

Abstract
Pd-catalyzed arylation or benzylation of nitroazoles using aryl sulfonates or benzyl acetates is described. Electronically varied aryl tosylates and mesylates, as well as benzyl acetates, afford the arylated and benzylated products. Arylation of nitrobenzene is also reported. The relative rate for the arylation of halides is greater than that of tosylates using the reported reaction parameters. These studies enhance the scope of electrophiles for nitroarene arylations and benzylations, which was hitherto limited to the use of halide electrophiles.
Graphical abstract
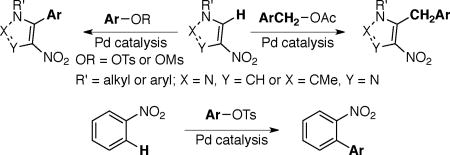
Nitroarenes are important building blocks for the synthesis of pharmaceuticals, agrochemicals, polymers and fine chemicals.1, 2 Many nitroarenes are readily available at low cost via nitration of arenes.2 Reduction of the nitro group serves as a versatile handle for structural elaboration through cross-coupling and functional group transformation of the resulting amino group.3 In lieu of these applications of nitroarenes, a few reports have detailed the arylation of nitroazoles (Schemes 1a, and 2a)4 and nitrobenzene derivatives (Scheme 4a)2, 5 using halide electrophiles.6, 7, 8 The allylation and benzylation of nitroazoles using allyl acetate and benzyl chloride, respectively has also been realized (Scheme 3a).9 Recently there have been increasing reports on the use of C–O electrophiles in place of halides for C–C bond formations.8, 10, 11 The use of C–O electrophiles instead of halides presents several advantages including: (i) the availability of alcohols from natural sources, (ii) ready access of various ortho-substituted phenol derivatives using directed ortho-metallation and (iii) requirement for different catalyst/ligand combinations than are used with halides, thereby presenting an opportunity to engage the C–X (X = halide) and C–O electrophiles at different stages in the context of multi-step synthesis.10, 11 As part of our program on transition metal catalyzed functionalizations using C–O electrophiles,12 we report herein the first general method for the direct arylation and benzylation of nitropyrazoles and nitroimidazoles using aryl sulfonates11, 13, 14 and benzyl acetates (Scheme 1b–3b). A brief scope for the arylation of nitrobenzene is also described (Scheme 4b). Notably, functionalized pyrazoles, imidazoles and nitrobenzene derivatives find applications in the synthesis of pharmaceuticals, agrochemicals, or ligands for transition metals.1, 9, 4c
Scheme 1.

Literature Precedent and Present Study.
Scheme 2. Scope of Nitropyrazole Arylations.

[a]conducted at 80 °C.[b] without CsOPiv.[c] Reaction set up outside the glove box under inert atmosphere.[d] p-xylene used as solvent.[e] 1.1 equiv of azole and 1.0 equiv of tosylate used.[f] General conditions with Pd(OAc)2 (0.15 equiv) and XPhos (0.45 equiv). [g]NMR yield against 1,4-dinitrobenzene as the standard.
Scheme 4. Scope of Nitroimidazole Arylations.

[a]Reaction set up outside the glove box under inert atmosphere.
Scheme 3. Heteroarylation of Nitropyrazolesa.

[a]Reaction conditions: Azole (1.0 equiv), tosylate (1.05-1.1 equiv), Pd(OAc)2 (0.1 equiv), XPhos (0.3 equiv), Cs2CO3 (1.5 equiv), CsOPiv (1.1 equiv), p-xylene (or toluene), 120 °C. [b]conducted at 80 °C. [c]General conditions but with Pd(OAc)2 (0.15 equiv), XPhos (0.45 equiv).
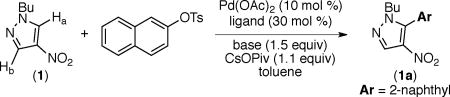
Our studies commenced with the optimization of the arylation of nitropyrazole 1 using 2-naphthyl tosylate. As shown in Table 1, product 1a can be obtained in the presence of Pd(OAc)2 with XPhos or SPhos as ligands (entries 4 and 5).
Table 1.
Optimization of Nitropyrazole Arylation
| ||||
|---|---|---|---|---|
|
| ||||
| entry | ligand | base | T(°C) | GC yield (%)a |
| 1 | PCy3 | Cs2CO3 | 80 | 0 |
| 2 | PtBu3 | Cs2CO3 | 80 | 0 |
| 3 | PPh3 | Cs2CO3 | 80 | 0 |
| 4 | X-Phos | Cs2CO3 | 80 | 89 |
| 5 | S-Phos | Cs2CO3 | 80 | 88 |
| 6 | dcype | Cs2CO3 | 80 | 0 |
| 7 | X-Phos | K2CO3 | 80 | 87 |
| 8 | X-Phos | Cs2CO3 | 120 | 56 |
| 9b | X-Phos | Cs2CO3 | 80 | 0 |
| 10 | none | Cs2CO3 | 80 | 0 |
Calibrated GC yields against hexadecane as the internal standard.
Reaction without Pd(OAc)2 catalyst.
In contrast to the analogous reaction with halide electrophiles4 the use of PPh3 does not lead to 1a. Additionally, the bidentate dcype ligand, which is effective for arylations of arenes using tosylates and mesylates12a,c does not promote the arylation of nitropyrazole (entry 6). Substrate 1 has two distinct C–H bonds (Ha and Hb) that can undergo arylation. Consistent with previous reports on arylation using aryl halides4 product 1a is derived via the preferential arylation of the more acidic C–Ha bond.15 Unlike arylations using aryl bromides,4 transformations using aryl tosylates do not require copper(I) additives. Carbonate bases are effective at promoting the transformation (entries 4 and 7). Increasing the temperature Pd-catalyzed arylation or benzylation of nitroazoles using aryl sulfonates or benzyl acetates is described. Electronically varied aryl tosylates and mesylates, as well as benzyl acetates, afford the arylated and benzylated products. Arylation of nitrobenzene is also reported. The relative rate for the arylation of halides is greater than that of tosylates using the reported reaction parameters. These studies enhance the scope of electrophiles for nitroarene arylations and benzylations, which was hitherto limited to the use of halide electrophiles. leads to diminished yield of 1a. Notably, 5–10% of diarylation product (1aa) is formed via functionalization of both C–Ha and C–Hb regardless of the temperature (entries 4, 5, 7 and 8).16 No product is obtained in the absence of the Pd(OAc)2 or the ligand (entries 9 and 10). However, comparable yields of 1a are obtained with or without CsOPiv (Scheme 2). Importantly, use of N-methyl pyrazole in these arylations affords only trace product, suggesting the importance of the nitro group for efficiency of arylations using nitroazoles.
Scheme 2 depicts the scope of nitropyrazole arylation with respect to the substrate and the electrophile. The optimal temperature for these transformations (80 °C versus 120 °C) varied based on the nature of the aryl tosylate. Arylated products are obtained in good to excellent yields using electronically varied aromatic tosylates. Furthermore, pyrazole substrates bearing various N-alkyl (N-butyl (1a-1g), N-Me (2c and 2h), and N-CH2CH2Ph (3i)) or N-aryl groups (4a) undergo arylation to afford the corresponding products in good yields.17
Heterocylic tosylates also couple with nitropyrazoles under the optimal conditions. For example, the use of tosylated pyridine, quinoline and quinoxaline affords products 1j (and 5j), 1k, and 1l, respectively in good yields (Scheme 3). Furthermore, 1m bearing a π-rich heterocycle is also obtained in good yield.18
The reaction conditions for pyrazole arylations are also effective for arylation of nitroimidazoles, with a similar substrate scope (Scheme 4). The higher yields for imidazole arylations are in part due to the absence of diarylation because these substrates contain only one acidic C–H bond.
A sequential tosylation/arylation protocol enhances the step economy of these arylations (Scheme 5).12 Tosylation of the phenol derivative affords a solution of the corresponding tosylate, which is employed directly in the subsequent Pd-catalyzed arylations without purification of the intermediate tosylate. Pyrazole and imidazole substrates undergo sequential tosylation/arylation to afford products (1a, 1j and 6a) in yields comparable to those obtained using preformed tosylates (Scheme 2–4).
Scheme 5.

Sequential Tosylation/Arylation.
We next investigated the use of the more atom economical mesylates in these reactions.14 As shown in Scheme 6, both nitropyrazole (1a, 4a, 1p, 1f, 1n) and nitroimidazoles (6a and 6c) are amenable to arylation with aromatic mesylates to afford the products in modest to good yields. Yields are generally higher with electron-neutral and electron-rich mesylates than with electron-deficient mesylates in part due to hydrolysis of the latter under the reaction conditions.
Scheme 6. Arylation of Nitroazoles using Mesylates.

[a]conducted at 80 °C. [b]Reaction set up outside the glove box under inert atmosphere. [c]NMR yield against 1,4-dinitrobenzene as the standard.
By employing benzyl acetates instead of aryl sulfonates pyrazoles and imidazoles can be benzylated in good yields under otherwise similar reaction conditions (Scheme 7).19, 20 Substituted benzyl acetates bearing electron-donating and electron-withdrawing groups are compatible with these transformations (2s and 2t). As observed for arylations described above, good yields of benzylated products are obtained using X-Phos as the ligand. Notably, previous reports on benzylation of nitroazoles were limited to the use of benzyl chlorides; the use of benzyl acetate with Pd(OAc)2/PPh3 catalyst combination was reported to give 0% yield of the desired product for the benzylation of N-benzylnitropyrazole.9
Scheme 7. Scope for Benzylation of Nitroazoles.

[a]Reaction set up outside the glove box under inert atmosphere using toluene as solvent. [b]CsOPiv (1.1 equiv) added and toluene used as solvent. [c]conducted at 120 °C.
We next explored these nitroazole arylations using electrophiles containing two different leaving groups (Scheme 8).
Scheme 8.

Mono- and Diarylation of Halotosylates.
