

Abstract
A dichloromethane extract of Trichospira verticillata from the Natural Products Discovery Institute was discovered to have good antiplasmodial activity (IC50 ~5 μg/mL). After purification by liquid-liquid partition and C18 reverse phase HPLC, four new germacranolide-type sesquiterpenoid lactones named trichospirolides A-D (1–4) were isolated. The structures of the new compounds were elucidated by analysis of their 1D and 2D NMR and MS data. The relative and absolute configurations were assigned based on a comparison of calculated and experimental ECD and UV spectra, specific rotations, internuclear distances, and coupling constants for all possible diastereomers for each compound. Among these four compounds, the conjugated dienone 1 displayed the most potent antiplasmodial activity, with an IC50 value of 1.5 μM.
Graphical abstract
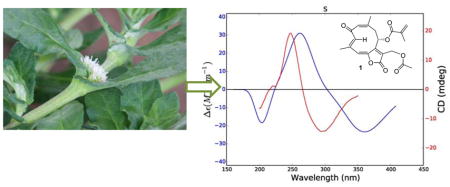
Comparison of experimental (red) and calculated (blue) ECD spectra of 1.
Malaria is a disease caused by infection with one of several species of protozoan parasites of the Plasmodium genus, usually transmitted by the bite of an infected female Anopheles mosquito. It continues to be one of the world’s most devastating diseases, with over 3.2 billion people at risk for contracting the disease, and an estimated 214 million malaria cases and over 400,000 deaths in 2015.1,2 The first effective antimalarial drug was the natural product quinine, and the current best drug is the natural product artemisinin,3,4 usually used as part of an artemisinin-based combination therapy (ACT).5 However, the malaria parasite P. falciparum has developed significant resistance to artemisinin in mainland Southeast Asia, although a 6-day course of treatment, as opposed to the normal 3-day course, is still effective in most cases.6 There is thus a continuing need for the discovery of new and effective antimalarial drugs, and the fact that arguably the two most important antimalarial drugs are both plant-derived natural products supports the hypothesis that a search for new antimalarial agents from plant sources will be fruitful. This paper reports the results of a study of Trichospira verticillata L. (S. F. Blake) (Asteraceae) for new antiplasmodial agents as part of collaboration between Virginia Tech and the Natural Products Discovery Institute (NPDI).7
Extracts from the NPDI library, which contains 22,000 samples from 7,500 plant species, were screened for antiplasmodial activity. A dichloromethane extract of Trichospira verticillata (Asteraceae) displayed moderate antiplasmodial activity against the drug resistant Dd2 strain of P. falciparum, with an IC50 value of approximately 5 μg/mL. The Asteraceae family is a large and widespread family of flowering plants,8 distributed from the polar regions to the tropics, colonizing a wide variety of habitats.9 This family is a rich source of medically active compounds, such as sesquiterpenes, lactones, flavonoids, and saponins, which are well-known for their broad range of bioactivities, including antimicrobial, antioxidant, cytotoxic, and anti-inflammatory activities.10–13 Trichospira verticillata is the only species in the genus Trichospira, and is found in wetlands and pine savannas in the Atlantic regions of tropical South America and Cuba.14 The phytochemistry of T. verticillata has not previously been investigated, thus, it was selected for bioassay-guided fractionation to isolate its antiplasmodial components.
RESULTS AND DISCUSSION
Isolation and Structure Elucidation
A CH2Cl2 extract of the whole plant of T. verticillata was subjected to liquid-liquid partition to afford active hexanes and MeOH fractions, with IC50 values against the drug-resistant Dd2 strain of Plasmodium falciparum of about 3 and 7 μg/mL, respectively. The hexanes fraction was directly separated by C18 reverse-phase HPLC to yield four novel germacranolide-type sesquiterpenoid lactones which were named trichospirolides A–D (1–4). Among these four compounds, the conjugated dienone 1 displayed the most potent antimalarial activity, with an IC50 value of 1.5 μM. Herein, we report the structural elucidation and stereochemical assignment of these four compounds.
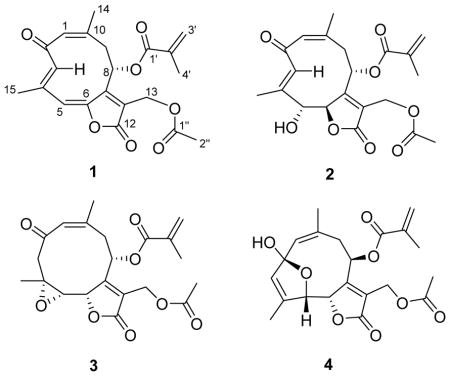
Trichospirolide A (1) was isolated as a yellow powder. Its positive ion HRESIMS data revealed a peak for a protonated molecular ion at m/z 387.1441 and for a sodium adduct ion at m/z 409.1273, both corresponding to a molecular formula of C21H22O7. Its IR spectrum exhibited bands at 1773, 1749, 1723, and 1639 cm−1, indicating the presence of three conjugated carbonyl groups, including two conjugated esters or lactones. Its UV spectrum, with absorptions at 298 and 252 nm, was consistent with dienone conjugation. The presence of an acetyl group was suggested by the 1H NMR signal at δH 2.08 (H-2″) and the 13C NMR signals at δC 170.4 (C-1″) and 20.9 (C-2″), and further confirmed by 2J-HMBC cross-peaks between H-2″ and C-1″. Similarly, the 1H NMR signals at δH 6.18 (H-3′a), 5.72 (H-3′b) and 1.96 (H-4′) and the 13C NMR signals at δC 165.6 (C-1′), 135.2 (C-2′), 127.5 (C-3′) and 20.9 (C-4′) indicated the presence of a methacryloyloxy group. The presence of this side group was also corroborated by 3J-HMBC cross-peak between H-3′a and C-1′, H-3′b and C-4′, H-4′ and C-1′, and 2J-HMBC correlation between H-3′b and C-2′ (Table 1).
Table 1.
