset. D) Plot of all log2 SILAC ratios in the phosphoproteomics dataset. 90% of unique phospho sites showed a log2 SILAC ratio within a range of >0.6±0.17 and <0.53±0.07 (mean±SD). Phosphosites showing log2 SILAC ratios outside this data interval were considered potentially regulated by PDE inhibitor stimulation.
Analysis of phosphopeptide-enriched samples led to the identification of 28336 unique phosphorylation sites on 14610 different peptide species derived from 4837 proteins. 93% of phosphorylated proteins were also identified in the background proteome (Figure 2B) and 63% of quantified phosphorylated proteins were also quantified in the background proteome in at least one experiment (Figure 2C). Hence, although the overlap in quantification between background proteins and corresponding phosphopeptides at various time points is low we still get a good estimate of protein abundance changes across the experiment. Further, 16732 of the phosphorylation sites could be quantified in at least one experiment and 4470 in more than 50% of the experiments (Figure 2D). Plotting the log2 SILAC ratios of the entire phosphoproteomic dataset revealed a narrow distribution of ratios with 90% of the data residing in a range of −0.60±0.17 to +0.53±0.07 (see Figure 2E). The most differential SILAC ratios occurred in PDE4 and 8 combination inhibition experiments, reaching a magnitude of up to ±4.7 on the log2 scale (see SI Table 1). Based on the narrow ratio distribution, we defined a cut-off of ±0.75 on the log2 scale as potentially “regulated” for further pathway analyses. To further distinguish true hits from false positives we only considered phosphosites that replicated at least twice (absolute log2 ratio ≥0.75) and with ratio changes consistent with our SILAC label swap experimental design. As other investigators before us, we further classified identified phosphosites by their localization probability on a certain S/T/Y residue in a peptide (Class I: >0.75; Class II: <0.75 and >0.5; Class III: <0.5) [16, 25]. Inspection of the phosphopeptide data shows that 67.7% were class I sites, 14.9% class II and 16.5% class III sites, indicating that the majority of sites were localized to a certain amino acid residue with high confidence. The following discussion considers only ratios found for singly phosphorylated peptides in our dataset.
Applying the aforementioned filters to our phosphoproteomics dataset, we found that 749 (2.7%) of the sites were reproducibly regulated by PDE inhibitor treatment. These sites were derived from 484 gene products and of these, 305 could be quantified in the background proteome. Consistent with our observation of synergy in steroid production, we found all of the sites modulated with the highest fold-change in the combined PDE4 and 8 inhibition experiment. PDE8 inhibition alone led to a robust regulation of 54 phosphorylation sites whereas PDE4 inhibition caused only minor effects with none of the phospho-regulated protein exceeding the threshold of a 0.75 SILAC log2 ratio. This confirms the notion that PDE8A and/or B play an important role in Leydig cell biology under basal conditions whereas PDE4 enzymes likely play a more important role only at higher cellular concentrations of cAMP [8].
3.3 Phosphoproteome dynamics in response to PDE inhibition
Clustering of time series data is a powerful means to identify regulatory patterns in biological processes and signaling pathways. We used fuzzy c-means clustering with Mfuzz [26] to cluster phosphorylation data obtained from a combined PDE4 and PDE8 inhibitor treatment of MA10 cells for 0, 1, 15, 60 and 180 minutes. We find two general temporal patterns (Figure 3 A), that are the bulk up-regulation (cluster 2, 3, 4) and down-regulation (cluster 1, 5, 6) of phosphorylation sites reaching a maximum fold-change at 1 h of inhibitor treatment. Closer inspection of the clusters 4, 5 and 6 revealed that many of the induced changes in phosphorylation are reversible over the time course of the experiment, returning to basal levels after 180 min. Clusters 1, 2 and 3 comprise sites with a continuous change in phosphorylation, possibly plateauing after 1 h. It is known that one major mechanism of feedback control of cAMP in many tissues is activation of PDEs, an effect that we do not expect to observe in an experiment using PDE inhibitors. Nevertheless, from these time course patterns we conclude that besides adaptive, long-term phosphorylation changes, PDE inhibition is able to trigger rapid phosphorylation changes that more closely resemble hormone/growth factor-initiated signaling events. Surprisingly, the data shows that the vast majority of sites (459 out of 749) were down-regulated significantly, suggesting either global phosphatase activation following PDE4 and PDE8 inhibitor treatment or inhibition of one or more non-PKA kinases responsible for phosphorylating these sites.
Figure 3.

A) Temporal clustering of phosphorylation changes quantified in the phosphoproteomics PDE4 and 8 inhibition time series experiment. Clustering was performed using the Mfuzz package in R with six clusters, only considering sites that are quantified in all experiments and that have a minimum abs log2 SILAC ratio of 0.2. B) and C) IceLogos created from the ±7 aa sequence window of significantly up- or down-regulated phosphorylation sites and pie charts of predicted upstream kinases targeting these phosphorylation sites. Sites were considered up- or down-regulated if increased or decreased in the 1 h PDE inhibition experiment or if they could be clustered to the corresponding temporal clusters (upregulated = cluster 2, 3, 4 and down-regulated = cluster 1, 5, 6) for sites derived from the time series experiment. Sequence logos were created using IceLogo 1.2 with the entire Mus musculus genome as background data set. Upstream kinases were predicted with the NetPhorest web tool (http://www.netphorest.info/index.shtml) using mouse protein sequences and human kinase substrate consensus sequences. Only predicted kinases were considered that showed a NetPhorest score of >0.1.
To confirm the involvement of PKA and obtain insights into what other kinase signaling pathways might be activated in response to PDE4+8 inhibition, we generated sequence logos of the surrounding 7 amino acids (aa) N- and C-terminal to the phosphorylated S/T/Y residue for up- and down-regulated phosphorylation sites using the IceLogo software (Figure 3 B and C) [22]. As expected, we found a strong enrichment of arginine (R) at positions −3/−2 and serine (S) at position 0 which corresponds to the X-(R/K)-(R/K)-X-S consensus substrate sequence of PKA. This clearly indicates the involvement of PKA/basophilic protein kinase activity for up-regulated sites (n = 290). Down-regulated sites (n = 459) showed a strong enrichment of proline (P) at position +1 and a less pronounced enrichment of P at other positions throughout the ±7 aa sequence window. This indicates that proline directed protein kinase sites might be a major target for phosphatase activity or cAMP inhibitory actions on P-directed kinases. Next, we used the NetPhorest algorithm [21] to predict putative upstream kinases as well as protein-protein interaction motifs for the 749 regulated sites that could yield mechanistic clues of protein regulation. Among up-regulated sites, 44.4% were predicted to be PKA sites, thus confirming the important role of the PDEs 4 and 8 in MA10 cAMP/PKA signaling (Figure 3 B). Further, putative CDK, Cdc-like kinase (CLK), casein kinase (CK) and protein kinase C (PKC) target sites constitute a major fraction of upregulated sites. Interestingly, we found that almost 50% of the down-regulated phosphosites showed a high probability of being cyclin-dependent kinase (CDK) target sites suggesting cell cycle regulation; 17% of down-regulated sites were putative MAPK targets, with other kinase target sites seemingly playing a minor role (Figure 3 C). Collectively, these data closely match the phosphorylated sequence motif analysis by IceLogo (Figure 3 B and C). Nonetheless, at this point it should be mentioned that differences in the NetPhorest score for related kinase family members, e.g. proline directed kinases like CDKs and MAPKs or basophilic kinases like PKA, PKB and PKC are often subtle; suggesting that our NetPhorest analysis may not be able to discriminate between related kinase family members in certain cases.
We next sought to gain insights into the biological processes regulated by PDE inhibition. We used Gene Ontology (GO) term enrichment analysis of biological processes (GOBP) with the GOrilla web application [23] to identify enriched GO terms for significantly phospho-regulated genes against the background of all phosphorylated but non-regulated genes. Table 1 shows examples from the list of GOBP terms found to be enriched in our dataset (p<0.01, for a full list of enriched terms see SI Excel 1). We found a large number of identifiable biological processes specifically associated with increased or decreased phosphorylation. Genes bearing up-regulated phosphosites were found to be involved in small GTPase signaling (Ras, Rho, Rac, Rab, etc.), including cell migration, vesicle transport and receptor mediated endocytosis. Tantalizingly, this suggests that PKA activity may coordinate the regulation of a broad range of processes, including endocytosis and transport of cholesterol (e.g. Ldlrap1, Gpr107 etc.; Table 1) to facilitate efficient steroidogenesis. Many of the processes specifically enriched among down-regulated sites can be associated with cytoskeletal dynamics including cell cycle-related processes, especially cytokinesis. This is in good agreement with the fact that 49.4% of downregulated phosphosites are predicted by NetPhorest to be CDK target sites. It has been shown before that ACTH treatment of mouse Y1 adrenocortical carcinoma cells can cause cell cycle arrest in G1 [27]. This leads us to speculate that PKA-triggered phosphatase activity (or decreased kinase activity) might induce ordered cell cycle arrest by specific mechanisms that are coordinated with other cellular processes to facilitate efficient steroidogenesis.
Table 1.
