

Abstract
UVB exposure at ambient outdoor levels triggers rapid K+ loss and apoptosis in human corneal limbal epithelial (HCLE) cells cultured in medium containing 5.5 mM K+, but considerably less apoptosis occurs when the medium contains the high K+ concentration that is present in tears (25 mM). Since Ba2+ blocks several K+ channels, we tested whether Ba2+-sensitive K+ channels are responsible for some or all of the UVB-activated K+ loss and subsequent activation of the caspase cascade and apoptosis. Corneal epithelial cells in culture were exposed to UVB at 80 or 150 mJ/cm2. Patch-clamp recording was used to measure UVB-induced K+ currents. Caspase-activity and TUNEL assays were performed on HCLE cells exposed to UVB followed by incubation in the presence or absence of Ba2+. K+ currents were activated in HCLE cells following UVB-exposure. These currents were reversibly blocked by 5 mM Ba2+. When HCLE cells were incubated with 5 mM Ba2+ after exposure to UVB, activation of caspases-9, -8, and -3 and DNA fragmentation were significantly decreased. The data confirm that UVB-induced K+ current activation and loss of intracellular K+ leads to activation of the caspase cascade and apoptosis. Extracellular Ba2+ inhibits UVB-induced apoptosis by preventing loss of intracellular K+ when K+ channels are activated. Ba2+ therefore has effects similar to elevated extracellular K+ in protecting HCLE cells from UVB-induced apoptosis. This supports our overall hypothesis that elevated K+ in tears contributes to protection of the corneal epithelium from adverse effects of ambient outdoor UVB.
Keywords: apoptosis, caspase, corneal epithelium, K+ channel, potassium, tears, ultraviolet
I. INTRODUCTION
Ultraviolet (UV) radiation at wavelengths in the UVB (peak 302 nm) or UVC (peak 254 nm) ranges causes cells to go into apoptosis. Upon exposure to UV, a common initiating factor for apoptosis appears to be activation of K+ channels in the cell membrane and loss of intracellular K+, which moves out of the cell down its concentration gradient.1–5 UV-induced activation of apoptotic signaling pathways then leads to activation of initiator and effector caspases, DNA fragmentation, formation of apoptosomes and finally, cell death.6,7
The cornea protects the lens and retina from damage by absorbing a majority of ambient UVB radiation.8–10 Although the corneal epithelium in vivo is exposed to outdoor UVB at levels that can cause apoptosis, it appears to be relatively resistant to damage by ambient UVB. In contrast, we have previously reported that exposure of human corneal limbal epithelial (HCLE) cells to UVB at 80–200 mJ/cm2 (a range relevant to ambient, outdoor exposure) in culture medium containing 5.5 mM K+ causes immediate activation of K+ channels, loss of 50% of intracellular K+ within 10 minutes after exposure11,12 and activation of initiator caspases-9 and -8 and the effector caspase-3. This demonstrates that corneal epithelial cells are, in fact, susceptible to damage by ambient levels of UVB. Based on this observation, the question arises, why are corneal epithelial cells acutely damaged by UVB in vitro but relatively protected from damage in vivo by ambient outdoor UVB?
The K+ concentration ([K+]) of tear fluid is 20–25 mM, a relatively high concentration compared to the [K+] in most other extracellular fluids.13–15 We have previously suggested that the high [K+] in tears may protect the corneal epithelium from UVB-induced apoptosis.11 High extracellular [K+] in tear fluid reduces the concentration gradient between the inside and outside of the epithelial cells. This may attenuate loss of intracellular [K+] when K+ channels are activated by ambient UVB, in turn inhibiting the activation of apoptotic mechanisms in the corneal epithelium. In previous studies using patch-clamp recording and measurement of intracellular [K+] by ion chromatography, we reported that UVB-induced loss of K+ from human corneal limbal epithelial (HCLE) cells is inhibited by incubation in medium with increased concentrations of K+ (25–100 mM).11,12 Incubation of HCLE cells in elevated levels of extracellular K+, including the 25 mM concentration found in tears, also inhibited UVB-induced caspase activation and DNA fragmentation.11,16,17 These observations support our overall hypothesis that the high concentration of K+ in tear fluid may contribute to protection of the in vivo corneal epithelium from the harmful effects of ambient UVB by reducing the electrochemical gradient for loss of intracellular K+ in response to UVB.
If UVB-induced K+ loss from cells causes apoptosis, then potassium channel blockers should also protect HCLE cells from UVB. We have previously reported that the Kv3.4 channel blocker, BDS-1, reduces UVB-induced K+ currents in HCLE cells by 50–75% and also attenuates apoptosis.11,17 BDS-1, however, blocks only one type of K+ channel and the residual UVB-induced current in the presence of BDS-1 suggests that other channel types may be present in HCLE cells. Blocking of additional K+ channels would be expected to cause a greater reduction in K+ currents and increased inhibition of apoptosis. Many types of K+ channels, both inward and outward rectifying, are blocked by Ba2+.18–20 Exposure of cells to Ba2+ should therefore stabilize intracellular [K+] in cells exposed to UVB. We previously observed, using ion chromatography, that Ba2+ at 0.1, 1 or 5 mM K+ inhibits UVB-induced loss of intracellular K+ ions after exposure to UVB in a dose-dependent manner.12 This suggests that Ba2+ should also inhibit UVB-induced apoptosis, and, if so, this would support our hypothesis that inhibiting loss of potassium can protect corneal epithelial cells from damage by UVB.
The purpose of the present study was to expand and complete our studies of the effects of blocking K+ channels on apoptosis of corneal epithelial cells exposed to UVB12,17 by using Ba2+ to achieve a complete inhibition of K+ currents. Patch-clamp recording was used to confirm that Ba2+ blocks outward rectifying UVB-induced K+ current. To investigate effects on apoptotic pathways, activation of caspases-3, -8 and -9 and also TUNEL staining were measured in cells incubated with Ba2+ following exposure to UVB.