For example the reaction of (1) with m-haloaryl tosylate affords product 1u via selective functionalization of the C–X (X=Cl, Br) bond (Scheme 8a).21 When the same reactions were conducted in the presence of excess azole, diarylated product 1v resulting from the sequential functionalization of the C–X and the C-OTs bonds is obtained in good yields (Scheme 8b). Similarly, and as expected, the arylation of azole 2 with m-chlorobenzyl acetate leads to 2w via preferential cleavage of the more reactive C–Cl bond (Scheme 9a). However, use of excess azole results in product 2x via sequential arylation and benzylation reactions (Scheme 9b). These results demonstrate the distinct reactivity of halides versus tosylates and benzyl acetates and support the potential for engaging these electrophiles selectively at different stages in a multi-step synthetic sequence. Furthermore, the result of the reaction in Scheme 9a is orthogonal to that reported previously using meta-chloro benzyl choride.9 As shown in Scheme 9c, the benzylated product (2y) is obtained preferentially for the arylation of azole 2 using meta-chloro benzyl chloride.
Scheme 9.

Sequential Arylation and Benzylations.
Having explored the arylations and benzylations of heteroaromatic nitroarenes with C–O electrophiles, we briefly examined the use of significantly less acidic nitrobenzene as a substrate for arylations using tosylates. K3PO4 is a more effective base (63% yield of 8a) than Cs2CO3 (40% yield of 8a) for arylation of nitrobenzene. Furthermore, CsOPiv is a necessary additive for these arylations to afford the products in significant yields. As shown in Scheme 10, electron-neutral (8a) and electron-rich aryl tosylates (8d, 8p, 8f and 8e) can be used to afford the ortho-arylated products. The site-selectivity for the arylation of the ortho C–H bond clearly implicates the directing effect of the nitro group (either as a directing ligand and/or enhancing the acidity of the ortho C–H bond) and is consistent with previous reports using aryl halides.2, 5 Unlike the azole reactions, an excess of the substrate (10 equivalents of nitrobenzene) and a higher temperature is required for these arylations.2, 5
Scheme 10.

Arylation of Nitrobenzene using Tosylates.
In summary, this manuscript describes the first report for the arylation and benzylation of nitroazoles using aryl sulfonates (tosylates and mesylates) and benzyl acetates, respectively. The products are obtained in good to excellent yields with electronically varied C–O electrophiles. The selective functionalization of halides in the presence of tosylates or benzyl acetates make these arylations amenable to sequential arylations and benzylations in the context of multi-step synthesis. The arylations using the more challenging nitrobenzene substrate also afford the desired biaryls albeit in lower yields than the corresponding azoles.
EXPERIMENTAL METHODS
Materials and Methods
NMR spectra were obtained on a Bruker 400 (399.96 MHz for 1H; 100.57 MHz for 13C) spectrometer. 1H NMR chemical shifts are reported in parts per million (ppm) relative to TMS, with the residual solvent peak used as an internal reference. Multiplicities are reported as follows: singlet (s), doublet (d), doublet of doublets (dd), triplet of doublets (td), triplet (t), doublet of triplets (dt), triplet of triplets (tt), multiplet (m), and broad resonance (br). IR spectra were obtained on a Thermo scientific Nicolet iS5 iD5 ATR spectrometer. Melting points were obtained on a Thomas Hoover melting point apparatus. HRMS data was obtained from the University of Illinois Urbana Champaign Mass Spectrometry Lab. Data were acquired either on VG 70-VSE (EI+) or TOF (ES+) instruments.
Cesium pivalate, 2-naphthol, and 3-pyridyl phenol were obtained from Aldrich and used as received. Pd(OAc)2 and XPhos were obtained from Strem chemical and used as received. Cesium carbonate was obtained from Acros and used as received. K2CO3 was obtained from JT Baker and used as received. The azoles,4 tosylates,12a mesylates12c and benzyl acetates were prepared using literature procedures. Anhydrous xylene was obtained from Aldrich and used as received. Toluene was purified using Glass Contour solvent purification system column composed of neutral alumina and a copper catalyst. Other solvents were obtained from Fisher Chemical or VWR Chemical and used without further purification. Flash chromatography was performed on EM Science silica gel 60 (0.040–0.063 mm particle size, 230–400 mesh) and thin layer chromatography was performed on Analtech TLC plates precoated with silica gel 60 F254.
General Procedures for Arylations
General Procedure A for azole arylations using solid electrophile
Substrate, electrophile (tosylate or mesylate) and Pd(OAc)2 were weighed into a 20 mL scintillation vial. The vial was taken into the glove box and XPhos, base (Cs2CO3 or K2CO3), CsOPiv and solvent (toluene or xylene) were added. The vial was sealed with a Teflon lined cap, taken out of the glove box and the reaction mixture was allowed to stir at the indicated temperature for the indicated time. The reaction mixture was cooled to room temperature and filtered through a 1.5 inch plug of silica gel, eluting with EtOAc (100 mL). The filtrate was concentrated and chromatographed on a silica gel column to afford the product.
General Procedure B for azole arylations using liquid electrophile
Substrate, and Pd(OAc)2 were weighed into a 20 mL scintillation vial. The vial was taken into the glove box and XPhos, base (Cs2CO3 or K2CO3), CsOPiv and a solution of the electrophile in the specified solvent (toluene or xylene) were added. The vial was sealed with a Teflon lined cap, taken out of the glove box and the reaction mixture was allowed to stir at the indicated temperature for the indicated time. The reaction mixture was cooled to room temperature and filtered through a 1.5 inch plug of silica gel, eluting with EtOAc (100 mL). The filtrate was concentrated and chromatographed on a silica gel column to afford the product.
General Procedure C for sequential tosylation/arylation
Phenol derivative and tosyl chloride were weighed into a scintillation vial. The vial was taken into the glove box and Cs2CO3 and toluene were added to it. The vial was sealed with a Teflon lined cap, taken out of the glove box and stirred at 120 °C for 3 h. The reaction vial cooled to rt, and taken into the glove box. The tosylate solution was added to a vial containing azole Pd(OAc)2 XPhos, Cs2CO3, and CsOPiv. The vial was sealed with a Teflon lined cap, taken out of the glove box and the reaction mixture was allowed to stir at the indicated temperature for the indicated time. The reaction mixture was cooled to room temperature and filtered through a 1.5 inch plug of silica gel, eluting with EtOAc (100 mL). The filtrate was concentrated and chromatographed on a silica gel column to afford the product.
General Procedure D for azole benzylations
Substrate, and Pd(OAc)2 were weighed into a 20 mL scintillation vial. The vial was taken into the glove box and XPhos, Cs2CO3, CsOPiv (if added) and a solution of the acetate in the specified solvent (toluene or xylene) were added. The vial was sealed with a Teflon lined cap, taken out of the glove box and the reaction mixture was allowed to stir at the indicated temperature for the indicated time. The reaction mixture was cooled to room temperature and filtered through a 1.5 inch plug of silica gel, eluting with EtOAc (100 mL). The filtrate was concentrated and chromatographed on a silica gel column to afford the product.
General Procedure E for nitrobenzene arylations
Pd(OAc)2 and tosylate was weighed into a 20 mL scintillation vial. The vial was taken into the glove box and nitrobenzene derivative, XPhos, K3PO4, and CsOPiv were added. Xylene and nitrobenzene were added sequentially, the vial was sealed with a Teflon lined cap, taken out of the glove box and the reaction mixture was allowed to stir at the indicated temperature for the indicated time. The reaction mixture was cooled to room temperature and filtered through a 1.5 inch plug of silica gel, eluting with EtOAc (100 mL). The filtrate was concentrated and chromatographed on a silica gel column to afford the product.
General Procedure F for azole arylations using solid electrophile
Substrate, electrophile (tosylate or mesylate) and Pd(OAc)2, XPhos, base (Cs2CO3 or K2CO3), and CsOPiv were weighed into a 20 mL scintillation vial. The vial was sealed with a Teflon lined cap having a septum. The headspace of the vial was purged with nitrogen. Anhydrous toluene was added and the reaction mixture was allowed to stir at the indicated temperature for the indicated time. The reaction mixture was cooled to room temperature and filtered through a 1.5 inch plug of silica gel, eluting with EtOAc (100 mL). The filtrate was concentrated and chromatographed on a silica gel column to afford the product.
General Procedure G for azole benzylations
Substrate, and Pd(OAc)2, XPhos, and Cs2CO3 were weighed into a 20 mL scintillation vial. The vial was sealed with a Teflon lined cap having a septum. The headspace of the vial was purged with nitrogen. A toluene solution of the acetate was added and the reaction mixture was allowed to stir at the indicated temperature for the indicated time. The reaction mixture was cooled to room temperature and filtered through a 1.5 inch plug of silica gel, eluting with EtOAc (100 mL). The filtrate was concentrated and chromatographed on a silica gel column to afford the product.
Synthesis and Characterization of Substrates
1-methyl-1H-indol-5-yl 4-methylbenzene-1-sulfonate22
1H NMR (CDCl3): δ 7.70 (d, J = 8.3 Hz, 2H), 7.28 (d, J = 8.5 Hz, 2H), 7.21 (d, J = 2.3 Hz, 1H), 7.17 (d, J = 8.5 Hz, 1H), 7.07 (d, J = 3.1 Hz, 1H), 6.83 (dd, J = 8.8, 2.4 Hz, 1H), 6.41 (dd, J = 3.1, 0.9 Hz, 1H), 3.77 (s, 3H), 2.44 (s, 3H). 13C NMR (CDCl3): δ 144.9, 143.2, 135.0, 132.6, 130.4, 129.5, 128.5, 128.3, 116.1, 114.0, 109.5, 101.3, 32.9, 29.6. IR (neat): 2924, 1486, 1337, 1237, 1215, 1189, 1086, 839, 816, 771, 756, 741, 734, 708, 698, 657 cm−1. mp = 135–137 °C. HRMS [EI+, M+] Calcd for C16H15NO3S: 301.0773; Found: 301.0766.