Examples from the GO term enrichment analysis (Biological Process, GOPB) for gene products found to be phosphoregulated in response to PDE4 and 8 inhibition. The analysis revealed that specific biological processes can be associated with proteins showing decreased or increased phosphorylation. Enrichment analysis was conducted with the GOrilla web application (http://cbl-gorilla.cs.technion.ac.il/) using all non-regulated, phosphorylated proteins as the background. p-value <0.01.
| Proteins Containing Upregulated Phosphosites (n = 236) | ||||
| GO Biological Process | P-Value | Enrichment | Count | Genes |
| regulation of small GTPase mediated signal transduction | 1.00E-05 | 3.65 | 15 | Als2, Arfgef2, Arhgef19, Arhgef2, Arhgef6, Arpp19, Camk2d, Dennd1a, Foxm1, Grlf1, Itpkb, Nf1, Plekhg3, Ralgapa1, Ralgps2 |
| regulation of cell migration | 2.45E-04 | 2.51 | 18 | Apc, Camk2d, Egfr, Gab1, Hdac7, Hspb1, Macf1, Mtus1, Nf1, Phldb2, Prkd2, Ptk2b, Rapgef2, Rreb1, Rtn4, Rufy3, Syne2, Vim |
| vesicle-mediated transport | 4.04E-04 | 1.93 | 29 | Arfgap1, Arfgef2, Arhgap17, Bet1, Ctnnb1, Dennd1a, Fam160a2, Fcho2, Flna, Fnbp1l, Golga5, Gpr107, Ldlrap1, Lrp4, Mapk1, Mapk3, Reps1, Rin2, Sec22b, Sgsm3, Snx1, Snx17, Snx2, Snx3, Spast, Stxbp5, Tbc1d10b, Ube2o, Usp20 |
| receptor-mediated endocytosis | 4.30E-04 | 4.25 | 8 | Fcho2, Gpr107, Ldlrap1, Mapk1, Mapk3, Reps1, Snx1, Snx17 |
| Proteins Containing Downregulated Phosphosites (n = 307) | ||||
| GO Biological Process | P-Value | Enrichment | Count | Genes |
| cytoskeleton organization | 2.82E-08 | 2.35 | 45 | Abl1, Ablim1, Ablim2, Agfg1, Arhgef17, Arhgef2, Atxn3, Bcl6, Camsap2, Clasp2, Dock1, Dock7, Dst, Enah, Epb4.1l1, Flna, Htt, Kif20a, Larp4, Map1b, Map2, Map4, Mapt, Nav1, Nde1, Numa1, Palm, Pdlim4, Pex14, Phactr4, Pkp2, Racgap1, Ranbp10, Rock2, Sgol1, Slain2, Slc9a3r1, Son, son, Sorbs1, Specc1l, Synpo, Tacc3, Tpx2, Trpm7, Vim |
| mitotic cytokinesis | 1.76E-04 | 6.39 | 6 | Anln, Daxx, Kif20a, Kif4, Racgap1, Sptbn1 |
| mitotic cell cycle process | 4.13E-04 | 1.85 | 32 | Ankle2, Anln, Arhgef2, Cdca3, Clasp2, Clspn, Daxx, Dis3l2, Dync1li1, E2f4, Flna, Gigyf2, Htt, Incenp, Kif20a, Kif4, Map4, Ncapd2, Ncaph, Nde1, Numa1, Pds5b, Ppp1r12a, Racgap1, Rps6kb1, Sgol1, Sptbn1, Top2a, Tpx2, Triobp, Ttk, Vcpip1 |
3.4 PDE4 and 8 regulation of cell cycle proteins
The high number of CDK target sites down-regulated in response to PDE4+8 inhibitor treatment, and the enrichment of cell cycle-related GO terms among phospho-regulated proteins prompted us to further investigate the extent of cell cycle regulation in response to PDE inhibition. Inspection of all phospho-regulated genes for the appearance of the terms “cell cycle”, “mitotic” and “cytokinesis” among the GOBP terms yielded 100 genes regulated on 152 phosphorylation sites. While only a small portion of these sites have been previously characterized, published findings suggest and support the notion of cell cycle regulation by PDE4 and 8 inhibition (Table 1). Strikingly, PKA and downstream kinases/phosphatases seem to specifically lead to the negative regulation of gene function involved in driving mitotic cell cycle progression (Table 2).
Table 2.
Biological processes highly regulated and corresponding genes that were found to be phospho-regulated on biologically characterized sites following MA10 cell treatment with PDE4 and 8 inhibitors. Arrows symbolize increased phosphorylation (↑) or decreased phosphorylation (↓) of a site.
| Process | Gene | Site | Reg. | Clustera) | Functionb) | Ref. |
|---|---|---|---|---|---|---|
| Cell Cycle | Arhgef2 | S885 | ↑ | 2 | interaction, inhibition | [28] |
| Clasp | S1004 | ↓ | 1 | localization, inhibition | [30] | |
| S1005 | ↓ | 1 | localization, inhibition | |||
| Flna | S2152 | ↑ | 2 | interaction | [42] | |
| Kif20a | T855 | ↓ | NA | localization, inhibition | [31] | |
| Ldlrap1 | S14 | ↓ | 5 | inhibition | [36] | |
| Mybl2 | T519 | ↓ | NA | inhibition | [40] | |
| Npm1 | T232 | ↓ | 6 | localization, inhibition | [37] | |
| T235 | ↓ | 6 | localization, inhibition | |||
| Ttk | T277 | ↓ | NA | localization, inhibition | [34, 35] | |
| T820 | ↓ | 6 | localization, inhibition | |||
| Vcip135 | S746 | ↓ | 1 | interaction, inhibition | [38] | |
| MAPK Signaling | Araf | S255 | ↓ | NA | inhibition | [47] |
| Cdc25b | S351 | ↑ | 3 | activation | [85] | |
| Egfr | T695 | ↑ | 2 | endocytosis, inhibition | [86] | |
| Gab1 | S552 | ↑ | 2 | activation | [53] | |
| Grb14 | S368 | ↑ | NA | inhibition | [56, 57] | |
| S370 | ↑ | 3 | inhibition | |||
| Ksr1 | S320 | ↓ | 5 | activation | [51, 52] | |
| T270 | ↓ | NA | activation | |||
| Map3k2 | S153 | ↑ | 2 | interaction | [87] | |
| S331 | ↑ | 2 | interaction | |||
| Map3k7 | S412 | ↑ | 2 | interaction | [88] | |
| Mapk1 | T183 | ↑ | 3 | activation | ||
| Y185 | ↑ | 2 | activation | |||
| Mapk3 | Y205 | ↑ | 2 | activation | ||
| Mapk14 | T180 | ↑ | 4 | activation | ||
| Y182 | ↑ | NA | activation | |||
| Raf1 | S43 | ↑ | NA | interaction | [48] | |
| Rps6kb1 | S447 | ↓ | 5 | inhibition | [54, 89] | |
| S452 | ↓ | 1 | inhibition | |||
| Regulation Transcription/Metbolism | Amotl2 | S752 | ↑ | NA | activation | [90] |
| Arhgap35 | S1150 | ↑ | NA | inhibition | [91] | |
| Crtc3 | S62 | ↓ | 1 | activation | [64] | |
| S329 | ↓ | 1 | activation | |||
| S370 | ↓ | 1 | activation | |||
| Ctnnb1 | S552 | ↑ | NA | activation | [70, 71] | |
| Esr1 | S298 | ↓ | NA | activation | [92] | |
| Itpr1 | S1588 | ↑ | NA | activation | [65] | |
| Lrrfip2 | S111 | ↑ | 2 | interaction | [73] | |
| Mlxipl | S196 | ↑ | 2 | inhibition | [66] | |
| S626 | ↑ | 2 | inhibition | |||
| Ptrf | S169 | ↑ | NA | NA | [93] | |
| S302 | ↑ | 2 | NA | |||
| T304 | ↓ | 6 | lipolysis, inhibition | |||
| Sik3 | S551 | ↑ | 2 | activation | [63] | |
| S674 | ↑ | NA | activation | |||
| Smad2 | T8 | ↓ | 1 | inhibition | [94] | |
| Yap1 | S46 | ↑ | 1 | inhibition | [68] | |
| S352 | ↓ | na | inhibition | [69] |
Affiliation of a site to a temporal cluster (see Figure 3A).
Functional role of the phosphorylation event in the context of the biological process discussed.
We observed, for example, highly up-regulated (~4-fold) phosphorylation of Arhgef2 (GEF-H1) on the known PKA target site pS885 upon PDE4 and 8 inhibition that could be validated by western blot analysis (SI Figure 2). This site is known to regulate 14-3-3 binding and sequestration of the Arhgef2 guanosine nucleotide exchange factor activity [28]. Arhgef2 specifically activates transforming protein RhoA during mitosis in which it acts as a key factor in formation of the cleavage furrow and successful cell division [29]. The negative regulation of RhoA function would thus be consistent with negative regulation of cell cycle progression.
Cap-Gly domain-containing linker protein(CLIP) associating protein 2 (Clasp2) stabilizes dynamic microtubules at the leading edge of migrating cells as well as at the kinetochore where its function is crucial for proper alignment of the mitotic spindle. We also found the important regulatory pS1005 to be significantly down-regulated in response to PDE4+8 inhibition; it has previously been shown that CDK-mediated phosphorylation of this site leads to recruitment of polo-like kinase 1 (PLK1) to the kinetochore and that this process is necessary for mitotic spindle assembly [30].
The motor protein kinesin-like protein 20A (Kif20A) is phosphorylated at pT855 by CDK to inhibit its activity and allow progression of early mitosis [31]. Our observation of significant down-regulation of this site therefore suggests a block of the cell cycle.
The dual specificity protein kinase Ttk is required for passage of the mitotic checkpoint and regulates the localization and activity of various cell cycle proteins at the mitotic spindle [32, 33]. We found two phosphorylated residues (pS820 and pS277) significantly down-regulated. These sites are proposed to be MAPK and Braf target sites, respectively. A Ttk S820A mutant has been previously shown to disrupt kinetochore localization of the kinase, thus suggesting that PDE4 and 8 inhibition abrogates mitotic Ttk function [34]. Further, phosphorylation at S277 has been suggested to regulate the ubiquitination-dependent degradation of Ttk leading to the conclusion that cAMP signaling might destabilize the kinase [35].
Further, Ldlrap1, first characterized as a clathrin-binding adaptor molecule regulating the endocytosis of low density lipoprotein (LDL) receptor also has been suggested to play a role in mitosis and cellular senescence. Thus, the putative CDK1 target site pS14 is up-regulated in mitosis and its deletion or mutation leads to defective mitotic spindle formation suggesting an important role for Ldlrap1 in the mitotic cell cycle [36]. We find pS14 robustly down-regulated again supporting the idea of cAMP-induced cell cycle arrest. Additionally, two uncharacterized sites pS194 and pS198 on Ldlrap1 were strongly up-regulated. S198 was predicted to be a PKA target site (NetPhorest score 0.32) with a high likelihood of being a 14-3-3 binding site (score 0.52; Table 2). It will be interesting to explore if regulation of this site has an impact on Ldlrap1 function in Ldl uptake and/or mitosis in this steroidogenic cell type.
