

Summary
The introduction of multiphoton microscopy has dramatically broadened the scope of intravital imaging studies and has allowed researchers to validate and refine basic mechanistic concepts in many areas of biology within the context of physiologically relevant tissue microenvironments. This has also led to new insights into the behavior of immune cells at steady state, and how their behaviors are altered during an immune response. At the same time, advances in the humanized mouse model have allowed for in vivo studies of strictly human pathogens, such as HIV-1. Here, we describe in detail an intravital microscope approach to visualize the dynamic behavior of HIV-infected T cells within the lymph nodes of live, anesthetized humanized mice.
Keywords: Multiphoton intravital microscopy, in vivo imaging, popliteal lymph node, lymphocyte motility, HIV, humanized mice, fluorescent protein, mean track velocity
1. Introduction
Multiphoton intravital microscopy (MP-IVM) is a powerful imaging modality that allows for the direct dynamic observation of biological processes in their physiological tissue environment at cellular and subcellular resolution. The physical principle of multiphoton excitation allows for deeper optical penetration into tissues, compared to conventional fluorescence techniques, as well as for prolonged, continuous observations due to minimized phototoxicity and photobleaching [1,2]. The study of the migratory behavior and function of immune cells, especially in the context of a developing immune response, is particularly amenable to this technology [3,4].
Current humanized mouse models of HIV infection closely recapitulate several key aspects of HIV-1 infection in humans, including high viral loads upon vaginal [5-7] and rectal transmission [8], CD4+ T cell depletion in the peripheral blood and mucosal tissues [9,5] and the generation of functional CTL responses [10]. Accordingly, the mouse models have been used to evaluate the efficacy of several preclinical vaccine and anti-retroviral strategies. Here, we describe a new in vivo imaging approach that utilizes fluorescent HIV reporter strains to visually track and characterize the dynamic behavior of infected cells in lymph nodes of humanized mice. The technique can be adapted to understand the cellular and molecular mechanisms that govern the efficient spread of HIV within lymphoid and non-lymphoid organs in real-time.
2. Materials
2.1 Construction and preparation of fluorescent reporter HIV
DNA plasmid containing a fluorescent protein-expressing, full-length HIV-1 proviral cDNA, such as the CCR5-using, NL4-3 IRES GFP reporter strain (HIV-GFP) [7] (see Note 1).
HEK 293T cell line (Human embryonic kidney cells expressing SV40 T antigen)
MAGI-CCR5 cell line (NIH AIDS Reagent Program)
DMEM supplemented with 10% FCS, 2mM L-glutamine, 1mM sodium pyruvate and 10mM HEPES
Calcium phosphate transfection kit
150 mm tissue culture plates
Ultracentrifuge with a swinging bucket rotor and buckets (e.g. sw32Ti)
Ultraclear ultracentrifuge tubes (e.g. 38.7 mL capacity tubes)
20% sucrose solution in phosphate buffered saline
50 mL conical tube
2.2 Human CD4+ T cell isolation and expansion into central memory-like T cells (Tcm)
Spleen and lymph node cells harvested from humanized mice
Immunomagnetic human CD4+ T cell positive selection kit
Anti-human CD3ε/CD28 antibody conjugated Dynabeads
Human recombinant IL-2
RPMI medium supplemented with 10% FCS, 2mM L-glutamine, 1mM sodium pyruvate and 10mM HEPES
CellTracker Orange (CMTMR; 5-(and-6-)-(((4-chloromethyl)benzoyl)amino) tetramethylrhodamine)
PBS supplemented with 1% FCS
2.3 In vitro infection of CD4+ Tcm with a fluorescent reporter HIV strain
96 well, flat-bottom culture plates
Polybrene (hexadimethrine bromide)
25 cm2 tissue culture flask
2.4 Surgical anesthesia
Ketamine HCl solution, 100 mg/mL (e.g. Ketaset)
Xylazine HCl solution, 20 mg/mL (e.g. Rompun)
NaCl 0.9% for injection
Insulin syringe (19G)
2.5 Microsurgical preparation of the mouse popliteal lymph node for intravital microscopy
Custom-built microscope stage (see Figure 1b, see Note 2)
Small animal clipper (e.g. Pocket Pro pet trimmer)
Depilation crème (e.g. Nair)
Cotton tip applicators
2 × 2 inch gauze sponges
Surgical instruments: Straight ‘tough cut’ iris scissors (1), Vannas spring scissors with 3-5 mm blades (1), Dumont #5 Forceps standard tip, straight, 0.1 mm × 0.6 mm, Inox, 11 cm total length (2), Dumont #5 Forceps, Biologie tip, 0.05 mm × 0.02 mm, Inox, 11 cm total length (2)
18 mm #1 round coverglasses
Ethyl-2-cyanoacrylate glue (e.g. Crazyglue)
Adhesive tape
Suture material (e.g. 5-0 braided silk)
High viscosity vacuum grease
Plasticine modeling clay
Miniature K type thermocouple
Figure 1. General overview of the methodological steps required to visualize HIV-infected cells in the lymph node.
