

Abstract
Background
Breast cancer patients who do not respond to neoadjuvant therapy have a poor prognosis. There is a pressing need for novel targets and models for preclinical testing. Here we report characterization of breast cancer patient derived xenografts (PDXs) largely generated from residual tumors following neoadjuvant chemotherapy.
Methods
PDXs were derived from surgical samples of primary or locally recurrent tumors. Normal and tumor DNA sequencing, RNASeq and Reverse Phase Protein Arrays (RPPA) were performed. Phenotypic profiling was performed by determining efficacy of a panel of standard and investigational agents.
Results
Twenty-six PDXs were developed from 25 patients. Twenty-two were generated from residual disease following neoadjuvant chemotherapy, and 24 were from triple negative breast cancer (TNBC). These PDXs harbored a heterogeneous set of genomic alterations and represented all TNBC molecular subtypes. On RPPA, PDXs varied in extent of PI3K and MAPK activation. PDX also varied in their sensitivity to chemotherapeutic agents. PI3K, mTOR and MEK inhibitors repressed growth but did not cause tumor regression. PARP inhibitor talazoparib caused dramatic regression in 5 of 12 PDXs. Notably 4 of 5 talazoparib-sensitive models did not harbor germline BRCA1/2 mutations, but several had somatic alterations in homologous repair pathways, including ATM deletion and BRCA2 alterations.
Conclusions
PDXs capture the molecular and phenotypic heterogeneity of TNBC. Here we show that PARP inhibition can have activity beyond germline BRCA1/2 altered tumors, causing regression in a variety of molecular subtypes. These models represent an opportunity for the discovery of rational combinations with targeted therapies and predictive biomarkers.
Keywords: Patient derived xenografts, PARP inhibitors, precision medicine, genomics, somatic mutation
Introduction
Approximately 10–15% of women with breast cancer have triple negative tumors which do not express estrogen and progesterone receptors (ER and PR) or harbor amplifications and/or overexpression of HER2, targets linked to approved breast cancer therapies (1). Patients presenting with operable Stage 2–3 triple negative breast cancer (TNBC) typically receive neoadjuvant chemotherapy. If these agents are unsuccessful in generating pathological complete response or in greatly reducing tumor burden prior to surgery, the patient has a decreased chance of long-term survival (2). Therefore, novel treatment strategies are needed for patients whose tumors are resistant to neoadjuvant chemotherapy (NeoCT).
There has been great interest in determining whether additional systemic therapy can improve outcomes in patients who have significant residual disease after NeoCT (3). Recently it was reported that in the Phase III trial CREATE-X, adjuvant capecitabine was associated with a significantly improved 2 year disease free survival, compared to observation in patients HER2 negative breast cancer with residual disease after NeoCT (4). Ongoing studies are assessing a variety of different strategies for adjuvant treatment of patients with TNBC and residual disease including platinums vs capecitabine (NCT02445391), immunotherapy (NCT02954874) as well as genomically-directed therapy (NCT02101385).
Since TNBC is simply defined by lack of ER/PR/HER2 (over)expression. Several studies have performed comprehensive molecular profiling of TNBC with elucidation of some potential therapeutic vulnerabilities (5,6). TNBC is a heterogeneous disease (7). Lehmann et al. identified six distinct TNBC subtypes, each displaying a unique biology, with recent studies merging these into four transcriptionally defined subtypes (8,9). These subtypes, have been found to be associated with response to chemotherapy (2,8,10). Balko et al have further evaluated the molecular profiles of residual disease in patients with TNBC who received NeoCT and reported a variety of genomic alterations (mutations and copy number changes) including alterations in cell cycle, phosphoinositide 3-kinase (PI3K)/mTOR alterations, growth factor receptor amplifications, Ras/mitogen-activated protein kinase (MAPK) alterations, DNA repair alterations (11). This suggests most patients with residual disease may have actionable genomic alterations; however the efficacy of targeting these alterations have not easily tested systematically in the pre-clinical setting in part due to a lack of linked diverse laboratory models of TNBC.
Efforts are therefore being taken to develop collections of heterogeneous patient-based models of TNBC that can be used to contrast therapeutic sensitivities and link antitumor responses to molecular traits that may only be present in subsets of patients (12–15). In particular, due to the poor outcomes for TNBC patients who harbor significant residual disease following neoadjuvant NeoCT, models derived from post-NeoCT, residual tumors are of particular importance. Patient-derived xenografts (PDXs) enable the generation of diverse models with characteristics similar to the originating tumor (12).
Here we report the molecular characterization (DNA, RNA and protein) of a panel of PDXs enriched for chemoresistant tumors (22 of 26). In addition, we compared the sensitivity of these PDXs to approved cytotoxic agents as well as promising molecularly-targeted agents against TNBC.
Methods
PDX Generation
Surgical samples and blood were collected with informed consent under an Institutional Review Board-Approved Protocol. Following collection, the first 13 PDXs were established by implanting tumor fragments to the flank of nude mice as previously described (13). The latter models were established using a similar surgical procedure but involved placing the fragment on the bilateral 4th mammary fat pads of female nude mice (Department of Radiation Oncology-MD Anderson Cancer Center) or NOD.Cg-Prkdcscid Il2rgtm1Sug/JicTac (Taconic). Estrogen supplementation was provided to one of the specimens BCX.066 as it was 60% ER+ during presurgery clinical pathological analysis. All animal experiments were approved by Institutional Animal Care and Use Committee.
DNA Sequencing Analysis-T200 Gene Panel
Small fragments of frozen tumor were placed in lysis buffer containing protease K and homogenized prior to DNA being extracted using the Qiagen DNA Mini Kit following manufacturer’s protocol. Normal DNA was isolated from whole blood using Qiagen Blood Mini Kit and following the manufacturer’s protocol. Sample preparation and targeted exome sequencing (T200 or T200.1) were performed by the Institute of Personalized Cancer Therapy Cancer Genomic Laboratory at MD Anderson Cancer Center. The sequencing procedure and genes included in the T200 panel have been previously reported (T200 panel contained 202 genes, and T200.1 included 264 genes) (16). T200 data for BCX.006, BCX.010, BCX.011, BCX.017, BCX.022, and BCX.024 has been previously published (13).
RNAseq
Small fragments of frozen tumor were placed in lysis buffer and quickly homogenized manually. RNA was isolated from the lysis using Norgen BIOTEK Total RNA Purification Plus Kit. RNA quality was assessed using Agilent Bioanalyzer. Genomic RNA was quantified by Picogreen (Invitrogen) and quality assessed using the 2200 Tapestation (Agilent). RNA from each sample was converted in double stranded cDNA using Ovation RNA-Seq System V2 kit from Nugen. cDNA was sonicated, libraries were made using KAPA kits and capture was performed using Nimblegen whole exome V3 probes according to manufacturer protocol. The captured libraries were sequenced on a HiSeq 2000 (Illumina) on a version 3 TruSeq paired end flowcell according to manufacturer’s instructions. RNA and DNA sequencing was paired end 2×100 bp. Data analysis was performed using tophat to align paired-end reads to the hg19 version of the reference genome, htseq-count and bedtools to obtain expression counts of genes and exons. Genetic variants were called using GATK unified genotyper. Fusions were detected using tophat-fusion and filtered using Oncofuse. Qualities of raw and aligned reads were assessed using FastQC and RSEQC.
