

Abstract
Background
After myocardial infarction (MI), mitral valve (MV) tethering stimulates adaptive leaflet growth, but counterproductive leaflet thickening and fibrosis augment mitral regurgitation (MR), doubling heart failure and mortality. MV fibrosis post-MI is associated with excessive endothelial-to-mesenchymal transition (EMT), driven by transforming growth factor (TGF)-β overexpression. In vitro, losartan-mediated TGF-β inhibition reduces EMT of MV endothelial cells.
Objectives
The authors tested the hypothesis that profibrotic MV changes post-MI are therapeutically accessible, specifically by losartan-mediated TGF-β inhibition.
Methods
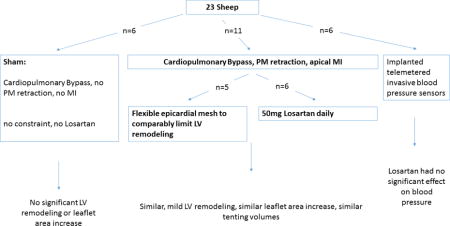
We studied 17 sheep, including 6 sham-operated controls and 11 with apical MI and papillary muscle retraction short of producing MR: 6 treated with daily losartan, and 5 untreated, with flexible epicardial mesh comparably limiting left ventricular (LV) remodeling. LV volumes, tethering, and MV area were quantified by 3-dimensional echocardiography at baseline and at 60 ± 6 days, and excised leaflets were analyzed by histopathology and flow cytometry.’
Results
Post-MI LV dilation and tethering were comparable in losartan-treated and untreated LV-constraint sheep. Telemetered sensors (n = 6) showed no significant losartan-induced arterial pressure changes. Losartan strongly reduced leaflet thickness (0.9 ± 0.2 mm vs. 1.6 ± 0.2 mm; p < 0.05; 0.4 ± 0.1 mm shams), TGF-β and downstream phosphorylated extracellular-signal–regulated kinase and EMT (27.2% ± 12.0% vs. 51.6% ± 11.7% α-smooth-muscle-actin-positive endothelial cells, p < 0.05; 7.2% ± 3.5% shams), cellular proliferation, collagen deposition, endothelial cell activation (vascular cell adhesion molecule-1 expression), neovascularization, and cells positive for cluster of differentiation (CD)45, a hematopoietic marker associated with post-MI valve fibrosis. Leaflet area increased comparably (17%) in constrained and Losartan-treated sheep.
Conclusions
Profibrotic changes of tethered MV leaflets post-MI can be modulated by losartan without eliminating adaptive growth. Understanding the cellular and molecular mechanisms could provide new opportunities to reduce ischemic MR.
Keywords: CD45, endothelial-to-mesenchymal transition, ischemic mitral regurgitation, losartan, TGF-β, VCAM-1
Graphical abstract
INTRODUCTION
Ischemic mitral regurgitation (MR) is a frequent complication of myocardial infarction (MI) that doubles mortality and heart failure (1). Papillary muscle (PM) displacement due to left ventricular (LV) remodeling results in leaflet tethering that restricts valve closure. Ischemic MR also reflects a mismatch between valve and ventricular size as the damaged heart enlarges but the MV fails to keep pace (2–9). Stiffening and fibrosis further limit valve closure (10–12). Mechanical stress imposed by tethering alone adaptively increases mitral valve (MV) leaflet area and is associated with endothelial-to-mesenchymal transformation (EMT) (2,13–15), an early developmental process: In EMT, the normally quiescent valve endothelial cells (VECs) loosen their cell contacts and express α-smooth muscle actin (SMA) while migrating into interstitial layers of the valve. Transforming growth factor beta (TGF-β) signaling, induced by tethering alone, augments EMT (16,17). In the ischemic setting (tethering plus myocardial infarction), however, excessive TGF-β signaling stimulates exuberant EMT, resulting in profibrotic changes of the leaflets, such as markedly increased thickness, cellular proliferation, and excessive matrix turnover with collagen deposition, along with increased cells expressing cluster of differentiation 45 (CD45), a marker associated with scar-forming cells, also influenced by TGF-β (18–21). These MV leaflet changes are likely precursors to the development of stiff and fibrotic human valves found at the late heart failure stage, and further impair closure by reducing systolic leaflet expansion and the flexibility needed to bend and seal effectively (10–12,18). Current therapeutic strategies based on revascularization and heart failure management often fail to relieve the restricted leaflet closure imposed by displaced leaflet attachments to the infarcted LV walls. Intrinsic valve changes likely persist after downsizing annuloplasty, and, along with persistent LV remodeling with increased heart failure, may also contribute to the MR recurrence evident in 32.6% of patients at 1 year and 58.8% at 2 years, as recently observed by the CardioThoracic Surgical Trials Network, with increased heart failure (22–24). The cellular and signaling changes seen in post-MI valves support the hypothesis that these profibrotic processes are therapeutically accessible. Specifically, as an initial approach, modulation by TGF-β inhibition seems reasonable based on the overexpression of TGF-β in tethered valves post-MI (2) and its stimulation of EMT (16,17,25). In vitro, TGF-β-induced EMT is effectively blocked in mitral valve endothelial cells (VECs) by losartan (17), a readily available, Food and Drug Administration–approved, and well-tolerated agent, which was therefore used to test the hypothesis. Losartan also blocks angiotensin-II receptors, and there is evidence for beneficial antifibrotic effects of angiotensin receptor blockers in aortic stenosis (26). In order to control for the losartan-mediated reduction in LV remodeling, we used our published tethering plus MI model with a flexible mesh sutured to the heart (LV constraint) to limit LV remodeling to a similar extent as losartan treatment so that the valve changes can be compared in hearts with comparable volumes and leaflet tethering (Figure 1), with tethering produced by papillary muscle retraction short of producing MR (2). Based on this experimental design, our aim was to test specifically for effects of losartan on MV morphology and cell biology, and not MR, while also avoiding potential confounding effects of MR jets on the MV itself (27).
Figure 1. Model and Examples of MV Leaflet Changes.
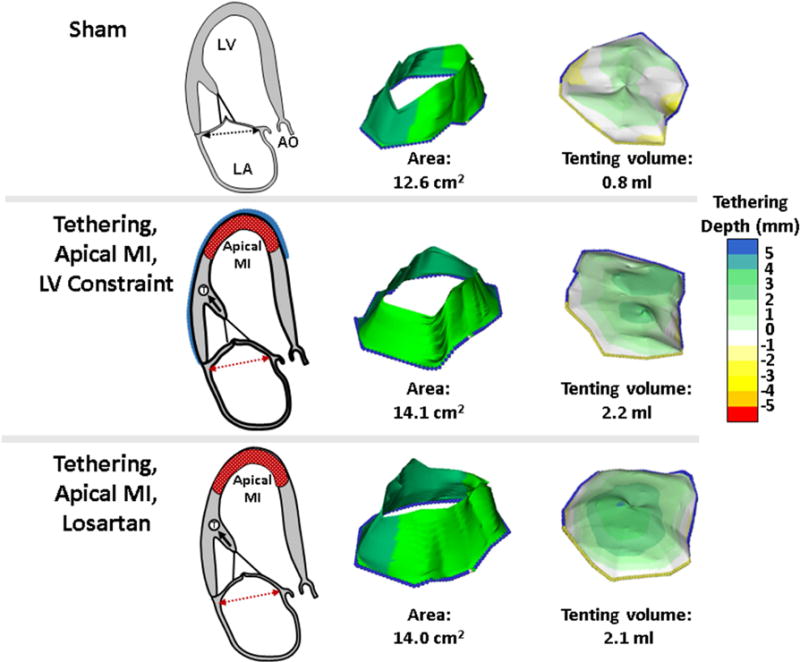
Both the losartan-treated and untreated sheep with LV constraint display mild LV remodeling and tethering of the MV leaflets relative to the annulus with resulting increased tenting volume relative to sham-operated controls (right panels). Post-MI leaflet growth over time results in comparably increased leaflet areas (middle panels). The model provides a controlled in vivo environment with standardized tethering and apical MI that is not directly adjacent to the subvalvular apparatus. Ao = ascending aorta; LA = left atrium; LV = left ventricle, MI = myocardial infarction; MV = mitral valve.