Nucleophosmin (NMP1) is another example of a mitotic regulator that translocates to the centrosome and midbody during mitosis in response to CDK phosphorylation. We found the regulatory site pT232 on NMP1 to be significantly down-regulated, possibly preventing its translocation (Table 2) [37].
During mitosis, the Golgi apparatus is disassembled and after successful cytokinesis reassembled to achieve functional integrity of the daughter cells. One of the regulators of Golgi assembly/reassembly during the cell cycle is the deubiquinating protein Vcip135 (Vcpip1). Phosphorylation of Vcpip1 at pT760 by CDK1 inhibits Golgi membrane fusion, a process which is reported to be a prerequisite for entry into mitosis. We find this site to have reduced phosphorylation after PDE inhibition [38, 39].
Besides regulation of the aforementioned proteins that directly modulate mitotic cell cycle processes, we also observed the inactivation of Myb-related protein 2 (Mybl2, phosphorylation decreased at 15 min; Table 2), a transcription factor that can drive the expression of cell cycle genes [40]. It has been shown previously that Mybl2 in complex with clathrin and filamin critically regulates mitotic spindle formation [41]. Interestingly, we also found an increased phosphorylation on Filamin A (FLNA) on pS2193, an important site linked to regulation of spindle formation [42]. The observation of differential regulation of phosphorylation on transcription factors and downstream cell cycle control genes further suggests an altered balance for cell cycle arrest in favor of steroid production in MA10 cells upon PDE inhibition.
We were able to observe 78% of all mouse genes reported to be involved in cell cycle regulation (by GOBP annotation) and 8% of these genes showed significant phospho-regulation, either by down-regulation of CDK target sites or increased phosphorylation by PKA (Figure 5 A). Interestingly, the majority of these regulated genes are functionally associated with mitosis/cytokinesis (M-phase), participating in processes like chromosome segregation, spindle formation and movement and cell division (Figure 5 A). By analyzing available data on the biological roles for some of the regulated phosphosites, we suggest that PDE4 and 8 inhibition and the resulting increase in cAMP would delay cell cycle progression in MA10 cells. We speculate that this “halt” signal for the mitotic cell cycle is coordinately timed with steroidogenesis and serves as a switch from cellular growth and proliferation to a state optimized for increased steroidogenic lipid metabolism. As both processes demand considerable cellular resources (ATP, lipids etc.) it presumably would not serve the cell well for both processes to be occurring concurrently.
Figure 5.

Regulated phosphoproteins after PDE4 and 8 inhibition that could be functionally associated with specific cellular processes. Only proteins associated with a given process by i) being tagged with a corresponding GOBP term and ii) for which confirmatory literature reports exist linking these genes to the process are considered. For simplicity, individual biological processes regulated are shown in separate tiles.
Further support for our finding that PDE4 and 8 inhibitor treatment possibly causes a delay of cell cycle progression comes from independent experiments in which we compared the effects of various cAMP elevating agents, e.g. PDE inhibitors and forskolin on the proliferation rate of MA10 cells (SI Figure 3). In good agreement with the results from our phosphoproteomics experiments we observed little growth inhibition by PDE4 or PDE8 inhibitor treatment alone but a robust growth arrest (80% reduction after 48 h treatment) when cells were treated with the PDE4 and 8 inhibitor combination.
Increases in cellular cAMP and PKA signaling have previously been shown to lead to cell cycle arrest and, in some cases, apoptosis in NIH3T3, HeLa, Y1 and S49 cells [27, 43, 44] Moreover, in yeast, cAMP signaling has been shown to facilitate synchronization between cell cycle progression and metabolism [45]. This and the fact that we observed a suppression of apoptotic signaling (e.g. strong upregulation of Bad pS155, see section 3.7) with concomitant cell cycle arrest in MA10 cells makes it reasonable to suspect that similar synchronization mechanisms between metabolism and the cell cycle might exist in mammalian cells.
3.5 PDE4 and 8 regulation of Erk1/2 and insulin signaling
Both MAPK families, extracellular signal-regulated kinase 1/2 (ERK1/2) and p38 have been shown previously to be activated downstream of the LHCGR and positively regulate steroidogenesis [46]. We found that PDE4 and 8 combination treatment robustly induced Erk1/2 as well as p38 MAPK activation (2.5-fold increase; Table 2) and this activation could be confirmed by western blotting for pERK1/2 (SI Figure 2).
We were also able to detect phosphorylation changes on several upstream components of the Erk1/2 cascade: the activating site pS255 on serine/threonine-protein kinase A-Raf (Araf) was found to be down-regulated (2-fold, 15min) and pS43 on Raf1 was up-regulated (11-fold, 1 h; Table 2)[47]. Robust regulation of Raf1 pS43 could also be confirmed by western blot analysis (SI Figure 3). This site has previously been reported as the major PKA target site in Raf1, but it was shown that two other PKA sites, pS233 and pS259, and not pS43 are crucial for 14-3-3 binding and abrogation of Raf1 binding to Ras [48, 49]. We quantified pS259 on Raf1 in 5 out of 8 experiments and yet it showed no regulation, suggesting that PDE4+8 inhibition/PKA failed to deactivate the kinase.
In other cells, it is known that the scaffolding protein, kinase suppressor of Ras 1 (KSR1), binds Raf, mitogen-activated protein kinase kinase (Meek) and Erk following dephosphorylation of pS392 and translocation to the peripheral membrane and thus activates MAPK signaling [50]. We detected a rapid decrease in phosphorylation (15 min, >2-fold) of two known Erk1/2 feedback sites, pS320 and pS260, on KSR1 [51]. The strikingly similar pattern of regulation for pS320, pS260, compared with pS392 and the fact that it has previously been shown that KSR1 mutants devoid of these feedback sites promote sustained ERK1/2 activation suggests that decreased phosphorylation of these sites might underlie an alternative route for MAPK activation [52].
Further, we observed significantly increased phosphorylation of growth factor receptor-bound protein 2 (GRB2)-associated-binding protein 1 (Gab1) at pS552, an ERK1/2 target site reported to facilitate Gab1 translocation and mitogenic signaling from the plasma membrane [53]. This also may lead to a reinforcement of the strong and sustained ERK induction observed in response to PDE4 and 8 inhibition.
Finally, we also found decreased phosphorylation of key enzymes downstream of MAPK that are known to be related to cell growth and mitosis. For example, pS447 on ribosomal protein S6 kinase b-1 (Rps6kb1) was found to be significantly down-regulated, suggesting an inactivation of the kinase as previously observed in other cells [54].
Similarly, the function of several components of the insulin signaling pathway was suppressed in response to an increase in cAMP levels. It is well known that activation of PDE3B by PKB or PKA can modulate insulin signaling in adipocytes [55]. Quantification of two key regulatory sites on Grb14 (pS368 and pS370; Table 2) an antagonist of Insr signaling, and the observation of other phospho-regulated components of the pathway may support this notion (Figure 5 C) [56, 57]. The fact that MA10 cells express Insr (SI Excel 2) suggests that this antagonism may play a physiological role in steroidogenic tissues [58]. Additionally, the reverse effect, suppression of LH induced steroidogenesis by insulin, was recently shown to be operative in MA10 cells [59].
Collectively, our dataset provides deep insights into the phosphorylation events regulating ERK activation and insulin receptor signaling (Figure 5 B and C). The exact mechanism(s) by which LH, CG and ACTH trigger ERK activation remains a matter of debate in the field of endocrinology [60]; although it has been proposed that agonist-occupied LHCGR trans-activates the EGFR and/or Src-family kinases further leading to ERK activation. Interestingly, we observed that Rapgef2 protein is expressed in substantial amounts in MA10 cells (log10 protein intensity 8.15, top 33 percentile in intensity). Rapgef2 is suggested to be a cAMP-activated guanine nucleotide exchange factor capable of activating Rap and Ras GTPases, thus leading to Raf-Meek-Erk activation [61]. Further, it has been shown previously that Gαs-coupled GPCRs can activate ERK via Rapgef2 [62]. Thus, it is highly likely that Rapgef2 is at least partially responsible for ERK activation in response to cAMP in MA10 cells. Additionally, our data also suggests that KSR1 Gab1 may amplify Ras signaling to Erk in MA10 cells. (Figure 5 B) [53].
3.6 PDE4 and 8 modulation of transcriptional/metabolic regulators
In the MA10 cell model we found changes in multiple phosphorylation sites that suggest PDE 4 and 8 inhibition modulated mRNA transcription in favor of steroidogenesis. We, for example, observed phosphorylation consistent with activation of the salt inducible kinase (SIK) – Creb-regulated transcription coactivator (Crtc) pathway. SIK3 was strongly up-regulated on two PKA target sites, pS551 and pS674, (2.5-fold and 8-fold, respectively), that are reported to result in cytosolic sequestration and its inability to phosphorylate Crtc3, thus preventing its nuclear translocation [63]. At the same time we also observed a rapid decrease in phosphorylation of Crtc3 (by 15 min) on the critical regulatory sites pS62, pS329 and pS370, indicating activation of Crtc3 transcriptional activity (Table 2) [64]. It was recently shown that PKA regulates the Sik2-Crtc2 signaling axis in the liver to promote gluconeogenesis under fasting conditions. PKA does this by means of activation of the inositol 1,4,5-triphosphate receptor type 1 (Itpr1), release of intracellular calcium and activation of calcineurin that in turn dephosphorylates and activates Crtc2 [65]. Tantalizingly we found that phosphorylation of Itpr1 was strongly up-regulated on its regulatory PKA target site, pS1588, suggesting that this signaling axis might also control Crtc3 dephosphorylation and activation in favor of steroidogenic gene expression in MA10 cells. However, it remains to be seen if the changes in phosphorylation of Sik3 and Ccrt3 seen in MA10 cells results in a similar regulation as seen in the liver with Sik2-Crtc2.
At the same time we observed the up-regulation of two key regulatory PKA sites, pS196 and pS626 (6-fold) on the Max-like protein X (MLX)-interacting protein-like (Mlxipl), also known as carbohydrate response element-binding protein (ChREBP) that were shown to lead to abrogation of its transcriptional activity and the repression of genes involved in glycolysis and lipogenesis [66]. This suggests that the de-novo synthesis of triglycerides in MA10 cells following PDE4 and 8 inhibition is suppressed.