Product Synthesis and Characterization (Scheme 2)
1-butyl-5-(naphthalen-2-yl)-4-nitro-1H-pyrazole (1a)
Following general procedure A, 1 (84.6 mg, 0.500 mmol, 1.0 equiv), 2-naphthyl tosylate (149 mg, 0.500 mmol, 1.0 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 80 °C for 16 h. Chromatography on a silica gel column using 95/5 hexanes/EtOAc (Rf = 0.18 in 95% hexanes/5% ethyl acetate) yielded product 1a as a yellow oil (122.8 mg, 83% yield). 1H NMR (CDCl3): δ 8.26 (s, 1H), 8.00 (d, J = 8.5 Hz, 1H), 7.95-7.90 (m, 2H), 7.87 (s, 1H), 7.64-7.57 (m, 2H), 7.43 (dd, J = 8.5, 1.7 Hz, 1H), 4.00 (t, J = 7.3 Hz, 2H), 1.80-1.73 (m, 2H), 1.25-1.15 (m, 2H), 0.81 (t, J = 7.3 Hz, 3H). 13C NMR (CDCl3): δ 141.4, 136.4, 133.7, 133.1, 132.8, 129.7, 128.7, 128.4, 128.0, 127.6, 127.0, 126.3, 124.1, 50.1, 31.8, 19.5, 13.4. IR (neat): 2959, 2932, 1555, 1505, 1471, 1400, 1320, 828 cm−1. HRMS [TOF, ES+, M+H] Calcd for C17H17N3O2: 296.1399; Found: 296.1402.
Procedure without the glove box
Following general procedure F, 1 (84.6 mg, 0.500 mmol, 1.0 equiv), 2-naphthyl tosylate (149 mg, 0.500 mmol, 1.0 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 80 °C for 16 h. Chromatography on a silica gel column using 95/5 hexanes/EtOAc (Rf = 0.18 in 95% hexanes/5% ethyl acetate) yielded product 1a as a yellow oil (131 mg, 89% yield). The spectroscopic data of the product was identical to that described above.
1-butyl-5-(4-methylphenyl)-4-nitro-1H-pyrazole (1b)
Following general procedure A, 1 (84.6 mg, 0.500 mmol, 1.0 equiv), para-methylphenyl tosylate (138 mg, 0.525 mmol, 1.05 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 16 h. Chromatography on a silica gel column using 93/7 hexanes/EtOAc (Rf = 0.23 in 93% hexanes/7% ethyl acetate) yielded product 1b as a yellow oil (77.4 mg, 60% yield). 1H NMR (CDCl3): δ 8.20 (s, 1H), 7.34 (d, J = 7.9 Hz, 2H), 7.25 (d, J = 8.3 Hz, 2H), 3.95 (t, J = 7.2 Hz, 2H), 2.45 (s, 3H), 1.78-1.71 (m, 2H), 1.25-1.16 (m, 2H), 0.83 (t, J = 7.3 Hz, 3H). 13C NMR (DMSO-d6): δ 141.3, 139.8, 136.1, 132.3, 129.7, 129.3, 123.6, 49.4, 30.9, 21.0, 18.9, 13.2. The spectroscopic data is consistent with that previously reported in the literature.4b
1-butyl-5-(3-methoxyphenyl)-4-nitro-1H-pyrazole (1c)
Following general procedure A, 1 (84.6 mg, 0.500 mmol, 1.0 equiv), meta-methoxyphenyl tosylate (153 mg, 0.550 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 16 h. Chromatography on a silica gel column using 93/7 hexanes/EtOAc (Rf = 0.19 in 93% hexanes/7% EtOAc) yielded product 1c as a yellow oil (83.0 mg, 60% yield). 1H NMR (CDCl3): δ 8.18 (s, 1H), 7.43 (t, J = 8.0 Hz, 1H), 7.08-7.05 (m, 1H), 6.92 (dt, J = 7.6, 1.4 Hz, 1H), 6.88-6.87 (m, 1H), 3.94 (t, J = 7.2 Hz, 2H), 3.83 (s, 3H), 1.78-1.71 (m, 2H), 1.25-1.16 (m, 2H), 0.82 (t, J = 7.3 Hz, 3H). 13C NMR (CDCl3): δ 159.6, 141.1, 136.3, 132.8, 129.9, 127.8, 121.7, 115.7, 115.2, 55.4, 50.0, 31.7, 19.5, 13.4. IR (neat): 1582, 1556, 1497, 1465, 1434, 1358, 1318, 1286, 1252, 1216, 1177, 1156, 1047, 1031, 855, 815, 786, 770, 743, 692 cm−1. HRMS [TOF, ES+, M+H] Calcd for C14H17N3O3: 276.1348 Found: 276.1347.
1-butyl-5-(7-methoxynaphthalen-2-yl)-4-nitro-1H-pyrazole (1d)
Following general procedure A, 1 (84.6 mg, 0.500 mmol, 1.0 equiv), 7-methoxynaphthyl tosylate (164 mg, 0.500 mmol, 1.0 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 80 °C for 16 h. Chromatography on a silica gel column using 85/15 hexanes/EtOAc (Rf = 0.21 in 85% hexanes/15% EtOAc) yielded product 1d as a yellow solid (153 mg, 94% yield), mp =84.7 °C. 1H NMR (CDCl3): δ 8.25 (s, 1H), 7.91 (d, J = 8.4 Hz, 1H), 7.83 (d, J = 9.0 Hz, 1H), 7.76 (s, 1H), 7.28-7.25 (m, 2H), 7.17 (d, J = 2.4 Hz, 1H), 4.00 (t, J = 7.2 Hz, 2H), 3.94 (s, 3H), 1.80-1.73 (m, 2H), 1.25-1.15 (m, 2H), 0.81 (t, J = 7.3 Hz, 3H). 13C{1H} NMR (CDCl3): δ 158.4, 141.6, 136.4, 134.1, 133.0, 129.4, 129.2, 128.43, 128.39, 124.5, 123.9, 120.6, 106.1, 55.4, 50.1, 31.8, 19.5, 13.4. IR (neat): 3138, 2976, 2958, 2926, 1607, 1509, 1496, 1466, 1437, 1397, 1367, 1335, 1244, 1229, 1216, 1176, 1028, 901, 850, 836, 828 cm−1. HRMS [TOF, ES+, M+H] Calcd for C18H19N3O3: 326.1505; Found: 326.1505.
5-(2H-1,3-benzodioxol-5-yl)-1-butyl-4-nitro-1H-pyrazole (1e)
Following general procedure A, 1 (84.6 mg, 0.500 mmol, 1.0 equiv), sesamol tosylate (161 mg, 0.550 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous xylene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 80 °C for 16 h. Chromatography on a silica gel column using 90/10 hexanes/EtOAc (Rf = 0.16 in 90% hexanes/10% ethyl acetate) yielded product 1e as an off white solid (105 mg, 73% yield) mp = 63.4 °C. 1H NMR (CDCl3): δ 8.18 (s, 1H), 6.95 (d, J = 8.5 Hz, 1H), 6.82 (dd, J = 6.4, 1.8 Hz, 2H), 6.08 (s, 2H), 3.96 (t, J = 7.3 Hz, 2H), 1.79-1.71 (m, 2H), 1.27-1.17 (m, 2H), 0.85 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3): δ 149.2, 148.0, 140.9, 136.2, 132.8, 123.7, 119.6, 109.9, 108.6, 101.7, 49.9, 31.7, 19.5, 13.4. IR (thin film, CH2Cl2): 2963, 2932, 1508, 1497, 1484, 1465, 1397, 1343, 1318, 1243, 1221, 1034, 976, 864, 826, 810, 763 cm−1. HRMS [TOF, ES+, M+H] Calcd for C14H15N3O4: 290.1141; Found: 290.1142.
1-butyl-4-nitro-5-(3,4,5-trimethoxyphenyl)-1H-pyrazole (1f)
Following general procedure A, 1 (93.1 mg, 0.550 mmol, 1.1 equiv), 3,4,5-trimethoxyphenyl tosylate (169 mg, 0.500 mmol, 1.0 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous xylene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 16 h. Chromatography on a silica gel column using 80/15/5 hexanes/CH2Cl2/acetone (Rf = 0.13 in 80% hexanes/15% CH2Cl2/5% acetone) yielded product 1f as a yellow oil (121 mg, 72% yield). 1H NMR (CDCl3): δ 8.20 (s, 1H), 6.55 (s, 2H), 3.97 (t, J = 7.2 Hz, 2H), 3.94 (s, 3H), 3.87 (s, 6H), 1.82-1.74 (m, 2H), 1.29-1.20 (m, 2H), 0.86 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3): δ 153.5, 141.2, 139.44, 139.42, 136.3, 132.7, 121.7, 106.89, 106.86, 60.9, 56.3, 50.1, 31.9, 19.6, 13.5. IR (thin film, CH2Cl2): 2959, 2936, 1584, 1557, 1505, 1463, 1400, 1341, 1302, 1239, 1124, 1002, 840 cm−1. HRMS [TOF, ES+, M+H] Calcd for C16H21N3O5: 336.1559; Found: 336.1558.
1-butyl-5-(2-methylphenyl)-4-nitro-1H-pyrazole (1g)
Following general procedure A, 1 (84.6 mg, 0.500 mmol, 1.0 equiv), ortho-tolyl tosylate (144 mg, 0.550 mmol, 1.1 equiv), Pd(OAc)2 (16.8 mg, 0.075 mmol, 0.15 equiv), XPhos (107 mg, 0.225 mmol, 0.45 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 16 h. Chromatography on a silica gel column using 95/5 hexanes/EtOAc (Rf = 0.18 in 95% hexanes/5% EtOAc) yielded product 1g as a yellow oil (54.7 mg, 42% yield). 1H NMR (CDCl3): δ 8.23 (s, 1H), 7.45 (td, J = 7.6, 1.5 Hz, 1H), 7.37 (d, J = 7.5 Hz, 1H), 7.33 (t, J = 7.5 Hz, 1H), 7.17 (dd, J = 7.7, 1.4 Hz, 1H), 3.93-3.75 (m, 2H), 2.12 (s, 3H), 1.76-1.68 (m, 2H), 1.25-1.16 (m, 2H), 0.83 (t, J = 7.3 Hz, 3H). 13C NMR (CDCl3): δ 140.9, 137.7, 136.2, 133.3, 130.5, 130.4, 129.3, 126.5, 126.1, 49.8, 31.5, 19.5, 19.4, 13.4. IR (neat): 2958, 2873, 1557, 1504, 1486, 1458, 1400, 1322, 828, 762, 730 cm−1. HRMS [EI+, M+] Calcd for C14H17N3O2: 259.1321; Found: 259.1316.