RNA-based classification
Normalized log transformed RNAseq data were uploaded to TNBCType(17) as csv files. TNBC tool provided predicted subtype, correlation coefficients, and the p-values.
RPPA
Reverse phase protein array (RPPA) was done in the MD Anderson Cancer Center Functional Proteomics Core Facility as described previously (18). Frozen tumor tissues were cut into fragments and placed in bead lysis tubes for protein extraction. The average of the triplicates was reported with the exception of BCX.060 and BCX.051 (duplicates) and BCX.017 (single). We only reported RPPA results from rabbit and goat antibodies (230 antibodies) since mouse antibodies may have significant background in PDX samples.
Immunohistochemistry (IHC)
We used the following monoclonal rabbit antibodies for ER, PR and HER2 staining: anti-estrogen receptor alpha (Abcam; ab16660), anti-progesterone receptor A/B (Cell Signaling; (#8757) and anti-HER2/ErbB2 Cell Signaling; #2165). We used CLIA-validated mouse antibodies (ER: Leica; NCL-L-ER-6F11 and PR: Dako; M3568) for IHC staining of surgical samples and PDXs. For the assessment of ER and PR status the percentage of positive tumor nuclei were estimated. HER2 status was assessed according to the HER2 guidelines (19).
In Vivo Treatment
Paclitaxel, eribulin, carboplatin, doxorubicin, and gemcitabine were purchased from the MD Anderson pharmacy. Trametinib, buparlisib, and talazoparib were provided by the Standup to Cancer Pharmacy. TAK228 (MLN0128) was purchased from ChemieTek. Doses of paclitaxel (10 mg/kg, iv, weekly), doxorubicin (8.3 mg/kg, iv, weekly), eribulin (1 mg/kg, iv, weekly), carboplatin (75 mg/kg, ip, weekly), and gemcitabine (10 mg/kg, iv, weekly) were diluted to appropriate volume in phosphate buffered saline prior to administering to mice. Trametinib (0.3 mg/kg, po, daily) was dissolved in dimethyl sulfoxide (DMSO) and diluted in 0.5% hydroxypropylmethylcellulose and 0.2% Tween-80 in water (pH 8.0). TAK228 was dissolved in NVP and diluted in 5% polyvinylpyrrolidone (PVP) in water. Buparlisib was dissolved in NMP and diluted in 50% PEG300. Talazoparib was dissolved in dimethylacetamide and diluted in 90% Solutol HS15 in PBS. Drug doses were selected based on the literature (20–25). Treatment testing was performed using subcutaneous implantation in female athymic nude mice. Tumor volume (TV) was calculated by the formula: TV (mm3) = ((width)2 × length)/2. Change TV from baseline was calculated as (TV DayX − TVDay0)/TVDay0.
Results
We generated a collection of 26 transplantable breast cancer PDXs from tumors resected from 25 patients, including one patient-matched primary tumor and subsequent local recurrence. Twenty-two out of 26 PDXs were derived from tumors following NeoCT. The other four PDXs were derived from patients that underwent surgery without first receiving therapy. The majority of PDXs were developed from patients whose tumors had low/no expression of ER, PR, and HER2 as assessed by clinical IHC either directly on the surgical specimen or needle biopsy prior to neoadjuvant chemotherapy (Table 1). Since some tumors that generated PDXs had low/variable ER expression at biopsy or surgery clinically, we reassessed ER and PR in residual tumors and early passage PDXs (Supplementary Table 1). One PDX was generated from a patient with 1+ HER2 expression by IHC and 4.17 gene copies by FISH prior to receiving neoadjuvant chemotherapy with HER2-targeted therapy. This patient’s post-neoadjuvant chemotherapy surgical sample and PDX did not display HER2 amplification (Supplemental Figure 1) or expression (Supplementary Table 1).
Table 1.
Patient clinical characteristics corresponding to PDXs.
| Source | ER (%) | PR (%) | HER2 | NeoCT | Overall Survival (months) | Distant Relapse | Follow-up (months) | Time to passage (months) | Comments | |
|---|---|---|---|---|---|---|---|---|---|---|
| BCX.006 | Primary | 0 | 0 | 0 | Yes | 9.6 | 4.3 | 1.5 | ||
| BCX.009 | cALN | 1 | 1 | 0 | Yes | Alive | 4.1 | 50 | >12 | IBC |
| BCX.010 | Primary | 0 | 0 | 0 | Yes | 1 | NAb | 2.1 | Metaplastic/IBC | |
| BCX.011 | Primary | 0 | 0 | 0 | Yes | 4.1 | 2.1 | 1.3 | Metaplastic | |
| BCX.017 | Primary | 25 | 0 | 1+a | Yes | Alive | Met free | 53.7 | 3.1 | |
| BCX.022 | Primary | 10 | 0 | 0 | Yes | Alive | Met free | 52.9 | 5.3 | |
| BCX.024 | Primary | 10 | 3 | 0 | No | Alive | Met free | 48.6 | 3.7 | |
| BCX.042 | Primary | 0 | 0 | 0 | No | Alive | Met free | 39.2 | 4.2 | |
| BCX.051 | LRR | 0 | 0 | 0 | Yes | 10 | 1.8 | 3.3 | ||
| BCX.055 | Primary | 1 | 0 | 0 | Yes | Alive | Met free | 36.0 | 6.3 | |
| BCX.060 | Primary | 5 | 0 | 0 | No | Alive | Met free | 4.8 | ||
| BCX.065 | cALN | 10 | 0 | 0 | Yes | Alive | Met free | 31.4 | 8.8 | |
| BCX.066 | LRR | 0 | 0 | 0 | Yes | Alive | 3.9 | 24.6 | 3.7 | |
| BCX.070 | Primary | 0 | 0 | 1+ | Yes | Alive | Met free | 29.7 | 2.1 | |
| BCX.080 | Primary | 10 | 0 | 1+ | Yes | Alive | Met free | 23.3 | 2.4 | |
| BCX.084 | Primary | 0 | 0 | 0 | Yes | 11.8 | 6.7 | 1.8 | ||
| BCX.087 | Primary | 2 | 0 | 1+ | Yes | 3.7 | 3.2 | 1.4 | IBC | |
| BCX.092 | Primary | 0 | 0 | 0 | No | Alive | Met free | 20.3 | 3.5 | |
| BCX.094 | Primary | 0 | 0 | 0 | Yes | Alive | 2.7 | 22.6 | 2.9 | |
| BCX.095 | LRR | 5 | 0 | 0 | Yes | 13.8 | 1 | 3.7 | IBC | |
| BCX.096 | LRR | 20 | 0 | 0 | Yes | Alive | 6.4 | 15.1 | 2.9 | |
| BCX.099 | LRR | 20 | 0 | 0 | Yes | Alive | NAb | 1.2 | ||
| BCX.100 | Primary | 20 | 0 | 0 | Yes | Alive | Met free | 20.0 | 2.8 | Squamous Cell |
| BCX.102 | LRR | 0 | 0 | 2+ | Yes | Alive | Met free | 19.5 | 3.2 | |
| BCX.104 | LRR | 40 | 40 | 0 | Yes | 15.9 | 2.6 | 4.4 | IBC | |
| BCX.105 | Primary | 5 | 0 | 1+ | No | Alive | 4.3 | 14.1 | 5.7 |
cALN-Contralateral Axillary Lymph Node Tumor; LRR-Locoregional Recurrence; ER-Estrogen Receptor; PR-Progesterone Receptor; IBC-Inflammatory Breast Cancer; NeoCT-Neoadjuvant chemotherapy (all PDX denoted as “Yes” were generated from the tumors collected after chemotherapy).