II. METHODS
A. Cell Line Preparation
HCLE cells were maintained as monolayers in 6-well plates in Keratinocyte-SFM (KSFM, Life Technologies, Grand Island, NY), as previously described.11,21 HCLE cells have the ability to stratify in specific culture conditions.21 We have shown that stratified and monolayer HCLE cells respond similarly to UVB exposure and incubation in medium with elevated [K+]o.16 For convenience in timing of caspase and TUNEL assays, and dispersal of cells for patch-clamp recording, monolayers were used in the present study.
B. UVB Exposure
HCLE cells were exposed to UVB radiation (302 nm) using a UVM-57 lamp, (Ultraviolet Products, Upland, CA), as previously described.11 The doses of UVB, 80 mJ/cm2 for patch-clamp studies or 150 mJ/cm2 for caspase and TUNEL experiments were chosen based on previous studies and are relevant to ambient outdoor exposure in less than 2 hr at midday in the summer at 42° north latitude.
C. Patch-clamp Recording
HCLE cells were removed from culture plates using TrypLE-Express and resuspended in a bath containing (in mM) 140 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, 10 glucose (pH 7.4). Pipettes were back-filled using bath solution with 0.25mg/ml amphotericin-B and standard amphotericin-B perforated patch techniques were employed to attain whole-cell voltage-clamp recordings. (Sigma, St. Louis, MO). Pipette solution was (in mM) 145 K-methanesulfonate, 2.5 MgCl2, 2.5 CaCl2, 5 HEPES (pH 7.3). Pipette resistances were 2–7 MΩ. Recordings began when membrane resistance exceeded 1GΩ and access resistance dropped below 20 MΩ. The holding potential of the cell was −80 mV and the recording protocol consisted of 250 ms duration voltage steps from −80 mV to 120 mV in 10 mV increments. Currents were recorded by an Axon Instruments Axopatch 200B (Molecular Devices, Sunnyvale, CA) and analyzed by accompanying software (Clampex/Clampfit 9.2). Access resistance, capacitance, and most leak resistance were compensated by amplifier circuitry, with remaining leakage currents subtracted offline.
Prior to UVB exposure, control K+ currents were recorded. The cell was then exposed to UVB while the recording pipette remained attached to the cell. After removal of the UVB lamp recording of UVB-induced K+ currents was continued within 2 minutes of UVB exposure. Subsequently, BaCl2 in external solution was applied to the cell using an AutoMate Scientific (Berkeley, CA) perfusion pencil. The currents were measured in the presence of 5 mM Ba2+, followed by washout of Ba2+ using clean external solution. K+ currents were again measured in the absence of Ba2+. In several experiments 0.1, 1 and 5 mM Ba2+ were sequentially applied to a cell followed by washout.
D. Caspase Activity Assays
Following exposure of cells to 150 mJ/cm2 UVB, cells were incubated in either standard KSFM medium or medium with 0.01–5 mM BaCl2 for 4–6 hours before measurement of caspase activity or TUNEL staining. Stock 1 M BaCl2 in deionized water was added to KSFM to achieve the desired Ba2+ concentrations. To control for possible adverse effects of Ba2+, cells that were not exposed to UVB were also incubated in medium with 5 mM Ba2+.
Activity of caspases-3, −8 and −9 was measured using fluorometric caspase assay kits (BioVision, Milpitas, CA) according to manufacturer’s instructions. Protein levels were measured using the Bio-Rad Protein Assay (Bio-Rad, Hercules, CA). After UVB exposure and incubation, the culture medium was removed from the wells and retained in a centrifuge tube so that apoptotic cells that had been released from the cell monolayer could be included in the analysis. Adherent cells were then removed from the plate using TrypLE-Express (Life Technologies) and combined with the cells in the retained medium.11 The cells were pelleted by centrifugation and resuspended in cell lysis buffer. The protein concentration of each sample was adjusted to 2 mg/ml using cell lysis buffer, and caspase activity assays were conducted according to manufacturer’s instructions. Samples were then transferred to a white-walled, 96-well assay plate and read on a Cary Eclipse Fluorescence Spectrometer (Varian, Inc., Palo Alto, CA). Caspase activity was expressed as relative fluorescence units (RFU)/mg protein.
E. TUNEL Assay
DNA fragmentation was measured using an APO-BrdU TUNEL assay kit (Invitrogen Molecular Probes, Carlsbad, CA). After exposure to UVB and incubation for 6 hours in medium with 5 mM Ba2+, floating and adherent cells were combined as described above. The cells were fixed in 1% paraformaldehyde/PBS, washed in PBS, and suspended in 70% EtOH. In preparation for analysis, the cells were washed in PBS and incubated in a DNA labeling solution from the kit consisting of TdT enzyme, BrdUTP and buffer for 90 minutes at 37°C. Cells were washed twice in PBS and incubated in an antibody staining solution and buffer for 30 min in the dark at room temperature. A propidium iodide/RNase A staining buffer was added to each sample and the cells were incubated for an additional 30 min in the dark at room temperature. The cells were analyzed on a BD FACSCaliber flow cytometer (BD Biosciences, San Jose, CA).
F. Statistical Analysis
Caspase and TUNEL assays were repeated at least 3 times. Statistical analysis was conducted using SigmaPlot 12 (Systat Software, Inc., San Jose, CA). Statistical comparisons were made using one-way analysis of variance (ANOVA) and Dunnett’s test. All data were tested for normal distribution by the Shapiro-Wilk test.
III. RESULTS
A. Effect of Ba2+ on UVB-induced K+ Current
It was confirmed that exposure of HCLE cells to UVB at 80 mJ/cm2 causes an immediate increase in K+ channel activation, identical to the effect of UVB previously reported.11,17 Application of 5 mM Ba2+ blocked UVB-activated outward K+ currents. Washout of the Ba2+ resulted in restoration of K+ currents to activated, post-UVB levels (Figures 1 and 2).
Figure 1.