Intersecting with the modulation of cell cycle and metabolic gene regulation our data also suggests a strong involvement of Wnt-signaling and the Hippo pathway in signaling events triggered by PDE4 and 8 inhibition. The induction of the Hippo pathway and the resulting phosphorylation and sequestration of Yes-associated protein 1 (Yap1) transcriptional activity in response to cAMP signaling has been described previously in other cells [67]. Very recently, interconnections between adenosine monophosphate (AMP)-activated protein kinase (AMPK) and the Hippo pathway have been revealed, linking Yap1 to metabolic processes regulated by these gene products [68]. We find a robust increase in Yap1 pS46 phosphorylation, a proposed AMPK target site responsible for disruption of Yap1 transcriptional activity. Interestingly, another site, pS352, which has been associated with Yap1 transcriptional regulation of genes necessary for mitotic spindle checkpoint passage, was found to be down-regulated [69]. This supports the notion that Yap1 might serve as an integrator of metabolic and cell cycle signaling downstream of PDE4 and 8/PKA in steroidogenic MA10 cells.
Further we observed a strong increase in β-catenin (Ctnnb1) phosphorylation on pS552, a site that has been shown to be phosphorylated by basophilic kinases like PKA, PKB and PKC. Up-regulation of this site leads to an enhanced interaction with Creb binding protein, increased nuclear localization of β-catenin and increased expression of Wnt-sensitive genes [70, 71]. In support of this finding, we detected significant phosphorylation of other Wnt-signaling pathway components, i.e. angiomotin-like protein 2 (Amotl2, Table2), adenomatous polyposis coli protein (Apc), low-density lipoprotein receptor-related protein 4 (Lrp4) and serine/threonine kinase Mark2, amongst others (SI Table 3). Of note, leucine-rich repeat Fli-I-interacting protein 2 (Lrrfip2), which has been proposed to be an upstream effector of β-catenin stability, was up-regulated on the regulatory phosphorylation site pS111 that modulates protein-protein interactions [72, 73].
The large impact of combined PDE inhibition and concomitant elevation of cellular cAMP on multiple levels of transcriptional regulation is apparent from our phosphoproteomics data. Conspicuously, the Sik3-Crtc3 signaling axis was found to be highly up-regulated, speaking to a robust transactivation of Creb via this pathway. Furthermore, the deactivation of carbohydrate-responsive element-binding protein (Mxlipl) is in good agreement with the idea that gene transcription is switched in favor of steroidogenic genes in response to cAMP [74]. Interestingly, the phosphorylation of several transcription factors modulating the expression of genes associated with the cell cycle, growth and survival were down-regulated. Examples include the estrogen receptor (ESR1), Yap1, Mybl2 and Smad2, all of which were modulated on sites that have previously been shown to inhibit their transcription factor function (Table 2). A number of other transcription factors also were found to be regulated on as yet uncharacterized sites (Figure 5 D). This further supports the idea that other cellular processes are “suspended/halted” in favor of steroidogenesis.
Of note, further support for the proposed regulation of cellular glucose and lipid metabolism from might come from our dataset. Thus, amongst others we found that glycogen synthase 1 (Gys1, pS711), phosphorylase b kinase regulatory subunit PHKA2 (pS729 and pS1015; both putative PKA target sites; SI Excel 3) as well as Polymerase I and transcript release factor (Ptrf; table 2) which was found to be involved in lipolysis highly regulated on unknown or poorly characterized sites.
3.7 PDE4+8 regulation endocytosis and vesicle transport
Supply of cholesterol is essential for the biosynthesis of steroid hormones and steroidogenic cells receive cholesterol by uptake of LDL or HDL from the bloodstream and/or de-novo synthesis [75]. It is therefore reasonable to expect that the tropic hormones regulating cAMP signaling and steroidogenesis also coordinate the uptake and intracellular distribution of lipids/cholesterol by modulating the phosphorylation and/or expression of gene products involved in receptor endocytosis and vesicle trafficking [76]. Indeed, we find a large number of proteins involved in these processes phospho-regulated in response to PDE inhibition (Figure 5 E and F). Gene products involved in receptor endocytosis include the lipolysis-stimulated lipoprotein receptor (Lsr) and Ldlrap1 (Ldl uptake), epidermal growth factor receptor substrate Eps15 and Ataxin-2 (Atxn2, EGFR internalization), among other key players in receptor-mediated endocytosis (Figure 5 E). We further traced phosphorylation changes on a number of proteins involved in endosome trafficking and vesicle movement between Golgi and ER, as well as from these organelles to the plasma membrane (Figure 5F). Since most of the regulated phosphosites quantified on proteins involved in endocytosis and vesicle trafficking are uncharacterized, we are not immediately able to determine which of these processes are actually activated or inhibited in response to PDE inhibition. Nonetheless the large number of these sites found to be highly modulated by the PDE4+8 inhibitors clearly indicate that cAMP may have a strong impact on intracellular transport processes with potential roles in delivering cholesterol and steroidogenic enzymes to their target organelles in MA10 cells. This phosphoproteomics dataset therefore suggests a large number of direct and indirect mechanisms by which PDE4 and 8 and PKA can regulate these processes on a molecular level.
3.7 Biological Pathways specifically regulated by PDE8
The combination treatment with PDE4 and 8 inhibitors revealed widespread phosphorylation changes in the MA10 proteome with ~750 phosphorylation sites modulated (Figure 4A). Surprisingly, treatment of the cells with either PDE4 inhibitor (Rolipram, 10 mM) or the PDE8 inhibitor PF-04957325 (200 nM) alone resulted in minimal changes in phosphorylation by the PDE4 inhibitor and only a few regulated sites with the PDE8 inhibitor alone. (0 and 54 significantly regulated sites for Rolipram and PF-04957325, respectively; Figure 4B).
Figure 4.

A) Pie chart showing predicted upstream kinases (NetPhorest) for phosphosites specifically regulated by PDE8. B) Venn diagram showing the overlap between PDE8 and PDE4 and 8 inhibition-regulated phosphorylation sites.
Table 3 shows all PDE8-specific phosphorylation sites regulated in our dataset. We found that some of the phosphorylation sites are regulated with a magnitude of SILAC ratio fold change comparable to PDE4+8 combination inhibition, suggesting that these phosphorylation events are specific to PDE8. For example, we find the aforementioned regulatory sites on Sik3 (pS551 and pS674) and Iptr1 (pS1588) up-regulated up to 4-fold (section 3.6). Also sites on Mxlipl (pS196 and pS626) were found to be up-regulated, suggesting a specific role of PDE8 in basal MA10 steroidogenic gene transcription. Phosphorylation of Grb14 was also found to be PDE8 inhibitor responsive suggesting that PDE8 activity prevents negative regulation of insulin receptor (Insr) signaling caused by high levels of intracellular cAMP as found in our dataset and implicated in previously published results [58].
Table 3.
Phosphorylation sites specifically regulated in response to PDE8 inhibition with 200 nM of PF-04957325. Arrows symbolize increased phosphorylation (↑) or decreased phosphorylation (↓) of a phosphorylation site.