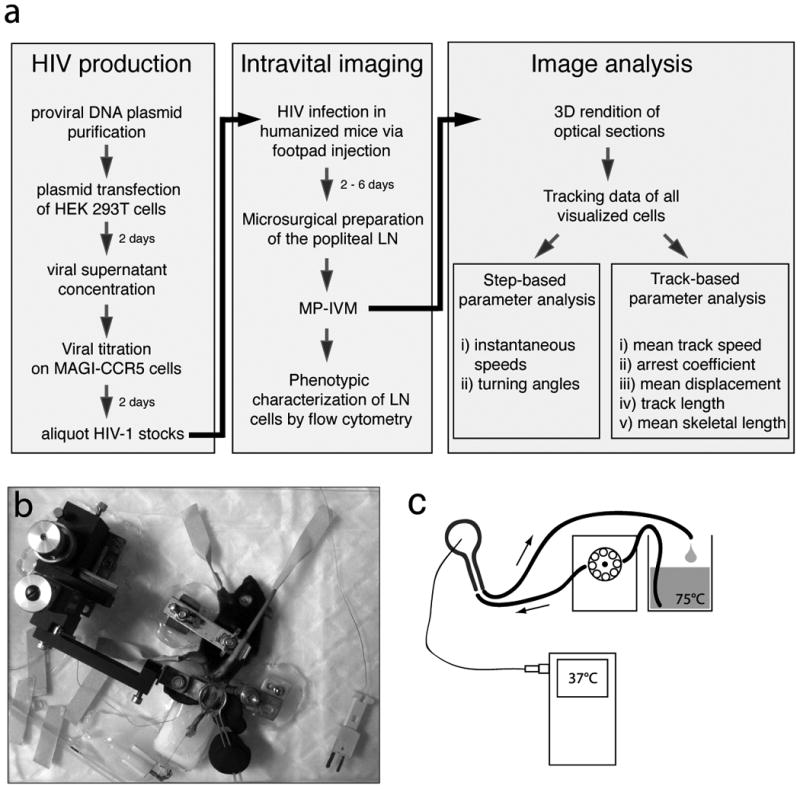
a: flowchart view of 1) HIV production, 2) intravital imaging and 3) image analysis. b: fully assembled popliteal LN preparation. C: schematic of the temperature control system.
2.6 Multiphoton intravital microscopy of the mouse popliteal lymph node (see Note 3)
Upright multiphoton microscope equipped with a) at least three non-descanned PMT detectors (e.g. Ultima IV, Bruker Corporation; TriMScope, LaVision Biotec; TCS SP8, Leica; LSM 7 MP Zeiss; FluoView MPE-RS, Olympus) b) a high numerical aperture objective lens (e.g. Olympus XLUMPLFL20XW 20X, 0.95 NA, water immersion, 2 mm working distance), and c) a femtosecond-pulsed infrared laser (e.g. Insight DS+, Spectra-Physics/Newport or Chameleon Vision II, Coherent).