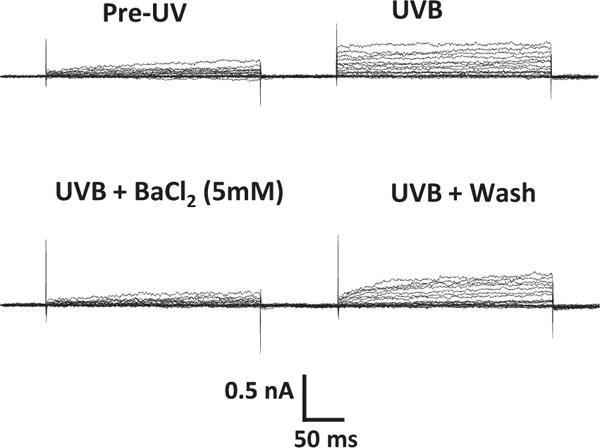
K+ channel activation in an HCLE cell following exposure to 80 mJ/cm2 UVB. Whole- cell currents were measured with 10 mV voltage steps given for 250 ms duration before exposure to UVB, after UVB exposure, during application of 5 mM BaCl2, and following washout of Ba2+.
Figure 2.
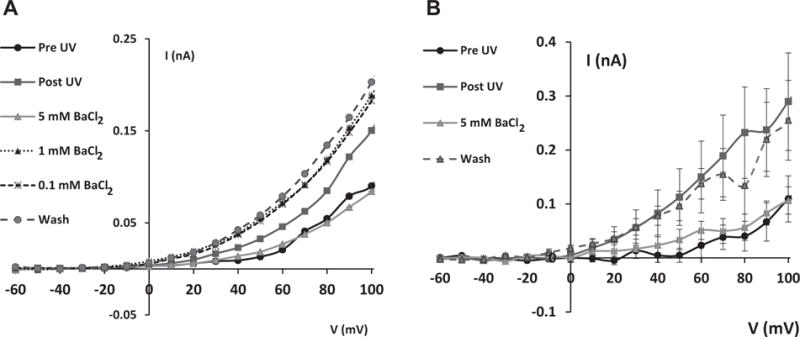
Normalized K+ channel current/voltage (I/V) relationships in HCLE cells exposed to UVB at 80 mJ/cm2. K+ currents were measured before UVB exposure, after UVB exposure, during treatment with Ba2+, and after washout of Ba2. UVB-activated K+ currents were reversibly inhibited by Ba2+. A. Response of a single cell to which 0.1, 1 and 5 mM Ba2+ were applied sequentially after exposure to UVB. This experiment was repeated in 3 cells with similar results. B. Effect of 5 mM Ba2+ alone on UVB-induced currents (n=9 cells).
UVB makes the plasma membrane fragile so that it is difficult to record for an extended time period. Therefore the effect of Ba2+ over several concentrations (0.1, 1 and 5 mM) on UVB-activated currents was successfully studied on only 3 cells. Data from one representative cell shows that Ba2+ at 0.1 or 1 mM had minimal effect on UVB-induced K+ current (Figure 2A). In an additional set of 9 cells that were exposed only to 5 mM Ba2+, the complete blocking of UVB-activated K+ currents is clearly shown in the averaged data (Fig. 2B).
B. Effect of Ba2+ on UVB-induced Caspase Activity
Exposure of HCLE cells to UVB at 150 mJ/cm2 led to activation of initiator caspase-9 which was maximal at 4 hours. Cells exposed to UVB and incubated for 4 hours in medium with various Ba2+ concentrations showed an inhibition of caspase-9 activation in a dose-dependent manner. UVB-induced caspase-9 activation was significantly decreased in the presence of 1 or 5 mM Ba2+ (Figure 3).
Figure 3.
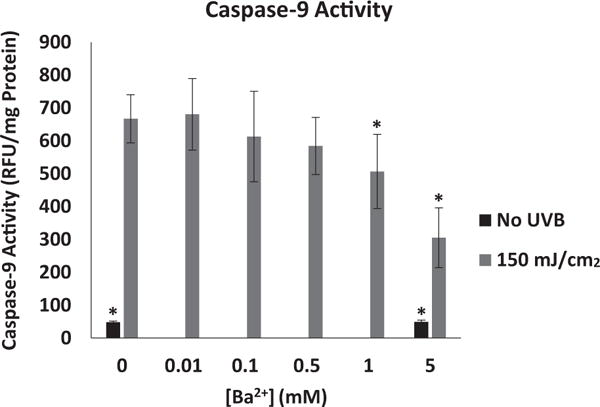
Effect of Ba2+ on UVB-induced activation of caspase-9 in HCLE cells. Cells were exposed to 150 mJ/cm2 UVB and incubated for 4 hr in the presence or absence of Ba2+. Increasing concentrations of Ba2+ caused a significant decrease in UVB-induced caspase-9 activity. *significantly different from cells exposed to UVB and incubated without Ba2+ (mean ± SD, n=6, One-way ANOVA and Dunnett’s test, P<.05).
Exposure of HCLE cells to UVB at 150 mJ/cm2 also led to activation of initiator caspase-8, with maximal activity at 6 hours post-UVB. Incubation of UVB-exposed HCLE cells for 6 hours in the presence of 0.1, 0.5, 1 or 5 mM Ba2+ significantly decreased UVB-induced caspase-8 activation compared to UVB-exposed cells not treated with Ba2+ (Figure 4).
Figure 4.
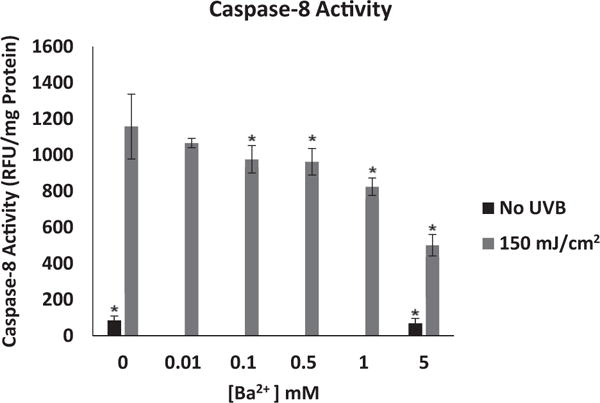
Effect of Ba2+ on UVB-induced activation of caspase-8 in HCLE cells. Cells were exposed to 150 mJ/cm2 UVB and incubated for 6 hr in the presence or absence of Ba2+. Increasing concentrations of Ba2+ caused a significant decrease in UVB-induced caspase-8 activity. *significantly different from cells exposed to UVB and incubated without Ba2+ (mean ± SD, n=6, One-way ANOVA and Dunnett’s test, P<.05).