NMR Spectroscopic Data for Compounds 1–4 in CDCl3 (500 MHz)
| 1 | 2 | 3 | 4 | |||||
|---|---|---|---|---|---|---|---|---|
| Posn. | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type |
| 1 | 6.12 s | 131.9, CH | 6.08 s | 131.6, CH | 6.07 p (1.0) | 130.6, CH | 5.71 s | 129.0, CH |
| 2 | 191.5, C | 194.0, C | 201.0, C | 108.6, C | ||||
| 3a | 6.57 s | 135.1, CH | 6.28 s | 131.8, CH | 2.93 d (11.4) | 52.9, CH2 | 5.53 dq (1.0, 2.0) | 127.6, CH |
| 3b | 2.41 dd (11.4, 1.0) | |||||||
| 4 | 137.7, C | 138.7, C | 56.9, C | 142.1, C | ||||
| 5 | 6.62 dd (1.0, 1.0) | 119.5, CH | 4.19 d (8.8) | 80.6, CH | 2.86 d (9.3) | 60.4, CH | 5.32 ddd (5.5, 2.0, 1.0) | 83.8, CH |
| 6 | 147.9, C | 5.43 d (8.8) | 86.3, CH | 4.94 d (9.3) | 81.0, CH | 5.55 d (5.5) | 82.2, CH | |
| 7 | 153.4, C | 163.2, C | 162.6, C | 171.1, C | ||||
| 8 | 5.95 d (8.0) | 73.0, CH | 4.98 d (8.0) | 73.1, CH | 5.10 t (6.7) | 67.8, CH | 5.51 t (3.3) | 73.2, CH |
| 9a | 3.48 d (16.0) | 40.5, CH2 | 3.37 dd (8.0, 15.2) | 38.9, CH2 | 3.18 dd (14.8, 6.7) | 36.3, CH2 | 3.12 ddd (15.6, 3.4, 1.5) | 35.4, CH2 |
| 9b | 2.86 dd (16.0, 8.0) | 2.76 d (15.2) | 2.70 dd (14.8, 6.7) | 2.35 dd (15.6, 3.4) | ||||
| 10 | 155.9, C | 155.6, C | 143.0, C | 141.9, C | ||||
| 11 | 126.6, C | 126.9, C | 126.4, C | 123.2, C | ||||
| 12 | 167.1, C | 170.4, C | 171.0, C | 167.2, C | ||||
| 13a | 5.01 d (12.5) | 55.6, CH2 | 4.87 d (12.4) | 55.4, CH2 | 4.87 s | 55.2, CH2 | 4.82 d (12.4) | 55.5, CH2 |
| 13b | 4.91 d (12.5) | 4.82 d (12.4) | 4.87 s | 4.75 d (12.4) | ||||
| 14 | 2.12 d (1.0) | 30.1, CH3 | 2.08 s | 28.5, CH3 | 1.86 d (1.0) | 25.7, CH3 | 1.89 d (1.6) | 26.2, CH3 |
| 15 | 2.08 dd (1.0, 1.0) | 17.3, CH3 | 1.94 d (1.0) | 13.5, CH3 | 1.70 s (1.0) | 19.6, CH3 | 1.80 dd (2.0, 1.0) | 12.9, CH3 |
| 1′ | 165.6, C | 166.7, C | 166.4, C | 166.2, C | ||||
| 2′ | 135.2, C | 135.1, C | 135.0, C | 135.3, C | ||||
| 3′a | 6.18 quint (1.0) | 127.5, CH2 | 6.14 quint (1.0) | 127.7, CH2 | 6.15 quint (1.0) | 127.8, CH2 | 6.17 quint (1.0) | 126.8, CH2 |
| 3′b | 5.72 quint (1.0) | 5.70 quint (1.0) | 5.70 quint (1.0) | 5.68 quint (1.0) | ||||
| 4′ | 1.96 dd (1.0, 1.0) | 20.9, CH3 | 1.93 dd (1.0, 1.0) | 18.2, CH3 | 1.93 dd (1.0, 1.0) | 20.9, CH3 | 1.96 dd (1.0, 1.0) | 18.2, CH3 |
| 1″ | 170.4, C | 170.5, C | 170.3, C | 170.3, C | ||||
| 2″ | 2.08 s | 20.9, CH3 | 2.06 s | 20.9, CH3 | 2.06 s | 20.9, CH3 | 2.05 s | 20.8, CH3 |
The remaining 15 carbons could be assigned to a germacranolide-type sesquiterpenoid skeleton by analysis of the 2D NMR spectroscopic data, including 1H-1H COSY, HSQC, and HMBC data.15,16 In particular, the presence of the 10-membered germacranolide ring was established by the HMBC cross-peaks between H-1 and C-3, H-3 and C-5, H-9 and C-1, H-5 and C-7, and H-8 and C-10. The C-14 and C-15 methyl carbons were assigned to C-10 and C-4, respectively, by observation of HMBC cross-peaks between H-1 and C-14, and H-3 and C-15. Moreover, HMBC cross-peaks between H-13 and C-7, as well as H-13 and the C-12 ester carbonyl carbon (δC 167.1) suggested the presence of an α,β-unsaturated lactone moiety located at C-6 (δC 147.9) and C-7. The acetoxy and methacryloyloxy substituents were determined to be attached to C-13 and C-8, respectively, based on the HMBC cross-peaks between H-13 and C-1″, and H-8 and C-1′ (Figure 1). The carbon-carbon double bonds in the 10-membered ring were assigned as 1(10)Z and 3E on the basis of a strong NOESY correlation between H-1 and H3-14 and the absence of a strong correlation between H-3 and H3-15. Strong correlations were also observed between H-3 and H-8 and between H-3 and H-9a (Figure 1).
Figure 1.
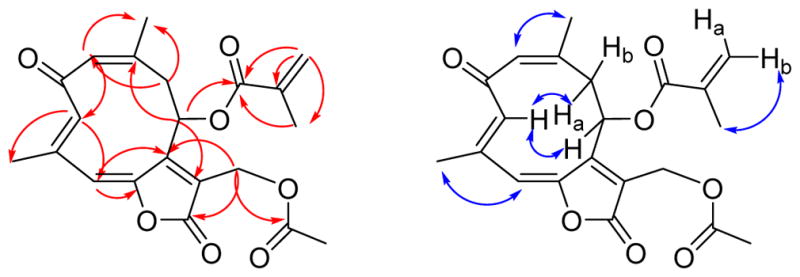
HMBC (red) and NOESY (blue) correlations for 1
Trichospirolide B (2) was isolated as a yellow powder. Its positive ion HRESIMS revealed a peak for a protonated molecular ion at m/z 405.1561 and for a sodium adduct ion at m/z 427.1389, both corresponding to a molecular formula of C21H24O8. Its IR spectrum exhibited an additional absorption at 3430 cm−1 as compared with the spectrum of 1, indicating the presence of a hydroxy group, and this coupled with the difference of 18 Daltons between the molecular weights of 1 and 2 suggested that 2 might be a hydration product of 1. The UV spectrum of 2 had its longest wavelength absorption at 247 nm, consistent with an enone function. Hydration of the C-5 double bond in 1 was confirmed by the shielding of the chemical shifts of C-5 and C-6 from δC 119.5 (CH) and 147.9 (C) in 1 to δC 80.6 (CH) and 86.3 (CH), respectively, in 2. The 1H and 13C NMR data of 2 (Table 1) were similar to those of 1 except as noted above, and were fully consistent with its structural assignment as a hydrate of 1 with the hydroxy group at C-5. This assignment was confirmed by the HMBC cross-peaks shown in Figure 2.