5-(4-methoxyphenyl)-1-methyl-4-nitro-1H-pyrazole (2h)
Following general procedure A, 1-methyl-4-nitro-1H-pyrazole (2) (63.6 mg, 0.500 mmol, 1.0 equiv), para-methoxyphenyl tosylate (146 mg, 0.525 mmol, 1.05 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 16 h. Chromatography on a silica gel column using 85/10/5 hexanes/CH2Cl2/acetone (Rf = 0.28 in 85% hexanes/10% methylene chloride/5% acetone) yielded product 2h as a light yellow solid (89.4 mg, 77% yield). 1H NMR (CDCl3): δ 8.19 (s, 1H), 7.33 (d, J = 8.8 Hz, 2H), 7.05 (d, J = 8.7 Hz, 2H), 3.89 (s, 3H), 3.75 (s, 3H). The spectroscopic data is consistent with that previously reported in the literature.4b
5-(3-methoxyphenyl)-1-methyl-4-nitro-1H-pyrazole (2c)
Following general procedure A, 2 (63.6 mg, 0.500 mmol, 1.0 equiv), meta-methoxyphenyl tosylate (153.1 mg, 0.55 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 80 °C for 16 h. Chromatography on a silica gel column using 80% hexanes/ 20% EtOAc (Rf = 0.20 in 80% hexanes/ 20% EtOAc) yielded product 2c as a yellow solid (104 mg, 89% yield). 1H NMR (CDCl3): δ 8.19 (s, 1H), 7.45 (t, J = 7.9 Hz, 1H), 7.08 (dd, J = 8.4, 2.6 Hz, 1H), 6.95 (dt, J = 7.6, 1.2 Hz, 1H), 6.90 (t, J = 2.1 Hz, 1H), 3.85 (s, 3H), 3.74 (s, 3H). 13C{1H} NMR (CDCl3): δ 159.6, 141.2, 136.2, 132.9, 129.9, 127.6, 121.7, 115.8, 115.2, 55.4, 37.9. The spectroscopic data is consistent with that previously reported in the literature.4b
4-nitro-1-(2-phenylethyl)-5-[3-(trifluoromethyl)phenyl]-1H-pyrazole (3i)
Following general procedure A, 4-nitro-1-(2-phenylethyl)-1H-pyrazole (3) (109 mg, 0.500 mmol, 1.0 equiv), meta-trifluoromethylphenyl tosylate (174 mg, 0.550 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 16 h. Chromatography on a silica gel column using 85/15 hexanes/EtOAc (Rf = 0.21 in 85% hexanes/15% EtOAc) yielded product 3i as a light yellow solid (115 mg, 64% yield). 1H NMR (CDCl3): δ 8.30 (s, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.48 (t, J = 7.8 Hz, 1H), 7.26-7.19 (m, 3H), 7.02 (s, 1H), 6.94 (d, J = 7.8 Hz, 1H), 6.79 (d, J = 6.6 Hz, 2H), 4.13 (t, J = 6.4 Hz, 2H), 3.14 (t, J = 6.5 Hz, 2H). 13C{1H} NMR (CDCl3): δ 140.5, 136.8, 136.7, 133.0, 132.9, 131.1 (J = 33 Hz), 129.1, 128.9, 128.6, 127.2, 127.1, 126.9 (J = 3.6 Hz), 126.4 (J = 3.8 Hz), 123.4 (J = 271 Hz), 51.6, 35.8. The spectroscopic data is consistent with that previously reported in the literature.4b
1-(4-methylphenyl)-5-(naphthalen-2-yl)-4-nitro-1H-pyrazole (4a)
Following general procedure A, 1-(4-methylphenyl)-4-nitro-1H-pyrazole (4) (50.8 mg, 0.250 mmol, 1.0 equiv), 2-naphthyl tosylate (74.6 mg, 0.250 mmol, 1.0 equiv), Pd(OAc)2 (5.6 mg, 0.025 mmol, 0.10 equiv), XPhos (35.8 mg, 0.075 mmol, 0.30 equiv), Cs2CO3 (122 mg, 0.375 mmol, 1.5 equiv), CsOPiv (64.4 mg, 0.275 mmol, 1.1 equiv), and anhydrous toluene (0.5 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 80 °C for 16 h. The reaction was filtered through a 1-inch pad of silica gel eluting with EtOAc (100 mL). The filtrate was concentrated. 1H NMR spectroscopic analysis of the crude product against 1,4-dinitrobenzene as the standard showed >99% yield of 4a.
1-butyl-3,5-bis(naphthalen-2-yl)-4-nitro-1H-pyrazole (1aa)
Following general procedure A, 1 (84.6 mg, 0.500 mmol, 1.0 equiv), 2-naphthyl tosylate (298 mg, 1.00 mmol, 2.0 equiv), Pd(OAc)2 (22.5 mg, 0.10 mmol, 0.20 equiv), XPhos (143 mg, 0.30 mmol, 0.60 equiv), Cs2CO3 (489 mg, 1.50 mmol, 3.0 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 16 h. Chromatography on a silica gel column using 95/5 hexanes/EtOAc (Rf = 0.15 in 95% hexanes/5% ethyl acetate) yielded product 1aa as a light yellow solid (159 mg, 76% yield). mp = 137–140 °C. 1H NMR (CDCl3): δ 8.24 (s, 1H), 8.03 (d, J = 8.4 Hz, 1H), 7.97-7.88 (m, 6H), 7.81 (dd, J = 8.6, 1.7 Hz, 1H), 7.66-7.59 (m, 2H), 7.56-7.50 (m, 3H), 4.06 (t, J = 7.4 Hz, 2H), 1.89-1.81 (m, 2H), 1.29-1.24 (m, 2H), 0.84 (t, J = 7.3 Hz, 3H). 13C NMR (CDCl3): δ 147.3, 142.9, 133.6, 133.5, 133.0, 132.8, 130.7, 129.6, 128.7, 128.6, 128.5, 128.3, 128.1, 127.9, 127.8, 127.7, 127.6, 126.9, 126.6, 126.5, 126.3, 126.2, 124.6, 50.2, 31.9, 19.6, 13.5. IR (neat): 2927, 1553, 1498, 1456, 1418, 1356, 926, 776, 766 cm−1. HRMS [EI+, M+] Calcd for C27H23N3O2: 421.1790; Found: 421.1789.
Product Synthesis and Characterization (Scheme 3)
3-(1-butyl-4-nitro-1H-pyrazol-5-yl)pyridine (1j)
Following general procedure A, 1 (120 mg, 0.710 mmol, 1.0 equiv), 3-pyridyl tosylate (187 mg, 0.750 mmol, 1.05 equiv), Pd(OAc)2 (15.9 mg, 0.071 mmol, 0.10 equiv), XPhos (101 mg, 0.213 mmol, 0.30 equiv), Cs2CO3 (347 mg, 1.065 mmol, 1.5 equiv), CsOPiv (183 mg, 0.781 mmol, 1.1 equiv), and anhydrous xylene (1.42 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 24 h. Chromatography on a silica gel column using 60/40 hexanes/EtOAc (Rf = 0.27 in 60%hexanes/40% EtOAc) yielded product 1j as a yellow oil (119 mg, 68% yield). 1H NMR (CDCl3): δ 8.80 (d, J = 4.9 Hz, 1H), 8.64 (s, 1H), 8.24 (s, 1H), 7.76 (d, J = 8.0 Hz, 1H), 7.52-7.49 (m, 1H), 3.97 (t, J = 7.3 Hz, 2H), 1.81-1.73 (m, 2H), 1.26-1.17 (m, 2H), 0.84 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3): δ 151.3, 149.8, 137.9, 137.6, 136.5, 133.5, 123.5, 123.3, 50.3, 31.8, 19.5, 13.4. IR (neat): 1502, 1477, 1460, 1398, 1322, 830, 761, 711 cm−1. HRMS [TOF, ES+, M+H] Calcd for C12H14N4O2: 247.1195; Found: 247.1195.
6-(1-butyl-4-nitro-1H-pyrazol-5-yl)quinoline (1k)
Following general procedure A, 1 (84.6 mg, 0.500 mmol, 1.0 equiv), 6-quinolyl tosylate (157 mg, 0.525 mmol, 1.05 equiv), Pd(OAc)2 (11.2 mg, 0.05 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous xylene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 80 °C for 24 h. Chromatography on a silica gel column using 50/50 hexanes/EtOAc (Rf = 0.27 in 50%hexanes/50% EtOAc) yielded product 1k as a yellow solid (93 mg, 63% yield). mp = 80–83 °C. 1H NMR (CDCl3): δ 9.06 (dd, J = 4.3, 1.7 Hz, 1H), 8.29-8.23 (m, 3H), 7.89 (d, J = 2.0 Hz, 1H), 7.68 (dd, J = 8.7, 2.0 Hz, 1H), 7.53 (dd, J = 8.3, 4.2 Hz, 1H), 4.00 (t, J = 7.2 Hz, 2H), 1.81-1.73 (m, 2H), 1.25-1.16 (m, 2H), 0.81 (t, J = 7.3 Hz, 3H). 13C NMR (CDCl3): δ 152.0, 148.3, 140.5, 136.4, 133.2, 130.4, 129.9, 129.8, 127.8, 124.9, 122.1, 50.2, 31.7, 19.5, 13.4 two carbon peaks are overlapping. IR (neat): 1556, 1502, 1470, 1401, 1374, 1329, 1310, 915, 861, 843, 825, 813, 776, 769, 759, 598 cm−1. HRMS [TOF, ES+, M+H] Calcd for C16H16N4O2: 297.1352; Found: 297.1357.