Small water bath
Roller pump (e.g. Masterflex L/S with Easyload II)
Digital thermometer (e.g. model 710, BK)
Polyethylene tubing (e.g. Intramedic PE160)
2.7 Image processing and quantitative analysis of HIV-infected cell motility
Image analysis computer workstation with high graphics processing capability
Image analysis software that allows processing and analysis of 4-dimensional (3-D and time) data sets (e.g. Volocity or Imaris). As a more cost-effective alternative to these commercial products, the freeware program ImageJ with its wide array of freely available plugins (http://rsb.info.nih.gov/ij/) can substitute for some of the most relevant functions of image processing and analysis.
3. Methods
We describe two approaches to visualize the behavior of HIV-infected cells in the lymph node of live, anesthetized humanized mice: (1) footpad injection with reporter HIV to visualize in situ infected cells (see Figure 2a), and (2) footpad injection with HIV-infected, CD4+ central memory-like T cells (Tcm) to visualize adoptively transferred cells (see Figure 2b). Each protocol is designed to address distinct questions, and the investigator can choose the approach that best suits their study objectives. The generation of humanized Bone marrow/ Liver/Thymus “BLT” mice will not be discussed in this chapter; a detailed description of how to generate these mice has been previously reported [9]. Notably, imaging studies were conducted using BLT mice reconstituted on the NOD.scid genetic background, where skin-draining lymph nodes (e.g. popliteal LNs) are well developed and suitable for intravital imaging. We found that mice with a minimum human peripheral blood reconstitution of >50% human CD45+ cells, and >30% T cells within the CD45+ gate, worked best for T cell imaging studies. A flowchart illustrating the overall methods described below is shown in Figure 1a.
Figure 2. Intravital Microscopy of HIV-infected cells in lymph nodes.

a: Visualization of in situ HIV-infected lymph node cells. The popliteal LN was prepared for microscopy at day 4 after footpad injection with HIV-GFP. Elapsed time in minutes:seconds. Migratory tracks are shown on the right. b: Visualization of adoptively transferred uninfected and HIV-infected Tcm in the lymph node. The popliteal LN was prepared at 12 hours after adoptive transfer of Tcm. Selected intravital micrographs of infected (GFP+; green) and uninfected (CMTMR+; red) in the lymph node show differences in the morphologies that these two populations adopt in the LN. Elapsed time in minutes:seconds. T cell migratory tracks are shown on the right.
3.1 Preparation of high-titer reporter HIV for footpad injection
The day before transfection, seed 7 × 106 HEK 293T cells in a 150 mm tissue culture plate in complete DMEM.
Referring to Table 1, add plasmid DNA (e.g. HIV-GFP) to nuclease-free water in a 50mL conical tube and gently mix by pipetting.
Again referring to Table 1, add the appropriate amount of 2M CaCl2 to the DNA/water mixture and thoroughly mix.
While gently vortexing the tube containing the DNA/water/CaCl2 mixture, add 2× HBS dropwise. Incubate the mixture for 3 minutes at room temperature.
Gently vortex the mixture and add dropwise directly onto the medium and gently rock the tissue culture plates. Incubate the cells at 37°C.
Replace the medium at 16-18 hours post-transfection after a single wash with PBS.
At 40-42 hours post-transfection, harvest the viral supernatant and centrifuge for 5 minutes at 400×g. This will remove cellular debris that may be present in the viral supernatant. An additional collection may be performed after incubating for another 24 hours with 20mL of fresh complete DMEM.
Fill 32mL of clarified viral supernatant into a 38.7 mL capacity ultracentrifuge tube, and underlay with 4mL of 20% sucrose solution.
Centrifuge the viral supernatant at 90,000×g for 2 hours at 4°C. Set the deceleration rate on low to avoid disruption of the viral pellet.
Decant the supernatant and resuspend the pellet with 32μL of PBS to achieve a concentration of approximately 1000× from the starting material.
Aliquot viral stocks into cryovials and keep in an -80°C or -152°C freezer for long-term storage. Repeated freeze-thaw cycles will result in reduced infectivity and should be avoided.