Figure 2.
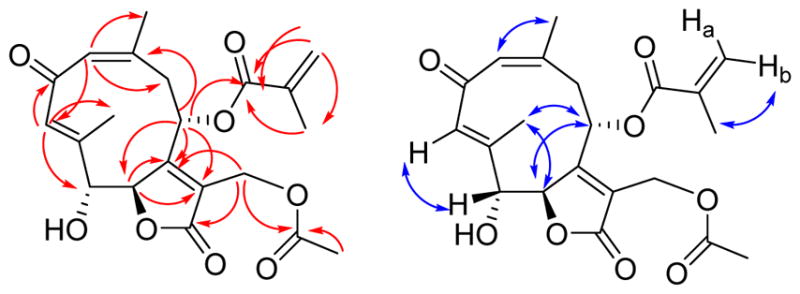
HMBC (red) and NOESY (blue) correlations for 2
The carbon-carbon double bonds in the 10-membered ring were assigned as 1(10)Z and 3E on the basis of NOESY crosspeaks between H-1 and H3-14, and H-3 and H-5, and the absence of a strong crosspeak between H-3 and H3-15 (Figure 2). The vicinal coupling constant 3J5,6 of 8.8 Hz was consistent with a H5-C5-C6-H6 dihedral angle in the 5º – 25º or the 130º – 150º range.17
Trichospirolide C (3) had the molecular formula C21H24O8 as determined by HRESIMS. Inspection of 1H and 13C NMR spectroscopic data of 3 in comparison with those of 2 (Table 1) indicated that 3 was also a germacranolide-type sesquiterpenoid lactone, with a similar structure to 2. However, the C-3 and C-4 signals were shifted from δC 131.8 (CH) and 138.7 (C) in 2 to δC 52.9 (CH2) and 56.9 (C) ppm in 3, indicating that the alkene group of 2 was not present in 3. The absence of an OH stretching frequency in the IR spectrum of 3, together with the 13C chemical shift of C-4, indicated the presence of an epoxide unit involving C-4 and C-5.18 The vicinal coupling constant 3J5,6 of 8.3 Hz was consistent with a H5-C5-C6-H6 dihedral angle in the 5º – 25º or the 130º – 150º range.17 The HMBC and NOESY cross-peaks of 3 (Figure 3) supported the structural assignment; in particular the 1(10) Z double bond configuration was assigned on the basis of the H-1/H3-14 NOESY cross-peak.
Figure 3.
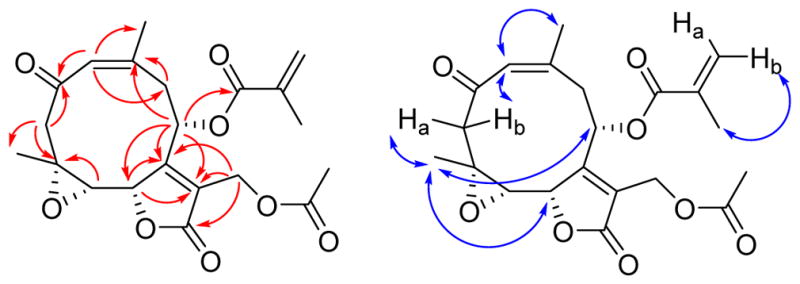
HMBC (red) and NOESY (blue) correlations for 3
Trichospirolide D (4) was also isolated as a yellow powder. Its HRESIMS data showed peaks for protonated and sodium adduct molecular ions corresponding to the molecular formula C21H24O8. Comparison of 1H and 13C NMR spectroscopic data of 4 with those of 2 (Table 1) indicated that both compounds had the same skeleton, as well as acetoxy and methacryloyloxy groups. However, the carbon signal for C-2 of 4 was shifted from 194.0 ppm in 2 to 108.6 ppm in 4, accompanied by a shielding of the 1H NMR signals for H-1, H-3 and H-5, suggesting the conversion of a carbonyl group to a hemiacetal group.15 HMBC cross-peaks (Figure 4) confirmed the formation of a five-membered ring, with the oxygen of the hemiacetal unit connecting C-5 and C-2 (Figure 4). The NOESY correlations of 4 (Figure 4) indicated that both the 1(10) and 3 double bonds were Z configured, based on the H-1/H3-14 and H-3/H3-15 NOESY cross-peaks.
Figure 4.
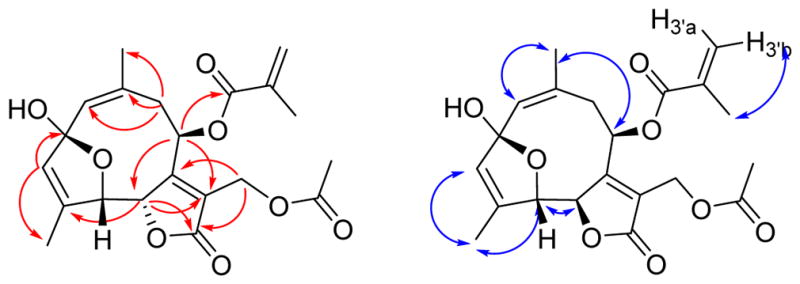
HMBC (red) and NOESY (blue) correlations for 4
The H5-C5-C6-H6 dihedral angle was estimated to be approximately 35º – 55º or 115º – 135º based on the observed 3J5,6 value of 4.5 Hz. Compound 4 proved to be moderately unstable, and samples decomposed on storage at −23 ºC.
Determination of Relative and Absolute Configurations
Determination of both the relative and absolute configurations of compounds 1 – 4 proved to be a challenging task because the flexibility of the side chains allowed many different conformations to contribute to the observed NOESY correlations, ECD and UV spectra, and specific rotations. We thus elected to address the problem by a direct comparison of experimental and calculated ECD and UV spectra and specific rotations for all possible stereoisomers of each compound.