| Gene | Site | Reg. | Average abs. log2 SILAC ratio |
NetPhorest Kinase Group (Score) |
NetPhorest Interaction (Score) |
±7 aa Window |
|---|---|---|---|---|---|---|
| Akap1 | S101 | ↓ | 0.67 | TGFBR (0.31) | 14-3-3 (0.35) | TRQVRRRsESSGNLP |
| Akap1 | S103 | ↓ | 0.82 | CLK (0.37) | 14-3-3 (0.52) | QVRRRSEsSGNLPSV |
| Als2 | S477 | ↑ | 0.78 | PKA (0.32) | 14-3-3 (0.5) | EGGSRRLsLPGLLSQ |
| Arfgap1 | S360 | ↑ | 1.24 | PKA (0.3) | ASTGRRSsDSWDVWG | |
| Bad | S155 | ↑ | 1.16 | PKA (0.28) | BRCT (0.21) | GRELRRMsDEFEGSF |
| Baiap2l1 | S281 | ↓ | 0.79 | CDK1/2/3/5 (0.28) | WW (0.13) | YDTLSKYsPKMPPAP |
| Cad | S1406 | ↑ | 1.50 | PKA (0.32) | 14-3-3 (0.12) | GAGGRRLsSFVTKGY |
| Casp8 | S188 | ↑ | 1.13 | PKA (0.31) | 14-3-3 (0.16) | SSTERRMsLEGREEL |
| Cherp | S868 | ↓ | 0.81 | CK2 (0.23) | 14-3-3 (0.11) | VKMGWSGsGGLGAKE |
| Eepd1 | S25 | ↑ | 2.50 | PKB (0.2) | 14-3-3 (0.12) | LSHNRKFsAACNFSN |
| Golga5 | S116 | ↑ | 1.48 | PKA (0.29) | 14-3-3 (0.54) | QFVRRKKsEPDDELL |
| Grb14 | S370 | ↑ | 1.19 | PKB (0.22) | 14-3-3 (0.49) | MSPMRSVsENSLVAM |
| Grb14 | T9 | ↑ | 1.61 | PKA (0.28) | 14-3-3 (0.48) | SLSARRVtLPAITPI |
| Hspb1 | S15 | ↑ | 0.57 | CLK (0.21) | 14-3-3 (0.29) | FSLLRSPsWEPFRDW |
| Itpr1 | S1588 | ↑ | 1.25 | PKA (0.32) | RNAARRDsVLAASRD | |
| Itpr3 | S1832 | ↑ | 1.34 | RCK (0.2) | 14-3-3 (0.12) | ATKGRVSsFSMPSSS |
| Kifap3 | S102 | ↑ | 0.86 | PKA (0.3) | 14-3-3 (0.5) | YLQNRRDsLPGKEKK |
| Ksr1 | T260 | ↓ | 0.79 | RCK (0.32) | SFITPPTtPQLRRHA | |
| Ldlrap1 | S198 | ↑ | 0.89 | PKA (0.32) | 14-3-3 (0.52) | VPGTRRDsTPSLKTL |
| Lrp4 | S1887 | ↑ | 0.97 | PKA (0.3) | 14-3-3 (0.46) | ATPERRGsLPDTGWK |
| Lrrfip2 | S111 | ↑ | 0.84 | RCK (0.32) | GNSSRRGsGDTSSLI | |
| Lrrfip2 | S111 | ↑ | 1.01 | CDK1/2/3/5 (0.35) | BRCT (0.21) | GNSSRRGsGDTSSLI |
| Map1b | S1403 | ↓ | 0.63 | MAPK1/3/7-NLK (0.18) | WW (0.12) | PPLLGSEsPYEDFLS |
| Matr3 | S188 | ↑ | 0.66 | PKA (0.31) | 14-3-3 (0.17) | KRHFRRDsFDDRGPS |
| Mlxipl | S195 | ↑ | 0.54 | PKC (0.39) | YKKRLRKsSREGDFL | |
| Mlxipl | S626 | ↑ | 1.62 | PKA (0.28) | BRCT (0.21) | SGSERRLsGDLNSIQ |
| Ncapd2 | S1320 | ↑ | 0.48 | RCK (0.14) | SRLQPLTsVDSDNDF | |
| Osbpl11 | S194 | ↑ | 2.14 | PKA (0.31) | PISQRRPsQNAMSFF | |
| Phldb2 | S510 | ↑ | 0.80 | PKA (0.27) | 14-3-3 (0.12) | YRCHRKGsLQDVDVA |
| Ppp1r13l | S394 | ↓ | 0.92 | CDK1/2/3/5 (0.25) | BRCT (0.21) | GRAVLPGsPIFSRAP |
| Ppp1r14a | S26 | ↓ | 0.61 | CDK1/2/3/5 (0.25) | RARGPGGsPSGLQKR | |
| Prrc2c | S755 | ↑ | 1.02 | CDK1/2/3/5 (0.15) | RRDQMEGsPNSSESF | |
| Ralgapa1 | S859 | ↑ | 1.60 | STMTRRGsSPGSLEI | ||
| Reps1 | S272 | ↑ | 1.39 | PKA (0.28) | 14-3-3 (0.1) | ANEIRRQsSSYEDPW |
| Rmdn3 | S46 | ↑ | 1.74 | CLK (0.28) | 14-3-3 (0.58) | QRHGRSHsLPNSLDY |
| Rtn4 | S165 | ↑ | 1.34 | PKA (0.3) | BRCT (0.21) | AAPKRRGsGSVDETL |
| Rtn4 | S167 | ↑ | 1.53 | CK2 (0.3) | 14-3-3 (0.11) | PKRRGSGsVDETLFA |
| Scap | S821 | ↑ | 1.31 | PKA (0.3) | 14-3-3 (0.22) | RPGPRRDsCGGGAFE |
| Serbp1 | S329 | ↑ | 0.43 | TGFBR (0.16) | 14-3-3 (0.27) | FVLHKSKsEEAHAED |
| Sh3pxd2a | S812 | ↑ | 2.27 | PKA (0.3) | 14-3-3 (0.16) | SEGSRRGsADIIPLT |
| Sik3 | S551 | ↑ | 0.77 | PKA (0.29) | 14-3-3 (0.34) | GPLGRRAsDGGANIQ |
| Sik3 | S674 | ↑ | 1.94 | PKA (0.27) | FSPVRRFsDGAASIQ | |
| Slc7a2 | S645 | ↑ | 0.92 | PKA (0.26) | 14-3-3 (0.5) | DHHQRNLsLPFILHE |
| Snx1 | S188 | ↑ | 0.98 | PKA (0.32) | 14-3-3 (0.12) | FAVKRRFsDFLGLYE |
| Sqstm1 | S207 | ↑ | 0.91 | CDK1/2/3/5 (0.28) | WW (0.28) | MGPPGNWsPRPPRAG |
| Srrm2 | S1343 | ↓ | 0.94 | CLK (0.28) | 14-3-3 (0.46) | RRSSSELsPEVVEKV |
| Stxbp5 | S760 | ↑ | 0.86 | PKA (0.27) | 14-3-3 (0.47) | AKMSRKLsLPTDLKP |
| Svil | S960 | ↑ | 0.71 | PKA (0.31) | 14-3-3 (0.11) | HVVLRRGsLELGNPS |
| Tbc1d10b | S673 | ↑ | 1.45 | CLK (0.32) | 14-3-3 (0.39) | PPPVRRAsAGPVPGA |
| Tnks1bp1 | S429 | ↑ | 1.93 | PKA (0.28) | 14-3-3 (0.28) | SLAQRRFsEGVLQPP |
| Top2a | S4 | ↑ | 0.77 | MAPK1/3/7-NLK (0.17) | _____MELsPLQPVNE | |
| Tpd52 | S170 | ↓ | 0.85 | CDK1/2/3/5 (0.23) | BRCT (0.21) | KLEDVKNsPTFKSFE |
| Ybx3 | S259 | ↑ | 1.83 | PKA (0.31) | 14-3-3 (0.47) | PYNYRRRsRPLNAVS |
| Znf609 | S1057 | ↓ | 1.35 | PKC (0.12) | PTLTKAPsLTDLVKS |
Beyond the biological relevant phosphorylation sites already discussed in the context of PDE4+8 inhibition, we found proteins involved in several other signaling pathways regulated by PDE8. We observed the major inhibitory PKA target site S155 on Bcl2-associated agonist of cell death (Bad) up-regulated by ~2.5-fold. In this context we also detected an uncharacterized putative PKA target site (NetPhorest score 0.31) on caspase 8 (Casp8) up-regulated 2.5-fold. This, together with the robust regulation of Reticulon-4 (Rtn4) and RelA-associated inhibitor (Ppp1r13l) strongly suggests a role for PDE8 in apoptosis signaling [77, 78].
Another biological process strongly represented amongst PDE8 regulated genes is receptor-mediated endocytosis and vesicle trafficking; we found the putative PKA target and 14-3-3-binding site pS198 on Ldldrap1 increased by PDE8 inhibition alone. Further, we quantified phosphorylation changes in putative PKA target sites on ADP-ribosylation factor GTPase-activating protein 1 (Arfgap1), and sorting nexin-1 (SNX1) - both of which are strongly indicated to be key players in regulation of endocytosis and vesicular trafficking [79–82].
It is apparent from our findings that PDE8s (A and/or B) play physiologically relevant roles even under basal conditions in MA10 cells. A role for PDE4 isoforms alone on phosphorylation is apparent only when the PDE8s are also inhibited [8]. Interestingly a large number of the regulated sites were predicted to be PKA target sites (51%) and most of these sites (44 of 54) were up-regulated after one hour of treatment. The remaining 11 sites were significantly down-regulated and predicted to be non-PKA target sites (Table 2).
Collectively, while we quantified only 54 differentially regulated phosphorylation sites upon PDE8 inhibition alone, the identity of the regulated genes yields a strong indication about the biological processes mediated by this phosphodiesterase family. None of the functional connections for PDE8A/B described above have been previously reported, and we expect that these data will greatly facilitate novel biological studies on likely new roles for cAMP and these PDEs as regulators of these processes.
5 Conclusion
Steroidogenesis is classically described as a linear sequence of cAMP/PKA/CREB signaling responsible for the transcriptional regulation and post-translational modification of a handful of key steroidogenic gene products (Hsl, StAR, CYPs etc.) [12]. We recently investigated the role of PDE4B and PDE8A/B activities in two different steroidogenic models (mouse Leydig MA10 cells and adrenocortical Y1 cells) and suggested that these PDEs play important roles in the physiology of steroidogenic tissues [8, 83]. We also found evidence for synergism between the signaling compartments regulated by PDE4 and PDE8 to facilitate maximal steroid output.
Using selective phosphodiesterase inhibitors against the PDE4 and PDE8 families and state-of-the-art MS-based phosphoproteomics, we analyzed global phosphoproteome dynamics in response to cAMP/PKA activation in the steroidogenic MA10 cell model. The assembled dataset covers the MA10 mouse proteome to a depth of 8234 proteins and comprises 28,336 phosphorylation sites, providing deep coverage of the MA10 cell (phospho)proteome. Collectively, our data suggest the regulation of these cells is highly complex and that cAMP regulates cellular function and steroidogenesis at multiple steps. These include pathways involved in the supply and trafficking of cholesterol and steroidogenic factors, the activation of cellular signaling pathways that regulate the expression and regulation of steroidogenic enzymes, and the switch between cellular metabolic and proliferative processes. A key question arising from our study is how the increase of cellular cAMP by PDE inhibition relates to native steroidogenic signaling, i.e. stimulation of Leydig cells with LH or CG. The stimulation of the LHCGR with its cognate ligands is thought to mainly cause an increase in cAMP by activation of Gαs, but other pathways may also play a significant role. Activation of phospholipase C (PLC)/PKC signaling by LHCGR coupled to other trimeric G-proteins and trimeric G-protein β and γ subunits has been suggested [9]. Further, arrestin-mediated endocytosis of GPCRs was found to be capable of triggering an array of signaling events that are as yet unexplored in the context of steroidogenesis [84]. We conclude that a key step for the elucidation of the roles of PDEs in the context of native steroidogenic signaling would be to analyze signaling processes triggered by tropic hormone treatment of steroidogenic cells.
Supplementary Material
Highlights.
Dataset covering the MA10 cell (phospho)proteome to a depth of 8,234 proteins and 28,336 phosphosites.
Dynamic regulation of 749 phosphorylation sites in response to selective PDE4 and PDE8 combination inhibition.
Wide-spread regulation of the cell cycle, transcription, endocytosis and vesicle trafficking by cAMP coordinated with steroidogenesis.
Identification of a subset the phosphoproteome selectively regulated by PDE8s.
Acknowledgments
We wish to thank members of the Ong lab and the Beavo lab as well as Prof. Dustin Maly for fruitful discussions. Research reported in this publication was supported the National Institute of General Medical Sciences and the National Cancer Institute of the National Institutes of Health under award numbers R01GM083926 and R21CA177402, respectively. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. M.G was supported by a post-doctoral fellowship of the DFG (German Research Foundation, GO 2358/1-1).