Viral titers are measured using MAGI-CCR5 cell lines, as described previously [11]. HIV titers obtained using this method are typically 5 × 106 – 5 × 107 infectious units/mL.
Inject HIV (typically 50,000 - 150,000 infectious particles) into the right footpad of humanized mice using an insulin syringe. The right popliteal lymph node is then prepared for intravital imaging at days 2-6 post-infection.
Table 1. Reagent and volumes required for calcium phosphate transfection.
| Reagent | Volume per 150mm culture dish |
|---|---|
| HIV plasmid | 36 μg |
| 2M CaCl2 | 174 μL |
| Nuclease-free water | Up to 1400 μL |
| 2× HBS, pH 7.05 | 1400 μL |
| Total | 2800 μL |
3.2 Human CD4+ central memory T cell expansion, infection and adoptive transfer into humanized mice
Spleen and lymph node cells are harvested from a humanized mouse (see Note 4). Naïve CD4+ T cells are isolated using an anti-CD4 microbead separation kit.
T cells are washed and resuspended at 1 × 106 cells/mL in complete RPMI medium. Plate 1 million cells into each well in a 24-well tissue culture plate and incubate with 75μL of anti-CD3ε/CD28 antibody-conjugated dynabeads to achieve a 3:1 ratio of beads to cells. Incubate at 37°C.
At day 2, remove the dynabeads by running the cell suspension through a magnetic column, and re-seed T cells at 2 × 105 cells/mL in complete RPMI medium supplemented with 50 IU/mL human recombinant IL-2. At days 4 and 6, centrifuge T cells again and re-seed at 2 × 105 cells/mL in fresh complete RPMI medium supplemented with 50 IU/mL IL-2 to gradually increase cell numbers.
At day 8, analyze T cell cultures for CD4, CCR7, CD45RO and CD62L cell surface expression. Central memory CD4+ T cells are CCR7+ CD45RO+ CD62L+, as described previously [7].
Pellet 5 million cells by centrifugation at 400×g and resuspend in a mixture of concentrated HIV-GFP viral stocks and complete RPMI medium to reach a final volume of 200 μL. Add polybrene to a final concentration of 8 μg/mL and plate the T cell/HIV suspension into a 96-well, flat-bottom tissue culture plate. Typically, a multiplicity of infection (MOI) of 0.5-1 is used to infected T cells.
Seal the lid onto the plate with paraffin wax and centrifuge the plate at 1200×g for 2 hours at 37°C.
After the spin, remove T cells, add 5mL of complete RPMI 1640 supplemented with 50 IU/mL hrIL-2 and plate in a 25 cm2 tissue culture flask, standed up.
At day 2 post-infection, T cells are collected and the proportion of GFP+ infected T cells is enumerated by flow cytometry.
Recipient humanized mice are given daily intraperitoneal injections of emtricitabine (FTC, 100mg/kg) and tenofovir (TDF, 150mg/kg) for two days prior to T cell transfer. (see Note 5)
Resuspend T cells in 50μL (or less) of PBS and inject into the right footpad of humanized mice using an insulin syringe. Injection of 250,000 – 500,000 GFP+ T cells typically seeds the draining lymph node with sufficient cell numbers for intravital imaging.
(Optional) Prepare and label 1 million uninfected T cells with 10μM CMTMR for 15 minutes in PBS + 1% FCS and wash with equal volume of 100% FCS. Adoptively transfer CMTMR+ T cells into the same right footpad to seed the draining popliteal lymph node with an uninfected, control T cell population.
Prepare the right popliteal lymph node for intravital microscopy 10-12 hours after adoptive cell transfer.
3.3. Surgical anesthesia
Mice are anaesthetized by initially intraperitoneal injection of a mixture of Ketamine, Xylazine, and NaCl 0.9% (to achieve a dose of 100 mg/kg body weight for Ketamine and 10 mg/kg for Xylazine). A surgical plane of anesthesia is achieved if the mouse does not react to firm pinching of the footpad. Repeat injections during the surgical procedure and later on during the imaging session can be carried out intramuscularly to achieve slower release and more even plasma levels.