HER2 gene amplification by FISH pre-neoadjuvant therapy. Following trastuzumab based neoadjuvant regimen, PDX was established. Both the residual tumor from the patient and PDX were found to have equivocal HER2 copy number (Supplementary Figure 1).
Presented with metastatic disease and underwent palliative surgery
Our collection of PDXs includes 2 metaplastic tumors and 1 squamous cell tumor. The morphology of the PDXs resembles those of the originating tumor (Supplementary Figure 2). In addition, PDXs were generated from five patients who clinically had inflammatory breast cancer, and one patient with a known deleterious germline BRCA1 mutation (Table 1). Combined with the fact that majority of these PDXs were generated from patients who had received NeoCT and were drug resistant, this collection represents a unique PDX set.
Integrated genomic and expression analysis
We analyzed 21 patient tumor-matched normal tissue-first passage PDX sets by ultradeep exome sequencing targeting 202 or 264 cancer-related genes (16). Sixteen (76%) of these PDXs had TP53 mutations and 12 (57%) had genomic phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) pathway alterations (Figure 1A and Table 2). We also found a high frequency of NOTCH2 amplifications (43%) and ATRX deletions (43%). Twenty of 21 PDXs had potentially actionable genomic alterations (Table 2; Supplementary Table 2). Notably BCX.066 and BCX.096 shared the same actionable alterations (FGFR2 AMP, NOTCH2 AMP) but were derived from subsequent local recurrences from the same patient.
Figure 1. RNAseq and targeted exome sequencing.
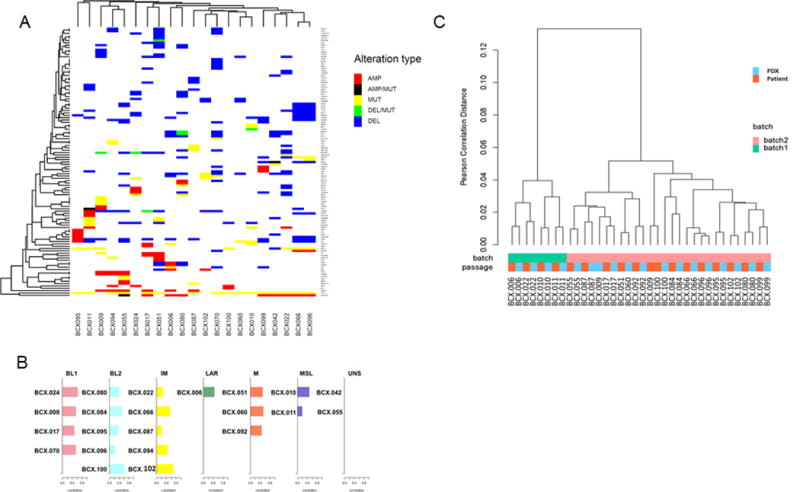
A. RNA sequencing of patient tumors and matched PDXs was performed in two batches. Unsupervised hierarchical clustering based on gene expression shows PDXs cluster with matching patient tumors. B. Subtypes of PDXs based on TNBCType. C. Unsupervised clustering of PDXs based on somatic genomic alterations identified by targeted exome sequencing. Gene amplification defined as >4 copies; gene deletion defined as <1 copy.
Table 2.
Selected Actionable Alterations.
| Model | Alteration |
|---|---|
| BCX.006 | PI3KCA H1047R, FGFR4 HAMP, NOTCH2 HAMP, BRCA2 HDEL |
| BCX.009 | AKT1 HAMP, AKT2 HAMP, CCNE1 HAMP, PIK3R1 HDEL |
| BCX.010 | PI3KCA H1047R, PTEN HDEL |
| BCX.011 | FGFR1 HAMP, PTEN N276S, ATM HDEL |
| BCX.017 | PIK3CA HAMP, CCNE1 HAMP, PIK3R1 HDEL |
| BCX.022 | ATM HDEL, BRCA2 HDEL, CDKN2A, FGFR1 HAMP, NOTCH2 HAMP, PIK3R1 HDEL |
| BCX.024 | PTEN HDEL |
| BCX.042 | NRAS Q61R, BRCA2 HDEL |
| BCX.051 | STK11 HDEL, PIK3R1 HDEL, PTEN HDEL |
| BCX.055 | PTEN HDEL |
| BCX.060 | PIK3R1 HDEL, CDKN2C HDEL, BRCA1 (germline) |
| BCX.066a | FGFR2 HAMP, NOTCH2 HAMP |
| BCX.070 | CCNE1 HAMP, AKT2 HAMP, NOTCH1 K2156*, ATM DEL, BRCA2 HDEL, STK11 HDEL |
| BCX.080 | CDK6 HAMP, BRCA2 splicing variant, CDKN2A, NOTCH2 HAMP |
| BCX.087 | None |
| BCX.094 | STK11 HDEL, BRCA2 HDEL |
| BCX.095 | RAF1 HAMP |
| BCX.096a | FGFR2 HAMP, NOTCH2 HAMP |
| BCX.099 | CDKN2A HDEL, NOTCH2 HAMP |
| BCX.100 | PTEN HDEL |
| BCX.102 | CCND1 HAMP |
HAMP≥4 gene copies; HDEL≤1 gene copies;
BCX.066 and BCX.096 were derived from primary tumor and local recurrence of the same patient
We analyzed RNA expression of 19 patient tumor-first passage PDX pairs by RNA sequencing. Unsupervised clustering based on global RNA expression found that the patient tumors and matched PDXs clustered together with the exception of BCX.009 (Figure 1B), which also had lower frequencies for genomic alterations compared to the PDX. This single patient-PDX discrepancy appears attributable to low cellularity in the tumor submitted for molecular analysis.