Abbreviations
- AC
adenylyl cyclase
- ACTH
adrenocorticotropic hormone
- AKAP
A-kinase anchor protein
- AMP
5´ adenosine monophosphate
- AMPK
AMP-activated protein kinase
- Arfgap1
ADP-ribosylation factor GTPase-activating protein 1
- Arhgef2
Rho guanine nucleotide exchange factor 2
- Bad
Bcl2-associated agonist of cell death
- cAMP
3´,5´-cyclic adenosine monophosphate
- Casp8
caspase 8
- CDK
cyclin-dependent kinase
- CG
choriogonadotropin
- cGMP
3´,5´-cyclic guanosine monophosphate
- ChREBP
carbohydrate responsive element-binding protein
- CK
casein kinase
- Clasp
CLIP-associating protein
- Clip
Cap-Gly domain-containing linker protein
- CLK
CDC-like kinase
- Creb
Cyclic AMP-responsive element-binding protein
- Crtc
CREB-regulated transcription coactivator
- Ctnnb1
Catenin beta-1
- Egfr
epidermal growth factor receptor
- Erk
extracellular signal-regulated kinase
- Esr1
estrogen receptor
- Flna
Filamin-A
- Gab1
GRB2-associated-binding protein 1
- Grb
Growth factor receptor-bound protein
- GPCR
G protein-coupled receptor
- GTP
guanosine triphosphate
- HSL
Hormone-sensitive lipase
- Insr
Insulin receptor
- Itpr1
Inositol 1,4,5-triphosphate receptor type 1
- Ksr1
Kinase suppressor of Ras 1
- Ldl
Low-density lipoprotein
- Ldlrap1
Low-density lipoprotein receptor adapter protein 1
- LH
Luteinizing hormone
- LHCGR
Lutropin-choriogonadotropic hormone receptor
- Lrrfip2
Leucine-rich repeat flightless-interacting protein 2
- MAPK
Mitogen-activated protein kinase
- MEK
Mitogen-activated protein kinase kinase
- Mlxipl
MLX-interacting protein-like
- MLX
Max-like protein X
- MS
Mass spectrometry
- Mybl2
Myb-related protein B
- Npm1
Nucleophosmin
- PDE
Phosphodiesterase
- PKA
Protein kinase A
- PKB
Protein kinase B
- PKC
Protein kinase C
- PKG
Protein kinase G
- PLK
Polo-like kinase
- PLC
Phospholipase C
- Ptrf
Polymerase I and transcript release factor
- Rapgef2
Rap guanine nucleotide exchange factor 2
- Reps1
RalBP1-associated Eps domain-containing protein1
- Rps6kb1
Ribosomal protein S6 kinase beta-1
- Rtn4
Reticulon-4
- Sik
Salt-inducible kinase
- SILAC
Stable isotope labeling by amino acids in cell culture
- Snx
Sorting nexin
- StAR
Steroidogenic acute regulatory protein
- Vcpip1
Valosin-containing protein p97/p47 complex-interacting protein 1
- Yap1
Yes-associated proten 1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
M.G., M.S., S.O. and J.B. designed experiments; M.G., M.S. and D.S. performed experiments; M.G., S.O. and J.B. analyzed and interpreted the data; M.G., S.O. and J.B. prepared the manuscript.
References
- 1.Beavo JA, Brunton LL. Cyclic nucleotide research -- still expanding after half a century. Nat Rev Mol Cell Biol. 2002;3:710–718. doi: 10.1038/nrm911. [DOI] [PubMed] [Google Scholar]
- 2.Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 2007;76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444. [DOI] [PubMed] [Google Scholar]
- 3.Conti M, Mika D, Richter W. Cyclic AMP compartments and signaling specificity: role of cyclic nucleotide phosphodiesterases. J. Gen. Physiol. 2014;143:29–38. doi: 10.1085/jgp.201311083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annu. Rev. Pharmacool. Toxicol. 2001;41:145–174. doi: 10.1146/annurev.pharmtox.41.1.145. [DOI] [PubMed] [Google Scholar]
- 5.Kritzer MD, Li J, Dodge-Kafka K, Kapiloff MS. AKAPs: the architectural underpinnings of local cAMP signaling. J. Mol. Cell. Cardiol. 2012;52:351–358. doi: 10.1016/j.yjmcc.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- 7.Maurice DH, Ke H, Ahmad F, Wang Y, Chung J, Manganiello VC. Advances in targeting cyclic nucleotide phosphodiesterases. Nat Rev Drug Discov. 2014;13:290–314. doi: 10.1038/nrd4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shimizu-Albergine M, Tsai LC, Patrucco E, Beavo JA. cAMP-specific phosphodiesterases 8A and 8B, essential regulators of Leydig cell steroidogenesis. Mol. Pharmacol. 2012;81:556–566. doi: 10.1124/mol.111.076125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ascoli M, Fanelli F, Segaloff DL. The lutropin/choriogonadotropin receptor, a 2002 perspective. Endocr. Rev. 2002;23:141–174. doi: 10.1210/edrv.23.2.0462. [DOI] [PubMed] [Google Scholar]
- 10.Beuschlein F, Fassnacht M, Klink A, Allolio B, Reincke M. ACTH-receptor expression, regulation and role in adrenocortial tumor formation. Eur. J. Endocrinol. 2001;144:199–206. doi: 10.1530/eje.0.1440199. [DOI] [PubMed] [Google Scholar]
- 11.Sassone-Corsi P. The cyclic AMP pathway. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a011148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manna PR, Dyson MT, Stocco DM. Regulation of the steroidogenic acute regulatory protein gene expression: present and future perspectives. Mol. Hum. Reprod. 2009;15:321–333. doi: 10.1093/molehr/gap025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manna PR, Cohen-Tannoudji J, Counis R, Garner CW, Huhtaniemi I, Kraemer FB, Stocco DM. Mechanisms of action of hormone-sensitive lipase in mouse Leydig cells: its role in the regulation of the steroidogenic acute regulatory protein. J. Biol. Chem. 2013;288:8505–8518. doi: 10.1074/jbc.M112.417873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tremblay JJ. Molecular regulation of steroidogenesis in endocrine Leydig cells. Steroids. 2015 doi: 10.1016/j.steroids.2015.08.001. [DOI] [PubMed] [Google Scholar]
- 15.Ascoli M. Characterization of several clonal lines of cultured Leydig tumor cells: gonadotropin receptors and steroidogenic responses. Endocrinology. 1981;108:88–95. doi: 10.1210/endo-108-1-88. [DOI] [PubMed] [Google Scholar]
- 16.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 17.Mertins P, Qiao JW, Patel J, Udeshi ND, Clauser KR, Mani DR, Burgess MW, Gillette MA, Jaffe JD, Carr SA. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat Methods. 2013;10:634–637. doi: 10.1038/nmeth.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blagoev B, Ong SE, Kratchmarova I, Mann M. Temporal analysis of phosphotyrosine-dependent signaling networks by quantitative proteomics. Nat. Biotechnol. 2004;22:1139–1145. doi: 10.1038/nbt1005. [DOI] [PubMed] [Google Scholar]
- 19.Villen J, Gygi SP. The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat Protoc. 2008;3:1630–1638. doi: 10.1038/nprot.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2007;2:1896–1906. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- 21.Horn H, Schoof EM, Kim J, Robin X, Miller ML, Diella F, Palma A, Cesareni G, Jensen LJ, Linding R. KinomeXplorer: an integrated platform for kinome biology studies. Nat Methods. 2014;11:603–604. doi: 10.1038/nmeth.2968. [DOI] [PubMed] [Google Scholar]
- 22.Colaert N, Helsens K, Martens L, Vandekerckhove J, Gevaert K. Improved visualization of protein consensus sequences by iceLogo. Nat Methods. 2009;6:786–787. doi: 10.1038/nmeth1109-786. [DOI] [PubMed] [Google Scholar]
- 23.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 25.Humphrey SJ, Yang G, Yang P, Fazakerley DJ, Stockli J, Yang JY, James DE. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 2013;17:1009–1020. doi: 10.1016/j.cmet.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar L, M EF. Mfuzz: a software package for soft clustering of microarray data. Bioinformation. 2007;2:5–7. doi: 10.6026/97320630002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kimura E, Armelin HA. Phorbol ester mimics ACTH action in corticoadrenal cells stimulating steroidogenesis, blocking cell cycle, changing cell shape, and inducing c-fos proto-oncogene expression. J. Biol. Chem. 1990;265:3518–3521. [PubMed] [Google Scholar]
- 28.Meiri D, Greeve MA, Brunet A, Finan D, Wells CD, LaRose J, Rottapel R. Modulation of Rho guanine exchange factor Lfc activity by protein kinase A-mediated phosphorylation. Mol. Cell. Biol. 2009;29:5963–5973. doi: 10.1128/MCB.01268-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Birkenfeld J, Nalbant P, Bohl BP, Pertz O, Hahn KM, Bokoch GM. GEF-H1 modulates localized RhoA activation during cytokinesis under the control of mitotic kinases. Dev Cell. 2007;12:699–712. doi: 10.1016/j.devcel.2007.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maia AR, Garcia Z, Kabeche L, Barisic M, Maffini S, Macedo-Ribeiro S, Cheeseman IM, Compton DA, Kaverina I, Maiato H. Cdk1 and Plk1 mediate a CLASP2 phospho-switch that stabilizes kinetochore-microtubule attachments. J. Cell Biol. 2012;199:285–301. doi: 10.1083/jcb.201203091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kitagawa M, Fung SY, Hameed UF, Goto H, Inagaki M, Lee SH. Cdk1 coordinates timely activation of MKlp2 kinesin with relocation of the chromosome passenger complex for cytokinesis. Cell Rep. 2014;7:166–179. doi: 10.1016/j.celrep.2014.02.034. [DOI] [PubMed] [Google Scholar]