3.4. Microsurgical preparation of the mouse popliteal lymph node for intravital microscopy (see Note 6)
We have previously described a detailed protocol for the lymph node preparation, which will not be discussed in this chapter [12] (Figure 1b). Notably, we found no differences in the positioning and size of the popliteal LN between conventional C57BL/6 and humanized mice. When performing surgeries on HIV-infected mice, biosafety measures approved by the relevant institutional agency must be strictly followed. Standard personal protective equipment in BSL 2+ facilities typically includes tyvek coveralls, hairnet, protective mask, goggles, shoe covers and double gloves. Additional safety measures such as the use of syringes with retractable needles, puncture-proof gloves and DecapiCone mouse restrainers are recommended.
3.5. Multiphoton intravital microscopy of the mouse popliteal lymph node
Through the eyepieces of the microscope, the popLN can be distinguished from the surrounding tissue structures by its oval shape and its green autofluorescence under mercury arc lamp illumination (see Note 7). Flow in negatively contrasted blood vessels can be assessed for robustness of perfusion. Any residual movement of the specimen that is noticeable at this point will preclude acquisition of high-resolution 3D image stacks and should be corrected by readjustment of the preparation under the dissecting microscope.
T cell migration is highly temperature dependent [13]. Since the water immersion objective lens functions as a heat sink, the ambient popLN temperature must be actively maintained close to 37°C. Perfusing the circular metal tube overlying the coverglass is a cost-effective and convenient approach, since the temperature can be quickly adjusted by changing the flow rate (Figure 1c).
The laser source is tuned to 920 nm and the microscope equipped with appropriate filter combinations to detect blue, green, and orange-red fluorescence emission (e.g. using 455/50, 525/50, and 590/50 nm bandpass filters). After switching to viewing by multiphoton fluorescence excitation, back-scattered second harmonic signals from the collagen-rich organ capsule and the lymph node reticulum are visible in the blue channel and serve as convenient landmark structures to assess the current location within the popLN. The green channel will show fluorescence of eGFP, and the red channel will show fluorescence of CMTMR. Any autofluorescence will be detected in all channels.
To test the viability of the preparation, the baseline migratory behavior of labeled lymphocytes is recorded. To this end, a field of view containing the T cell area (typically 150-200 μm away from the LN capsule) is chosen and a stack of optical sections (e.g. 11 sections spaced 4 μm apart in the z-axis, corresponding to 40 μm of depth) repetitively recorded (e.g. at 15 s intervals) for 15 minutes without optical zoom (when using a 20× lens this typically results in a field size of 500 - 700 μm). Most commercial image acquisition software allows subsequent browsing through the acquired image sequence along the time axis to visually assess cell motility. At this point attention should also be paid to the stability of the preparation. In particular, a high frequency jitter caused by the respiratory movement of the mouse or the pulsation of a nearby arterial vessel, as well as a gradual drift of the specimen need to be corrected by readjustment of the preparation under the dissection microscope. At this point, the optimal microscope settings (laser power, PMT gain, offset etc.) to obtain balanced signals of GFP and CMTMR is determined and loaded.
Once a representative location with a sufficient number of fluorescently labeled cells is identified, begin image acquisition. Ideally, a continuous recording of several hours should be generated, but because of practical limitations, typically multiple sequential recordings have to be obtained (see Note 8).