2-(1-butyl-4-nitro-1H-pyrazol-5-yl)quinoxaline (1l)
Following general procedure A, 1 (84.6 mg, 0.500 mmol, 1.0 equiv), quinoxalin-2-yl 4-methylbenzene-1-sulfonate (157 mg, 0.525 mmol, 1.05 equiv), Pd(OAc)2 (16.8 mg, 0.075 mmol, 0.15 equiv), XPhos (107 mg, 0.225 mmol, 0.45 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous xylene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 80 °C for 24 h. Chromatography on a silica gel column using 85/15 hexanes/EtOAc (Rf = 0.20 in 85%hexanes/15% EtOAc) yielded product 1l as a yellow solid (92 mg, 62% yield). mp = 43–46 °C. 1H NMR (CDCl3): δ 9.09 (s, 1H), 8.28 (s, 1H), 8.24 (dd, J = 8.0, 1.8 Hz, 1H), 8.17 (dd, J = 7.9, 1.9 Hz, 1H), 7.95-7.87 (m, 2H), 4.24 (t, J = 7.4 Hz, 2H), 1.88-1.80 (m, 2H), 1.30-1.21 (m, 2H), 0.84 (t, J = 7.4 Hz, 3H). 13C NMR (CDCl3): δ 146.2, 142.1, 141.9, 141.6, 136.5, 136.1, 133.8, 131.7, 130.9, 129.55, 129.54, 51.1, 31.9, 19.5, 13.4. IR (neat): 1537, 1508, 1492, 1478, 1461, 1452, 1402, 1388, 1368, 1335, 1315, 1271, 1225, 1197, 1131, 1049, 980, 964, 954, 923, 850, 825, 799, 757, 747, 731, 623, 589 cm−1. HRMS [TOF, ES+, M+H] Calcd for C15H15N5O2: 298.1304; Found: 298.1295.
5-(1-butyl-4-nitro-1H-pyrazol-5-yl)-1-methyl-1H-indole (1m)
Following general procedure A, 1 (42.3 mg, 0.25 mmol, 1.0 equiv), 1-methyl-1H-indol-5-yl 4-methylbenzene-1-sulfonate (83.3 mg, 0.275 mmol, 1.1 equiv), Pd(OAc)2 (5.6 mg, 0.025 mmol, 0.10 equiv), XPhos (35.6 mg, 0.075 mmol, 0.30 equiv), Cs2CO3 (122 mg, 0.375 mmol, 1.5 equiv), CsOPiv (64.4 mg, 0.275 mmol, 1.1 equiv), and anhydrous toluene (0.5 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 16 h. Chromatography on a silica gel column using 80/20 hexanes/EtOAc (Rf = 0.23 in 80%hexanes/20% EtOAc) yielded product 1m as a viscous yellow oil (63.7 mg, 85% yield). 1H NMR (CDCl3): δ 8.23 (s, 1H), 7.62 (s, 1H), 7.46 (d, J = 8.4 Hz, 1H), 7.18 (dd, J = 8.4, 1.7 Hz, 1H), 7.16 (d, J = 3.3 Hz, 1H), 6.57 (d, J = 3.2 Hz, 1H), 3.98 (t, J = 7.3 Hz, 2H), 3.86 (s, 3H), 1.79-1.72 (m, 2H), 1.24-1.15 (m, 2H), 0.81 (t, J = 7.3 Hz, 3H). 13C NMR (CDCl3): δ 142.9, 137.1, 136.4, 132.8, 130.2, 128.3, 122.7, 122.5, 117.2, 109.7, 101.7, 49.9, 33.0, 31.8, 19.6, 13.5. IR (neat): 2958, 1734, 1555, 1502, 1459, 1398, 1372, 1337, 1318, 1285, 1242, 1177, 1045, 828, 803, 761, 724 cm−1. HRMS [EI+, M+] Calcd for C16H18N4O2: 298.1429; Found: 298.1424.
3-{1-[(4-methylphenyl)methyl]-4-nitro-1H-pyrazol-5-yl}pyridine (5j)
Following general procedure A, 1-[(4-methylphenyl)methyl]-4-nitro-1H-pyrazole (5) (109 mg, 0.500 mmol, 1.0 equiv), 3-pyridyl tosylate (131 mg, 0.525 mmol, 1.05 equiv), Pd(OAc)2 (11.2 mg, 0.05 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous xylene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 24 h. Chromatography on a silica gel column using 60/40 hexanes/EtOAc (Rf = 0.18 in 60%hexanes/40% EtOAc) yielded product 5j as a yellow solid (117 mg, 80% yield). 1H NMR (CDCl3): δ 8.77 (d, J = 5.0 Hz, 1H), 8.54 (s, 1H), 8.28 (s, 1H), 7.61 (dt, J = 7.9, 2.0 Hz, 1H), 7.43 (dd, J = 7.8, 4.9 Hz, 1H), 7.09 (d, J = 7.8 Hz, 2H), 6.89 (d, J = 8.0 Hz, 2H), 5.15 (s, 2H), 2.31 (s, 3H). 13C NMR (CDCl3): δ 151.3, 149.9, 138.5, 138.2, 137.6, 136.6, 133.9, 131.7, 129.6, 127.3, 123.3, 123.2, 54.4, 21.1. The spectroscopic data is consistent with that previously reported in the literature.4b
Product Synthesis and Characterization (Scheme 4)
2-methyl-1-[(4-methylphenyl)methyl]-5-(naphthalen-2-yl)-4-nitro-1H-imidazole (6a)
Following general procedure A, 2-methyl-1- [(4-methylphenyl)methyl]-4-nitro-1H-imidazole (6) (116 mg, 0.500 mmol, 1.0 equiv), 2-naphthyl tosylate (167 mg, 0.550 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.05 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (224 mg, 0.690 mmol, 1.4 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 14 h. Chromatography on a silica gel column using 42/55/3 hexanes/EtOAc/acetone (Rf = 0.30 in 42%hexanes/55%EtOAc/3%acetone) yielded product 6a as a yellow solid (165 mg, 93% yield). mp = 128–131 °C. 1H NMR (CDCl3): δ 7.89 (t, J = 8.2 Hz, 2H), 7.77 (s, 1H), 7.76 (d, J = 7.0 Hz, 1H), 7.58-7.50 (m, 2H), 7.37 (dd, J = 8.9, 1.8 Hz, 1H), 7.12 (d, J = 7.9 Hz, 2H), 6.78 (d, J = 7.9 Hz, 2H), 4.97 (s, 2H), 2.41 (s, 3H), 2.33 (s, 3H). 13C NMR (CDCl3): 144.3, 143.3, 137.9, 133.4, 133.0, 132.5, 131.6, 129.9, 129.6, 128.4, 128.2, 127.7, 127.3, 126.8, 126.6, 125.7, 124.4, 47.8, 20.9, 13.7. δ. IR (neat): 1569, 1534, 1506, 1433, 1400, 1384, 1356, 1329, 1310, 1287, 1274, 1236, 1227, 1197, 1184, 1144, 1123, 1110, 1036, 1019, 938, 923, 870, 840, 830, 813, 801, 772, 765, 750, 716, 694, 663, 630, 613, 577, 567, 555 cm−1. HRMS [TOF, ES+, M+H] Calcd for C22H19N3O2: 358.1556; Found: 358.1553.
Procedure without the glove box
Following general procedure F, 6 (116 mg, 0.500 mmol, 1.0 equiv), 2-naphthyl tosylate (167 mg, 0.550 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.05 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (224 mg, 0.690 mmol, 1.4 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 14 h. Chromatography on a silica gel column using 42/55/3 hexanes/EtOAc/acetone (Rf = 0.30 in 42%hexanes/55%EtOAc/3%acetone) yielded product 6a as a yellow solid (164 mg, 92% yield).
5-(3-methoxyphenyl)-2-methyl-1-[(4-methylphenyl)methyl]-4-nitro-1H-imidazole (6c)
Following general procedure A, 6 (116 mg, 0.500 mmol, 1.0 equiv), meta-methoxyphenyl tosylate (150 mg, 0.540 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.05 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (224 mg, 0.690 mmol, 1.4 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 14 h. Chromatography on a silica gel column using 42/55/3 hexanes/EtOAc/acetone (Rf = 0.13 in 42%hexanes/55%EtOAc/3%acetone) yielded product 6c as a light yellow solid (130 mg, 77% yield). 1H NMR (CDCl3): δ 7.34 (t, J = 7.9 Hz, 1H), 7.13 (d, J = 7.9 Hz, 2H), 7.01–6.98 (m, 1H), 6.87 (d, J = 7.3 Hz, 1H), 6.79-6.77 (m, 3H), 4.92 (s, 2H), 3.69 (s, 3H), 2.37 (s, 3H), 2.33 (s, 3H). 13C NMR (CDCl3): 159.4, 144.1, 143.0, 137.9, 132.8, 131.7, 129.7, 129.6, 128.2, 125.6, 122.1, 115.8, 115.2, 55.0, 47.8, 20.9, 13.6.4c
5-(4-methoxyphenyl)-2-methyl-1-[(4-methylphenyl)methyl]-4-nitro-1H-imidazole (6h)
Following general procedure A, 6 (116 mg, 0.500 mmol, 1.0 equiv), para-methoxyphenyl tosylate (209 mg, 0.750 mmol, 1.5 equiv), Pd(OAc)2 (11.2 mg, 0.05 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (224 mg, 0.690 mmol, 1.4 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 14 h. Chromatography on a silica gel column using 42/55/3 hexanes/EtOAc/acetone (Rf = 0.28 in 42%hexanes/55%EtOAc/3%acetone) yielded product 6h as a yellow solid (153 mg, 91% yield). 1H NMR (CDCl3): δ 7.22 (d, J = 8.8 Hz, 2H), 7.13 (d, J = 7.9 Hz, 2H), 6.94 (d, J = 8.8 Hz, 2H), 6.78 (d, J = 7.9 Hz, 2H), 4.92 (s, 2H), 3.83 (s, 3H), 2.35 (s, 3H), 2.33 (s, 3H). 13C NMR (CDCl3): 160.7, 144.0, 143.1, 137.9, 133.1, 131.7, 131.5, 129.7, 125.6, 118.8, 114.1, 55.2, 47.7, 21.0, 13.7.4c
2-methyl-1-[(4-methylphenyl)methyl]-4-nitro-5-[3-(trifluoromethyl)phenyl]-1H- imidazole (6i)
Following general procedure A, 6 (116 mg, 0.500 mmol, 1.0 equiv), meta-trifluoromethylphenyl tosylate (174 mg, 0.550 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.005 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (224 mg, 0.690 mmol, 1.4 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 14 h. Chromatography on a silica gel column using 42/55/3 hexanes/EtOAc/acetone (Rf = 0.30 in 42%hexanes/55%EtOAc/3%acetone) yielded product 6i as a light yellow solid (160 mg, 84% yield). 1H NMR (CDCl3): δ 7.72 (d, J = 7.8 Hz, 1H), 7.55 (t, J = 7.9 Hz, 1H), 7.46 (d, J = 6.2 Hz, 1H), 7.46 (s, 1H), 7.12 (d, J = 7.8 Hz, 2H), 6.72 (d, J = 7.8 Hz, 2H), 4.91 (s, 2H), 2.45 (s, 3H), 2.33 (s, 3H). 13C NMR (CDCl3): 144.7, 143.4, 138.3, 133.4, 131.2, 130.97, 130.96 (J = 33 Hz), 129.7, 129.2, 128.1, 127.1 (J = 3.7 Hz), 126.6 (J = 3.6 Hz), 125.6, 123.4 (J = 271 Hz), 48.1, 20.9, 13.6.4c
2-methyl-1-[(4-methylphenyl)methyl]-4-nitro-5-[4-(trifluoromethyl)phenyl]-1H-imidazole (6n)
Following general procedure A, 6 (116 mg, 0.500 mmol, 1.0 equiv), para-trifluoromethylphenyl tosylate (174 mg, 0.550 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (224 mg, 0.689 mmol, 1.4 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 14 h. Chromatography on a silica gel column using 42/55/3 hexanes/EtOAc/acetone (Rf = 0.28 in 42%hexanes/55%EtOAc/3%acetone) yielded product 6n as a yellow solid (176 mg, 94% yield). mp = 131–134 °C. 1H NMR (CDCl3): δ 7.68 (d, J = 7.7 Hz, 2H), 7.40 (d, J = 8.3 Hz, 2H), 7.13 (d, J = 7.9 Hz, 2H), 6.74 (d, J = 7.8 Hz, 2H), 4.91 (s, 2H), 2.41 (s, 3H), 2.33 (s, 3H). 13C NMR (CDCl3): δ 144.8, 143.3, 138.2, 131.7 (q, J = 33 Hz), 131.17, 131.16, 130.89, 130.88, 130.6, 129.8, 125.5 (m), 123.5 (q, J = 271 Hz), 47.9, 20.9, 13.6. IR (neat): 1513, 1493, 1402, 1385, 1320, 1294, 1258, 1166, 1108, 1066, 1027, 1007, 859, 809, 781, 752, 723, 698, 673, 619 cm−1. HRMS [TOF, ES+, M+H] Calcd for C19H16F3N3O2: 376.1273; Found: 376.1262.