Thus, all possible ring conformations for compounds 1 – 4 were established using chemical intuition due to the limited number of possible structural variations. Additional conformers involving fluctuations of the side chains on each ring conformer were generated by systematically rotating torsions within the side chains to generate diverse low-energy conformers. This conformational search was conducted for all enantiomerically unique stereoisomers of compounds 1 – 4, resulting in a total of 2,590 structures to be optimized. Optimized geometries with relative absolute energies less than 10−6 Hartree were considered to be duplicate geometries and were not included in subsequent calculations. Of the 2,590 optimized conformer geometries, 1,100 proved to be unique. Boltzmann populations were also computed for all 1,100 unique conformer geometries in order to determine individual contributions to the simulated weighted average spectra and specific rotations. Conformers with a Boltzmann population < 2% were deemed insignificant and were not included in any further calculations. Of the 1,100 unique optimized geometries, only 185 had significant contributions to the weighted average spectra and optical rotations. For each of these conformers with significant Boltzmann populations, weighted average ECD and UV spectra, specific rotations, and internuclear distances were calculated for comparison to experimental data. The specified calculations were conducted for all enantiomerically unique stereoisomers, yielding the associated weighted average spectra and specific rotations for all possible stereoisomers.
Although the calculated and experimental data did not always agree completely, the differences between the data for the assigned structures and those with different configurations were large enough to give confidence in the correctness of the assigned structures.
In the case of compound 1 the positive π–π* Cotton effect at 248 nm (Δε237 = +19.2) could not be used to assign the absolute configuration of its single stereocenter directly, since the simple rules proposed by Uchida and Kuriyame for the circular dichroism of α,β-unsaturated-γ-lactones19 are not applicable in cases where there is an allylic oxygen substituent. Calculations of its ECD and UV spectra and optical rotations were carried out for the R enantiomer of 1. The weighted average distances between hydrogens for the lowest energy conformers (Boltzmann population > 2%) of the R enantiomer were calculated for comparison with the observed NOESY correlations, and the ECD and UV spectra and specific rotation were calculated from additional data.
The results of these calculations are shown in Figure 5 and Table 2. The calculated and experimental ECD spectra for 1 agree reasonably well, although the calculated maxima and minima are shifted to longer wavelengths as compared with the experimental data; this may be a result of the extended conjugation in 1. The calculated internuclear distances also agree well with the observed NOESY signal strengths, giving added assurance to the correctness of the calculated conformations (Table 2). In a final comparison, the experimental and calculated specific rotations of 1 (− 360 and −1,141 respectively) agreed with each other in sign, although not in magnitude. The reason for this can be understood from the UV spectra shown in Figure 5, which show significantly longer wavelength absorptions than observed for 2 – 4. The DFT absorption around 367 nm lies close enough to the polarized light wavelength of 589 nm such that the computed rotation is overestimated. This phenomenon has been described previously.20 Based on these data the structure of trichospirolide A (1) was assigned as 1(10)Z,3E,5E,7(12)Z-(8S)-13-acetoxy-8-methacryloyloxy-2-oxogermacra-1,3,5,7-tetraen-6,12-olide.
Figure 5.

Comparison of experimental (red) and calculated (blue) ECD (left) and UV (right) spectra of 1.
Table 2.
Calculated Internuclear Distances and NOESY Signal Strength for 1a
| Proton Pair | Weighted average distance (Å) | NOESY intensity |
|---|---|---|
| H5-H15 | 3.19 | medium |
| H3-H9a | 1.94 | strong |
| H3-H8 | 2.65 | strong |
| H1-H14 | 3.01 | medium |
| H8-H15 | 3.85 | medium |
See SI for details of calculations.
The relative and absolute configurations of 2 were established by comparison of experimental and calculated ECD and UV spectra (Figure 6). Calculations for 2 were carried out for its RRR, RSR, SRR, and SSR diastereomers. The calculated ECD and UV spectra for the RRS diastereomer of 2 gave the best agreement with the experimental data (Figure 6) and with one exception the calculated internuclear distances also agree well with the observed NOESY signal strengths (Table 3). The coupling constants for three key dihedral angles of the RRS enantiomer were also in agreement with this configuration (Table 4), while the experimental and calculated specific rotations of this diastereomer (− 140 and − 221 respectively) agreed with each other in sign and reasonably well in magnitude. Based on these data, trichospirolide B (2) is assigned the structure 1(10)Z,3E,7(11)Z-(5R,6R,8S)-13-acetoxy-5-hydroxy-8-methacryloyloxy-2-oxogermacra-1(10),3,7(11)-trien-6,12-olide.
Figure 6.

Comparison of experimental (red) and calculated (blue) ECD (left) and UV (right) spectra of 2.
Table 3.
Calculated Internuclear Distances and NOESY Signal Strength for 2a
| Proton Pair | Weighted average distance (Å) | NOESY intensity |
|---|---|---|
| H3-H9a | 2.16 | strong |
| H3-H9b | 3.86 | medium |
| H3-H5 | 3.71 | strong |
| H3-H8 | 3.26 | v. weak |
| H1-H14 | 3.02 | medium |
| H3′b-H4′ | 3.15 | medium |
| H6-H15 | 4.72 | medium |
| H6-H8 | 3.35 | medium |
| H8-H15 | 3.01 | strong |
| H5-H15 | 2.93 | weak |
See SI for details of calculations.
Table 4.
Calculated Dihedral Angles and J-values for 2a
| Dihedral | Weighted average dihedral angle | Observed J value |
|---|---|---|
| H5-C5-C6-H6 | −164.3 | 8.8 |
| H8-C8-C9-H9a | −88.6 | 0 |
| H8-C8-C9-H9b | 156.9 | 8.0 |
See SI for details of calculations.
Using the same procedure as above, the distance and energy data of the lowest energy conformations (Boltzmann population > 2%) of the RRRR, RRSR, RSRR, RSSR, SRRR, SRSR, SSRR, and SSSR diastereomers of trichosporolide C (3) were used to calculate weighted average internuclear distances and ECD and UV spectra; the resulting data were only consistent with the RSSS diastereomer of 3. The experimental and calculated ECD and UV spectra for this diastereomer are shown in Figure 7, and the calculated key internuclear distances and NOESY data are shown in Table 5. The calculated internuclear distances agree reasonably well with the observed NOESY correlations, although two correlations were observed with greater intensity than would have been expected from the calculated distances. The 3J5,6 value of 9.3 Hz (Table 6) supports the assigned structure, which has a calculated dihedral angle of − 24.6 º. The experimental and calculated specific rotations of 3 (− 55.2 and − 58.0 respectively) agreed with each other in sign and in magnitude.
Figure 7.

Comparison of experimental (red) and calculated (blue) ECD (left) and UV (right) spectra of 3.