3.6. Image processing and quantitative analysis of cell migration behavior
Image analysis software that allows for processing and analysis of 4-dimensional (3-D and time) data sets is used (e.g. Volocity, Improvision or Imaris, Bitplane). Each lymphocyte track is visually inspected, and those that track artifacts (e.g. autofluorescent structures) are manually deleted from the analysis (see Note 9). Information on the calibrated spatial position in the x, y and z dimension and on the green (infected) and red (uninfected) integrated fluorescence for all individual time points of measured T lymphocyte tracks are exported as tab-delimited .txt-files. These can then be computed to obtain parameters describing cell motility (e.g. instantaneous migratory velocity, turning angles) in a spreadsheet calculation program such as Excel (Microsoft corporation) or a matrix-based computing environment such as Matlab (Mathworks). The speed of cellular movement is a useful parameter describing cell motility, and is often reported. The instantaneous migratory velocity is measured by dividing the distance travelled between two subsequent time frames by the time between frames, and provides information on the short-term fluctuations of cell motility of individual cells. Track velocity, on the other hand, describes the average velocity of individual cells. The arrest co-efficient, finally, describes the fraction of time an individual cells is ‘arrested’ defined by moving slower than an arbitrarily defined threshold velocity, e.g. 4 μm/min. Only determining the mean arrest co-efficent or track velocity of a population can obscure the behavior of minor, but biological relevant subpopulations, which may be revealed by analyzing the distribution of these parameters.
The turning angle is also an important feature describing the directional persistence or bias of migrating cells and can be used to detect subtle changes in movement behavior. A cell displaying random migration would exhibit an even distribution of turning angles, whereas a preferred range of turning angles may be over-represented in biased random or non-random movement. It is important to note that the speed and turning angle are dependent on the acquisition parameters, and larger time intervals between frames will generally lead to a larger error. Another important parameter used to describe the type of cell motion is derived by plotting the mean displacement of cells by the square root of time. A linear relationship indicates random migration, whereas directed migration will yield an upward-curved plot and a downward-curved plot is indicative of confined motion. The motility coefficient is then calculated from the linear portion of the mean displacement plot to approximate the rate of random cell migration and displacement. Taken together, the speed, turning angle and mean displacement of a cell population are all powerful measures to describe cell motility and to characterize subtle changes in movement behavior in vivo.
4. Notes
Note 1
HIV encodes only a small number of genes, whose open reading frames overlap. This makes it difficult to insert fluorescent reporter genes or tags within the genome without negatively affecting multiple genes or critical regulatory elements required for viral infection and replication. The HIV-GFP reporter virus used in our experiments was derived by replacing the V3 loop region of env in the originally CXCR4-tropic NL4-3-IRES-GFP [14] with that of HIV BaL and was validated for in vitro and in vivo infectivity[7].
All newly constructed reporters must be rigorously validated to ensure that the introduced reporter sequence does not negatively impact the infectivity, replication kinetics and budding/release of HIV in primary cells. If a fluorescent protein-based reporter is used [7,15,16], additional considerations include the brightness, photostability, oligomerization property and maturation time of the fluorescent protein (FP), the impact of FP fusions on the function of the protein they are fused to, and whether the FP used is optimally excited and detected by the imaging setup being employed [17] need to be taken into account. When non-fluorescent protein-based reporters are used [18,19], potential toxicity of the labeling procedure and the low signal-to-noise ratio in many tissues must be considered.
Note 2
Our microscope stage is manufactured using a Plexiglas plate as the base, onto which various elements for the fixation of the mouse are mounted. These elements for the most part consist of materials available in any hardware store that are shaped to suit their particular function. We found small steel corner brackets, angle style steel shelf support pegs, screws and nuts of various calibers, silicone-based windshield sealer, and the casting resin, to be very useful items. The only expensive element is a 3D-micromanipulator, which allows for the controlled positioning of the coverglass on the popLN.
Note 3
The microscope must be situated in the appropriate biosafety containment level facility for the handling of HIV-infected mice. All required biosafety and animal care protocols must be approved by the relevant institutional agencies in order to conduct all imaging studies described in this chapter.
Note 4
Typically, 10 - 20 humanized mice are implanted with tissue/cells from a single human donor and are immunologically tolerant to each other within the same “batch”. Donor (source of T cells) and recipient (host receiving adoptive T cell transfers) humanized mice from the same batch are used to prevent immune rejection and/or inappropriate alloreactivity of transferred T cells in all experiments.
Note 5
The pretreatment of recipient mice with a combination of antiretroviral drugs prevents the infection of recipient mice to ensure that only the transferred HIV-infected T cells are visualized.