2-{2-methyl-1-[(4-methylphenyl)methyl]-4-nitro-1H-imidazol-5-yl}quinoline (6o)
Following general procedure A, 6 (116 mg, 0.500 mmol, 1.0 equiv), 2-quinolyl tosylate (299 mg, 1.00 mmol, 2.0 equiv), Pd(OAc)2 (11.2 mg, 0.05 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (224 mg, 0.690 mmol, 1.4 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 14 h. Chromatography on a silica gel column using 42/55/3 hexanes/EtOAc/acetone (Rf = 0.28 in 42%hexanes/55%EtOAc/3%acetone) yielded product 6o as a light yellow solid (174 mg, 97% yield). mp = 137 −139 °C. 1H NMR (CDCl3): δ 8.21 (d, J = 8.5 Hz, 1H), 8.07 (d, J = 8.4 Hz, 1H), 7.88 (d, J = 8.3 Hz, 1H), 7.77 (t, J = 7.3 Hz, 1H), 7.65-7.61 (m, 2H), 7.00 (d, J = 7.8 Hz, 2H), 6.78 (d, J = 7.7 Hz, 2H), 5.30 (s, 2H), 2.46 (s, 3H), 2.27 (s, 3H). 13C NMR (CDCl3): δ 147.4, 147.3, 145.1, 143.6, 137.7, 136.3, 131.5, 130.8, 130.0, 129.29, 129.26, 127.7, 127.6, 127.3, 126.3, 123.9, 48.2, 20.8, 13.7. IR (neat): 2921, 1495, 1402, 1389, 1334, 1304, 1292, 868, 842, 831, 815, 808, 792, 773, 768, 757 cm−1. HRMS [EI+, M+] Calcd for C21H18N4O2: 358.1429; Found: 358.1422.
5-(3-methoxyphenyl)-2-methyl-4-nitro-1-(3-phenylpropyl)-1H-imidazole (7c)
Following general procedure A, 2-methyl-4-nitro-1-(3-phenylpropyl)-1H-imidazole (7) (124 mg, 0.506 mmol, 1.0 equiv), meta-methoxyphenyl tosylate (150 mg, 0.540 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.05 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (224 mg, 0.690 mmol, 1.4 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 14 h. Chromatography on a silica gel column using 40/55/5 hexanes/EtOAc/acetone (Rf = 0.29 in 40%hexanes/55%EtOAc/5%acetone) yielded product 7c as a light yellow solid (178 mg, >99% yield). mp = 113–114 °C. 1H NMR (CDCl3): δ 7.39 (t, J = 8.0 Hz, 1H), 7.26-7.17 (m, 3H), 7.04 (dd, J = 8.2, 2.6 Hz, 1H), 6.99-6.97 (m, 2H), 6.89 (d, J = 7.4 Hz, 1H), 6.83 (t, J = 2.2 Hz, 1H), 3.82 (s, 3H), 3.74-3.70 (m, 2H), 2.50 (t, J = 7.4 Hz, 2H), 2.39 (s, 3H), 1.89-1.82 (m, 2H). 13C NMR (CDCl3): δ 159.5, 143.3, 143.0, 139.4, 132.0, 129.9, 128.51, 128.46, 127.9, 126.3, 121.9, 115.37, 115.34, 55.2, 44.0, 32.3, 31.0, 13.3. IR (neat): 1604, 1588, 1566, 1535, 1496, 1447, 1403, 1381, 1329, 1300, 1280, 1261, 1249, 1222, 1044, 1023, 904, 849, 837, 822, 800, 747, 695 cm−1. HRMS [TOF, ES+, M+H] Calcd for C20H21N3O3: 352.1661; Found: 352.1665.
2-methyl-4-nitro-1-(3-phenylpropyl)-5-[3-(trifluoromethyl)phenyl]-1H-imidazole (7i)
Following general procedure A, 7 (124 mg, 0.506 mmol, 1.0 equiv), meta-trifluoromethylphenyl tosylate (174 mg, 0.550 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.05 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (224 mg, 0.690 mmol, 1.4 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 14 h. Chromatography on a silica gel column using 40/55/5 hexanes/EtOAc/acetone (Rf = 0.29 in 40%hexanes/55%EtOAc/5%acetone) yielded product 7i as a cream solid (186 mg, 94% yield). 1H NMR (CDCl3): δ 7.77 (d, J = 7.9 Hz, 1H), 7.60 (t, J = 7.8 Hz, 1H), 7.58 (s, 1H), 7.51 (d, J = 7.6 Hz, 1H), 7.26-7.18 (m, 3H), 6.95 (d, J = 7.3 Hz, 2H), 3.71-3.67 (m, 2H), 2.51 (t, J = 7.2 Hz, 2H), 2.42 (s, 3H), 1.88-1.81 (m, 2H). 13C NMR (CDCl3): 143.9, 143.2, 139.1, 133.4, 131.1 (J = 33 Hz), 130.2, 129.4, 128.5, 128.3, 127.7, 126.6 (J = 4.2 Hz), 126.5 (J = 3.6 Hz), 126.3, 123.4 (J = 271 Hz), 44.0, 32.2, 31.1, 13.2. The spectroscopic data is consistent with that previously reported in the literature.4c
Product Synthesis and Characterization (Scheme 5)
1-butyl-5-(naphthalen-2-yl)-4-nitro-1H-pyrazole (1a)
Following general procedure C, 2-naphthol (79.3 mg, 0.550 mmol, 1.1 equiv), para-toluenesulfonyl chloride (107 mg, 0.560 mmol, 1.12 equiv), Cs2CO3 (269 mg, 0.825 mmol, 1.6 equiv), and anhydrous toluene (2.0 mL) were combined in a 20 mL scintillation vial for step 1. For step 2, the reaction mixture from step 1 was combined with 1 (84.6 mg, 0.500 mmol, 1.0 equiv), Pd(OAc)2 (11.2 mg, 0.05 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.550 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 80 °C for 24 h. Chromatography on a silica gel column using 85/15 hexanes/EtOAc (Rf = 0.34 in 85%hexanes/15%EtOAc) yielded product 1a as a yellow oil (146 mg, 99% yield). The spectroscopic data is consistent with that of the product isolated using 2-naphthyl tosylate.
3-(1-butyl-4-nitro-1H-pyrazol-5-yl)pyridine (1j)
Following general procedure C, 3-hydroxy pyridine (52.3 mg, 0.550 mmol, 1.1 equiv), para-toluenesulfonyl chloride (107 mg, 0.560 mmol, 1.12 equiv), Cs2CO3 (269 mg, 0.8250 mmol, 1.6 equiv), and anhydrous toluene (2.0 mL) were combined in a 20 mL scintillation vial for step 1. For step 2, the reaction mixture from step 1 was combined with 1 (84.6 mg, 0.500 mmol, 1.0 equiv), Pd(OAc)2 (11.2 mg, 0.05 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.550 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 24 h. Chromatography on a silica gel column using 40/60 hexanes/EtOAc (Rf = 0.15 in 40%hexanes/60%EtOAc) yielded product 1j as a yellow oil (94.1 mg, 76% yield). The spectroscopic data is consistent with that of the product isolated using 3-pyridyl tosylate.
2-methyl-1-[(4-methylphenyl)methyl]-5-(naphthalen-2-yl)-4-nitro-1H-imidazole (6a)
Following general procedure C, 2-naphthol (79.3 mg, 0.550 mmol, 1.1 equiv), para-toluenesulfonyl chloride (107 mg, 0.560 mmol, 1.12 equiv), Cs2CO3 (269 mg, 0.8250 mmol, 1.6 equiv), and anhydrous toluene (2.0 mL) were combined in a 20 mL scintillation vial for step 1. For step 2, the reaction mixture from step 1 was combined with 6 (116 mg, 0.500 mmol, 1.0 equiv), Pd(OAc)2 (11.2 mg, 0.05 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.550 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 14 h. Chromatography on a Biotage purification system silica gel column using 45/55 hexanes/EtOAc (Rf = 0.36 in 45%hexanes/55%EtOAc) yielded product 6a as a yellow solid (176 mg, 98% yield). The spectroscopic data is consistent with that of the product isolated using 2-naphthyl tosylate.