Table 5.
Calculated Internuclear Distances and NOESY Signal Strength for 3a
| Proton Pair | Weighted average distance (Å) | NOESY Intensity |
|---|---|---|
| H1-H14 | 3.03 | medium |
| H1-H3a | 3.65 | medium |
| H1-H3b | 3.81 | medium |
| H8-H15 | 4.65 | strong |
| H6-H15 | 4.44 | strong |
| H3a-H15 | 3.16 | strong |
| H3b-H5 | 3.81 | strong |
See SI for details of calculations.
Table 6.
Calculated Dihedral Angle and J-value for 3a
| Dihedral | Weighted average dihedral angle | Observed J value |
|---|---|---|
| H5-C5-C6-H6 | −24.6 | 9.3 |
See SI for details of calculations.
Trichospirolide C (3) was thus assigned as 1(10)Z,7(11)Z-(4R,5S,6S,8S)-13-acetoxy-4,5-epoxy-8-methacryloyloxy-2-oxogermacra-1,7-dien-6,12-olide. It is structurally related to glaucolide E, whose structure was confirmed by X-ray crystallography.21 It differs from glaucolide E by the configurations of the 1(10) double bond and the 4,5-epoxide, and by having a carbonyl group at C-2 instead of the acetoxy group of glaucolide E.
The ECD and UV spectra and optical rotation of 4 were calculated for its RRRR, RRSR, RSRR, RSSR, SRRR, SRSR, SSRR, SSSR diastereomers. In the event only the SSSR diastereomers gave data consistent with the experimental data (Figure 8). Calculation of the weighted averages of key internuclear distances and comparison with NOESY data (Table 7) also supported this assignment, as did the 3J5,6 value of 5.5 Hz through the calculated H5-C5-C6-H6 dihedral angle of 54.3 º. The experimental and calculated specific rotations of 4 (− 232.2 and − 280.4 respectively) also agreed with each other in sign and magnitude.
Figure 8.

Comparison of experimental (red) and calculated (blue) ECD (left) and UV (right) spectra of 4.
Table 7.
Calculated Internuclear Distances and NOESY Signal Strength for 4a
| Proton Pair | Weighted average distance (Å) | NOESY Intensity |
|---|---|---|
| H1-H14 | 3.03 | medium |
| H3′b-H4′ | 3.15 | medium |
| H3-H15 | 3.32 | medium |
| H3-H9a | 4.99 | medium |
| H5-H15 | 3.35 | medium |
See SI for details of calculations.
Based on these data, the structure of compound 4 was assigned as 1(10)Z,3Z,7(11)Z-(2S,5S,6S,8R)-13-acetoxy-2,5-epoxy-2-hydroxy-8-O-methacryloyloxygermacra-1(10),3,7(11)-trien-6,12-olide, based on the good agreement of the spectroscopic data for this structure with the calculated data.
It is interesting to note that compounds 1 – 4 belong to two different stereochemical families, with 1 – 3 and 4 having the (8S) and (8R) configurations, respectively. In many cases a particular plant produces only one stereochemical series, as appears to be the case for instance with similar sesquiterpenoid lactones from Eupatorium kiirunense.15 On the other hand, the existence of enantiomeric natural products is well documented,22 and in particular both the R and S enantiomers of germacrene D are produced by Goldenrod (Solidago canadensis),23 hence the production of both (8R) and (8S) isomers by T. verticillata is not an unprecedented event.
Biological Activities
All the isolated compounds were tested for antiplasmodial activity against the Dd2 strain of Plasmodium falciparum and for toxicity to HEK293 cells. Trichospirolide A (1) showed moderate antiplasmodial activity, with an IC50 value of 1.5 μM, but it also showed similar toxicity to HEK293 cells and significant antiproliferative activity to A2780 ovarian cancer cells. Trichospirolides B–D (2–4) showed weak antiplasmodial activities, with IC50 values of 37.0, 12.1, and 44.3 μM, respectively, and correspondingly weak toxicity to HEK293 cells (Table 8).
Table 8.
Bioactivities of Compounds 1–4
| Compound | ||||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | Artemisinin | Paclitaxel | |
| Dd2 (μM) | 1.49 ± 0.29 | 37.05 ± 3.22 | 12.10 ± 0.45 | 44.33 ± 3.64 | 0.007 | NT |
| HEK293 (μM) | 3.5 ± 0.3 | 56.0 ± 7.8 | 23.8 ± 2.5 | 58.8 ± 1.6 | NT | NT |
| A2780 (μM) | 0.210 ± 0.003 | NT | NT | NT | NT | 0.028 ± 0.002 |
Sesquiterpenoid lactones are a series of well-known compounds, which have been reported to have various bioactivities, including antiplasmodial, antiproliferative, and antifungal activities.24–26 Several bioactive germacranolide-type sesquiterpenoid lactones have been isolated recently, most often from the Asteraceae family.27,28 Previous structure-activity relationship studies indicated that the α,β-unsaturated γ-lactone unit, which could target cysteine residues in proteins by acting as a Michael acceptor, is important for the bioactivity of germacranolides.29 This α,β-unsaturated γ-lactone unit is present in all four trichospirolides A-D, but their activities are varied due to modifications in the 10-membered ring. Trichospirolide A (1, P. falciparum IC50 1.49 μM) was 25-fold more potent than trichospirolide B, and showed the highest antiplasmodial activity among the four compounds. Trichospirolide D (4), with an IC50 value of 44 μM, displayed the least antiplasmodial activity, and this may be due to the formation of a hemiacetal accompanied by the loss of an α,β-unsaturated carbonyl unit, since the presence of this system in germacranolides has been reported to be essential for bioactivity, regardless of the presence of a α,β-unsaturated γ-lactone moiety.30 Moreover, the presence of the 4,5-epoxy group may be responsible for the fact that trichospirolide C (3, IC50 12.1 μM) displayed a 3-fold higher potency to P. falciparum than that of trichospirolide B (2, IC50 37.1 μM).31
In spite of the promising antiplasmodial activity of trichospirolide A, it is not likely to be an antimalarial drug candidate because of its toxicity to mammalian cells, in common with most other germacranolide-type sesquiterpenoid lactones. Its relatively potent antiproliferative activity to A2780 ovarian cancer cells, only 10-fold less potent than paclitaxel, might indicate it to be a potential anticancer lead, but this also is unlikely since its activity is most probably linked to its α,β-unsaturated carbonyl group acting as a Michael acceptor.