Following activation of the initiator caspase cascade by UVB, the effector caspase-3 was also activated, reaching maximal activation at 6 hours. Incubation of UVB-exposed cells for 6 hours in the presence of 1 or 5 mM Ba2+ led to a significant decrease in UVB-induced caspase-3 activity compared to UVB-exposed cells not treated with Ba2+ (Figure 5). Ba2+ did not have an effect on baseline caspase activity in cells that were not exposed to UVB (Figures 3–5).
Figure 5.
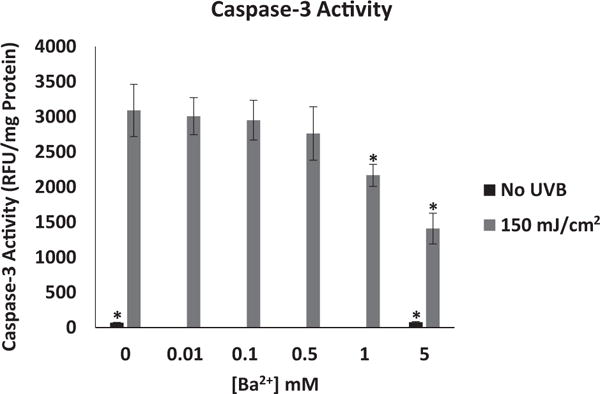
Effect of Ba2+ on UVB-induced activation of caspase-3 in HCLE cells. Cells were exposed to 150 mJ/cm2 UVB and incubated for 6 hr in the presence or absence of Ba2+. Increasing concentrations of Ba2+ caused a significant decrease in UVB-induced caspase-3 activity. *significantly different from cells exposed to UVB and incubated without Ba2+ (mean ± SD, n=6, One-way ANOVA and Dunnett’s test, P<.05).
C. Effect of Ba2+ on UVB-induced DNA Fragmentation
Since Ba2+ inhibited UVB-induced K+ currents and activation of caspases in HCLE cells, then ultimately apoptosis, as indicated by DNA fragmentation, should also be inhibited by Ba2+. In cell cultures exposed to UVB, the number of cells with fragmented DNA, as measured by the TUNEL assay, increased markedly. In HCLE cells incubated with 5 mM Ba2+ for 6 hr after UVB exposure, the number of apoptotic cells remained at nearly control levels. (Figures 6 and 7). Ba2+ did not affect numbers of apoptotic cells in cultures that were not exposed to UVB.
Figure 6.
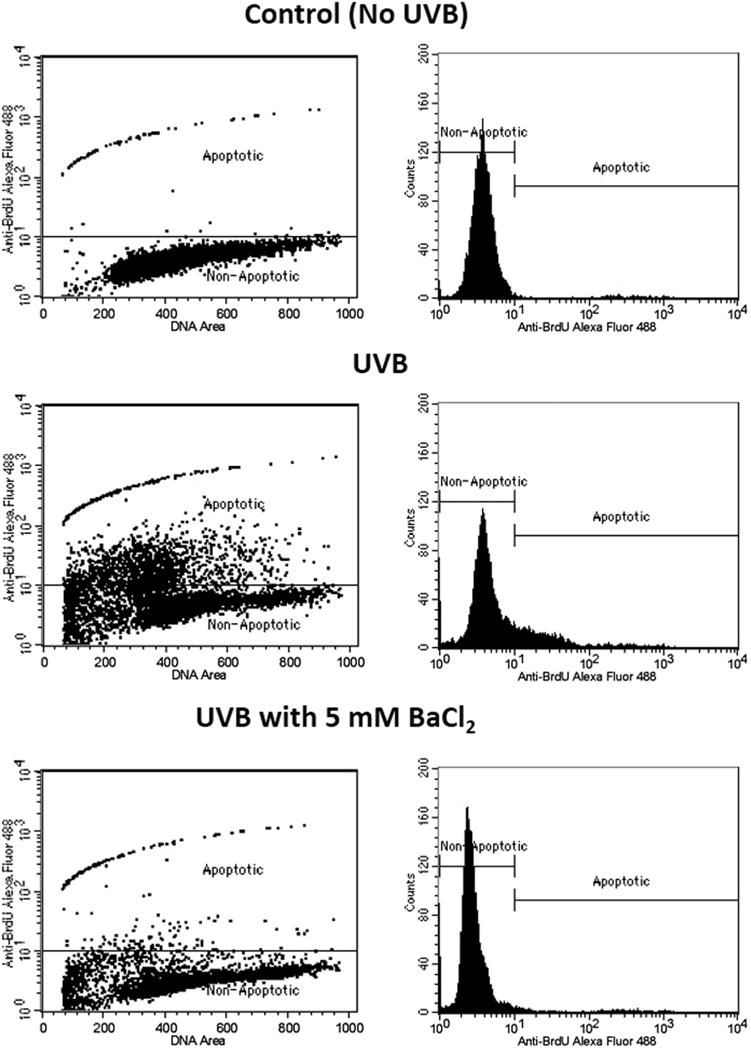
Protection of HCLE cells from UVB-induced apoptosis by Ba2+. Cells were exposed to 150 mJ/cm2 UVB, incubated for 6 hours and analyzed by the TUNEL assay and flow cytometry. Control cells were not exposed to UVB. Non-apoptotic cells have low fluorescence while UVB-induced apoptotic cells are shifted into a higher fluorescence window. During incubation in medium with 5 mM Ba2+ cells exposed to UVB remain the non-apoptotic window.
Figure 7.