Product Synthesis and Characterization (Scheme 6)
1-butyl-5-(naphthalen-2-yl)-4-nitro-1H-pyrazole (1a)
Following general procedure A, 1 (84.6 mg, 0.500 mmol, 1.0 equiv), 2-naphthyl mesylate (124 mg, 0.556 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 80 °C for 16 h. Chromatography on a silica gel column using 95/5 hexanes/EtOAc (Rf = 0.24 in 95% hexanes/5% ethyl acetate) yielded product 1a as a yellow oil (148 mg, >99% yield). The spectroscopic data is consistent with that of the product isolated using 2-naphthyl tosylate.
Procedure outside the glove box
Following general procedure F, 1 (42.3 mg, 0.250 mmol, 1.0 equiv), 2-naphthyl mesylate (61.1 mg, 0.275 mmol, 1.1 equiv), Pd(OAc)2 (5.6 mg, 0.025 mmol, 0.10 equiv), XPhos (35.8 mg, 0.075 mmol, 0.30 equiv), Cs2CO3 (122 mg, 0.375 mmol, 1.5 equiv), CsOPiv (64.4 mg, 0.275 mmol, 1.1 equiv), and anhydrous toluene (0.5 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 80 °C for 16 h. The reaction was filtered through a 1-inch pad of silica gel eluting with EtOAc (100 mL). The filtrate was concentrated. 1H NMR spectroscopic analysis of the crude product against 1,4-dinitrobenzene as the standard showed 30% yield of the desired product and 70% of azole 1.
1-(4-methylphenyl)-5-(naphthalen-2-yl)-4-nitro-1H-pyrazole (4a)
Following general procedure A, 4 (50.8 mg, 0.25 mmol, 1.0 equiv), 2-naphthyl mesylate (61.1 mg, 0.275 mmol, 1.1 equiv), Pd(OAc)2 (5.6 mg, 0.025 mmol, 0.10 equiv), XPhos (35.8 mg, 0.075 mmol, 0.30 equiv), Cs2CO3 (122 mg, 0.375 mmol, 1.5 equiv), CsOPiv (64.4 mg, 0.275 mmol, 1.1 equiv), and anhydrous toluene (0.5 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 80 °C for 16 h. Chromatography on a silica gel column using 95/5 hexanes/EtOAc (Rf = 0.15 in 95% hexanes/5% ethyl acetate) yielded product 4a as a light yellow solid (79.5 mg, 97% yield). mp = 129–134 °C 1H NMR (CDCl3): δ 8.43 (s, 1H), 7.87-7.83 (m, 3H), 7.79 (d, J = 7.8 Hz, 1H), 7.59-7.50 (m, 2H), 7.33 (dd, J = 8.6, 1.7 Hz, 1H), 7.12-7.05 (m, 4H), 2.29 (s, 3H). 13C NMR (CDCl3): δ 140.8, 138.9, 137.4, 136.1, 134.0, 133.5, 132.6, 130.8, 129.7, 128.5, 128.3, 127.8, 127.6, 126.71, 126.65, 125.0, 123.8, 21.0. IR (thin film, CH2Cl2): 2923, 1549, 1512, 1499, 1393, 1318, 821 cm−1. HRMS [EI+, M+] Calcd for C20H15N3O2: 329.1164; Found: 329.1161.
1-butyl-5-(3,4-dimethoxyphenyl)-4-nitro-1H-pyrazole (1p)
Following general procedure A, 1 (84.6 mg, 0.500 mmol, 1.0 equiv), 3,4-dimethoxyphenyl mesylate (128 mg, 0.550 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 16 h. Chromatography on a silica gel column using 85/15 hexanes/EtOAc (Rf = 0.18 in 85% hexanes/15% ethyl acetate) yielded product 1p as a viscous yellow oil (97.0 mg, 64% yield). 1H NMR (CDCl3): δ 8.20 (s, 1H), 7.00 (d, J = 8.2 Hz, 1H), 6.93 (dd, J = 8.2, 2.0 Hz, 1H), 6.85 (d, J = 1.9 Hz, 1H), 3.97 (t, J = 7.2 Hz, 2H), 3.96 (s, 3H), 3.90 (s, 3H), 1.80-1.72 (m, 2H), 1.27-1.18 (m, 2H), 0.84 (t, J = 7.3 Hz, 3H). 13C NMR (CDCl3): δ 150.5, 149.1, 141.2, 136.4, 132.7, 122.5, 118.6, 112.6, 111.1, 56.1, 55.9, 50.0, 31.8, 19.6, 13.5. IR (thin film, CH2Cl2): 2958, 2934, 1508, 1463, 1398, 1320, 1262, 1249, 1218, 1174, 1140, 1023, 857, 826, 759 cm−1. HRMS [TOF, ES+, M+H] Calcd for C15H19N3O4: 306.1454; Found: 306.1454.
1-butyl-4-nitro-5-(3,4,5-trimethoxyphenyl)-1H-pyrazole (1f)
Following general procedure A, 1 (84.6 mg, 0.500 mmol, 1.0 equiv), 3,4,5-trimethoxyphenyl mesylate (144 mg, 0.550 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 16 h. Chromatography on a silica gel column using 80/20 hexanes/EtOAc (Rf = 0.20 in 80% hexanes/20% EtOAc) yielded product 1f as a yellow oil (111 mg, 66% yield). The spectroscopic data is consistent with that of the product isolated using 3,4,5-trimethoxyphenyl tosylate.
1-butyl-4-nitro-5-[4-(trifluoromethyl)phenyl]-1H-pyrazole (1n)
Following general procedure B, 1 (84.6 mg, 0.500 mmol, 1.0 equiv), para-trifluoromethylphenyl mesylate (132 mg, 0.550 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 16 h. Chromatography on a silica gel column using 90/10 hexanes/EtOAc (Rf = 0.18 in 90% hexanes/10% EtOAc) yielded product 1n as a yellow oil (80 mg, 51% yield). 1H NMR (CDCl3): δ 8.23 (s, 1H), 7.81 (d, J = 8.1 Hz, 2H), 7.52 (d, J = 8.1 Hz, 2H), 3.94 (t, J = 7.2 Hz, 2H), 1.79-1.72 (m, 2H), 1.27-1.16 (m, 2H), 0.84 (t, J = 7.3 Hz, 3H). 13C NMR (CDCl3): δ 139.7, 136.4, 133.1, 132.3 (q, J = 33 Hz), 130.5, 130.3, 125.8 (q, J = 3.7 Hz), 123.6 (q, J = 271 Hz), 50.3, 31.8, 19.5, 13.4. IR (neat): 2962, 1567, 1514, 1498, 1461, 1400, 1320, 1166, 1125, 1109, 1066, 1025, 847, 826, 762 cm−1. HRMS [TOF, ES+, M+H] Calcd for C14H14F3N3O2: 314.1116; Found: 314.1115.
2-methyl-1-[(4-methylphenyl)methyl]-5-(naphthalen-2-yl)-4-nitro-1H-imidazole (6a)
Following general procedure A, 6 (116 mg, 0.500 mmol, 1.0 equiv), 2-naphthyl mesylate (122 mg, 0.550 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (224 mg, 0.690 mmol, 1.4 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 14 h. Chromatography on a silica gel column using 42/55/3 hexanes/EtOAc/acetone (Rf = 0.28 in 42%hexanes/55%EtOAc/3%acetone) yielded product 6a as a light yellow solid (176 mg, 98% yield). The spectroscopic data is consistent with that of the product isolated using 2-naphthyl tosylate.
5-(3-methoxyphenyl)-2-methyl-1-[(4-methylphenyl)methyl]-4-nitro-1H-imidazole (6c)
Following general procedure B, 6 (116 mg, 0.500 mmol, 1.0 equiv), meta-methoxyphenyl mesylate (111 mg, 0.550 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (224 mg, 0.690 mmol, 1.4 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 14 h. Chromatography on a silica gel column using 42/55/3 hexanes/EtOAc/acetone (Rf = 0.28 in 42%hexanes/55%EtOAc/3%acetone) yielded product 6c as a light yellow solid (153 mg, 91% yield). The spectroscopic data is consistent with that of the product isolated using 3-methoxy phenyl tosylate.
Product Synthesis and Characterization (Scheme 7)
5-benzyl-1-methyl-4-nitro-1H-pyrazole (2q)
Following general procedure D, 2 (63.6 mg, 0.500 mmol, 1.0 equiv), benzyl acetate (82.6 mg, 0.550 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), and anhydrous xylene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 80 °C for 16 h. Chromatography on a silica gel column using 55/45 hexanes/EtOAc (Rf = 0.45 in 55% hexanes/45% EtOAc) yielded product 2q as a light yellow solid (106 mg, 97% yield). 1H NMR (CDCl3): δ 8.13 (s, 1H), 7.34-7.24 (m, 3H), 7.13 (d, J = 7.5 Hz, 2H), 4.49 (s, 2H), 3.77 (s, 3H). 13C NMR (CDCl3): δ 140.7, 136.1, 134.8, 133.1, 129.0, 128.0, 127.3, 37.6, 29.9. The spectroscopic data is consistent with that previously reported in the literature.9
Procedure outside the glove box
Following general procedure G, 2 (63.6 mg, 0.500 mmol, 1.0 equiv), benzyl acetate (82.6 mg, 0.550 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 80 °C for 16 h. Chromatography on a silica gel column using 55/45 hexanes/EtOAc (Rf = 0.45 in 55% hexanes/45% EtOAc) yielded product 2q as a light yellow solid (77.1 mg, 71% yield).