Note 6
Our choice of the popLN for MP-IVM studies over other skin-draining LNs was based on two observations: i) The quantity of footpad-injected reagents that drains to the LN can be controlled more accurately with the popLN than with other skin-draining LNs; ii) Due the popLN's remote location to the animal's trunk, the respiratory movements, which would inevitably cause motion artifacts during the slow image acquisition achieved by laser-scanning microscopy, could be shielded off effectively without applying pressure to the surrounding tissues.
Note 7
If a conventional epifluorescence unit is unavailable, marking the position of the lymph node by drawing a circle around it on the dry coverglass with a fine marker pen can help with locating and focusing of the tissue under the multiphoton microscope. Oblique brightfield epi-illumination with a handheld flashlight then allows viewing of the specimen through the eyepieces of the microscope.
Note 8
The duration of the recording is limited by: i) The requirement to obtain access to the animal in order to maintain surgical anesthesia, ii) Evaporation of the immersion water, iii) Restrictions of either the acquisition software or the computer hardware on the maximum size of the recorded data files. Besides the total length of the recording, the file size can typically be adjusted through the pixel resolution, the number of z planes, and the cycle time. We typically use a pixel resolution of 512 × 512, 11 optical sections, and a cycle time of 15 s, since this provides us with a good compromise between file size, image detail, and the ability to follow individual motile cells for sufficient time to obtain meaningful data.
Note 9
Specific points to consider when visually inspecting tracks: i) Tracks with very low meandering indices (large displacement values between the first and last track despite a low length of distance travelled) may indicate non-cellular artifacts, ii) Tracks that contain few time points may indicate cells leaving the field of view (FOV) and possibly not suitable for analysis (this should be recorded and kept constant for the entire data set), iii) Tracks with low displacement values may be indicative of non-motile auto-florescent artifacts, iv) Two intersecting tracks leading to a switching of the two tracks should be manually corrected, v) Tracks that border the FOV where only a portion of the cell is visible should be deleted. It is important to note that these are guidelines, and manual inspection of all tracks is essential not to over/under-estimate cellular motility.
References
- 1.Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248(4951):73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- 2.Helmchen F, Denk W. Deep tissue two-photon microscopy. Nat Methods. 2005;2(12):932–940. doi: 10.1038/nmeth818. [DOI] [PubMed] [Google Scholar]
- 3.Cahalan MD, Parker I, Wei SH, Miller MJ. Two-photon tissue imaging: seeing the immune system in a fresh light. Nature Reviews Immunology. 2002;2(11):872–880. doi: 10.1038/nri935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pittet MJ, Mempel TR. Regulation of T-cell migration and effector functions: insights from in vivo imaging studies. Immunol Rev. 2008;221:107–129. doi: 10.1111/j.1600-065X.2008.00584.x. [DOI] [PubMed] [Google Scholar]
- 5.Denton PW, Estes JD, Sun Z, Othieno FA, Wei BL, Wege AK, Powell DA, Payne D, Haase AT, Garcia JV. Antiretroviral pre-exposure prophylaxis prevents vaginal transmission of HIV-1 in humanized BLT mice. PLoS Med. 2008;5(1):e16. doi: 10.1371/journal.pmed.0050016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denton PW, Othieno F, Martinez-Torres F, Zou W, Krisko JF, Fleming E, Zein S, Powell DA, Wahl A, Kwak YT, Welch BD, Kay MS, Payne DA, Gallay P, Appella E, Estes JD, Lu M, Garcia JV. One percent tenofovir applied topically to humanized BLT mice and used according to the CAPRISA 004 experimental design demonstrates partial protection from vaginal HIV infection, validating the BLT model for evaluation of new microbicide candidates. J Virol. 