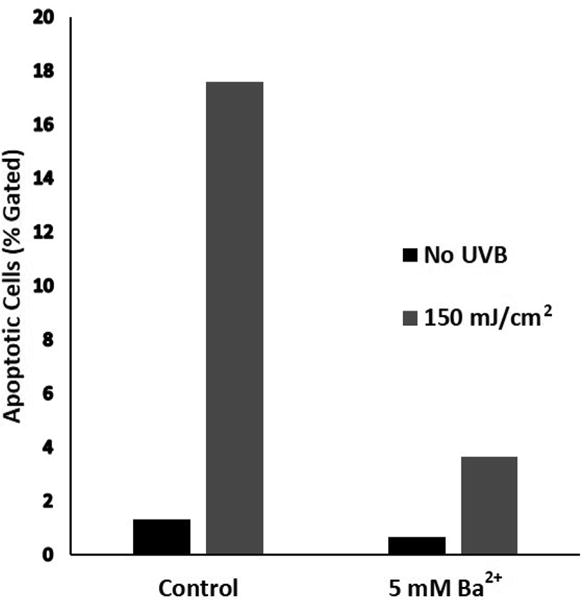
Decrease in UVB-induced apoptosis of HCLE cells, treated with Ba2+. HCLE cells were exposed to 150 mJ/cm2 UVB and incubated in KSFM with 5 mM Ba2+ for 6 hr followed by measurement of apoptosis by the TUNEL assay and flow cytometry. Control cells were maintained for 6 hr in KSFM without Ba2+. Each bar represents an average of 2 samples of 10,000 cells. This experiment was repeated 3 times with similar results.
IV. DISCUSSION
This study shows that treatment of HCLE cells with Ba2+ after exposure to a dose of UVB known to cause apoptosis blocks UVB-activated K+ currents, decreases activation of caspases-9, −8 and −3 and markedly reduces DNA fragmentation. Since Ba2+ blocks K+ channels, these observations are in agreement with the previous reports showing that loss of intracellular K+ is an initiating event in apoptosis,1–7 supporting our hypothesis that preventing loss of K+ can protect the corneal epithelial cells from damage caused by UVB.11,12,17
In our previous studies of activation of K+ channels in HCLE cells by UVB, we focused on the Kv3.4 channel. It has been shown by immunohistochemistry that this channel is strongly expressed in the corneal epithelium of rats and rabbits,22 and we have confirmed its expression in HCLE cells by western blot (data not shown). BDS-1 is a specific blocker of Kv3.4 channels. When applied to HCLE cells after they are exposed to UVB, BDS-1 blocks only 50–75% of the UVB-activated current.11,17 In contrast, as shown in the present study, 5 mM Ba2+ completely blocks the UVB-induced current. Ba2+ at 5 mM also strongly inhibits UVB-induced loss of intracellular K+ from HCLE cells and does so more effectively than BDS-1.12 As will be discussed in more detail below, this suggests that several types of K+ channels are activated by UVB in HCLE cells.
The effect of Ba2+ as a blocker of K+ channels is well known, as briefly reviewed by Hurst et al, who also demonstrated by mutation of amino acids in the selectivity pore of the Shaker H4 (Shaker B, KCNA2) channel that Ba2+ blocks channels by binding to K+ binding sites in the pore.23 More recent papers have also characterized the interactions of Ba2+ with several types of K+ channels. Cheng et al reported that Ba2+ binds to a low affinity superficial site and a deeper, high affinity site of the Kv1.5 channel (KCNA5), which is abundant in myocardial cells.24 The KCa1.1, or Maxi K, channel (KCNMA1), is blocked by 5 mM Ba2+,25 and the Ca2+-independent Shal channel in Drosophila is blocked by 1.8 mM Ba2+.26 This channel corresponds to the mammalian Kv4 channel sub-family (KCND1-3). Kehl et al, using HEK293 cells transfected with rat Kv4 channels, have shown that not only are these channels blocked by Ba2+ but that this binding is competitively inhibited by external K+, which binds in the pore, displacing Ba2+.19 The fact that Ba2+ can interact with a wide variety of K+ channels is further demonstrated by the observations that it also blocks the TWIK-1 (KCNK1) and TREK-1 (KCNK2) leak channels, as well as inward rectifying K+ channels in Müller cells.18,20,27
These observations in various cell types and organisms are important to the present study for several reasons. First, our observation that 0.1 or 1 mM Ba2+ had little effect on UVB-activated currents, while 5 mM Ba2+ blocked these currents completely, is in agreement with the effective concentration ranges in all of the studies cited above.
Second, the complete block of UVB-activated K+ currents by Ba2+ in our study, and the effects of Ba2+ on the channel types discussed above, provide some clues as to the channels expressed by HCLE cells. Several K+ channels have been characterized in rabbit corneal epithelial cells.28–30 However, little is known about K+ channels in human corneal epithelium, except for a K+ conductance identified by Bockman et al31 and Griffeth et al32 and our identification of Kv3.4 channels in HCLE cells. We have, however, recently conducted a preliminary screen of K+ channel expression in HCLE cells using a Human Neuronal Ion Channels RT2 Profiler PCR Array (Qiagen, Valencia, CA). This screen indicated that HCLE cells may express the Ba2+-sensitive KCNA2, KCNMA1, KCND2 and 3 and KCNK1 channels discussed above, suggesting that UVB may increase the open probability of these channels types in addition Kv3.4. We mention these data with caution, since expression of these channels by HCLE cells has not yet been confirmed by immunoblotting or immunohistochemistry.
The evidence that extracellular K+ competitively inhibits binding of Ba2+ in the selectivity pore is intriguing because of its implications for our understanding of the inhibitory effects of elevated [K+]o on loss of intracellular K+,12 and activation of apoptosis in response to UVB that we have reported.11,16,33 Inhibition of K+ loss from HCLE cells in the presence of elevated [K+]o may not simply be due to a reduction in the concentration gradient that decreases simple diffusion, but rather due to binding of K+ in the selectivity pore from the extracellular side of the membrane that blocks K+ efflux. This would explain why an extracellular [K+] as low as 25 mM, the concentration in tear fluid, can prevent UVB-induced loss of intracellular K+ and inhibit caspase activation and DNA fragmentation. Indeed, Kehl et al. showed that 20 mM extracellular K+ can displace Ba2+ from Kv4.2 (KCND2) channels.19
Finally, the electrical gradient across the membrane must also be considered. K+ leaves the cell down its concentration gradient but against its electrical gradient. Given [K+]i = 140 mM, the current-voltage curve given by the electrochemical gradient shifts as [K+]o changes. As shown by Gray et al,34 shifting K+o from 5.5 mM to 25 mM would change the reversal potential for the current-voltage curve to close to the resting potential, which we have measured as about −35 mV in HCLE cells (unpublished data), with the result that K+ efflux would be greatly reduced in the presence of 25 mM external K+. This offers an additional explanation for the effectiveness of 25 mM K+o in inhibiting UVB-induced K+ loss and activation of apoptosis.