METHODS
EXPERIMENTAL MODELS
Seventeen adult Dorsett hybrid sheep (weight > 45 kg) studied over 60 ± 6 days were compared: 6 sham-operated sheep and 11 with leaflet tethering plus MI as described later and in Online Figure 1. Of the11 tethered plus MI sheep, 6 were treated with daily losartan (50 mg by mouth), shown to be effective in reducing LV remodeling in sheep (28), and 5 had a flexible Prolene surgical mesh (Ethicon, Somerville, New Jersey). The mesh was loosely sutured circumferentially over the LV walls from base to apex to control post-MI LV remodeling and limit it to a degree comparable to that in the losartan-treated group (2). Our approach allows independent production of PM displacement and MI (2,15). After pericardial cradle construction and initiation of cardiopulmonary bypass, the left atrium was opened and suture loops were inserted via the MV orifice into the exposed PM tips. The loops were buttressed by Teflon felt pledgets, exteriorized to the epicardium overlying the PMs, and pulled through a Dacron anchoring patch. Retracting these sutures parallel to the PM axis pulled the PM tips apically, creating an adjustable and standardized degree of mechanical leaflet tethering. The heart was restarted and a limited apical infarct produced by distal left anterior descending coronary artery ligation, avoiding interference with PM tethering from inferior wall bulging. Final suture length was then adjusted in the beating heart under echocardiographic guidance just short of producing MR, and the suture was knotted against an anchoring patch. Losartan (Teva Pharmaceuticals, Sellersville, Pennsylvania) treatment was initiated post-MI on the day of MI creation. Sham controls underwent cardiopulmonary bypass without MI, leaflet tethering, or LV constraint.
Animals were cared for over 60 ± 6 days and euthanized after thoracotomy, with echocardiographic follow-up including 3-dimensional (3D) mitral leaflet area. These studies conformed to National Institutes of Health animal care guidelines and had institutional care approval.
IMAGING
Standard 2-dimensional and full-volume echocardiographic data sets were obtained epicardially at baseline (before and after model creation) and 60 days later, before euthanasia. Data were collected with an ie33 scanner (Philips, Andover, Massachusetts) using 3.5- to 5-MHz probes (S5 and X3). Volumetric datasets were ECG-gated from 4 to 7 consecutive beats. MR was assessed by vena contracta immediately after MI creation with PM tethering and before euthanasia. Digital data were analyzed with validated custom Software (Omni 4D, Mark Handschumacher) (5,7). Total leaflet area was measured in the unstretched, noncoapting position (diastole) to factor out superimposed systolic tethering forces, a validated method with reasonable inter- and intra-observer variability (5,7).
BLOOD PRESSURE MEASUREMENTS
To determine the effect of losartan on blood pressure and heart rate continuously (29), 6 sheep not included in the 17 study animals described previously underwent implantation of a blood pressure probe attached to a wireless transmitting system (TSE systems, Inc., Chesterfield, Missouri) (Online Appendix).
TISSUE HARVESTING, HISTOLOGY, AND FLOW CYTOMETRY
The left atrium was opened and the LV wall was dissected starting from the anterolateral commissure in a sterile manner, with irrigation of pre-cooled phosphate buffered saline. Both leaflets were divided for histopathology (frozen in optimal cutting temperature compound at −80°C), and cell isolation and flow cytometry (transported in pre-cooled physiological collecting medium). Blocks were sectioned (6-μm) and stained with hematoxylin-eosin for overall morphology. Masson’s trichrome and picrosirius red staining assessed collagen content, orientation, and fibrosis. Picrosirius red was visualized under circular polarized light (Eclipse 50i polarizing microscope, Nikon, Melville, New York). Elastin was detected using the van Gieson method. Immunohistochemistry was performed with the avidin-biotin-peroxidase method, as previously described (4). Leaflet thickness was determined by measuring the 10 thickest areas across the midportion of the anterior and posterior leaflets. Endothelial cells were identified with an anti-CD31 antibody (Santa Cruz Biotechnology, Santa Cruz, California). Activated valvular interstitial cells were determined by staining for α-SMA (anti-α-SMA antibody, clone 1A4; Sigma, St. Louis, Missouri) (30,31). Vascular cell adhesion molecule-1 (VCAM-1; VCAM-1 antibody, Bioss, Woburn, Massachusetts) staining assessed endothelial cell activation. CD45, considered a pan-hematopoietic marker, was identified by mouse anti-sheep CD45 (AbD Serotec); anti-Ki67 antibody (Abcam, Cambridge, Massachusetts) determined cellular proliferation. To explore mechanisms of cellular changes, sections were stained with anti-TGF-β-1 (R&D Systems, Minneapolis, Minnesota) for latent and activated protein (2), and anti-phosphorylated extracellular-signal–regulated kinase (p-ERK) antibody (Cell Signaling, Danvers, Massachusetts).
For flow-cytometry, MV leaflet tissue was minced into 2–3mm pieces and incubated with 3 ml Cell Dissociation Buffer (ThermoFisher Scientific, Waltham, Massachusetts), an enzyme-free, ethylenediaminetetraacetic acid–containing solution, for 4 min at 37°C. Repeated pipetting produced a single-cell suspension. Endothelial cells were detected and quantified with murine anti-sheep CD31 antibody conjugated to fluorescein isothyocyanate (AbD Serotec); activated cells were detected with a murine anti-human α-SMA (clone 1A4) conjugated to phycoerythrin (R&D Systems) (2,15).
STATISTICAL ANALYSIS
Continuous variables are expressed as mean ± SD. One-way analysis of variance with Tukey-Kramer post hoc tests were used to compare multiple groups of interest. Paired Student t-tests were used to compare baseline and euthanasia results within groups, as well as blood pressures and heart rates on and off losartan. Statistical significance was set at p < 0.05 (2-sided).
RESULTS
Post-MI LV dilation and dysfunction were modest and comparable in the sheep with tethered MVs in the losartan-treated and untreated (LV constraint) groups (Tables 1 and 2). At baseline, there were no significant differences in left ventricular volumes or mitral valve tenting volumes (Table 2). Volumes at euthanasia were comparable in these 2 groups and greater than in shams (Table 2, Figure 2A upper left). Tenting volumes and tethering distances increased in both groups, and were not significantly different at euthanasia between groups (Figure 1, right, Table 1). By study design (tethering short of MR), MR remained trace-to-mild in both groups, with vena contracta of 1.6 ± 0.9 mm versus 2.1 ± 1.1 mm in the losartan versus constraint groups, p = 0.58. In the sheep studied by implanted telemetry, systolic, diastolic, and mean arterial blood pressure and heart rate were not significantly different on or off losartan, averaged over 3 to 4 days each (Figure 2A, lower right).
Table 1.
Echocardiographic Baseline and Euthanasia Measurements: Tethered + MI + Losartan Model
| Baseline | Euthanasia | p Value | |
|---|---|---|---|
| LVEDV (ml) | 45.4 ± 8.3 | 62.4 ± 6 | 0.0011 |
| LVESV (ml) | 16.9 ± 3.1 | 30.6 ± 6.6 | 0.0005 |
| LVEF (%) | 62.1 ± 7.4 | 51 ± 8.9 | 0.0184 |
| Posteromedial PM to lateral MA trigone (systole), mm | 34.3 ± 3.3 | 38.7 ± 4.6 | 0.0070 |
| Anterolateral PM to medial MA trigone (systole), mm | 35.9 ± 2.4 | 40 ± 1.6 | 0.0044 |
| Anterolateral to posteromedial PM distance (systole), mm | 20 ± 1.4 | 21.6 ± 1.9 | 0.0095 |
| MV leaflet area (cm2) | 11.4 ± 1 | 13.3 ± 0.7 | 0.0003 |
| Annular area (diastole, cm2) | 6.3 ± 0.8 | 8.5 ± 0.4 | 0.0688 |
| Tenting volume (cm3) (early systolic) | 1.2 ± 0.5 | 2 ± 0.6 | 0.0002 |
| Tenting volume (cm3) (late systolic) | 0.7 ± 0.2 | 1.7 ± 0.4 | 0.0002 |
Values are mean ± SD.