- 32.Abrieu A, Magnaghi-Jaulin L, Kahana JA, Peter M, Castro A, Vigneron S, Lorca T, Cleveland DW, Labbe JC. Mps1 is a kinetochore-associated kinase essential for the vertebrate mitotic checkpoint. Cell. 2001;106:83–93. doi: 10.1016/s0092-8674(01)00410-x. [DOI] [PubMed] [Google Scholar]
- 33.Jelluma N, Brenkman AB, van den Broek NJ, Cruijsen CW, van Osch MH, Lens SM, Medema RH, Kops GJ. Mps1 phosphorylates Borealin to control Aurora B activity and chromosome alignment. Cell. 2008;132:233–246. doi: 10.1016/j.cell.2007.11.046. [DOI] [PubMed] [Google Scholar]
- 34.Zhao Y, Chen RH. Mps1 phosphorylation by MAP kinase is required for kinetochore localization of spindle-checkpoint proteins. Curr. Biol. 2006;16:1764–1769. doi: 10.1016/j.cub.2006.07.058. [DOI] [PubMed] [Google Scholar]
- 35.Liu J, Cheng X, Zhang Y, Li S, Cui H, Zhang L, Shi R, Zhao Z, He C, Wang C, Zhao H, Zhang C, Fisk HA, Guadagno TM, Cui Y. Phosphorylation of Mps1 by BRAFV600E prevents Mps1 degradation and contributes to chromosome instability in melanoma. Oncogene. 2013;32:713–723. doi: 10.1038/onc.2012.94. [DOI] [PubMed] [Google Scholar]
- 36.Sun XM, Patel DD, Acosta JC, Gil J, Soutar AK. Premature senescence in cells from patients with autosomal recessive hypercholesterolemia (ARH): evidence for a role for ARH in mitosis. Arterioscler. Thromb. Vasc. Biol. 2011;31:2270–2277. doi: 10.1161/ATVBAHA.111.232223. [DOI] [PubMed] [Google Scholar]
- 37.Cha H, Hancock C, Dangi S, Maiguel D, Carrier F, Shapiro P. Phosphorylation regulates nucleophosmin targeting to the centrosome during mitosis as detected by cross-reactive phosphorylation-specific MKK1/MKK2 antibodies. Biochem. J. 2004;378:857–865. doi: 10.1042/BJ20031173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Totsukawa G, Matsuo A, Kubota A, Taguchi Y, Kondo H. Mitotic phosphorylation of VCIP135 blocks p97ATPase-mediated Golgi membrane fusion. Biochem. Biophys. Res. Commun. 2013;433:237–242. doi: 10.1016/j.bbrc.2013.02.090. [DOI] [PubMed] [Google Scholar]
- 39.Zhang X, Zhang H, Wang Y. Phosphorylation regulates VCIP135 function in Golgi membrane fusion during the cell cycle. J. Cell Sci. 2014;127:172–181. doi: 10.1242/jcs.134668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson TK, Schweppe RE, Septer J, Lewis RE. Phosphorylation of B-Myb regulates its transactivation potential and DNA binding. J. Biol. Chem. 1999;274:36741–36749. doi: 10.1074/jbc.274.51.36741. [DOI] [PubMed] [Google Scholar]
- 41.Yamauchi T, Ishidao T, Nomura T, Shinagawa T, Tanaka Y, Yonemura S, Ishii S. A B-Myb complex containing clathrin and filamin is required for mitotic spindle function. EMBO J. 2008;27:1852–1862. doi: 10.1038/emboj.2008.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vadlamudi RK, Li F, Adam L, Nguyen D, Ohta Y, Stossel TP, Kumar R. Filamin is essential in actin cytoskeletal assembly mediated by p21-activated kinase 1. Nature Cell Biology. 2002;4:681–690. doi: 10.1038/ncb838. [DOI] [PubMed] [Google Scholar]
- 43.Yan L, Herrmann V, Hofer JK, Insel PA. beta-adrenergic receptor/cAMP-mediated signaling and apoptosis of S49 lymphoma cells. Am J Physiol Cell Physiol. 2000;279:C1665–1674. doi: 10.1152/ajpcell.2000.279.5.C1665. [DOI] [PubMed] [Google Scholar]
- 44.Kotani S, Tugendreich S, Fujii M, Jorgensen PM, Watanabe N, Hoog C, Hieter P, Todokoro K. PKA and MPF-activated polo-like kinase regulate anaphase-promoting complex activity and mitosis progression. Mol. Cell. 1998;1:371–380. doi: 10.1016/s1097-2765(00)80037-4. [DOI] [PubMed] [Google Scholar]
- 45.Muller D, Exler S, Aguilera-Vazquez L, Guerrero-Martin E, Reuss M. Cyclic AMP mediates the cell cycle dynamics of energy metabolism in Saccharomyces cerevisiae. Yeast. 2003;20:351–367. doi: 10.1002/yea.967. [DOI] [PubMed] [Google Scholar]
- 46.Manna PR, Stocco DM. The role of specific mitogen-activated protein kinase signaling cascades in the regulation of steroidogenesis. J Signal Transduct. 2011;2011:821615. doi: 10.1155/2011/821615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baljuls A, Schmitz W, Mueller T, Zahedi RP, Sickmann A, Hekman M, Rapp UR. Positive regulation of A-RAF by phosphorylation of isoform-specific hinge segment and identification of novel phosphorylation sites. J. Biol. Chem. 2008;283:27239–27254. doi: 10.1074/jbc.M801782200. [DOI] [PubMed] [Google Scholar]
- 48.Dumaz N, Marais R. Protein kinase A blocks Raf-1 activity by stimulating 14-3-3 binding and blocking Raf-1 interaction with Ras. J. Biol. Chem. 2003;278:29819–29823. doi: 10.1074/jbc.C300182200. [DOI] [PubMed] [Google Scholar]
- 49.Sidovar MF, Kozlowski P, Lee JW, Collins MA, He Y, Graves LM. Phosphorylation of serine 43 is not required for inhibition of c-Raf kinase by the cAMP-dependent protein kinase. J. Biol. Chem. 2000;275:28688–28694. doi: 10.1074/jbc.M909351199. [DOI] [PubMed] [Google Scholar]
- 50.Muller J, Ory S, Copeland T, Piwnica-Worms H, Morrison DK. C-TAK1 regulates Ras signaling by phosphorylating the MAPK scaffold, KSR1. Mol. Cell. 2001;8:983–993. doi: 10.1016/s1097-2765(01)00383-5. [DOI] [PubMed] [Google Scholar]
- 51.Cacace AM, Michaud NR, Therrien M, Mathes K, Copeland T, Rubin GM, Morrison DK. Identification of constitutive and ras-inducible phosphorylation sites of KSR: implications for 14-3-3 binding, mitogen-activated protein kinase binding, and KSR overexpression. Mol. Cell. Biol. 1999;19:229–240. doi: 10.1128/mcb.19.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Canal F, Palygin O, Pankratov Y, Correa SA, Muller J. Compartmentalization of the MAPK scaffold protein KSR1 modulates synaptic plasticity in hippocampal neurons. FASEB J. 2011;25:2362–2372. doi: 10.1096/fj.10-173153. [DOI] [PubMed] [Google Scholar]
- 53.Wolf A, Eulenfeld R, Bongartz H, Hessenkemper W, Simister PC, Lievens S, Tavernier J, Feller SM, Schaper F. MAPK-induced Gab1 translocation to the plasma membrane depends on a regulated intramolecular switch. Cell. Signal. 2015;27:340–352. doi: 10.1016/j.cellsig.2014.11.017. [DOI] [PubMed] [Google Scholar]
- 54.Weng QP, Kozlowski M, Belham C, Zhang A, Comb MJ, Avruch J. Regulation of the p70 S6 kinase by phosphorylation in vivo. Analysis using site-specific anti-phosphopeptide antibodies. J. Biol. Chem. 1998;273:16621–16629. doi: 10.1074/jbc.273.26.16621. [DOI] [PubMed] [Google Scholar]
- 55.Degerman E, Ahmad F, Chung YW, Guirguis E, Omar B, Stenson L, Manganiello V. From PDE3B to the regulation of energy homeostasis. Curr Opin Pharmacol. 2011;11:676–682. doi: 10.1016/j.coph.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taira J, Higashimoto Y. Phosphorylation of Grb14 BPS domain by GSK-3 correlates with complex forming of Grb14 and insulin receptor. J Biochem. 2014;155:353–360. doi: 10.1093/jb/mvu011. [DOI] [PubMed] [Google Scholar]
- 57.Goenaga D, Hampe C, Carre N, Cailliau K, Browaeys-Poly E, Perdereau D, Holt LJ, Daly RJ, Girard J, Broutin I, Issad T, Burnol AF. Molecular determinants of Grb14-mediated inhibition of insulin signaling. Mol. Endocrinol. 2009;23:1043–1051. doi: 10.1210/me.2008-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Graves LM, Lawrence JC., Jr Insulin, growth factors, and cAMP: antagonism in the signal transduction pathways. Trends Endocrinol Metab. 1996;7:43–50. doi: 10.1016/1043-2760(95)00204-9. [DOI] [PubMed] [Google Scholar]
- 59.Ahn SW, Gang GT, Kim YD, Ahn RS, Harris RA, Lee CH, Choi HS. Insulin Directly Regulates Steroidogenesis via Induction of the Orphan Nuclear Receptor DAX-1 in Testicular Leydig Cells. J. Biol. Chem. 2013;288:15937–15946. doi: 10.1074/jbc.M113.451773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Light A, Hammes SR. Membrane receptor cross talk in steroidogenesis: recent insights and clinical implications. Steroids. 2013;78:633–638. doi: 10.1016/j.steroids.2012.12.016. [DOI] [PubMed] [Google Scholar]
- 61.Pham N, Cheglakov I, Koch CA, de Hoog CL, Moran MF, Rotin D. The guanine nucleotide exchange factor CNrasGEF activates ras in response to cAMP and cGMP. Curr. Biol. 2000;10:555–558. doi: 10.1016/s0960-9822(00)00473-5. [DOI] [PubMed] [Google Scholar]