We have recently reported that UVB-induced activation of caspases-8 and -3 in HCLE cells is primarily mediated by caspase-9 in the intrinsic pathway.33 Ba2+ had the predicted effect on apoptosis in HCLE cells in that it reduced UVB-induced activation of these caspases and inhibited DNA fragmentation. It is important to emphasize that this is not a direct effect of Ba2+ on apoptotic pathways, since it does not enter the cell, but binds in the K+ channels, as discussed above. Rather, the protective effect of extracellular Ba2+ is that it prevents the loss of intracellular K+. Loss of K+i appears to be a common initiating event in apoptosis in many cell types, including lymphocytes, neurons, epithelial cells and smooth muscle, to name but a few, and this loss of K+, due to channel activation, is common to all methods of inducing apoptosis, including dexamethasone, staurosporine, serum withdrawal and UV exposure.1,3–5,7,35,36 Early reports showed that when apoptosis of lymphocytes is induced by dexamethasone or staurosporin, resulting in cell shrinkage, activation of caspase-3, and DNA laddering, these effects can be prevented by incubation of the cells in medium with isosmotic 100 mM K+.1,2 Subsequent studies using channel blockers and elevated [K+]o have confirmed that preventing K+ loss inhibits apoptosis.2–3,11–12,16–17,36 The present study corroborates these findings with specific relevance to the corneal epithelium.
The degree of inhibition of UVB-induced caspase activity by 5 mM Ba2+ is in agreement with the effects of high extracellular K+ that we have previously reported.11,16,33 The relative lack of effect of lower concentrations of Ba2+ on caspase activity is in agreement the minimal effect of 0.1 and 1 mM Ba2+ on UVB-activated K+ currents. Of particular interest is our observation that while 5 mM Ba2+ reduces caspase activity to about 45% of UVB-induced activity in the absence of Ba2+ (Figures 3–5), the number of apoptotic cells is reduced to about 21% of levels in the absence of Ba2+ (Figure 7). This suggests that in addition to the effects of UVB and loss of K+i on the caspase-9, -8 and -3 signaling cascade that has been the focus of our research,33 the UVB-induced reduction in levels of intracellular K+ also effects other enzymes and signaling molecules in the various highly complex signaling pathways that lead to DNA fragmentation.
This study must be placed into the context of the voluminous literature on UV-induced apoptosis, since a great many reports on mechanisms of apoptosis, effects of UV on various cell types and role of K+ loss have preceded our work. In lymphocytes apoptosis is part of the normal system that controls numbers of lymphocytes in the context of responses to infection and presence of various antigens, and errors in this system underlie abnormalities that lead to leukemia. As such, the use of UV exposure is a tool in the study of apoptosis of lymphocytes, since these cells are not normally exposed to UV. In dermatology, the damage to skin by ambient UVB is well documented. In this case apoptosis is a protective mechanism that eliminates damaged cells, reducing the risks of carcinoma.37,38
In contrast, in the cornea, apoptosis in response to ambient levels of UVB carries both benefit and risk. Rapid turnover of corneal epithelial cells continually renews this cell layer. Studies measuring turnover of 3H-thymidine labeled cells in animal models and in vitro human corneas or loss of sex chromatin from corneal grafts indicate that the epithelium turns over completely every 1–2 weeks through a process of mitosis in the basal layer, differentiation in the wing cell layer, and sloughing of senescent cells from the corneal surface.39–42 Wilson and coworkers directly measured the rate of cell sloughing from human corneas in vivo and reported that increasing the K+ concentration in solutions bathing the ocular surface decreases sloughing of epithelial cells.43,44 The balance among proliferation, differentiation, and cell sloughing must be tightly controlled to prevent corneal epithelial erosions.45 As such, we propose that corneal epithelial cells must be protected from apoptosis induced by ambient UVB to a greater extent than keratinocytes. It is evident that maintaining the normal intracellular [K+] is very important in inhibiting apoptotic mechanisms, but we have clearly shown that ambient levels of UVB cause activation of K+ channels in corneal epithelial cells. Thus, preventing K+ loss from corneal epithelial cells in the face of outdoor exposure to UVB would be an important mechanism which, along with absorption of UVB by the tryptophan and ascorbic acid in corneal epithelial cells,8,9 would contribute to protection of the cornea from adverse effects of ambient UVB.
V. CONCLUSION
The results of this study show that blocking UVB-activated K+ channels by Ba2+ prevents loss of intracellular K+ and inhibits apoptosis in a manner similar to incubating UVB-treated cells in high extracellular K+ concentrations.11,17 In contrast to BDS-1, which blocks only Kv3.4 channels, Ba2+ completely blocks UVB-induced currents, suggesting that multiple K+ channel types are activated by UVB in HCLE cells. Ba2+ also is more effective than BDS-1 in inhibiting caspases.17 This shows that maintaining high intracellular [K+] is required for inhibition of the apoptosis cascade. The data support our hypothesis that the relatively high level of K+ in tears may contribute to protection of the corneal epithelium from ambient outdoor UVB by reducing the gradient for UVB-induced loss of intracellular K+.
Acknowledgments
We thank Dr. Ilene K. Gipson, Department of Ophthalmology, Harvard Medical School, for the HCLE cells and Dr. Mitchell A. Watsky, Department of Cellular Biology and Anatomy, Augusta University, for consultation on patch-clamp recording.
Supported by NIH grant R15 EY023836 (JLU and LDH), the Joseph C. Stevens Faculty Research Fellowship in the Natural Sciences (JLU), the West Michigan Optometric Scholarship (CDG), the Arnold and Mabel Beckman Foundation (PMB), and a gift to the Calvin College Department of Biology from Robert and Anita Huizenga.