LVEDV = left ventricular end-diastolic volume; LVESV = left ventricular end-systolic volume; LVEF = left ventricular ejection fraction; MA = mitral annulus; MV = mitral valve; PM = papillary muscle
Table 2.
Echocardiographic, Histological, and Flow Cytometric Results of LV Constraint Versus Losartan-Treated Sheep
| Sham | Tethered + MI LV Constraint | Tethered + MI Losartan | |
|---|---|---|---|
| Baseline LVEDV, ml | 47.0 ± 4.4 | 47.9 ± 2.7 | 45.6 ± 8.3 |
| Euthanasia LVEDV, ml | 47.5 ± 6.7 | 57.7 ± 11.6 | 62.4 ± 6* |
| Baseline LVESV, ml | 16.6 ± 3.4 | 18.7 ± 2.9 | 16.9 ± 3.1 |
| Euthanasia LVESV, ml | 19.6 ± 3.9 | 33.7 ± 8.1* | 30.6 ± 6.6* |
| Baseline tenting volume, cm3 | 1.2 ± 0.5 | 1.2 ± 0.3 | 1.2 ± 0.5 |
| Euthanasia tenting volume, cm3 | 1.3 ± 0.2 | 2.3 ± 0.4* | 2 ± 0.6* |
| Leaflet area increase, % | −0.9 ± 4.1 | 17.8 ± 8.1* | 16.7 ± 5.4* |
| Leaflet thickness, mm | 0.4 ± 0.05 | 1.6 ± 0.2* | 0.8 ± 0.2*‡ |
| VECs coexpressing α-SMA, % | 7.2 ± 3.4 | 51.6 ± 11.7* | 27.2 ± 12‡ |
| CD45-positive cells/HPF | 3.3 ± 1.2 | 21.9 ± 3.5* | 10.7 ± 1.3*‡ |
| Ki67-positive cells/HPF | 4.7 ± 1 | 32.5 ± 14* | 4.3 ± 1.2‡ |
| Microvessels/HPF | 0.9 ± 1.6 | 4.9 ± 1.9* | 0.3 ± 0.8‡ |
Values are mean ± SD.
p < 0.05 sham vs. LV constraint vs. losartan
p < 0.05 tethered + MI LV constraint vs. tethered + MI losartan.
HPF = high-powered field, LV = left ventricular; LVEDV = left ventricular end-diastolic volume; LVESV = left ventricular end-systolic volume; MI = apical myocardial infarction; SMA = smooth muscle actin; VECs = valvular endothelial cells
Figure 2. LV, MV, and Hemodynamic Changes.
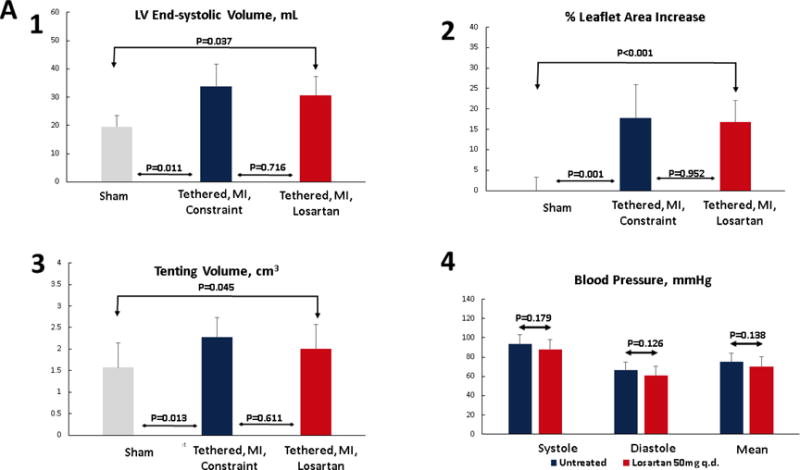
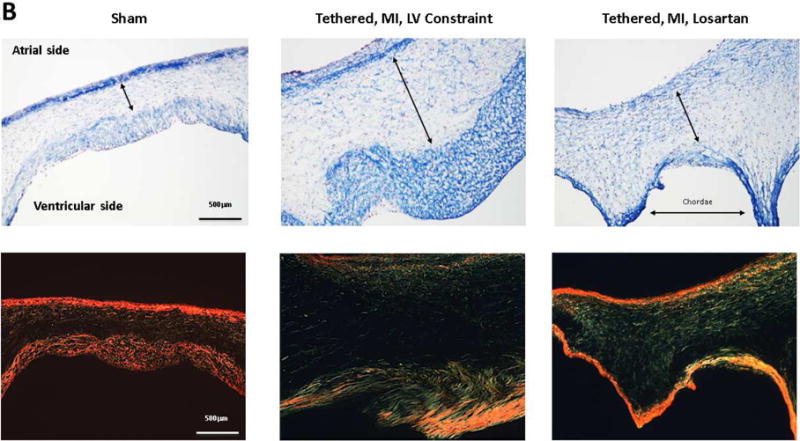
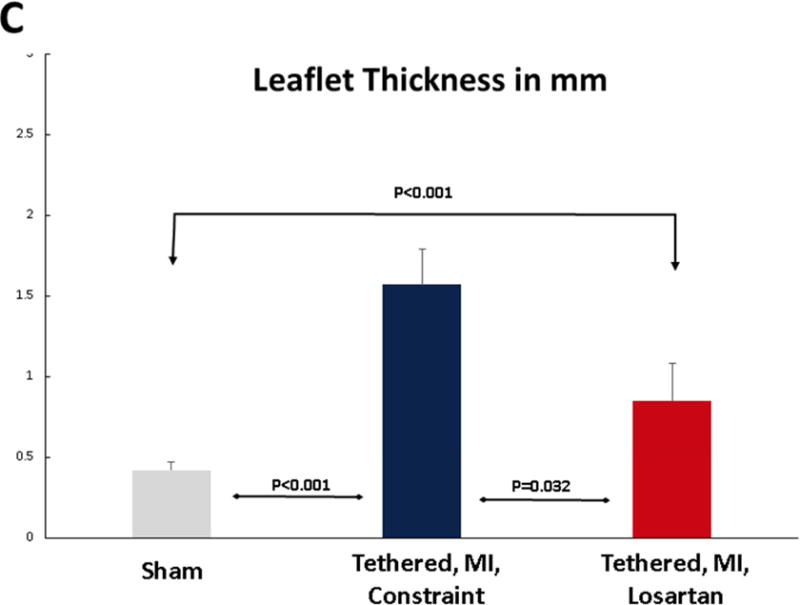
(A) Post-MI LV dilation was comparable in sheep with tethered MVs treated with losartan and untreated with LV constraint (upper left), with similar tenting volumes (lower left). Invasively monitored blood pressures were not significantly different in 6 losartan-treated or untreated sheep (lower right panel). Leaflet area increased comparably in losartan-treated and untreated LV constraint sheep post-MI (upper right). (B) Tethering and myocardial infarction (MI) result in leaflet thickening via expansion of the spongiosa layer (arrows, Masson, upper panels; picrosirius red, lower panels) and the collagen-rich valvular fibrosa near the ventricular leaflet surface. Valve thickening was reduced in losartan-treated animals (right panels), with decreased fibrosa thickness (upper right). Picrosirius red showed poorly organized collagen in the spongiosa layer predominantly in the tethered plus MI LV constraint group without losartan (center panels) with a combination of compacted and less well-organized collagen in the untreated valves. (C) Leaflets were thicker in the LV constraint versus losartan-treated sheep. Abbreviations as in Figure 1.
Open diastolic mitral leaflet surface increased to a comparable extent in both losartan-treated and LV constraint groups (17% to 18%) compared with no change over time in sham controls (p < 0.05) (Tables 1 and 2, Figure 2A upper right; illustrated in Figure 1, right). However, 60 days post-MI, leaflets from sheep without losartan were roughly twice as thick as valves from losartan-treated animals (1.57 ± 0.22 mm vs. 0.85 ± 0.23 mm, respectively, vs. 0.42 ± 0.05 mm in shams, p < 0.05) (Figures 2B and 2C). The collagen-rich valvular fibrosa, near the ventricular leaflet surface, was thicker in the untreated versus losartan-treated valves (Figure 2B, center vs. right), with a combination of collagen compaction and disorganization, suggesting abnormal matrix remodeling, in the untreated valves, which was well-visualized in Picrosirius red images (Figure 2B, lower panel).