Lehmann et al have developed a seven-subtype classification system based on gene expression profile for TNBC, and these subtypes have been shown to have clinical significance and the potential to inform treatment (2,8). The six major subtypes are basal-like (BL1), Basal-like 2 (BL2), Mesenchymal (M), Mesenchymal Stem-like (MSL), Immune (IM), and Luminal Androgen (LAR). The seventh subtype is comprised of tumors that are Unstable (UNS) and do not significantly represent a single subtype. Using TNBCType (17), we identified the TNBC subtype of 21 PDXs using RNA expression data (Figure1C). PDXs representing all 6 subtypes were identified, including two UNS (BCX.055-M/BL1; BCX.042-MSL/BL2). BCX.099 patient specimen and PDX were rejected by TNBCType potentially due to its ER positivity. In the cases with available patient expression data, the subtype of the patient was exhibited by the PDX tumor in 63% of cases. For those that exhibited divergent subtypes between PDX and patient, the patient tumor TNBC subtype exhibited the second best fit of the PDX in 66% of cases. The most divergence between patient and PDX was BCX.009 which took over a year to expand in mice, and the patient sample is suspected to have low cellularity due to low allelic frequency of genomic alterations. Interestingly, this was the only patient sample to have high correlation to both IM and MSL subtypes, suggesting normal cell contribution to this models signature as supported by new reports (9). Two additional patients switched from IM subtype in the patient to BL2 in the PDX, potentially reflecting the loss of human immune cells in the microenvironment in the PDXs.
More recently, these TNBC subtypes have been further narrowed to 4 major subtypes by excluding the IM and MSL subtypes due to their strong overlap with immune cells (IM) and tumor stroma (MSL) gene expression, respectively (13). We reclassified the PDXs based on this report by classifying those models in the IM or MSL subtype to the subtype with the next highest correlation (Supplementary Table 3). While most IM models did have relatively high correlation with an additional subtype, BCX.102 only had correlation with the IM subtype. The two PDXs originally classified as MSL were unique from the other PDXs in that they were derived from metaplastic tumors, and maintained this histopathology in mice. Metaplastic tumors are known to have mesenchymal traits and exhibit stem-like gene expression similar to the MSL subtype. For this reason, we believe that these 2 PDXs indeed represent a specific gene expression subtype (MSL) different than the other PDXs that is driven by the tumor rather than by infiltrating stroma. Moreover, while normal stromal interference may result in MSL subtype calls in the original tumor, these PDXs maintain the MSL subtype as determined by human specific RNAseq in the mouse stroma background. Thus we feel that metaplastic tumors have unique gene expression that are best classified into the MSL subtype though indeed most similar to normal stromal cells.
Response to Standard Chemotherapy
We sought to determine whether the PDXs differed in their sensitivity to standard of care chemotherapy agents used for breast cancer treatment. We assessed the effect of 5 chemotherapeutic agents on 7 PDXs using cohorts of 2 or 3 mice similar to Gao et al (26) (Figure 2). The agents tested included microtubule inhibitors (paclitaxel, eribulin), DNA damaging agents (carboplatin), anti-metabolite (gemcitabine), and anthracycline/antibiotic (doxorubicin). We defined three PDX response categories for this study: Progressive disease as >50% median change in volume from start of treatment; Stable disease as <50% median increase from baseline and <50% decrease from baseline; Responsive disease as 100% of engraftments showing >50% median decrease in volume from baseline. All treatments were done in parallel with untreated control PDXs, which all fell into the progressive category. Responses were assessed either at 28 days or once the control tumors reached maximum allowable size.
Figure 2. PDX responses to approved therapies.
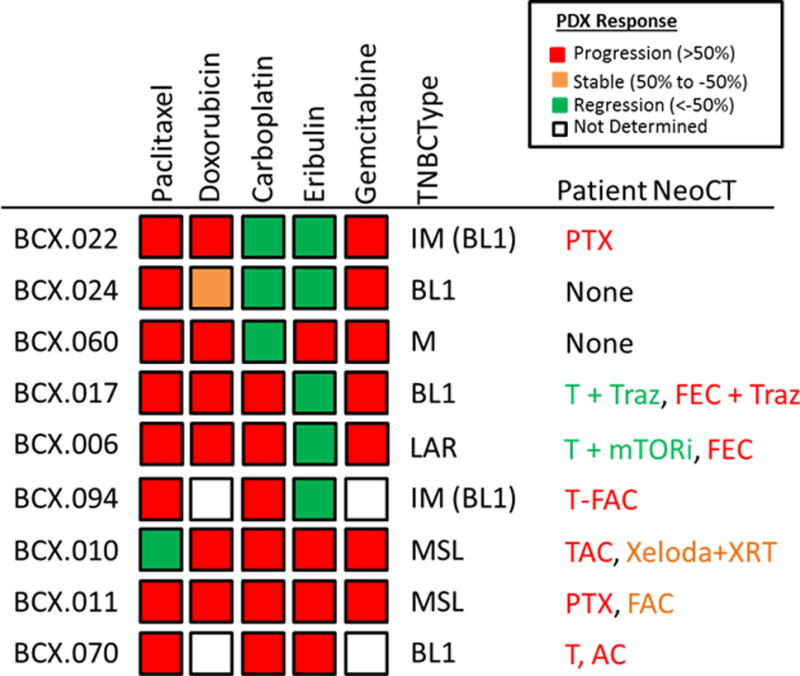
The response of PDXs is shown on the left. PDXs were treated with paclitaxel, doxorubicin, carboplatin, eribulin or gemcitabine weekly for up to 28 days. Median response is shown as defined in the key. All models defined as regressive had 100% of engraftments regress. No progressive models showed interval regression. TNBCtype shown in center; the subtypes in parenthesis are based on the revised TNBCType-4 scheme. The neoadjuvant therapy that the patient received is shown on the right. Radiographic responses were color-coded: red: progression, orange: stable disease, and green: partial response. PTX-Paclitaxel; T-paclitaxel (Taxol), Traz-trastuzumab; FEC-fluorouracil+epirubicin+cyclophosphamide; FAC-Fluorouracil+doxorubicin (Adriamycin)+cyclophosphamide;TAC-docetaxel (Taxotere)+ doxorubicin + cyclophosphamide.
Five of these PDX models were developed from residual disease after chemotherapy and 2 PDXs from treatment-naive tumors (BCX.024 and BCX.060). Six patients had progressed on anthracycline based therapy, and all six PDXs progressed on doxorubicin. The best PDX response to doxorubicin was BCX.024, a chemonaive tumor, with stable disease over 28 days (BCX.024). All but one PDX model progressed on paclitaxel; two of the PDXs tested were from tumors that had interval responses to taxane in combination with a targeted agent. Three PDXs regressed with carboplatin treatment. Five out of 9 models tested regressed when treated with eribulin. It is notable that the PDXs differed from each other in sensitivity. Previous work had suggested differential sensitivity to different chemotherapeutics such as platinum agents based on TNBC gene expression subtype (8) Sin contrast in our limited series sensitivity to specific agents did not have a clear association with transcriptional TNBCtype.