EXPERIMENTAL SECTION
General Experimental Procedures
Optical rotations were recorded on a JASCO P-2000 polarimeter. UV and IR spectroscopic data were measured on a Shimadzu UV-1201 spectrophotometer and a MIDAC M-series FTIR spectrophotometer, respectively. ECD spectra were obtained on a JASCO J-815 spectrometer. NMR spectra were recorded in CDCl3 on Bruker Avance 500 or 600 spectrometers. The chemical shifts are given in δ (ppm), and coupling constants (J) are reported in Hz. Mass spectra were obtained on an Agilent 6220 LC-TOF-MS in the positive ion mode.
Antiplasmodial Bioassays
The effect of each fraction and pure compound on parasite growth of the P. falciparum Dd2 strain was measured in a 72 h growth assay in the presence of drug as described previously with minor modifications. Briefly, ring stage parasite cultures (200 μL per well, with 1% hematocrit and 1% parasitemia) were grown for 72 h in the presence of increasing concentrations of the drug in a 5.05% CO2, 4.93% O2, and 90.2% N2 gas mixture at 37 °C. After 72 h in culture, parasite viability was determined by DNA quantitation using SYBR Green I (50 μL of SYBR Green I in lysis buffer at 0.4 μL of SYBR Green I/mL of lysis buffer). The half-maximum inhibitory concentration (IC50) calculation was performed with GraFit software using nonlinear regression curve fitting. IC50 values are the average of three independent determinations with each determination in duplicate and are expressed ± SEM.32
In vitro Cytotoxicity Against HEK293 Cells
Compounds were evaluated for their cytotoxicity against the HEK293 (Human Embryonic Kidney) normal cell line. Briefly, 10,000 HEK cells per well were plated in a clear bottom 96 well plate. After allowing the cells to adhere, the media was replaced with 100 μL of media containing varying amounts of the test compound and incubated for 24 hours. Later, 10 μL of resazurin sodium salt (Sigma) at 0.125 mg/mL was added to each well and incubated for 2 h. Cell viability was determined by measuring the fluorescence at 585 nm after excitation at 540 nm.
In vitro Antiproliferative Activity Against A2780 Cells
The A2780 ovarian cancer cell line antiproliferative bioassay was performed at Virginia Tech as previously reported.33,34 The A2780 cell line is a drug-sensitive ovarian cancer cell line.35 Paclitaxel was used as the positive control.
Plant Material
Whole plant specimens of Trichospira verticillata L. (S.F. Blake) were collected by Alexander Rodriguez alongside the main road in La Lagartera, Los Chiles, Alajuela Province, Costa Rica under the auspices of the National Biodiversity Institute of Costa Rica (INBIO). A herbarium sample is on deposit at this Institute under the accession number AR03477.
Extraction and Isolation
The dried and powdered whole plant of T. verticillata (731 g) was exhaustively extracted with EtOH in two 24-hour percolation steps; successive partition of the concentrated extract with hexanes and DCM gave an active DCM extract (about 10 g). For purposes of fractionation and purification, 0.517 g of the DCM extract designated 39114-5H was shipped to Virginia Tech for bioassay-guided isolation. A 0.35 g sample of 39114-5H (IC50 ~5 μg/mL) was suspended in aqueous MeOH [MeOH-H2O (9:1), 100 mL], and extracted with hexane (3 × 100 mL portions). The hexanes fraction was evaporated in vacuo to afford 85 mg of material with IC50 2.5~5 μg/mL. The remaining aqueous MeOH fraction was centrifuged to give a supernatant (270 mg) with an IC50 value of >10 μg/mL.
The hexanes fraction was directly applied on C18 HPLC, and eluted by 40 to 80% MeCN in H2O gradient in 60 min to yield compound 2 (6.3 mg), compound 4 (4.3 mg), compound 3 (5.2 mg) and compound 1 (4.7 mg), with retention times of 19.5, 26.5, 31.5 and 34.7 minutes, respectively.
Trichospirolide A (1): yellow powder; [α]D21 −360.3 (c 7.16×10−4 g/mL, MeOH); UV (c 0.030 mM, MeOH) λmax (ε) 298 nm (9464) 252 nm (13939); IR υmax 2930, 1773, 1749,1723, 1639, 1231, 1149, 1030 cm−1; ECD (c 0.030 mM, MeOH) Δε295 −14.3, Δε248 +19.2; HRESIMS m/z 409.1273 [M+Na]+ (calcd. for C21H22NaO7+, 409.1258) and 387.1441 [M+H]+ (calcd. for C21H23O7+, 387.1438); 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3), see Table 1.
Trichospirolide B (2): yellow powder; [α]D21 −140.1 (c 3.14×10−4 g/mL, MeOH); UV (c 0.034 mM, MeOH) λmax (ε) 247 nm (7353); IR υmax 3430, 2930, 1767, 1723, 1639, 1231, 1153 cm−1; ECD (c 0.034 mM, MeOH) Δε255 −4.2, Δε237 +3.7, Δε207 −13.2; HRESIMS m/z 427.1389 [M+Na]+ (calcd for C21H24NaO8+, 427.1363) and 405.1561 [M+H]+ (calcd for C21H25O8+, 405.1544); 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3), see Table 1.
Trichospirolide C (3): yellow powder; [α]D21 −55.2 (c 5.62×10−4 g/mL, MeOH); UV (c 0.030 mM, MeOH) λmax (ε) 229 nm (13134); IR υmax 2930, 1773, 1749, 1723, 1689, 1233, 1153 cm−1; ECD (c 0.030 mM, MeOH) Δε224 +10.1; HRESIMS m/z 427.1367 [M+Na]+ (calcd. for C21H24NaO8+, 427.1363) and 405.1540 [M+H ]+ (calcd. for C21H25O8+, 405.1544); 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3), see Table 1.
Trichospirolide D (4): yellow powder; [α]D21 −232.3 (c 3.1×10−4 g/mL, MeOH); UV (c 0.030 mM, MeOH) λmax (ε) 234 nm (4227); IR υmax 3427, 2930, 1767, 1721, 1233, 1154, 1041 cm−1; ECD (c 0.030 mM, MeOH) Δε251 −3.4, Δε217 +14.0; HRESIMS m/z 427.1360 [M+Na]+ (calcd. for C21H24NaO8+, 427.1363) and 405.1554 [M+H]+ (calcd. for C21H25O8+, 405.1544); 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3), see Table 1.