EMT was markedly increased in post-MI sheep not treated with losartan compared with shams. Flow cytometry showed that 51.6% ± 11.7% of endothelial cells expressing CD31 coexpressed α-SMA, indicating EMT, versus 27.2% ± 12.1% in losartan-treated sheep and 7.2% ± 3.4% in shams (p < 0.05) (Figure 3A). Consistent with this, extensive α-SMA staining was seen in the endothelium extending into the underlying interstitium under the atrial surface of the valve in post-MI sheep, but was notably reduced in losartan-treated sheep (Figure 3B, top row). Staining for TGF-β and downstream p-ERK, not evident in shams, was substantial in the untreated post-MI valves, extending into the interstitial area. Losartan considerably reduced this staining and its interstitial extent (Figure 3B, lower 2 rows).
Figure 3. MV EMT, TGF-β, and Downstream p-ERK Signaling.
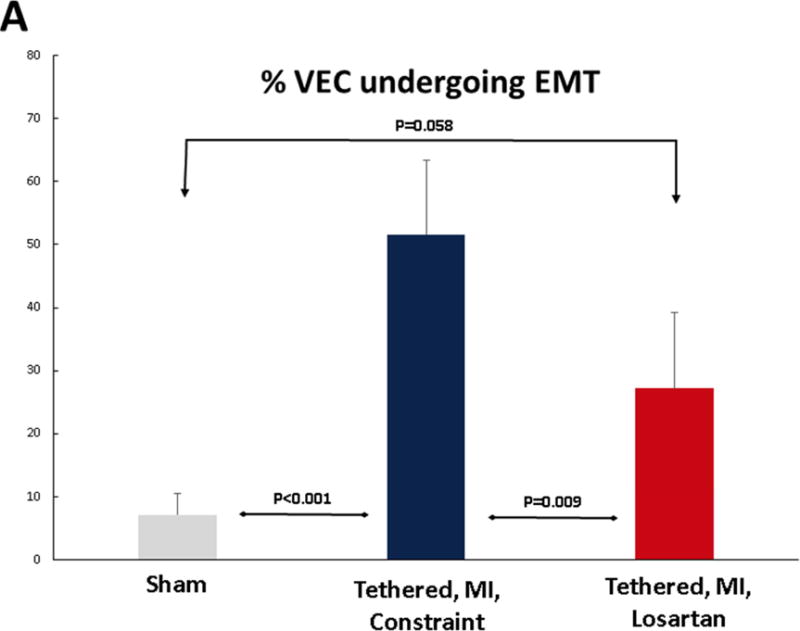
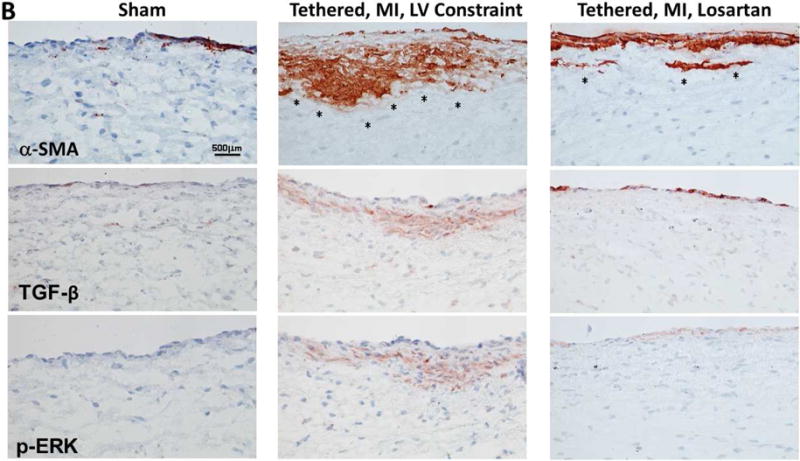
(A) Quantitative flow-cytometry analysis of SMA-positive valvular endothelial cells (VECs), indicating a marked endothelial-to-mesenchymal transition (EMT) in LV constraint sheep, which was significantly blunted in losartan-treated sheep. (B) Staining for the interstitial marker SMA (upper panels) shows increasing extension of SMA-positive cells into the leaflet interstitium (asterisks), extensively in untreated LV constraint (upper panel, center) and only mildly in the losartan-treated sheep (upper panel, right). Transforming growth factor-beta (TGF-β) (middle panels) and downstream phosphorylated-extracellular-signal regulated kinase (p-ERK) staining (lower panels) demonstrate excessive signaling spreading into subendothelial layers of the LV constraint valves (center panels) versus only mild, mainly endothelial activation in losartan-treated valves (right). SMA = α-smooth muscle actin. Other abbreviations as in Figure 1.
CELL POPULATIONS AND PROLIFERATION
Post-MI valves showed extensive cell staining for CD45, a protein tyrosine phosphatase typically expressed in hematopoietic cells and recently associated with post-MI MV fibrosis and EMT (18). CD45 was negligible in sham valves. CD45 staining was markedly reduced by losartan from 21.9 ± 3.5 positive cells per high-power field (hpf) to 10.7 ± 1.3 cells/hpf (p < 0.05) (Table 2, Figures 4A and 4B). Cellular proliferation, assessed by Ki67 staining, was also markedly reduced by losartan, from 32.5 ± 14 cells/hpf to 4.3 ± 1.2 cells/hpf (Table 2, Figures 4A and 4B). Microvessels, a potential route of entry for hematopoietic cells, were also increased post-MI and substantially reduced in losartan-treated sheep (Table 2, Figures 4A and B). VEC activation, evident by expression of VCAM-1, a leukocyte adhesion molecule normally absent in quiescent valve endothelium or tethered valves without MI (2), was increased in post-MI valves, but not detected in sham or losartan-treated valves (Figures 4A and 4B). Further evidence of altered extracellular matrix remodeling in post-MI valves was provided by poorly organized collagen in the spongiosa layer, predominantly in the untreated tethered plus MI LV constraint valves and thicker collagen-rich valvular fibrosa, with areas of compacted and less well-organized collagen in the untreated valves (Figure 2B).
Figure 4. MV CD45-Positive Cells, Cellular Proliferation, Neovascularization, and Endothelial Activation.