Footnotes
Ali Djalilian, MD, Editor
The authors have no proprietary or commercial interest in any concept or product discussed in this article.
References
- 1.Bortner CD, Hughes FM, Cidlowski JA. A primary role for K+ and Na+ efflux in the activation of apoptosis. J Biol Chem. 1997;272:32436–542. doi: 10.1074/jbc.272.51.32436. [DOI] [PubMed] [Google Scholar]
- 2.Hughes FM, Bortner CD, Purdy GD, Cidlowski JA. Intracellular K+ suppresses the activation of apoptosis in lymphocytes. J Biol Chem. 1997;272:30567–76. doi: 10.1074/jbc.272.48.30567. [DOI] [PubMed] [Google Scholar]
- 3.Wang L, Xu D, Dai W, Lu L. An ultraviolet-activated K+ channel mediates apoptosis of myeloblastic leukemia cells. J Biol Chem. 1999;274:3678–85. doi: 10.1074/jbc.274.6.3678. [DOI] [PubMed] [Google Scholar]
- 4.Wang L, Li T, Lu L. UV-induced corneal epithelial cell death by activation of potassium channels. Invest Ophthalmol Vis Sci. 2003;44:5095–101. doi: 10.1167/iovs.03-0590. [DOI] [PubMed] [Google Scholar]
- 5.Arrebola F, Fernández-Segura E, Campos A, et al. Changes in intracellular electrolyte concentrations during apoptosis induced by UV irradiation of human myeloblastic cells. Am J Physiol Cell Physiol. 2006;290:C638–49. doi: 10.1152/ajpcell.00364.2005. [DOI] [PubMed] [Google Scholar]
- 6.Vu CC, Bortner CD, Cidlowski JA. Differential involvement of initiator caspases in apoptotic volume decrease and potassium efflux during Fas-and UV-induced cell death. J Biol Chem. 2001;276:37602–11. doi: 10.1074/jbc.M104810200. [DOI] [PubMed] [Google Scholar]
- 7.Yu SP, Yeh CH, Sensi SL, et al. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science. 1997;278:114–7. doi: 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]
- 8.Kolozvari L, Nogradi A, Hopp B, Bor Z. UV absorbance of the human cornea in the 240- to 400-nm range. Invest Ophthalmol Vis Sci. 2002;43:2165–8. [PubMed] [Google Scholar]
- 9.Ringvold A. Corneal epithelium and UV-protection of the eye. Acta Ophthalmol Scand. 1998;76:149–53. doi: 10.1034/j.1600-0420.1998.760205.x. [DOI] [PubMed] [Google Scholar]
- 10.Podskochy A. Protective role of corneal epithelium against ultraviolet radiation damage. Acta Ophthalmol Scand. 2004;82:714–7. doi: 10.1111/j.1600-0420.2004.00369.x. [DOI] [PubMed] [Google Scholar]
- 11.Singleton KR, Will DS, Schotanus MP, et al. Elevated extracellular potassium inhibits apoptosis of corneal epithelial cells exposed to UV-B radiation. Exp Eye Res. 2009;89:140–51. doi: 10.1016/j.exer.2009.02.023. [DOI] [PubMed] [Google Scholar]
- 12.Ubels JL, Van Dyken RE, Louters JR, et al. Potassium ion fluxes in corneal epithelial cells exposed to UVB. Exp Eye Res. 2011;92:425–31. doi: 10.1016/j.exer.2011.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Atta AG, Maringoni RL, Borsio Netto JB, Silveira Filho B. Potassium and sodium levels in the initial saliva of the dog’s submandibular and parotid glands. Rev Bras Pesqui Med Biol. 1975;3:37–43. [PubMed] [Google Scholar]
- 14.Botelho SY, Martinez EV. Electrolytes in lacrimal gland fluid and in tears at various flow rates in the rabbit. Am J Physiol. 1973;225:606–9. doi: 10.1152/ajplegacy.1973.225.3.606. [DOI] [PubMed] [Google Scholar]
- 15.Rismondo V, Osgood TB, Leering P, et al. Electrolyte composition of lacrimal gland fluid and tears of normal and vitamin A-deficient rabbits. CLAO J. 1989;15:222–9. [PubMed] [Google Scholar]
- 16.Schotanus MP, Koetje LR, Van Dyken RE, Ubels JL. Stratified corneal limbal epithelial cells are protected from UVB-induced apoptosis by elevated extracellular K+ Exp Eye Res. 2011;93:735–40. doi: 10.1016/j.exer.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ubels JL, Schotanus MP, Bardolph SL, et al. Inhibition of UV-B induced apoptosis in corneal epithelial cells by potassium channel modulators. Exp Eye Res. 2010;90:216–22. doi: 10.1016/j.exer.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Solessio E, Linn DM, Perlman I, Lasater EM. Characterization with barium of potassium currents in turtle retinal Müller cells. J Neurophysiol. 2000;83:418–30. doi: 10.1152/jn.2000.83.1.418. [DOI] [PubMed] [Google Scholar]
- 19.Kehl SJ, Fedida D, Wang Z. External Ba2+ block of Kv4.2 channels is enhanced in the closed-inactivated state. Am J Physiol Cell Physiol. 2013;304:C370–81. doi: 10.1152/ajpcell.00267.2012. [DOI] [PubMed] [Google Scholar]
- 20.Ma XY, Yu JM, Zhang SZ, et al. External Ba2+ block of the two-pore domain potassium channel TREK-1 defines conformational transition in its selectivity filter. J Biol Chem. 2011;286:39813–22. doi: 10.1074/jbc.M111.264788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gipson IK, Spurr-Michaud S, Argüeso P, et al. Mucin gene expression in immortalized human corneal-limbal and conjunctival epithelial cell lines. Invest Ophthalmol Vis Sci. 2003;44:2496–506. doi: 10.1167/iovs.02-0851. [DOI] [PubMed] [Google Scholar]
- 22.Wang L, Fyffe RE, Lu L. Identification of a Kv3.4 channel in corneal epithelial cells. Invest Ophthalmol Vis Sci. 2004;45:1796–803. doi: 10.1167/iovs.03-1056. [DOI] [PubMed] [Google Scholar]