2011;85(15):7582–7593. doi: 10.1128/JVI.00537-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murooka TT, Deruaz M, Marangoni F, Vrbanac VD, Seung E, von Andrian UH, Tager AM, Luster AD, Mempel TR. HIV-infected T cells are migratory vehicles for viral dissemination. Nature. 2012;490(7419):283–287. doi: 10.1038/nature11398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun Z, Denton PW, Estes JD, Othieno FA, Wei BL, Wege AK, Melkus MW, Padgett-Thomas A, Zupancic M, Haase AT, Garcia JV. Intrarectal transmission, systemic infection, and CD4+ T cell depletion in humanized mice infected with HIV-1. J Exp Med. 2007;204(4):705–714. doi: 10.1084/jem.20062411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brainard DM, Seung E, Frahm N, Cariappa A, Bailey CC, Hart WK, Shin HS, Brooks SF, Knight HL, Eichbaum Q, Yang YG, Sykes M, Walker BD, Freeman GJ, Pillai S, Westmoreland SV, Brander C, Luster AD, Tager AM. Induction of robust cellular and humoral virus-specific adaptive immune responses in human immunodeficiency virus-infected humanized BLT mice. J Virol. 2009;83(14):7305–7321. doi: 10.1128/JVI.02207-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dudek TE, No DC, Seung E, Vrbanac VD, Fadda L, Bhoumik P, Boutwell CL, Power KA, Gladden AD, Battis L, Mellors EF, Tivey TR, Gao X, Altfeld M, Luster AD, Tager AM, Allen TM. Rapid evolution of HIV-1 to functional CD8(+) T cell responses in humanized BLT mice. Sci Transl Med. 4(143):143ra198. doi: 10.1126/scitranslmed.3003984. doi:4/143/143ra98 [pii] 10.1126/scitranslmed.3003984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pirounaki M, Heyden NA, Arens M, Ratner L. Rapid phenotypic drug susceptibility assay for HIV-1 with a CCR5 expressing indicator cell line. J Virol Methods. 2000;85(1-2):151–161. doi: 10.1016/s0166-0934(99)00163-9. [DOI] [PubMed] [Google Scholar]
- 12.Murooka TT, Mempel TR. Multiphoton intravital microscopy to study lymphocyte motility in lymph nodes. Methods Mol Biol. 2012;757:247–257. doi: 10.1007/978-1-61779-166-6_16. [DOI] [PubMed] [Google Scholar]
- 13.Miller MJ, Wei SH, Parker I, Cahalan MD. Two-photon imaging of lymphocyte motility and antigen response in intact lymph node. Science. 2002;296(5574):1869–1873. doi: 10.1126/science.1070051. [DOI] [PubMed] [Google Scholar]
- 14.Schindler M, Munch J, Kirchhoff F. Human immunodeficiency virus type 1 inhibits DNA damage-triggered apoptosis by a Nef-independent mechanism. J Virol. 2005;79(9):5489–5498. doi: 10.1128/JVI.79.9.5489-5498.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gelderblom HC, Vatakis DN, Burke SA, Lawrie SD, Bristol GC, Levy DN. Viral complementation allows HIV-1 replication without integration. Retrovirology. 2008;5:60. doi: 10.1186/1742-4690-5-60. doi:1742-4690-5-60 [pii] 10.1186/1742-4690-5-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown A, Gartner S, Kawano T, Benoit N, Cheng-Mayer C. HLA-A2 down-regulation on primary human macrophages infected with an M-tropic EGFP-tagged HIV-1 reporter virus. J Leukoc Biol. 2005;78(3):675–685. doi: 10.1189/jlb.0505237. [DOI] [PubMed] [Google Scholar]
- 17.Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2(12):905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- 18.Arhel N, Genovesio A, Kim KA, Miko S, Perret E, Olivo-Marin JC, Shorte S, Charneau P. Quantitative four-dimensional tracking of cytoplasmic and nuclear HIV-1 complexes. Nat Methods. 2006;3(10):817–824. doi: 10.1038/nmeth928. doi:nmeth928 [pii] 10.1038/nmeth928. [DOI] [PubMed] [Google Scholar]
- 19.Eckhardt M, Anders M, Muranyi W, Heilemann M, Krijnse-Locker J, Muller B. A SNAP-tagged derivative of HIV-1--a versatile tool to study virus-cell interactions. PLoS One. 6(7):e22007. doi: 10.1371/journal.pone.0022007. PONE-D-11-06412 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]