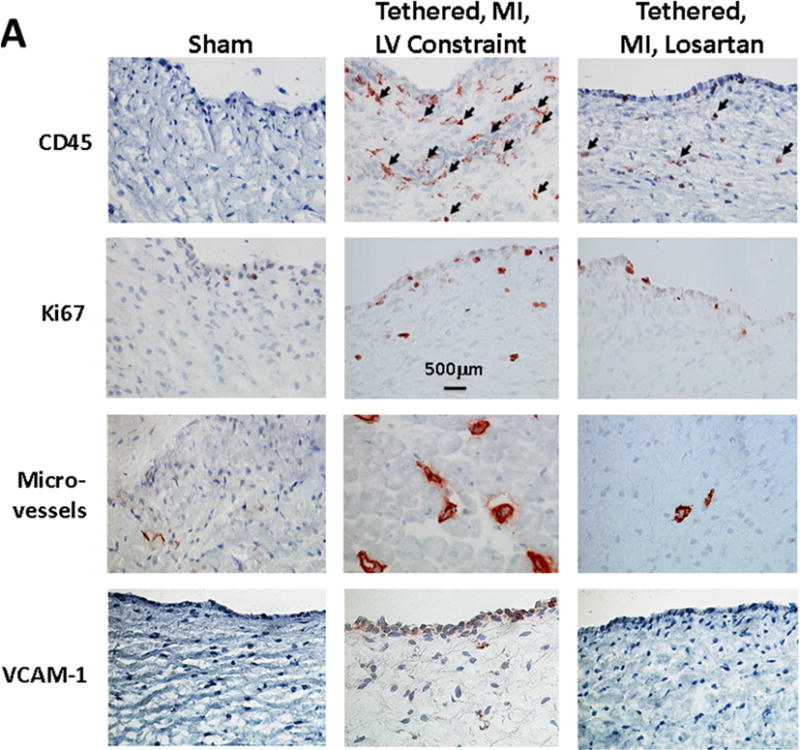
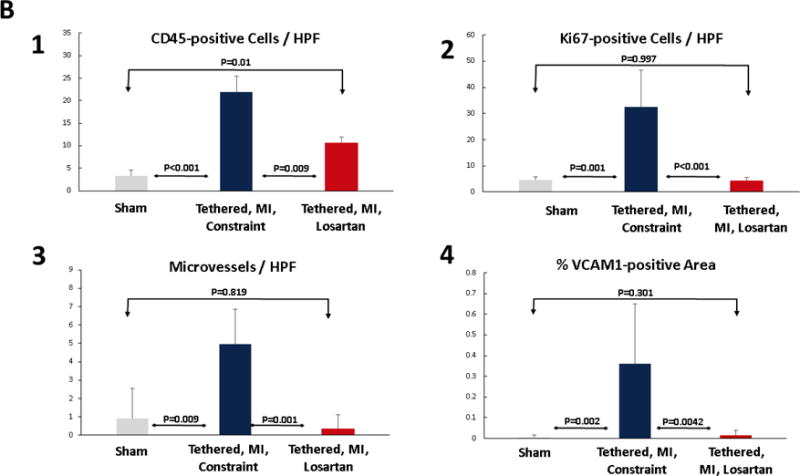
(A) Immunohistochemistry shows CD45-positive cells (arrows) are frequent in the untreated post-MI LV constraint valves (center) with marked increase in cellular proliferation indicated by Ki67 staining, significantly increased number of microvessels and prominent endothelial activation indicated by VCAM-1, all markedly reduced by losartan (right panels) and largely absent in the quiescent sham group valves (no tethering or MI). (B) Histological quantification confirms these losartan effects. CD = cluster of differentiation; HPF = high-powered field; VCAM-1 = vascular cell adhesion molecule-1. Other abbreviations as in Figure 1.
DISCUSSION
The MV actively adapts to mechanical tethering alone through EMT and matrix remodeling, which facilitate leaflet growth (15). In the absence of MI, as in aortic regurgitation, the MV enlarges to match the dilating LV without MR (32). After MI, however, potentially maladaptive profibrotic valve changes can ultimately produce stiff, fibrotic leaflets (10,11) that can accelerate the vicious cycle of MR and LV remodeling (Central Illustration). These profibrotic valve changes persist even if LV dilation is limited (2), suggesting mechanisms other than purely mechanical stress. The possibility of therapeutic modulation is suggested by increased TGF-β and its downstream signaling, and processes associated with TGF-β: EMT; cellular hyperproliferation; and infiltration of CD45-positive cells (2). These CD45+ cells can transform into fibrocytes and myofibroblasts that release more TGF-β (33). Microvascularization might facilitate blood-borne cell recruitment and energy–demanding proliferation and matrix turnover. Profibrotic leaflet changes promote leaflet thickening (2,10,11), which was shown to impair coaptation (12) and increase MR (unpublished data, J.M. Beaudoin, April 2017).
Central Illustration. Losartan Reduces Post-MI Profibrotic Mitral Valve Changes Without Eliminating Adaptive Leaflet Growth.
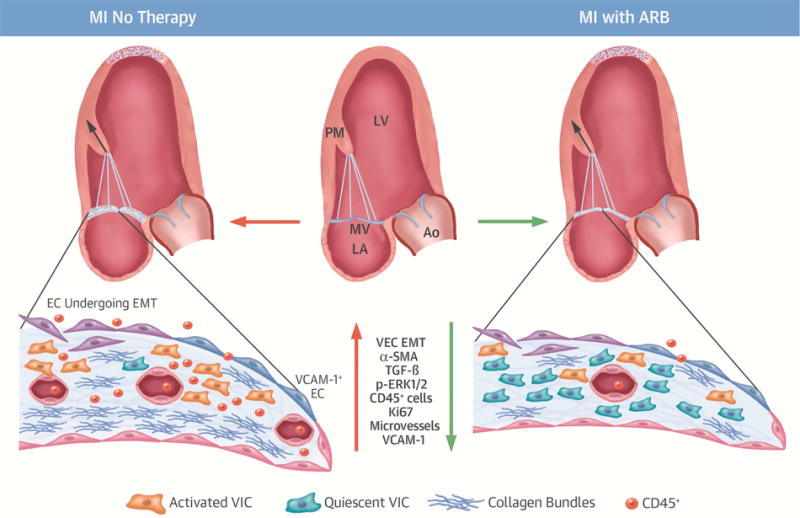
In a controlled myocardial infarction (MI) model, treatment with the angiotensin receptor blocker (ARB) losartan reduces mitral valve (MV) fibrosis, with far less collagen, decreased endothelial-to-mesenchymal transition (EMT) as fibrotic substrate, and less inflammatory endothelial activation (VCAM-1). Compared with untreated MVs of comparable left ventricular (LV) volume, there are far fewer activated valvular interstitial cells (VICs, α-SMA) and fibrosis-associated CD45-positive cells, along with significantly less TGF-β and p-ERK1/2 signaling. Leaflet cell turnover (Ki67) is lower, with fewer microvessels. ARB therapy in the post-MI LV directly modulates MV leaflet adaptation with less fibrosis, but maintained leaflet area growth. This opens the possibility of leaflet-specific therapy to improve MV adaptation and reduce regurgitation and heart failure after MI. Ao = aorta; CD = cluster of differentiation; EC = endothelial cells; LA = left atrium; p-ERK = phosphorylated-extracellular-signal regulated kinase; PM = papillary muscle; SMA = α-smooth muscle actin; TGF = transforming growth factor; VCAM = vascular cell adhesion molecule; VEC = valvular endothelial cells.
Our results show that a pharmacological intervention can reduce profibrotic MV changes in vivo. Despite similar LV volumes, tethering distances, and tenting volumes, MVs in animals treated with losartan showed markedly decreased EMT, cellular proliferation and thickness, (2,15) endothelial activation (VCAM-1 expression), presence of CD45+ cells, neovascularization, and collagen deposition. The abundant post-MI expression of TGF-β, a key driver of fibrosis, was markedly reduced, but not absent, in losartan-treated valves, modulating EMT. Extensive EMT sets the stage for fibrosis (34), yet EMT remains associated with leaflet growth (2,15). TGF-β might therefore be a double-edged sword: beneficial by increasing leaflet area to match LV remodeling, but also counterproductively causing fibrosis and impaired coaptation. Importantly, losartan-modulated EMT still allowed a comparable increase in total leaflet surface area while inhibiting profibrotic leaflet changes and excessive collagen deposition. TGF-β can also drive fibrosis by activating valve interstitial cells to become contractile SMA-positive myofibroblasts that compact and remodel the extracellular matrix (33,35–37), further augmenting ischemic MR by limiting the increase in valve area. TGF-β may originate from the valve itself (2,15,18), and is released by the ischemic myocardium (35) together with other proinflammatory cytokines (38). Sustained cytokine up-regulation may contribute to chronic post-MI remodeling (39,40). Cytokine-induced endothelial activation (38) can recruit circulating cells into the tissue, further fueling cytokine production and fibrosis (39–42). Losartan might therefore not only reduce TGF-β in the valve, but also disrupt cytokine crosstalk among cardiac tissues.
CD45+ CELLS
One hypothesis is that these are fibrocytes, circulating bone marrow– derived cells that increase in response to inflammatory cytokines and exit the blood at sites of injury, where they adopt a myofibroblast phenotype, express α-SMA, and produce collagens 1 and 3 and cytokines (21,43,44). Beneficial in wound healing, fibrocytes produce sclerosis in fibrosing pulmonary, renal, and cutaneous diseases. Fibrocyte inhibition reduces LV remodeling (19). It is therefore reasonable to propose that the CD45+ cells represent fibrocytes recruited by MI-induced cytokines, endothelial activation, and TGF-β (45,46), which promote their differentiation to myofibroblasts (33,44) that secrete more TGF-β (47).