Computational Details
Conformational searches involving fluctuations of the side chains on each ring conformer were generated using the Open Babel software36 in conjunction with the MMFF94 force field37 and the Confab systematic rotor conformer generator.38 An energy cutoff was employed such that all diverse side chain conformers with a relative energy < 10 kcal/mol were kept. Geometries were optimized at the DFT/B3LYP/6-31G* level of theory39–42 within a MeOH solvent simulated using the polarizable continuum model (PCM).43 Harmonic vibrational frequencies were also computed at the same level of theory to ensure that no imaginary values were present, thus confirming that all of the structures were minima on their respective potential energy surfaces. Thermal Gibbs free energies were obtained using partition functions computed within the harmonic oscillator/rigid rotor approximations,44,45 permitting the calculation of room temperature equilibrium Boltzmann populations. Excitation energies, rotatory strengths, and oscillator strengths for each transition (in the velocity representation) were calculated for the first 40 electronic states at the TDDFT/B3LYP/aug-cc-pVDZ level of theory,39,41,42,46,47 again including the PCM description of the MeOH solvent. All quantum chemical calculations were performed using the Gaussian 09 electronic structure package.48 The ECD spectra were subsequently simulated by overlapping Gaussian functions for each transition according to49
where σ is defined as half the bandwidth at 1/e peak height, and ν̃0a and R0a are the excitation energy (in wavenumbers) and rotatory strength for transition 0 → a, respectively. The σ value is an empirical parameter, and we chose a value of 0.40 eV in agreement with the resolution of the experimental ECD bandwidths. The UV spectra were simulated by overlapping Gaussian functions for each transition according to
where σ is defined as half the bandwidth at 1/e peak height, and ν̃0a and fi are the excitation energy (in wavenumbers) and oscillator strength for transition 0 → a, respectively. The σ value is an empirical parameter, and we chose a value of 0.40 eV in agreement with the resolution of the experimental UV bandwidths.
Individual proton distances were calculated and averaged (using the Boltzmann populations above) to permit comparison with experimental NOESY data. In the case of methyl hydrogens, simple averages of the three individual Boltzmann averaged distances are reported.
Relative thermal Gibbs free energies and associated room temperature equilibrium Boltzmann populations of all unique conformers as well as weighted average ECD and UV spectra for all possible stereoisomers are included in Supporting Information.
Supplementary Material
Acknowledgments
This project was supported by the National Center for Complementary and Integrative Health under award 1 R01 AT008088, and this support is gratefully acknowledged. This work was also supported by the National Science Foundation under Grant No. CHE-0619382 for purchase of the Bruker Avance 500 NMR spectrometer and Grant No. CHE-0722638 for the purchase of the Agilent 6220 mass spectrometer. We thank Mr. B. Bebout for obtaining the mass spectra, Dr. N. Shanaiah for assistance with the NMR spectra, and Dr. T. Grove for the use of the JASCO J-815 spectrometer. We gratefully acknowledge Alexander Rodriguez of INBIO for the collection of plant material. TDC was supported by the National Science Foundation under grant CHE-1465149 and KCP by a graduate fellowship from the Virginia Tech Institute for Critical Technology and Applied Science. The authors acknowledge Advanced Research Computing at Virginia Tech for providing computational resources and technical support that have contributed to the results reported within this paper.
Footnotes
The authors declare no competing financial interest.
1H, 13C, gHMBC, gHSQC, and NOESY spectra for spectra for compounds 1 – 4, and thermal Gibbs free energies and Boltzmann populations for all unique conformers of 1 – 4, and weighted average ECD and UV spectra, optical rotations, and internuclear distances for all possible stereoisomers of 1 – 4. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES AND NOTES
- 1.World Health Organization. [accessed June 9, 2016];10 facts on malaria. http://www.who.int/features/factfiles/malaria/en/
- 2.Anonymous. World Malaria Report 2014. World Health Organization; Geneva, Switzerland: 2014. pp. 1–227. [Google Scholar]
- 3.Kong LY, Tan RX. Nat Prod Rep. 2015;32:1617–1621. doi: 10.1039/c5np00133a. [DOI] [PubMed] [Google Scholar]
- 4.Balint G. Pharmacol Ther. 2001;90:261–5. doi: 10.1016/s0163-7258(01)00140-1. [DOI] [PubMed] [Google Scholar]
- 5.Eastman RT, Fidock DA. Nature. 2009;7:864–874. doi: 10.1038/nrmicro2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, Sopha C, Chuor CM, Nguon C, Sovannaroth S, Pukrittayakamee S, Jittamala P, Chotivanich K, Chutasmit K, Suchatsoonthorn C, Runcharoen R, Hien TT, Thuy-Nhien NT, Thanh NV, Phu NH, Htut Y, Han KT, Aye KH, Mokuolu OA, Olaosebikan RR, Folaranmi OO, Mayxay M, Khanthavong M, Hongvanthong B, Newton PN, Onyamboko MA, Fanello CI, Tshefu AK, Mishra N, Valecha N, Phyo AP, Nosten F, Yi P, Tripura R, Borrmann S, Bashraheil M, Peshu J, Faiz MA, Ghose A, Hossain MA, Samad R, Rahman MR, Hasan MM, Islam A, Miotto O, Amato R, MacInnis B, Stalker J, Kwiatkowski DP, Bozdech Z, Jeeyapant A, Cheah PY, Sakulthaew T, Chalk J, Intharabut B, Silamut K, Lee SJ, Vihokhern B, Kunasol C, Imwong M, Tarning J, Taylor WJ, Yeung S, Woodrow CJ, Flegg JA, Das D, Smith J, Venkatesan M, Plowe CV, Stepniewska K, Guerin PJ, Dondorp AM, Day NP, White NJ. New Engl J Med. 2014;371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jarvis LM. Chem Eng News. 2012;90:30. [Google Scholar]