Response to PI3K/mTOR or MEK/ERK Inhibition
Due to the interest in targeting the PI3K and mitogen-activated protein kinase (MAPK) pathways for breast cancer therapy, we assessed activation of the PI3K and MAPK pathways by RPPA (Figure 3A). We calculated the PI3K pathway activation score as previously reported (18). As expected, PIK3CA, PTEN, AKT1/2 alterations were frequent among the top half of PDXs when stratified on PI3K activation score (Figure 3A). Some PDXs had high PI3K activation scores but no genomic alterations currently linked to PI3K pathway activation. The three metaplastic or splindloid PDXs (BCX.010; BCX.011; BCX.100) had high expression of phospho-S6 and low expression of PTEN protein (Figure 3A; Supplementary Figure 3). BCX.010 and BCX.100 harbor PTEN deletions, and BCX.011 has a rare PTEN alteration (N276S) linked to decreased protein stability (27). Indeed BCX.011 has relatively high PTEN mRNA expression and relatively low PTEN protein expression (Supplementary Figure 3). Notably PI3K activation score and MAPK/MEK activation showed a converse pattern, suggesting that PDXs are differentially driven by one pathway or the other. PDXs harboring RAS (BCX.042), and RAF (BCX.095) alterations had low PI3K activation and relatively high MAPK/ERK activation.
Figure 3. Response of PDXs that differ in baseline genomic alterations and pathway activations to PI3K or mTOR or MEK inhibitors.

A. Genomic alterations related to PI3K pathway activation are shown on the left: purple=activating/inactivating mutation: orange=gene amplification (≥4 copies); blue: gene deletion (≤1 copy). Functional proteomic analysis by RPPA is on the right=red: higher expression, green=lower expression. PI3K/mTOR activation score was calculated for each PDX using RPPA data as previously described. B. Growth curves of PDXs treated with buparlisib, trametinib, or TAK228. TV: Tumor Volume
We next assessed the antitumor efficacy of agents targeting PI3K/mTOR, MEK and PARP, molecular targets that are actively pursued in clinical trials and representing pathways dysregulated in TNBC. We tested seven PDXs for response to buparlisib, a pan-PI3K inhibitor, eight PDXs for response to TAK228, a catalytic mTOR inhibitor, and eight PDXs for response to trametinib, a MEK1/2 inhibitor, and 13 PDXs for response to talazoparib, a PARP1/2 inhibitor. These tested PDXs exhibited a range of PI3K/mTOR activation and range of MEK/ERK phosphorylation and included PDXs with and without genomic alterations linked to PI3K/mTOR activation (Figure 3A). No PDXs regressed with buparlisib, trametinib, or TAK228 (Figure 3B). However, buparlisib, trametinib, or TAK228 did have broad growth inhibitory effects on the PDXs tested.
Five models regressed when treated with talazoparib and one model had stable disease for at least 28 days (Figure 4). Four of the talazoparib-sensitive PDXs had genomic alterations linked to homologous repair deficiency and PARP inhibitor sensitivity: BCX.060-deleterious germline BRCA1 mutation; BCX.022-ATM deletion; BCX.024-PTEN deletion; BCX.080-Somatic BRCA2 mutation (25,28–30). Of the non-BRCA1 germline models that regressed with talazoparib treatment, 2 were BL1 and two were BL2 based on the TNBCtype-4 classification scheme. The model that displayed stable disease for at least 28 days (BCX.087) was also BL1 subtype. The BRCA1 germline altered model was M subtype. Two M PDXs, two MSL PDXs, one LAR, two BL1, and one BL2 models progressed on talazoparib. Four out of five regressive models had relatively low expression of BRCA1/2 RNA (Supplementary Figure 4). Three of the PDX models that regressed with PARP inhibition, were also tested for carboplatin sensitivity and all three demonstrated regression when treated with carboplatin (Supplementary Figure 5).
Figure 4. Response of PDXs to PARP inhibitor Talazoparib.
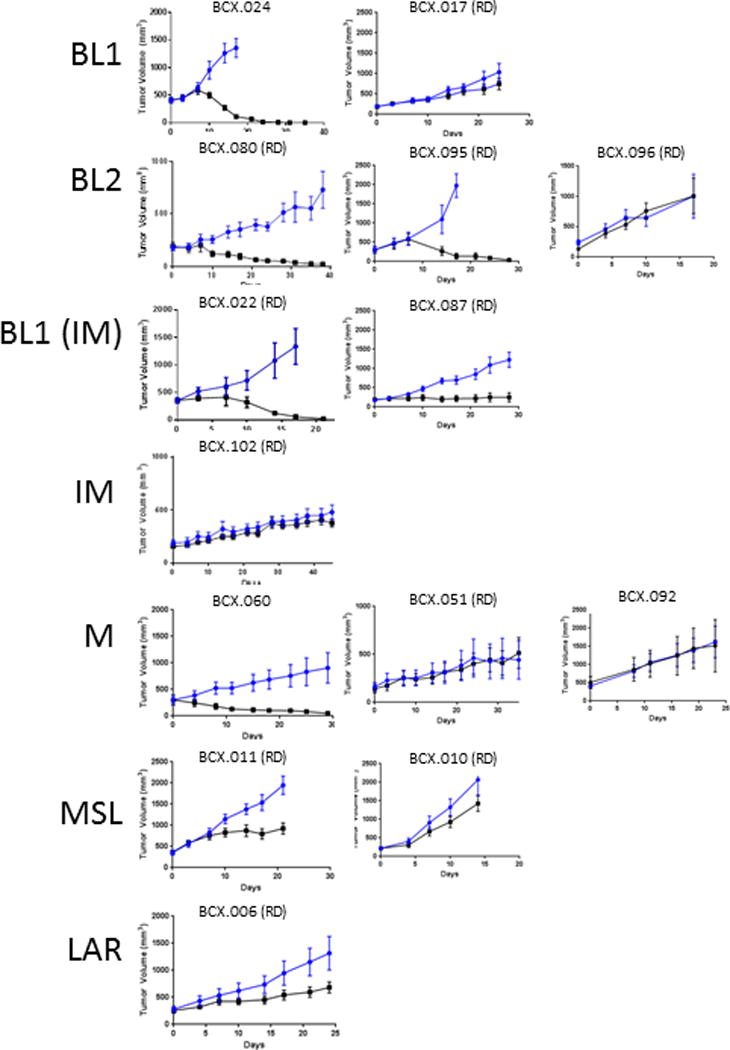
A. Growth curves of PDXs treated with Talazoparib (0.3 mg/kg/day). TNBC subtype is shown on the left; subtype in parenthesis in based on revised classification scheme.