Our recent study also unexpectedly implicates the MV endothelium as a source of fibrotic CD45+ cells post-MI: mitral VECs express CD45 in response to TGF-β in vitro, undergoing EMT with a fibrotic gene expression profile that includes collagens 1 and 3, TGF-β, and the EMT markers α-SMA and Slug (18). These changes are blocked by a CD45 phosphatase inhibitor and are specific to MV endothelial cells (18). A substantial proportion of post-MI CD45+ MV cells also express markers for endothelial cells undergoing EMT, and this proportion correlates well with valve fibrosis (18). In the current study, losartan markedly reduced cells expressing this fibrosis-associated marker.
MECHANISMS OF LOSARTAN ACTION
In many tissues, losartan decreases production of TGF-β and its receptor and angiotensin (AgII)-induced release of latent TGF-β (48–51). It blocks the interaction of AgII with its AT1 receptor, decreasing TGF-β signaling through phosphorylated Smad2 (37,52). Importantly, losartan inhibits phosphorylation of ERK1/2 for noncanonical TGF-β signaling (53,54), thereby inhibiting TGF-β–driven EMT in cultured mitral VECs (17). Losartan inhibits AgII-induced expression of endoglin, which promotes the fibrogenic effects of TGF-β (55), as well as AT1 receptor up-regulation by TNF-α (56) and AgII-mediated cardiac fibroblast activation. It can potentially decrease TGF-β–induced fibrocyte recruitment and myofibroblast differentiation in many tissues (33,44). Through such effects on the MV, losartan can potentially inhibit fibrosis (36), while maintaining adaptive leaflet growth with flexible leaflet closure. Although angiotensin receptor inhibitors are part of the antiremodeling approach to post-MI patients (57), it is of interest that ~37% of patients in the CardioThoracic Surgical Trials Network severe ischemic MR study (22) were neither on such medication nor on angiotensin-converting enzyme (ACE) inhibitors (CTSN Data Coordinating Center, personal data communication, February 2017). Demonstrating MR reduction might strengthen indications for such therapies.
LIMITATIONS AND FUTURE DIRECTIONS
Early treatment with angiotensin receptor blockers reduces LV post-infarct remodeling (58), but we limited LV remodeling by constraint to a similar degree, providing comparable tethering distances and tenting volumes. There is no evidence indicating blood pressure effects on MV remodeling and blood pressure did not change in nonhypertensive patients treated with losartan long-term (59) or in invasively monitored pressures in our treated and untreated sheep. The present model allows us to study the drug effect in a controlled in vivo environment with standardized tethering and an apical MI not adjacent to the subvalvular apparatus. Whether the drug reduces maladaptation and MR in the clinical scenario of inferior MI remains to be demonstrated, as our current design controlled for LV volume and MR to isolate the drug’s effect on the valve (27). The likelihood of treatment benefit with MR is suggested by the elevation of fibrosis-driving cytokines in chronic MR and the inadequate MV leaflet adaptation after inferior MI (4,5) as losartan targets, and by suggestive evidence for reduced progression of MV leaflet thickness in ARB-treated post-MI patients (unpublished data, J.M. Beaudoin, April 2017). Whether maladaptive fibrosis and need for treatment extend beyond the 2 months studied is beyond the scope of the present work, as is testing other potential therapies suggested by the inflammatory and profibrotic processes. Future studies can also test whether ACE inhibition reduces profibrotic valve changes less than AT1 receptor blockade by losartan because ACE-inhibition cannot, in principle, selectively block p-ERK signaling through AT2 receptors as losartan does (53,54). Losartan reduces CD45+ cell recruitment and fibrosis (60,61). However, there is no current mechanism for testing losartan on the post-MI valve without also treating the infarcted LV. Importantly, our underlying hypothesis is that the MV is caught in the inflammatory process triggered by MI; therefore, if losartan acts on the myocardial changes (60–62) and that benefits the valve, the hypothesis is still confirmed.
CONCLUSIONS
Profibrotic changes in the mitral valve occurring after MI, such as excess cellular proliferation and valve thickening, the presence of CD45-positive cells, endothelial activation, and excess matrix remodeling, can be modulated using losartan without eliminating the capacity for adaptive EMT and leaflet growth. Such leaflet-directed pharmacological intervention suggests a novel therapeutic approach to reduce ischemic MR as a driving force for ventricular remodeling.
Supplementary Material
Condensed Abstract.
Mitral regurgitation (MR) doubles heart failure and mortality after myocardial infarction (MI). This MR is caused by mitral leaflet tethering, compounded by intrinsic fibrotic changes in the leaflets themselves in the setting of excessive transforming growth factor beta (TGF-β) signaling. The angiotensin receptor blocker losartan, which inhibits TGF-β, reduces these profibrotic cellular and matrix changes without eliminating adaptive leaflet growth compensatory for left ventricular remodeling. This opens the possibility for leaflet-specific therapy to improve mitral valve adaptation and reduce regurgitation and heart failure after MI.
PERSPECTIVES.
COMPETENCY IN MEDICAL KNOWLEDGE
The tethered MV leaflets have the potential for growth to adapt to LV cavity dilation though expression of embryonic pathways such as endothelial-to-mesenchymal transition. After MI, TGF-β signaling promotes cellular proliferation, matrix turnover, fibrosis, and leaflet thickening that limits valve closure and exacerbates regurgitation. Treatment with losartan modulates these changes without eliminating adaptive leaflet growth.
TRANSLATIONAL OUTLOOK
Further research is needed to elucidate the cellular and molecular mechanisms underlying progression of ischemic mitral regurgitation and expose potential avenues of leaflet-directed therapy to favorably influence ventricular remodeling post-MI.
Acknowledgments
Funding: This work was supported in part by grant 07CVD04 from the Fondation Leducq, Paris, France, for the Transatlantic MITRAL Network of Excellence. Research reported in this manuscript was also supported by the NHLBI of the National Institutes of Health under award number R01HL109506 to Drs. Levine, Aikawa, and Bischoff. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Additional support was from grants R01 HL128099 and K24 HL67434 from the National Institutes of Health, an American Society of Echocardiography Career Development Award, and an Erwin-Schrödinger Stipend (FWF Austrian Science Fund).