- 23.Hurst RS, Toro L, Stefani E. Molecular determinants of external barium block in Shaker potassium channels. FEBS Lett. 1996;388:59–65. doi: 10.1016/0014-5793(96)00516-9. [DOI] [PubMed] [Google Scholar]
- 24.Cheng YM, Fedida D, Kehl SJ. External Ba2+ block of human Kv1.5 at neutral and acidic pH: evidence for Ho+-induced constriction of the outer pore mouth at rest. Biophys J. 2008;95:4456–68. doi: 10.1529/biophysj.108.133165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sørensen MV, Matos JE, Sausbier M, et al. Aldosterone increases KCa1.1 (BK) channel-mediated colonic K+ secretion. J Physiol. 2008;586:4251–64. doi: 10.1113/jphysiol.2008.156968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ryglewski S, Duch C. Shaker and Shal mediate transient calcium-independent potassium current in a Drosophila flight motoneuron. J Neurophysiol. 2009;102:3673–88. doi: 10.1152/jn.00693.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou M, Xu G, Xie M, et al. TWIK-1 and TREK-1 are potassium channels contributing significantly to astrocyte passive conductance in rat hippocampal slices. J Neurosci. 2009;29:8551–64. doi: 10.1523/JNEUROSCI.5784-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farrugia G, Rae JL. Regulation of a potassium-selective current in rabbit corneal epithelium by cyclic GMP, carbachol and diltiazem. J Membr Biol. 1992;129:99–107. doi: 10.1007/BF00232058. [DOI] [PubMed] [Google Scholar]
- 29.Rae JL, Farrugia G. Whole-cell potassium current in rabbit corneal epithelium activated by fenamates. J Membr Biol. 1992;129:81–97. doi: 10.1007/BF00232057. [DOI] [PubMed] [Google Scholar]
- 30.Farrugia G, Rae J. Effect of volume changes on a potassium current in rabbit corneal epithelial cells. Am J Physiol. 1993;264:C1238–45. doi: 10.1152/ajpcell.1993.264.5.C1238. [DOI] [PubMed] [Google Scholar]
- 31.Bockman CS, Griffith M, Watsky MA. Properties of whole-cell ionic currents in cultured human corneal epithelial cells. Invest Ophthalmol Vis Sci. 1998;39:1143–51. [PubMed] [Google Scholar]
- 32.Griffith M, Osborne R, Munger R, et al. Functional human corneal equivalents constructed from cell lines. Science. 1999;286:2169–72. doi: 10.1126/science.286.5447.2169. [DOI] [PubMed] [Google Scholar]
- 33.Ubels JL, Glupker CD, Schotanus MP, Haarsma LD. Involvement of the extrinsic and intrinsic pathways in ultraviolet B-induced apoptosis of corneal epithelial cells. Exp Eye Res. 2015;145:26–35. doi: 10.1016/j.exer.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gray DA, Frindt G, Palmer LG. Quantification of K secretion through apical low-conductance K channels in the CCD. Am J Physiol Renal Physiol. 2005;289:F117–26. doi: 10.1152/ajprenal.00471.2004. [DOI] [PubMed] [Google Scholar]
- 35.Redman PT, He K, Hartnett KA, et al. Apoptotic surge of potassium currents is mediated by p38 phosphorylation of Kv2.1. Proc Natl Acad Sci U S A. 2007;104:3568–73. doi: 10.1073/pnas.0610159104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valencia-Cruz G, Shabala L, Delgado-Enciso I, et al. Kbg and Kv1.3 channels mediate potassium efflux in the early phase of apoptosis in Jurkat T lymphocytes. Am J Physiol Cell Physiol. 2009;297:C1544–53. doi: 10.1152/ajpcell.00064.2009. [DOI] [PubMed] [Google Scholar]
- 37.Aragane Y, Kulms D, Metze D, et al. Ultraviolet light induces apoptosis via direct activation of CD95 (Fas/APO-1) independently of its ligand CD95L. J Cell Biol. 1998;140:171–82. doi: 10.1083/jcb.140.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kulms D, Schwarz T. Molecular mechanisms of UV-induced apoptosis. Photodermatol Photoimmunol Photomed. 2000;16:195–201. doi: 10.1034/j.1600-0781.2000.160501.x. [DOI] [PubMed] [Google Scholar]
- 39.Hanna C, Bicknell DS, O’Brien JE. Cell turnover in the adult human eye. Arch Ophthalmol. 1961;65:695–8. doi: 10.1001/archopht.1961.01840020697016. [DOI] [PubMed] [Google Scholar]
- 40.Sharma A, Coles WH. Kinetics of corneal epithelial maintenance and graft loss: A population balance model. Invest Ophthalmol Vis Sci. 1989;30:1962–71. [PubMed] [Google Scholar]
- 41.Cenedella RJ, Fleschner CR. Kinetics of corneal epithelium turnover in vivo: Studies of lovastatin. Invest Ophthalmol Vis Sci. 1990;31:1957–62. [PubMed] [Google Scholar]
- 42.Gipson IK. Anatomy of the conjunctiva, cornea and limbus. In: Smolin G, Thoft RA, editors. The Cornea: Scientific Foundations and Clinical Practice. 3rd. Boston: Little, Brown and Co; 1994. pp. 3–24. [Google Scholar]
- 43.Bachman WG, Wilson G. Essential ions for maintenance of the corneal epithelial surface. Invest Ophthalmol Vis Sci. 1985;26:1484–8. [PubMed] [Google Scholar]
- 44.Fullard RJ, Wilson GS. Investigation of sloughed corneal epithelial cells collected by non-invasive irrigation of the corneal surface. Curr Eye Res. 1986;5:847–56. doi: 10.3109/02713688609029236. [DOI] [PubMed] [Google Scholar]
- 45.Thoft RA, Friend J. The X, Y, Z hypothesis of corneal epithelial maintenance. Invest Ophthalmol Vis Sci. 1983;24:1442–3. [PubMed] [Google Scholar]