Discussion
There is growing interest in using PDX models to test novel therapeutics and for predictive biomarker discovery. Here we report a panel of PDXs, enriched for chemoresistant TNBC. The models captured many of the actionable targets currently being pursued for drug development. The PDXs demonstrate the molecular as well as phenotypic heterogeneity of TNBC.
There has been increasing recognition of the interpatient molecular heterogeneity in TNBC. In a recent study we reported that of 112 TNBC patients who underwent genomic profiling on a commercial next generation sequencing platform, none had the same genomic profile (31). Similarly, in our study, PDXs showed marked heterogeneity of underlying genomic alterations. Of the PDXs, few had clinically relevant recurrent genomic alterations. Our PDX panel displayed multiple actionable alterations currently being pursued in drug development including PI3K pathway alterations (PTEN mutations/loss, AKT1/2 amplification, PIK3CA mutations), FGFR1-4 alterations, mutations/loss in homologous repair genes (ATM), and NOTCH mutations/amplifications. The heterogeneity of TNBC clearly adds to the challenge of precision oncology; however our models provide a representative panel for testing of novel agents and biomarker-sensitivity associations.
Selected cohort studies have suggested that PDXs recapitulate responses seen in corresponding patients (32,33). Our study was not designed to address the question of concordance of PDX responses with patient tumor in vivo responses. However, in our study, all but one PDX tested with patient-matched (same class) therapy had a similar response as the tumor in the patient. The divergent response was seen in BCX.010, which drastically underwent regression in the first 14 days of paclitaxel treatment in the mice while this patient progressed after 2 cycles of docetaxel (another taxane), adriamycin and cyclophosphamide (TAC). The diversity of responses in PDXs to different therapies, highlights the need for predictors of response. There has been growing interest in using PDXs as “avatars” to select more effective therapy for individual patients. Although the rate of PDX engraftment is modest in our series, engraftment is higher in chemoresistant disease (13), and the timeline of PDX engraftment may allow for generation of PDX from residual disease to facilitate drug sensitivity testing in case of systemic relapse. However, extensive further study would be needed to determine if indeed post-neoCT PDX models predict therapy response in the metastatic setting. The utility of PDXs may be greater in the context of drug development. PDXs are a powerful discovery platform, and can allow us to interrogate drug sensitivity of established as well as novel agents across a variety of cancer subtypes, and can expedite biomarker-phenotype correlations.
Breast cancer profiling done by The Cancer Genome Atlas (TCGA) has demonstrated that TNBC is associated with PI3K pathway activation (5). PI3K and mTOR inhibitors had broad growth inhibitory effects across most models, including those with PI3K/mTOR pathway activation. However, lack of regression in TNBC PDXs with these agents even in the presence of pathway activation by RPPA or genomic alterations in PI3K genes suggests that the single agent efficacy of targeting the PI3K/MTOR pathway may be limited in chemoresistant TNBC. These findings support these agents being better aligned with combination strategies. Recently, in a Phase 1 trial doxorubicin, bevacizumab, and everolimus (mTORi) or liposomal doxorubicin, bevacizumab (VEGFi) and temsirolimus (mTORi) showed objective responses in metaplastic breast cancer patients with PI3K pathway aberrations (34). The growth inhibitory effect of buparlisib and TAK228 on the 2 metaplastic PDXs with PTEN alterationssupports the clinical findings of the utility of targeting PI3K/MTOR pathway in metaplastic breast cancer (34,35) and offers an opportunity to find combinations that will further enhance this effect. These findings support the idea that these combinations are well-suited for tumors with aberrant PI3K/MTOR signaling but do not rule out similar effects in non-PI3K/mTOR activated TNBC, potentially by non-cancer cell autonomous effects such as inhibiting angiogenesis (36,37).
Recently Litton and colleagues have shown that talazoparib has marked antitumor efficacy in patients with germline BRCA mutations (38). Of the targeted agents tested in our PDX models, talazoparib seems to have the most potential as a single agent, and its utility appears to extend beyond BRCA1/2 germline deleterious mutations. While one of the talazoparib-sensitive models had a germline BRCA1 mutation, three others arose from tumors without germline BRCA alterations but had somatic alterations: PTEN loss and ATM loss and a BRCA2 splice variant. While the molecular traits of four out of five PARP inhibitor-sensitive PDXs are in line with prior hypotheses regarding PARP inhibitor response, BCX.095 was divergent in expressing relatively high BRCA1/2 mRNA and having no genomic alteration directly linked to PARP inhibitor sensitivity. However, this model did display loss of heterozygosity and a truncating mutation in the last codon of BRCA2 (K3326*). The functional impact of this alteration is somewhat controversial but is believed to be non-pathogenic (39). However, the patient has a strong family history of cancer suggesting either a functional role in this context or that there may be another alteration that plays a role in cancer predisposition and therapeutic sensitivity.
Using cell lines in vitro, basal-like tumors (BL1 and BL2) have been shown to more frequently be sensitive to PARP inhibitors compared to M and MSL subtypes (8). Indeed, we have yet to identify M or MSL PDXs without germline BRCA1/2 mutations that are sensitive to talazoparib. Several BL1 and BL2 PDXs did however progress on talazoparib, suggesting the need to better define a PARP inhibitor sensitive molecular profile. This question is especially timely given recent report of efficacy of PARP inhibitor olaparib in patients with HER2-negative metastatic breast cancer and germline BRCA1 or BRCA2 mutations (40).
There is recognition that tumors with DNA Damage repair defects are more sensitive to platinum agents. In the TNT trial, carboplatin demonstrated significantly increased activity versus docetaxel in BRCA carriers, but not among unselected TNBC patients (41). In our study PDXs tested with both carboplatin and talazoparib were sensitive to both agents, suggesting common mechanisms of action. This finding is also aligned with the finding that PARP inhibitors have been shown to be effective as maintenance therapy in patients with platinum-sensitive tumors (42). Ongoing studies are assessing whether addition of PARP inhibitors to platinum agents or other approved or investigational agents can further enhance antitumor efficacy.
In summary, we here report a series of PDXs primarily derived from chemoresistant TNBC, thus addressing an area of urgent need for novel therapeutics. These PDXs have been extensively characterized at the level of DNA, RNA, and protein, expanding on existing datasets such as those from the TCGA, that have been primarily derived from chemonaive disease. We also performed a preliminary characterization of sensitivity to standard or emerging investigational therapies.We expect our molecularly annotated PDX set will be an invaluable resource for drug development and biomarker discovery. We here also report the potential utility of broader use of PARP inhibitors for TNBC without BRCA1/2 germline alterations. Further study is needed to determine if there is preferential implantation of cancer clones and further clonal selection upon establishment of PDXs that could have contributed to our results. In future studies we will expand our PDX panel to facilitate discovery of predictive and pharmacodynamics markers of response to PARP inhibitors as well as other novel therapeutics.
Supplementary Material
Translational Relevance.