ABBREVIATIONS AND ACRONYMS
- CD
cluster of differentiation
- EMT
endothelial-to-mesenchymal transition
- LV
left ventricle/ventricular
- MI
myocardial infarction
- MMP
matrix metalloproteinase
- MR
mitral regurgitation
- MV
mitral valve
- PM
papillary muscle
- SMA
α-smooth muscle actin
- TGF-β
transforming growth factor beta
- VCAM
vascular cell adhesion molecule
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: None
References
- 1.Grigioni F, Enriquez-Sarano M, Zehr KJ, Bailey KR, Tajik AJ. Ischemic mitral regurgitation: long-term outcome and prognostic implications with quantitative Doppler assessment. Circulation. 2001;103:1759–64. doi: 10.1161/01.cir.103.13.1759. [DOI] [PubMed] [Google Scholar]
- 2.Dal-Bianco JP, Aikawa E, Bischoff J, et al. Leducq Transatlantic Mitral Network Myocardial infarction alters adaptation of the tethered mitral valve. J Am Coll Cardiol. 2016;67:275–87. doi: 10.1016/j.jacc.2015.10.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Timek TA, Lai DT, Dagum P, et al. Mitral leaflet remodeling in dilated cardiomyopathy. Circulation. 2006;114:I518–23. doi: 10.1161/CIRCULATIONAHA.105.000554. [DOI] [PubMed] [Google Scholar]
- 4.Chaput M, Handschumacher MD, Guerrero JL, et al. Leducq Foundation Transatlantic MITRAL Network Mitral leaflet adaptation to ventricular remodeling: prospective changes in a model of ischemic mitral regurgitation. Circulation. 2009;120:S99–103. doi: 10.1161/CIRCULATIONAHA.109.844019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaput M, Handschumacher MD, Tournoux F, et al. Mitral leaflet adaptation to ventricular remodeling: occurrence and adequacy in patients with functional mitral regurgitation. Circulation. 2008;118:845–52. doi: 10.1161/CIRCULATIONAHA.107.749440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rausch MK, Tibayan FA, Miller DC, Kuhl E. Evidence of adaptive mitral leaflet growth. J Mech Behav Biomed Mater. 2012;15:208–17. doi: 10.1016/j.jmbbm.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beaudoin J, Thai WE, Wai B, Handschumacher MD, Levine RA, Truong QA. Assessment of mitral valve adaptation with gated cardiac computed tomography: validation with three-dimensional echocardiography and mechanistic insight to functional mitral regurgitation. Circ Cardiovasc Imaging. 2013;6:784–9. doi: 10.1161/CIRCIMAGING.113.000561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Debonnaire P, Al Amri I, Leong DP, et al. Leaflet remodelling in functional mitral valve regurgitation: characteristics, determinants, and relation to regurgitation severity. Eur Heart J Cardiovasc Imaging. 2015;16:290–9. doi: 10.1093/ehjci/jeu216. [DOI] [PubMed] [Google Scholar]
- 9.Obase K, Weinert L, Hollatz A, et al. Elongation of chordae tendineae as an adaptive process to reduce mitral regurgitation in functional mitral regurgitation. Eur Heart J Cardiovasc Imaging. 2016;17:500–9. doi: 10.1093/ehjci/jev314. [DOI] [PubMed] [Google Scholar]
- 10.Grande-Allen KJ, Barber JE, Klatka KM, et al. Mitral valve stiffening in end-stage heart failure: evidence of an organic contribution to functional mitral regurgitation. J Thorac Cardiovasc Surg. 2005;130:783–90. doi: 10.1016/j.jtcvs.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 11.Grande-Allen KJ, Borowski AG, Troughton RW, et al. Apparently normal mitral valves in patients with heart failure demonstrate biochemical and structural derangements: an extracellular matrix and echocardiographic study. J Am Coll Cardiol. 2005;45:54–61. doi: 10.1016/j.jacc.2004.06.079. [DOI] [PubMed] [Google Scholar]
- 12.Kunzelman KS, Cochran RP. Stress/strain characteristics of porcine mitral valve tissue: parallel versus perpendicular collagen orientation. J Card Surg. 1992;7:71–8. doi: 10.1111/j.1540-8191.1992.tb00777.x. [DOI] [PubMed] [Google Scholar]
- 13.Levine RA, Hagége AA, Judge DP, et al. Leducq Mitral Transatlantic Network Mitral valve disease–morphology and mechanisms. Nat Rev Cardiol. 2015;12:689–710. doi: 10.1038/nrcardio.2015.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kovacic JC, Mercader N, Torres M, Boehm M, Fuster V. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: from cardiovascular development to disease. Circulation. 2012;125:1795–808. doi: 10.1161/CIRCULATIONAHA.111.040352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dal-Bianco JP, Aikawa E, Bischoff J, et al. Active adaptation of the tethered mitral valve: insights into a compensatory mechanism for functional mitral regurgitation. Circulation. 2009;120:334–42. doi: 10.1161/CIRCULATIONAHA.108.846782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balachandran K, Alford PW, Wylie-Sears J, et al. Cyclic strain induces dual-mode endothelial-mesenchymal transformation of the cardiac valve. Proc Natl Acad Sci U S A. 2011;108:19943–8. doi: 10.1073/pnas.1106954108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wylie-Sears J, Levine RA, Bischoff J. Losartan inhibits endothelial-to-mesenchymal transformation in mitral valve endothelial cells by blocking transforming growth factor-β–induced phosphorylation of ERK. Biochem Biophys Res Comm. 2014;446:870–5. doi: 10.1016/j.bbrc.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bischoff J, Casanovas G, Wylie-Sears J, et al. CD45 expression in mitral valve endothelial cells after myocardial infarction. Circ Res. 2016;119:1215–25. doi: 10.1161/CIRCRESAHA.116.309598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haudek SB, Xia Y, Huebener P, et al. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc Natl Acad Sci U S A. 2006;103:18284–9. doi: 10.1073/pnas.0608799103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Visconti RP, Ebihara Y, LaRue AC, et al. An in vivo analysis of hematopoietic stem cell potential: hematopoietic origin of cardiac valve interstitial cells. Circ Res. 2006;98:690–6. doi: 10.1161/01.RES.0000207384.81818.d4. [DOI] [PubMed] [Google Scholar]
- 21.Keeley EC, Mehrad B, Strieter RM. Fibrocytes: bringing new insights into mechanisms of inflammation and fibrosis. Int J Biochem Cell Biol. 2010;42:535–42. doi: 10.1016/j.biocel.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Acker MA, Parides MK, Perrault LP, et al. CTSN Mitral-valve repair versus replacement for severe ischemic mitral regurgitation. N Engl J Med. 2014;370:23–32. doi: 10.1056/NEJMoa1312808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kron IL, Hung J, Overbey JR, et al. CTSN Investigators Predicting recurrent mitral regurgitation after mitral valve repair for severe ischemic mitral regurgitation. J Thorac Cardiovasc Surg. 2015;149:752–61.e1. doi: 10.1016/j.jtcvs.2014.10.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldstein D, Moskowitz AJ, Gelijns AC, et al. CTSN Two-year outcomes of surgical treatment of severe ischemic mitral regurgitation. N Engl J Med. 2016;374:344–53. doi: 10.1056/NEJMoa1512913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Markwald RR, Norris RA, Moreno-Rodriguez R, Levine RA. Developmental basis of adult cardiovascular diseases: valvular heart diseases. Ann N Y Acad Sci. 2010;1188:177–83. doi: 10.1111/j.1749-6632.2009.05098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Capoulade R, Clavel MA, Mathieu P, et al. Impact of hypertension and renin-angiotensin system inhibitors in aortic stenosis. Eur J Clin Invest. 2013;43:1262–72. doi: 10.1111/eci.12169. [DOI] [PubMed] [Google Scholar]
- 27.Fornes P, Heudes D, Fuzellier JF, Tixier D, Bruneval P, Carpentier A. Correlation between clinical and histologic patterns of degenerative mitral valve insufficiency: a histomorphometric study of 130 excised segments. Cardiovasc Pathol. 1999;8:81–92. doi: 10.1016/s1054-8807(98)00021-0. [DOI] [PubMed] [Google Scholar]
- 28.Mankad S, d’Amato TA, Reichek N, et al. Combined angiotensin II receptor antagonism and angiotensin-converting enzyme inhibition further attenuates postinfarction left ventricular remodeling. Circulation. 2001;103:2845–50. doi: 10.1161/01.cir.103.23.2845. [DOI] [PubMed] [Google Scholar]
- 29.Ohtawa M, Takayama F, Saitoh K, Yoshinaga T, Nakashima M. Pharmacokinetics and biochemical efficacy after single and multiple oral administration of losartan, an orally active nonpeptide angiotensin II receptor antagonist, in humans. Br J Clin Pharmacol. 1993;35:290–7. doi: 10.1111/j.1365-2125.1993.tb05696.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aikawa E, Whittaker P, Farber M, et al. Human semilunar cardiac valve remodeling by activated cells from fetus to adult: implications for postnatal adaptation, pathology, and tissue engineering. Circulation. 2006;113:1344–52. doi: 10.1161/CIRCULATIONAHA.105.591768. [DOI] [PubMed] [Google Scholar]