- 62.Emery AC, Eiden MV, Mustafa T, Eiden LE. Rapgef2 connects GPCR-mediated cAMP signals to ERK activation in neuronal and endocrine cells. Sci Signal. 2013;6:ra51. doi: 10.1126/scisignal.2003993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Berggreen C, Henriksson E, Jones HA, Morrice N, Goransson O. cAMP-elevation mediated by beta-adrenergic stimulation inhibits salt-inducible kinase (SIK) 3 activity in adipocytes. Cell. Signal. 2012;24:1863–1871. doi: 10.1016/j.cellsig.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 64.Clark K, MacKenzie KF, Petkevicius K, Kristariyanto Y, Zhang J, Choi HG, Peggie M, Plater L, Pedrioli PG, McIver E, Gray NS, Arthur JS, Cohen P. Phosphorylation of CRTC3 by the salt-inducible kinases controls the interconversion of classically activated and regulatory macrophages. Proc. Natl. Acad. Sci. U. S. A. 2012;109:16986–16991. doi: 10.1073/pnas.1215450109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Y, Li G, Goode J, Paz JC, Ouyang K, Screaton R, Fischer WH, Chen J, Tabas I, Montminy M. Inositol-1,4,5-trisphosphate receptor regulates hepatic gluconeogenesis in fasting and diabetes. Nature. 2012;485:128–132. doi: 10.1038/nature10988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kawaguchi T, Takenoshita M, Kabashima T, Uyeda K. Glucose and cAMP regulate the L-type pyruvate kinase gene by phosphorylation/dephosphorylation of the carbohydrate response element binding protein. Proc. Natl. Acad. Sci. U. S. A. 2001;98:13710–13715. doi: 10.1073/pnas.231370798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu FX, Zhang Y, Park HW, Jewell JL, Chen Q, Deng Y, Pan D, Taylor SS, Lai ZC, Guan KL. Protein kinase A activates the Hippo pathway to modulate cell proliferation and differentiation. Genes Dev. 2013;27:1223–1232. doi: 10.1101/gad.219402.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang W, Xiao ZD, Li X, Aziz KE, Gan B, Johnson RL, Chen J. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat Cell Biol. 2015;17:490–499. doi: 10.1038/ncb3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang S, Zhang L, Chen X, Chen Y, Dong J. Oncoprotein YAP regulates the spindle checkpoint activation in a mitotic phosphorylation-dependent manner through up-regulation of BubR1. J. Biol. Chem. 2015;290:6191–6202. doi: 10.1074/jbc.M114.624411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Taurin S, Sandbo N, Qin Y, Browning D, Dulin NO. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase. J. Biol. Chem. 2006;281:9971–9976. doi: 10.1074/jbc.M508778200. [DOI] [PubMed] [Google Scholar]
- 71.Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H, Mills GB, Kobayashi R, Hunter T, Lu Z. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J. Biol. Chem. 2007;282:11221–11229. doi: 10.1074/jbc.M611871200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu J, Bang AG, Kintner C, Orth AP, Chanda SK, Ding S, Schultz PG. Identification of the Wnt signaling activator leucine-rich repeat in Flightless interaction protein 2 by a genome-wide functional analysis. Proc. Natl. Acad. Sci. U. S. A. 2005;102:1927–1932. doi: 10.1073/pnas.0409472102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gunawardena HP, Huang Y, Kenjale R, Wang H, Xie L, Chen X. Unambiguous characterization of site-specific phosphorylation of leucine-rich repeat Fli-I-interacting protein 2 (LRRFIP2) in Toll-like receptor 4 (TLR4)-mediated signaling. J. Biol. Chem. 2011;286:10897–10910. doi: 10.1074/jbc.M110.168179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Takemori H, Kanematsu M, Kajimura J, Hatano O, Katoh Y, Lin XZ, Min L, Yamazaki T, Doi J, Okamoto M. Dephosphorylation of TORC initiates expression of the StAR gene. Mol. Cell. Endocrinol. 2007;265–266:196–204. doi: 10.1016/j.mce.2006.12.020. [DOI] [PubMed] [Google Scholar]
- 75.Azhar S, Reaven E. Scavenger receptor class BI and selective cholesteryl ester uptake: partners in the regulation of steroidogenesis. Mol. Cell. Endocrinol. 2002;195:1–26. doi: 10.1016/s0303-7207(02)00222-8. [DOI] [PubMed] [Google Scholar]
- 76.Wojtal KA, Hoekstra D, van Ijzendoorn SC. cAMP-dependent protein kinase A and the dynamics of epithelial cell surface domains: moving membranes to keep in shape. Bioessays. 2008;30:146–155. doi: 10.1002/bies.20705. [DOI] [PubMed] [Google Scholar]
- 77.Bergamaschi D, Samuels Y, O'Neil NJ, Trigiante G, Crook T, Hsieh JK, O'Connor DJ, Zhong S, Campargue I, Tomlinson ML, Kuwabara PE, Lu X. iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat. Genet. 2003;33:162–167. doi: 10.1038/ng1070. [DOI] [PubMed] [Google Scholar]
- 78.Tagami S, Eguchi Y, Kinoshita M, Takeda M, Tsujimoto Y. A novel protein, RTN-XS, interacts with both Bcl-XL and Bcl-2 on endoplasmic reticulum and reduces their anti-apoptotic activity. Oncogene. 2000;19:5736–5746. doi: 10.1038/sj.onc.1203948. [DOI] [PubMed] [Google Scholar]
- 79.Cullis DN, Philip B, Baleja JD, Feig LA. Rab11-FIP2, an adaptor protein connecting cellular components involved in internalization and recycling of epidermal growth factor receptors. J. Biol. Chem. 2002;277:49158–49166. doi: 10.1074/jbc.M206316200. [DOI] [PubMed] [Google Scholar]
- 80.Bai M, Gad H, Turacchio G, Cocucci E, Yang JS, Li J, Beznoussenko GV, Nie Z, Luo R, Fu L, Collawn JF, Kirchhausen T, Luini A, Hsu VW. ARFGAP1 promotes AP-2-dependent endocytosis. Nat Cell Biol. 2011;13:559–567. doi: 10.1038/ncb2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nisar S, Kelly E, Cullen PJ, Mundell SJ. Regulation of P2Y1 receptor traffic by sorting Nexin 1 is retromer independent. Traffic. 2010;11:508–519. doi: 10.1111/j.1600-0854.2010.01035.x. [DOI] [PubMed] [Google Scholar]
- 82.Mari M, Bujny MV, Zeuschner D, Geerts WJ, Griffith J, Petersen CM, Cullen PJ, Klumperman J, Geuze HJ. SNX1 defines an early endosomal recycling exit for sortilin and mannose 6-phosphate receptors. Traffic. 2008;9:380–393. doi: 10.1111/j.1600-0854.2007.00686.x. [DOI] [PubMed] [Google Scholar]
- 83.Tsai LC, Shimizu-Albergine M, Beavo JA. The high-affinity cAMP-specific phosphodiesterase 8B controls steroidogenesis in the mouse adrenal gland. Mol. Pharmacol. 2011;79:639–648. doi: 10.1124/mol.110.069104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sorkin A, von Zastrow M. Endocytosis and signalling: intertwining molecular networks. Nat Rev Mol Cell Biol. 2009;10:609–622. doi: 10.1038/nrm2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dutertre S, Cazales M, Quaranta M, Froment C, Trabut V, Dozier C, Mirey G, Bouche JP, Theis-Febvre N, Schmitt E, Monsarrat B, Prigent C, Ducommun B. Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G2-M transition. J. Cell Sci. 2004;117:2523–2531. doi: 10.1242/jcs.01108. [DOI] [PubMed] [Google Scholar]
- 86.Tong J, Taylor P, Moran MF. Proteomic analysis of the epidermal growth factor receptor (EGFR) interactome and post-translational modifications associated with receptor endocytosis in response to EGF and stress. Mol Cell Proteomics. 2014;13:1644–1658. doi: 10.1074/mcp.M114.038596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pearson GW, Earnest S, Cobb MH. Cyclic AMP selectively uncouples mitogen-activated protein kinase cascades from activating signals. Mol. Cell. Biol. 2006;26:3039–3047. doi: 10.1128/MCB.26.8.3039-3047.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kobayashi Y, Mizoguchi T, Take I, Kurihara S, Udagawa N, Takahashi N. Prostaglandin E2 enhances osteoclastic differentiation of precursor cells through protein kinase A-dependent phosphorylation of TAK1. J. Biol. Chem. 2005;280:11395–11403. doi: 10.1074/jbc.M411189200. [DOI] [PubMed] [Google Scholar]
- 89.Shah OJ, Iniguez-Lluhi JA, Romanelli A, Kimball SR, Jefferson LS. The activated glucocorticoid receptor modulates presumptive autoregulation of ribosomal protein S6 protein kinase, p70 S6K. J. Biol. Chem. 2002;277:2525–2533. doi: 10.1074/jbc.M105935200. [DOI] [PubMed] [Google Scholar]
- 90.Artinian N, Cloninger C, Holmes B, Benavides-Serrato A, Bashir T, Gera J. Phosphorylation of the Hippo Pathway Component AMOTL2 by the mTORC2 Kinase Promotes YAP Signaling, Resulting in Enhanced Glioblastoma Growth and Invasiveness. J. Biol. Chem. 2015;290:19387–19401. doi: 10.1074/jbc.M115.656587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mori K, Amano M, Takefuji M, Kato K, Morita Y, Nishioka T, Matsuura Y, Murohara T, Kaibuchi K. Rho-kinase contributes to sustained RhoA activation through phosphorylation of p190A RhoGAP. J. Biol. Chem. 2009;284:5067–5076. doi: 10.1074/jbc.M806853200. [DOI] [PubMed] [Google Scholar]
- 92.Williams CC, Basu A, El-Gharbawy A, Carrier LM, Smith CL, Rowan BG. Identification of four novel phosphorylation sites in estrogen receptor alpha: impact on receptor-dependent gene expression and phosphorylation by protein kinase CK2. BMC Biochem. 2009;10:36. doi: 10.1186/1471-2091-10-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Aboulaich N, Chui PC, Asara JM, Flier JS, Maratos-Flier E. Polymerase I and transcript release factor regulates lipolysis via a phosphorylation-dependent mechanism. Diabetes. 2011;60:757–765. doi: 10.2337/db10-0744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Funaba M, Zimmerman CM, Mathews LS. Modulation of Smad2-mediated signaling by extracellular signal-regulated kinase. J. Biol. Chem. 2002;277:41361–41368. doi: 10.1074/jbc.M204597200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.