- 8.Nielsen I. Nord J Bot. 1994;14:462–462. [Google Scholar]
- 9.Panero J. [accessed March 21, 2017];Asteraceae. Sunflowers, Daisies. Version 27. 2012 Jan; http://tolweb.org/Asteraceae/20780/2012.01.27 in The Tree of Life Web Project, http://tolweb.org/
- 10.Aljančić I, Vajs V, Menković N, Karadžić I, Juranić N, Milosavljević S, Macura S. J Nat Prod. 1999;62:909–911. doi: 10.1021/np980536m. [DOI] [PubMed] [Google Scholar]
- 11.Huang G, Yang Y, Wu W, Zhu Y. J Nat Prod. 2010;73:1954–1957. doi: 10.1021/np1004505. [DOI] [PubMed] [Google Scholar]
- 12.Milosavljević S, Macura S, Stefanović M, Aljanćić I, Milinković D. J Nat Prod. 1994;57:64–67. [Google Scholar]
- 13.Zidorn C, Stuppner H, Tiefenthaler M, Konwalinka G. J Nat Prod. 1999;62:984–987. doi: 10.1021/np990058v. [DOI] [PubMed] [Google Scholar]
- 14.Blake S. Torreya. 1915;15 [Google Scholar]
- 15.Shen YC, Lo K, Kuo Y, Khalil A. J Nat Prod. 2005;68:745–750. doi: 10.1021/np040214k. [DOI] [PubMed] [Google Scholar]
- 16.Spring O, Zipper R, Reeb S, Vogler B, Da Costa F. Phytochemistry. 2001;57:267–272. doi: 10.1016/s0031-9422(01)00019-x. [DOI] [PubMed] [Google Scholar]
- 17.Karplus M. J Am Chem Soc. 1963;85:2870–2871. [Google Scholar]
- 18.Catalán C, del Cuenca RM, Hernández L, Joseph-Nathan P. J Nat Prod. 2003;66:949–953. doi: 10.1021/np030055p. [DOI] [PubMed] [Google Scholar]
- 19.Uchida I, Kuriyama K. Tetrahedron Lett. 1974;15:3761–3764. [Google Scholar]
- 20.Crawford TD, Stephens PJ. J Phys Chem A. 2008;112:1339–1345. doi: 10.1021/jp0774488. [DOI] [PubMed] [Google Scholar]
- 21.Van Calsteren MR, Jankowski CK, Reyes-Chilpa R, Jimenez-Estrada M, Campos MG, Zarazua-Lozada MO, Lesage D. Can J Chem. 2008;86:1077–1084. [Google Scholar]
- 22.Finefield JM, Sherman DH, Kreitman MMWR. Angew Chem Int Ed. 2012;51:4802–4836. doi: 10.1002/anie.201107204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prosser I, Altug IG, Phillips AL, Konig WA, Bouwmeester HJ, Beale MH. Arch Biochem Biophys. 2004;432:136–44. doi: 10.1016/j.abb.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 24.Barrero AF, Oltra JE, Álvarez M, Raslan DS, Saúde DA, Akssira M. Fitoterapia. 2000;71:60–64. doi: 10.1016/s0367-326x(99)00122-7. [DOI] [PubMed] [Google Scholar]
- 25.Chung IM, Moon HI. J Enzyme Inhib Med Chem. 2009;24:131–135. doi: 10.1080/14756360801936274. [DOI] [PubMed] [Google Scholar]
- 26.Li XW, Weng L, Gao X, Zhao Y, Pang F, Liu JH, Zhang HF, Hu JF. Bioorg Med Chem Lett. 2011;21:366–372. doi: 10.1016/j.bmcl.2010.10.138. [DOI] [PubMed] [Google Scholar]
- 27.Chea A, Hout S, Long C, Marcourt L, Faure R, Azas N, Elias R. Chem Pharm Bull. 2006;54:1437–1439. doi: 10.1248/cpb.54.1437. [DOI] [PubMed] [Google Scholar]
- 28.Pillay P, Vleggaar R, Maharaj V, Smith P, Lategan C. J Ethnopharmacol. 2007;112:71–76. doi: 10.1016/j.jep.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 29.Chaturvedi D. Sesquiterpene Lactones : Structural Diversity and Their Biological Activities. Opportunity, Challenge and Scope of Natural Products in Medicinal Chemistry. 2011:313. [Google Scholar]
- 30.François G, Passreiter C, Woerdenbag H, Van Looveren M. Planta Med. 1996;62:126–129. doi: 10.1055/s-2006-957833. [DOI] [PubMed] [Google Scholar]
- 31.Ortet R, Prado S, Mouray E, Thomas O. Phytochemistry. 2008;69:2961–2965. doi: 10.1016/j.phytochem.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 32.Dai Y, Harinantenaina L, Bowman J, Da Fonseca IO, Brodie P, Goetz M, Cassera M, Kingston DGI. Bioorg Med Chem. 2014;22:269–276. doi: 10.1016/j.bmc.2013.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cao S, Brodie PJ, Miller JS, Randrianaivo R, Ratovoson F, Birkinshaw C, Andriantsiferana R, Rasamison VE, Kingston DGI. J Nat Prod. 2007;70:679–681. doi: 10.1021/np060627g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan E, Harinantenaina L, Brodie PJ, Miller JS, Callmander MW, Rakotonandrasana S, Rakotobe E, Rasamison VE, Kingston DGI. J Nat Prod. 2010;73:1792–1795. doi: 10.1021/np100411d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Louie KG, Behrens BC, Kinsella TJ, Hamilton TC, Grotzinger KR, McKoy WM, Winker MA, Ozols RF. Cancer Res. 1985;45:2110–2115. [PubMed] [Google Scholar]
- 36.O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Halgren TA. J Comput Chem. 1996;17:490–519. [Google Scholar]
- 38.O’Boyle NM, Vandermeersch T, Flynn CJ, Maguire AR, Hutchison GR. J Cheminf. 2011;3:8. doi: 10.1186/1758-2946-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 40.Ditchfield R, Hehre WJ, Pople JA. J Chem Phys. 1971;54:724–728. [Google Scholar]
- 41.Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. J Phys Chem. 1994;98:11623–11627. [Google Scholar]
- 42.Lee C, Wang W, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 43.Miertuš S, Scrocco E, Tomasi J. Chem Phys. 1981;55:117–129. [Google Scholar]
- 44.McQuarrie DA. Statistical Mechanics. Harper and Row; New York: 1975. p. 356. [Google Scholar]
- 45.Hehre WJ, Radom L, von R, Schleyer PAPJ. Ab Initio Molecular Orbital Theory. Wiley-Interscience; New York: 1986. [Google Scholar]
- 46.Casida ME, Jamorski C, Bohr F, Guan J, Salahub DR. Optical Properties from Density-Functional Theory. In: Karna SP, Yeates AT, editors. Theoretical and Computational Modeling of NLO and Electronic Materials. Vol. 628. American Chemical Society; Washington DC: 1996. p. 145. [Google Scholar]
- 47.Kendall RA, Dunning TH, Jr, Harrison RJ. J Chem Phys. 1992;96:6796–6806. [Google Scholar]
- 48.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JJA. Gaussian 09 Revision E.1. Gaussian Inc; Wallingford CT: 2009. [Google Scholar]
- 49.Stephens PJ, Harada N. Chirality. 2010;22:229–233. doi: 10.1002/chir.20733. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.