- 31.Rabkin E, Aikawa M, Stone JR, Fukumoto Y, Libby P, Schoen FJ. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation. 2001;104:2525–32. doi: 10.1161/hc4601.099489. [DOI] [PubMed] [Google Scholar]
- 32.Beaudoin J, Handschumacher MD, Zeng X, et al. Mitral valve enlargement in chronic aortic regurgitation as a compensatory mechanism to prevent functional mitral regurgitation in the dilated left ventricle. J Am Coll Cardiol. 2013;61:1809–16. doi: 10.1016/j.jacc.2013.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walker GA, Masters KS, Shah DN, Anseth KS, Leinwand LA. Valvular myofibroblast activation by transforming growth factor-β: implications for pathological extracellular matrix remodeling in heart valve disease. Circ Res. 2004;95:253–60. doi: 10.1161/01.RES.0000136520.07995.aa. [DOI] [PubMed] [Google Scholar]
- 34.Zeisberg EM, Tarnavski O, Zeisberg M, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–61. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 35.Bujak M, Frangogiannis NG. The role of TGF-β signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74:184–95. doi: 10.1016/j.cardiores.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leask A. Potential therapeutic targets for cardiac fibrosis: TGFβ, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res. 2010;106:1675–80. doi: 10.1161/CIRCRESAHA.110.217737. [DOI] [PubMed] [Google Scholar]
- 37.Rosenkranz S. TGF-β1 and angiotensin networking in cardiac remodeling. Cardiovasc Res. 2004;63:423–32. doi: 10.1016/j.cardiores.2004.04.030. [DOI] [PubMed] [Google Scholar]
- 38.Kakio T, Matsumori A, Ono K, Ito H, Matsushima K, Sasayama S. Roles and relationship of macrophages and monocyte chemotactic and activating factor/monocyte chemoattractant protein-1 in the ischemic and reperfused rat heart. Lab Invest. 2000;80:1127–36. doi: 10.1038/labinvest.3780119. [DOI] [PubMed] [Google Scholar]
- 39.Deten A, Volz HC, Briest W, Zimmer HG. Cardiac cytokine expression is upregulated in the acute phase after myocardial infarction. Experimental studies in rats. Cardiovasc Res. 2002;55:329–40. doi: 10.1016/s0008-6363(02)00413-3. [DOI] [PubMed] [Google Scholar]
- 40.Irwin MW, Mak S, Mann DL, et al. Tissue expression and immunolocalization of tumor necrosis factor-α in postinfarction dysfunctional myocardium. Circulation. 1999;99:1492–8. doi: 10.1161/01.cir.99.11.1492. [DOI] [PubMed] [Google Scholar]
- 41.Dabek J, Kulach A, Monastyrska-Cup B, Gasior Z. Transforming growth factor β and cardiovascular diseases: the other facet of the ‘protective cytokine’. Pharmacol Rep. 2006;58:799–805. [PubMed] [Google Scholar]
- 42.Mahler GJ, Farrar EJ, Butcher JT. Inflammatory cytokines promote mesenchymal transformation in embryonic and adult valve endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33:121–30. doi: 10.1161/ATVBAHA.112.300504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herzog EL, Bucala R. Fibrocytes in health and disease. Exp Hematol. 2010;38:548–56. doi: 10.1016/j.exphem.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hong KM, Belperio JA, Keane MP, Burdick MD, Strieter RM. Differentiation of human circulating fibrocytes as mediated by transforming growth factor-β and peroxisome proliferator-activated receptor γ. J Biol Chem. 2007;282:22910–20. doi: 10.1074/jbc.M703597200. [DOI] [PubMed] [Google Scholar]
- 45.Fava RA, Olsen NJ, Postlethwaite AE, et al. Transforming growth factor beta 1 (TGF-beta 1) induced neutrophil recruitment to synovial tissues: implications for TGF-beta-driven synovial inflammation and hyperplasia. J Exp Med. 1991;173:1121–32. doi: 10.1084/jem.173.5.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wahl SM, Hunt DA, Wakefield LM, et al. Transforming growth factor type β induces monocyte chemotaxis and growth factor production. Proc Natl Acad Sci U S A. 1987;84:5788–92. doi: 10.1073/pnas.84.16.5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chesney J, Metz C, Stavitsky AB, Bacher M, Bucala R. Regulated production of type I collagen and inflammatory cytokines by peripheral blood fibrocytes. J Immunol. 1998;160:419–25. [PubMed] [Google Scholar]
- 48.Campbell SE, Katwa LC. Angiotensin II stimulated expression of transforming growth factor-β1 in cardiac fibroblasts and myofibroblasts. J Mol Cell Cardiol. 1997;29:1947–58. doi: 10.1006/jmcc.1997.0435. [DOI] [PubMed] [Google Scholar]
- 49.Peng H, Carretero OA, Vuljaj N, et al. Angiotensin-converting enzyme inhibitors: a new mechanism of action. Circulation. 2005;112:2436–45. doi: 10.1161/CIRCULATIONAHA.104.528695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gray MO, Long CS, Kalinyak JE, Li HT, Karliner JS. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-beta 1 and endothelin-1 from fibroblasts. Cardiovasc Res. 1998;40:352–63. doi: 10.1016/s0008-6363(98)00121-7. [DOI] [PubMed] [Google Scholar]
- 51.Yu CM, Tipoe GL, Wing-Hon Lai K, Lau CP. Effects of combination of angiotensin-converting enzyme inhibitor and angiotensin receptor antagonist on inflammatory cellular infiltration and myocardial interstitial fibrosis after acute myocardial infarction. J Am Coll Cardiol. 2001;38:1207–15. doi: 10.1016/s0735-1097(01)01518-2. [DOI] [PubMed] [Google Scholar]
- 52.Rodríguez-Vita J, Sánchez-López E, Esteban V, Rupérez M, Egido J, Ruiz-Ortega M. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-β–independent mechanism. Circulation. 2005;111:2509–17. doi: 10.1161/01.CIR.0000165133.84978.E2. [DOI] [PubMed] [Google Scholar]
- 53.Habashi JP, Doyle JJ, Holm TM, et al. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science. 2011;332:361–5. doi: 10.1126/science.1192152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Holm TM, Habashi JP, Doyle JJ, et al. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332:358–61. doi: 10.1126/science.1192149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen K, Mehta JL, Li D, Joseph L, Joseph J. Transforming growth factor β receptor endoglin is expressed in cardiac fibroblasts and modulates profibrogenic actions of angiotensin II. Circ Res. 2004;95:1167–73. doi: 10.1161/01.RES.0000150369.68826.2f. [DOI] [PubMed] [Google Scholar]
- 56.Peng J, Gurantz D, Tran V, Cowling RT, Greenberg BH. Tumor necrosis factor-α-induced AT1 receptor upregulation enhances angiotensin II-mediated cardiac fibroblast responses that favor fibrosis. Circ Res. 2002;91:1119–26. doi: 10.1161/01.res.0000047090.08299.d5. [DOI] [PubMed] [Google Scholar]
- 57.Solomon SD, Skali H, Anavekar NS, et al. Changes in ventricular size and function in patients treated with valsartan, captopril, or both after myocardial infarction. Circulation. 2005;111:3411–9. doi: 10.1161/CIRCULATIONAHA.104.508093. [DOI] [PubMed] [Google Scholar]
- 58.Møller JE, Dahlström U, Gøtzsche O, et al. OPTIMAAL Study Group Effects of losartan and captopril on left ventricular systolic and diastolic function after acute myocardial infarction: results of the Optimal Trial in Myocardial Infarction with Angiotensin II Antagonist Losartan (OPTIMAAL) echocardiographic substudy. Am Heart J. 2004;147:494–501. doi: 10.1016/j.ahj.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 59.Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC., III Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med. 2008;358:2787–95. doi: 10.1056/NEJMoa0706585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.González GE, Seropian IM, Krieger ML, et al. Effect of early versus late AT1 receptor blockade with losartan on postmyocardial infarction ventricular remodeling in rabbits. Am J Physiol Heart Circ Physiol. 2009;297:H375–86. doi: 10.1152/ajpheart.00498.2007. [DOI] [PubMed] [Google Scholar]
- 61.De Carvalho Frimm C, Sun Y, Weber KT. Angiotensin II receptor blockade and myocardial fibrosis of the infarcted rat heart. J Lab Clin Med. 1997;129:439–46. doi: 10.1016/s0022-2143(97)90077-9. [DOI] [PubMed] [Google Scholar]
- 62.Leuschner F, Panizzi P, Chico-Calero I, et al. Angiotensin-converting enzyme inhibition prevents the release of monocytes from their splenic reservoir in mice with myocardial infarction. Circ Res. 2010;107:1364–73. doi: 10.1161/CIRCRESAHA.110.227454. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.