Patients with triple negative breast cancer (TNBC) that do not respond to neoadjuvant therapy have a poor prognosis indicating a need for novel therapies and better patient selection. TNBC is a heterogeneous disease driven by a diverse set of alterations and exhibiting varied potential molecular intervention points. Here we report characterization of heterogeneous patient-derived xenografts (PDXs) largely generated from residual support broad utility of talazoparib in TNBC following neoadjuvant chemotherapy. We established robust sensitivities to approved cytotoxic agents and promising targeted agents. While cell signaling targeting agents did not cause regression as single agents in any PDXs, the poly(ADP-ribose)polymerase inhibitor talazoparib caused marked regression in 5 out of 13 PDXs, including 3 of 10 PDXs that arose from tumors that did not respond to neoadjuvant therapy. Notably four of five talazoparib-sensitive models did not harbor known deleterious germline BRCA1/2 mutations. These results support broad utility of talazoparib in TNBC and the potential for PDXs for in vivo screening and defining novel phenotype/genotype correlations.
Acknowledgments
Funding: This work was supported by National Institutes of Health T32 CA009599 (NPA, FMB), MD Anderson Women’s Cancers Moonshot Program (KE, FMB), the Nellie B. Connally Breast Cancer Research Endowment (AA, KE, EY, SS, FMB), National Cancer Institute Patient Derived Xenograft Administrative Supplement CA186688-03S2, Susan G. Komen Foundation for the Cure grant SAC10006 (FMB), The Cancer Prevention and Research Institute of Texas (RP150535), the Barr funds, and the MD Anderson Cancer Center support grant (P30 CA016672), and by a Stand Up To Cancer Dream Team Translational Research Grant (Grant Number: SU2C-AACR-DT0209). Stand Up To Cancer is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C. STR DNA fingerprinting was done by the CCSG-funded Characterized Cell Line Core, NCI # CA016672. Reverse protein arrays done in core funded by NCI # CA16672
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Anders CK, Carey LA. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin Breast Cancer. 2009;9(Suppl 2):S73–81. doi: 10.3816/CBC.2009.s.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Masuda H, Baggerly KA, Wang Y, Zhang Y, Gonzalez-Angulo AM, Meric-Bernstam F, et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin Cancer Res. 2013;19(19):5533–40. doi: 10.1158/1078-0432.CCR-13-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gonzalez-Angulo AM, Lei X, Alvarez RH, Green MC, Murray JL, Valero V, et al. Phase II Randomized Study of Ixabepilone Versus Observation in Patients With Significant Residual Disease After Neoadjuvant Systemic Therapy for HER2-Negative Breast Cancer. Clin Breast Cancer. 2015;15(5):325–31. doi: 10.1016/j.clbc.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toi M, Lee S-J, Lee E, Ohtani S, Im Y-H, Im S-A, et al. Abstract S1-07: A phase III trial of adjuvant capecitabine in breast cancer patients with HER2-negative pathologic residual invasive disease after neoadjuvant chemotherapy (CREATE-X, JBCRG-04) Cancer Research. 2016;76(4 Supplement) doi: 10.1158/1538-7445.sabcs15-s1-07. S1-07-S1. [DOI] [Google Scholar]
- 5.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vaca-Paniagua F, Alvarez-Gomez RM, Maldonado-Martinez HA, Perez-Plasencia C, Fragoso-Ontiveros V, Lasa-Gonsebatt F, et al. Revealing the Molecular Portrait of Triple Negative Breast Tumors in an Understudied Population through Omics Analysis of Formalin-Fixed and Paraffin-Embedded Tissues. PLoS One. 2015;10(5):e0126762. doi: 10.1371/journal.pone.0126762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mayer IA, Abramson VG, Lehmann BD, Pietenpol JA. New strategies for triple-negative breast cancer–deciphering the heterogeneity. Clin Cancer Res. 2014;20(4):782–90. doi: 10.1158/1078-0432.CCR-13-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–67. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lehmann BD, Jovanovic B, Chen X, Estrada MV, Johnson KN, Shyr Y, et al. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS One. 2016;11(6):e0157368. doi: 10.1371/journal.pone.0157368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jovanovic B, Mayer IA, Mayer EL, Abramson VG, Bardia A, Sanders M, et al. A randomized phase II neoadjuvant study of cisplatin, paclitaxel with or without everolimus in patients with stage II/III triple-negative breast cancer (TNBC) Clin Cancer Res. 2017 doi: 10.1158/1078-0432.CCR-16-3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balko JM, Giltnane JM, Wang K, Schwarz LJ, Young CD, Cook RS, et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014;4(2):232–45. doi: 10.1158/2159-8290.CD-13-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whittle JR, Lewis MT, Lindeman GJ, Visvader JE. Patient-derived xenograft models of breast cancer and their predictive power. Breast Cancer Res. 2015;17:17. doi: 10.1186/s13058-015-0523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McAuliffe PF, Evans KW, Akcakanat A, Chen K, Zheng X, Zhao H, et al. Ability to Generate Patient-Derived Breast Cancer Xenografts Is Enhanced in Chemoresistant Disease and Predicts Poor Patient Outcomes. PLoS One. 2015;10(9):e0136851. doi: 10.1371/journal.pone.0136851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bruna A, Rueda OM, Greenwood W, Batra AS, Callari M, Batra RN, et al. A Biobank of Breast Cancer Explants with Preserved Intra-tumor Heterogeneity to Screen Anticancer Compounds. Cell. 2016;167(1):260–74 e22. doi: 10.1016/j.cell.2016.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goetz MP, Kalari KR, Suman VJ, Moyer AM, Yu J, Visscher DW, et al. Tumor Sequencing and Patient-Derived Xenografts in the Neoadjuvant Treatment of Breast Cancer. J Natl Cancer Inst. 2017;109(7) doi: 10.1093/jnci/djw306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen K, Meric-Bernstam F, Zhao H, Zhang Q, Ezzeddine N, Tang LY, et al. Clinical actionability enhanced through deep targeted sequencing of solid tumors. Clin Chem. 2015;61(3):544–53. doi: 10.1373/clinchem.2014.231100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen X, Li J, Gray WH, Lehmann BD, Bauer JA, Shyr Y, et al. TNBCtype: A Subtyping Tool for Triple-Negative Breast Cancer. Cancer Inform. 2012;11:147–56. doi: 10.4137/CIN.S9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meric-Bernstam F, Akcakanat A, Chen H, Sahin A, Tarco E, Carkaci S, et al. Influence of biospecimen variables on proteomic biomarkers in breast cancer. Clin Cancer Res. 2014;20(14):3870–83. doi: 10.1158/1078-0432.CCR-13-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolff AC, Hammond ME, Hicks DG, Dowsett M, McShane LM, Allison KH, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013;31(31):3997–4013. doi: 10.1200/JCO.2013.50.9984. [DOI] [PubMed] [Google Scholar]
- 20.Holder AM, Akcakanat A, Adkins F, Evans K, Chen H, Wei C, et al. Epithelial to mesenchymal transition is associated with rapamycin resistance. Oncotarget. 2015;6(23):19500–13. doi: 10.18632/oncotarget.3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamaguchi T, Kakefuda R, Tajima N, Sowa Y, Sakai T. Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int J Oncol. 2011;39(1):23–31. doi: 10.3892/ijo.2011.1015. [DOI] [PubMed] [Google Scholar]
- 22.Hassan B, Akcakanat A, Sangai T, Evans KW, Adkins F, Eterovic AK, et al. Catalytic mTOR inhibitors can overcome intrinsic and acquired resistance to allosteric mTOR inhibitors. Oncotarget. 2014;5(18):8544–57. doi: 10.18632/oncotarget.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burger MT, Pecchi S, Wagman A, Ni ZJ, Knapp M, Hendrickson T, et al. Identification of NVP-BKM120 as a Potent, Selective, Orally Bioavailable Class I PI3 Kinase Inhibitor for Treating Cancer. ACS Med Chem Lett. 2011;2(10):774–9. doi: 10.1021/ml200156t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bieniasz M, Radhakrishnan P, Faham N, De La OJ, Welm AL. Preclinical Efficacy of Ron Kinase Inhibitors Alone and in Combination with PI3K Inhibitors for Treatment of sfRon-Expressing Breast Cancer Patient-Derived Xenografts. Clin Cancer Res. 2015;21(24):5588–600. doi: 10.1158/1078-0432.CCR-14-3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I, Elliott R, et al. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin Cancer Res. 2013;19(18):5003–15. doi: 10.1158/1078-0432.CCR-13-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015;21(11):1318–25. doi: 10.1038/nm.3954. [DOI] [PubMed] [Google Scholar]
- 27.Spinelli L, Black FM, Berg JN, Eickholt BJ, Leslie NR. Functionally distinct groups of inherited PTEN mutations in autism and tumour syndromes. J Med Genet. 2015;52(2):128–34. doi: 10.1136/jmedgenet-2014-102803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McEllin B, Camacho CV, Mukherjee B, Hahm B, Tomimatsu N, Bachoo RM, et al. PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors. Cancer Res. 2010;70(13):5457–64. doi: 10.1158/0008-5472.CAN-09-4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hennessy BT, Timms KM, Carey MS, Gutin A, Meyer LA, Flake DD, 2nd, et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol. 2010;28(22):3570–6. doi: 10.1200/JCO.2009.27.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gilardini Montani MS, Prodosmo A, Stagni V, Merli D, Monteonofrio L, Gatti V, et al. ATM-depletion in breast cancer cells confers sensitivity to PARP inhibition. J Exp Clin Cancer Res. 2013;32:95. doi: 10.1186/1756-9966-32-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wheler JJ, Atkins JT, Janku F, Moulder SL, Stephens PJ, Yelensky R, et al. Presence of both alterations in FGFR/FGF and PI3K/AKT/mTOR confer improved outcomes for patients with metastatic breast cancer treated with PI3K/AKT/mTOR inhibitors. Oncoscience. 2016;3(5–6):164–72. doi: 10.18632/oncoscience.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marangoni E, Vincent-Salomon A, Auger N, Degeorges A, Assayag F, de Cremoux P, et al. A new model of patient tumor-derived breast cancer xenografts for preclinical assays. Clin Cancer Res. 2007;13(13):3989–98. doi: 10.1158/1078-0432.CCR-07-0078. [DOI] [PubMed] [Google Scholar]
- 33.Zhang X, Claerhout S, Prat A, Dobrolecki LE, Petrovic I, Lai Q, et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013;73(15):4885–97. doi: 10.1158/0008-5472.CAN-12-4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basho RK, Gilcrease M, Murthy RK, Helgason T, Karp DD, Meric-Bernstam F, et al. Targeting the PI3K/AKT/mTOR Pathway for the Treatment of Mesenchymal Triple-Negative Breast Cancer: Evidence From a Phase 1 Trial of mTOR Inhibition in Combination With Liposomal Doxorubicin and Bevacizumab. JAMA Oncol. 2016 doi: 10.1001/jamaoncol.2016.5281. [DOI] [PubMed] [Google Scholar]
- 35.Moulder S, Helgason T, Janku F, Wheler J, Moroney J, Booser D, et al. Inhibition of the phosphoinositide 3-kinase pathway for the treatment of patients with metastatic metaplastic breast cancer. Ann Oncol. 2015;26(7):1346–52. doi: 10.1093/annonc/mdv163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matas-Cespedes A, Rodriguez V, Kalko SG, Vidal-Crespo A, Rosich L, Casserras T, et al. Disruption of follicular dendritic cells-follicular lymphoma cross-talk by the pan-PI3K inhibitor BKM120 (Buparlisib) Clin Cancer Res. 2014;20(13):3458–71. doi: 10.1158/1078-0432.CCR-14-0154. [DOI] [PubMed] [Google Scholar]
- 37.Gokmen-Polar Y, Liu Y, Toroni RA, Sanders KL, Mehta R, Badve S, et al. Investigational drug MLN0128, a novel TORC1/2 inhibitor, demonstrates potent oral antitumor activity in human breast cancer xenograft models. Breast Cancer Res Treat. 2012;136(3):673–82. doi: 10.1007/s10549-012-2298-8. [DOI] [PubMed] [Google Scholar]
- 38.Litton JK, Scoggins M, Ramirez DL, Murthy RK, Whitman GJ, Hess KR, et al. A pilot study of neoadjuvant talazoparib for early-stage breast cancer patients with a BRCA mutation. Annals of Oncology. 2016;27(suppl 6) doi: 10.1093/annonc/mdw364.10. [DOI] [Google Scholar]
- 39.Meeks HD, Song H, Michailidou K, Bolla MK, Dennis J, Wang Q, et al. BRCA2 Polymorphic Stop Codon K3326X and the Risk of Breast, Prostate, and Ovarian Cancers. J Natl Cancer Inst. 2016;108(2) doi: 10.1093/jnci/djv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robson ME, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. Abstract LBA4: OlympiAD: Phase III trial of olaparib monotherapy versus chemotherapy for patients (pts) with HER2-negative metastatic breast cancer (mBC) and a germline BRCA mutation (gBRCAm) J Clin Oncol. 2017;35 (Suppl, abstr LBA4) [Google Scholar]
- 41.Tutt A, Ellis P, Kilburn L, Gilett C, Pinder S, Abraham J, et al. Abstract S3-01: The TNT trial: A randomized phase III trial of carboplatin (C) compared with docetaxel (D) for patients with metastatic or recurrent locally advanced triple negative or <em>BRCA1/2</em> breast cancer (CRUK/07/012) Cancer Research. 2015;75(9 Supplement) doi: 10.1158/1538-7445.sabcs14-s3-01. S3-01-S3. [DOI] [Google Scholar]
- 42.Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15(8):852–61. doi: 10.1016/S1470-2045(14)70228-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.