1-methyl-5-[(4-methylphenyl)methyl]-4-nitro-1H-pyrazole (2r)
Following general procedure D, 2 (63.6 mg, 0.500 mmol, 1.0 equiv), para-methylbenzyl acetate (90.3 mg, 0.550 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), and anhydrous xylene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 80 °C for 16 h. Chromatography on a silica gel column using 80/20 hexanes/EtOAc (Rf = 0.22 in 80% hexanes/20% EtOAc) yielded product 2r as a yellow solid (86.1 mg, 74% yield). 1H NMR (CDCl3): δ 8.12 (s, 1H), 7.11 (d, J = 7.9 Hz, 2H), 7.02 (d, J = 7.9 Hz, 2H), 4.44 (s, 2 H), 3.77 (s, 3H), 2.32 (s, 3H). 13C NMR (CDCl3): δ 140.9, 136.9, 136.1, 133.0, 131.6, 129.6, 127.9, 37.5, 29.5, 20.9. The spectroscopic data is consistent with that previously reported in the literature.9
5-[(3-fluorophenyl)methyl]-1-methyl-4-nitro-1H-pyrazole (2s)
Following general procedure D, 2 (63.6 mg, 0.500 mmol, 1.0 equiv), meta-fluorobenzyl acetate (92.5 mg, 0.550 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), and anhydrous xylene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 80 °C for 16 h. Chromatography on a silica gel column using 75/25 hexanes/EtOAc (Rf = 0.20 in 75% hexanes/25% EtOAc) yielded product 2s as a light yellow solid (84.8 mg, 72% yield). 1H NMR (CDCl3): δ 8.14 (s, 1H), 7.32-7.26 (m, 1H), 6.97 (t, J = 8.5 Hz, 1H), 6.91 (d, J = 7.8 Hz, 1H), 6.84 (d, J = 9.3 Hz, 1H), 4.49 (s, 2H), 3.79 (s, 3H). 13C NMR (CDCl3): δ 163.0 (J = 246 Hz), 139.9, 137.1 (J = 7.4 Hz), 136.2, 133.2, 130.5 (J = 8.1 Hz), 123.7 (J = 2.9 Hz), 115.1 (J = 22 Hz), 114.3 (J = 21 Hz), 37.6, 29.6 (J = 2.1 Hz). The spectroscopic data is consistent with that previously reported in the literature.9
5-[(3-methoxyphenyl)methyl]-1-methyl-4-nitro-1H-pyrazole (2t)
Following general procedure D, 2 (63.6 mg, 0.500 mmol, 1.0 equiv), meta-methoxybenzyl acetate (99.1 mg, 0.550 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), and anhydrous xylene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 80 °C for 16 h. Chromatography on a silica gel column using 60/40 hexanes/EtOAc (Rf = 0.35 in 60% hexanes/40% EtOAc) yielded product 2t as a yellow viscous oil (95.7 mg, 78% yield). 1H NMR (CDCl3): δ 8.12 (s, 1H), 7.23 (t, J = 7.9 Hz, 1H), 6.79 (dd, J = 8.0, 2.5 Hz, 1H), 6.70 (d, J = 7.7 Hz, 1H), 6.67 (t, J = 2.2 Hz, 1H), 4.46 (s, 2H), 3.77 (s, 6H). The spectroscopic data is consistent with that previously reported in the literature.9
5-benzyl-1-(4-methylphenyl)-4-nitro-1H-pyrazole (4q)
Following general procedure D, 4 (50.8 mg, 0.25 mmol, 1.0 equiv), benzyl acetate (41.3 mg, 0.275 mmol, 1.1 equiv), Pd(OAc)2 (5.6 mg, 0.025 mmol, 0.10 equiv), XPhos (35.8 mg, 0.075 mmol, 0.30 equiv), Cs2CO3 (122 mg, 0.375 mmol, 1.5 equiv), CsOPiv (64.4 mg, 0.275 mmol, 1.1 equiv), and anhydrous toluene (0.5 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 80 °C for 16 h. Chromatography on a silica gel column using 90/10 hexanes/EtOAc (Rf = 0.26 in 90% hexanes/10% EtOAc) yielded product 4q as a clear viscous oil (39.1 mg, 53% yield). 1H NMR (CDCl3): δ 8.31 (s, 1H), 7.27-7.15 (m, 7H), 6.96 (dd, J = 7.6, 2.1 Hz, 2H), 4.42 (s, 2H), 2.42 (s, 3H). 13C NMR (CDCl3): δ 141.5, 140.1, 137.1, 135.8, 135.5, 133.8, 130.0, 128.7, 128.0, 127.0, 125.7, 30.6, 21.2. IR (neat): 2922, 1544, 1504, 1494, 1463, 1401, 1318, 1178, 829, 821, 804, 778, 757, 723, 712, 694 cm−1. HRMS [EI+, M+] Calcd for C17H15N3O2: 293.1164; Found: 293.1162.
1-benzyl-5-[(3-methoxyphenyl)methyl]-2-methyl-4-nitro-1H-imidazole (6t)
Following general procedure D, 6 (115.6 mg, 0.500 mmol, 1.0 equiv), meta-methoxybenzyl acetate (99.1 mg, 0.550 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 14 h. Chromatography on a silica gel column using 12/30/58 acetone/CH2Cl2/hexanes (Rf = 0.36 in 12% acetone/30% CH2Cl2/58% hexanes) yielded product 6t as a viscous oil (88.0 mg, 50% yield). 1H NMR (CDCl3): δ 7.19 (t, J = 7.9 Hz, 1H), 7.14 (d, J = 7.9 Hz, 2H), 6.77 (d, J = 8.4 Hz, 1H), 6.76 (d, J = 8.0 Hz, 2H), 6.68 (d, J = 7.8 Hz, 1H), 6.64 (t, J = 2.1 Hz, 1H), 4.93 (s, 2H), 4.34 (s, 2H), 3.75 (s, 3H), 2.35 (s, 3H), 2.34 (s, 3H). 13C NMR (CDCl3): δ 160.0, 144.2, 144.0, 138.2, 137.2, 132.1, 130.8, 129.92, 129.89, 125.5, 120.3, 114.0, 112.2, 55.2, 47.6, 30.0, 21.0, 13.5. IR (neat): 1599, 1584, 1567, 1540, 1488, 1455, 1435, 1406, 1386, 1338, 1281, 1257, 1161, 1145, 1043, 858, 792, 764, 740, 690 cm−1. HRMS [TOF, ES+, M+H] Calcd for C20H21N3O3: 352.1661; Found: 352.1657.
Product Synthesis and Characterization (Scheme 8)
3-(1-butyl-4-nitro-1H-pyrazol-5-yl)phenyl 4-methylbenzene-1-sulfonate (1u)
Following general procedure A, 1 (42.3 mg, 0.25 mmol, 1.0 equiv), 3-chloro phenyl tosylate (106 mg, 0.375 mmol, 1.5 equiv), Pd(OAc)2 (2.80 mg, 0.012 mmol, 0.05 equiv), XPhos (11.8 mg, 0.025 mmol, 0.10 equiv), K2CO3 (51.8 mg, 0.375 mmol, 1.5 equiv), and anhydrous toluene (0.25 mL) were combined in a 4 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 24 h. Chromatography on a silica gel column using 80/20 hexanes/EtOAc (Rf = 0.28 in 80% hexanes/20% EtOAc) yielded product 1u as a yellow oil (81.3 mg, 78% yield). 1H NMR (CDCl3): 8.18 (s, 1H), 7.72 (d, J = 8.3 Hz, 2H), 7.48 (t, J = 8.0 Hz, 1H), 7.32 (d, J = 8.0 Hz, 2H), 7.27-7.25 (m, 1H), 7.21 (ddd, J = 8.3, 2.4, 1.1 Hz, 1H), 7.03 (t, J = 1.7 Hz, 1H), 3.88 (t, J = 7.2 Hz, 2H), 2.44 (s, 3H), 1.74-1.67 (m, 2H), 1.23-1.15 (m, 2H), 0.83 (t, J = 7.3 Hz, 3H). 13C NMR (CDCl3): δ 149.6, 145.8, 139.4, 136.1, 132.9, 131.7, 130.2, 129.9, 128.43, 128.38, 128.2, 124.3, 123.8, 50.1, 31.6, 21.6, 19.4, 13.3. IR (neat): 1557, 1505, 1481, 1463, 1400, 1374, 1324, 1257, 1192, 1180, 1147, 1120, 1091, 890, 828, 814, 804, 763, 748, 732, 689, 661 cm−1. HRMS [EI+, M+] Calcd for C20H21N3O5S: 415.1202; Found: 415.1206.
3-(1-butyl-4-nitro-1H-pyrazol-5-yl)phenyl 4-methylbenzene-1-sulfonate (1u)
Following general procedure A, 1 Following general procedure A, 1 (42.3 mg, 0.25 mmol, 1.0 equiv), 3-bromo phenyl tosylate (123 mg, 0.375 mmol, 1.5 equiv), Pd(OAc)2 (2.80 mg, 0.012 mmol, 0.05 equiv), XPhos (11.8 mg, 0.025 mmol, 0.10 equiv), K2CO3 (51.8 mg, 0.375 mmol, 1.5 equiv), and anhydrous toluene (0.25 mL) were combined in a 4 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 24 h. Chromatography on a silica gel column using 80/20 hexanes/EtOAc (Rf = 0.28 in 80% hexanes/20% EtOAc) yielded product 1u as a yellow oil (92.3 mg, 89% yield). The spectroscopic data is identical to that of the product obtained using 3-chlorophenyl tosylate.
1-butyl-5-[3-(1-butyl-4-nitro-1H-pyrazol-5-yl)phenyl]-4-nitro-1H-pyrazole (1v)
Following general procedure A, 1 (169 mg, 1.00 mmol, 2.0 equiv), 3-chloro phenyl tosylate (141 mg, 0.500 mmol, 1.0 equiv), Pd(OAc)2 (5.6 mg, 0.002 mmol, 0.05 equiv), XPhos (23.5 mg, 0.05 mmol, 0.10 equiv), K2CO3 (415 mg, 3.00 mmol, 6.0 equiv), and anhydrous toluene (0.5 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 24 h. Chromatography on a silica gel column using 80/20 hexanes/EtOAc (Rf = 0.25 in 80% hexanes/20% EtOAc) yielded product 1v as a yellow oil (165 mg, 80% yield). 1H NMR (CDCl3): 8.23 (s, 2H), 7.73 (t, J = 7.8 Hz, 1H), 7.56 (dd, J = 7.7, 1.7 Hz, 2H), 7.45 (t, J = 1.8 Hz, 1H), 4.05 (t, J = 7.3 Hz, 4H), 1.78-1.72 (m, 4H), 1.28-1.18 (m, 4H), 0.83 (t, J = 7.3 Hz, 6H). 13C NMR (CDCl3): δ 139.8, 136.3, 133.0, 131.6, 131.3, 129.4, 127.6, 50.4, 31.7, 19.5, 13.4. IR (neat): 1551, 1502, 1480, 1464, 1398, 1321, 1267, 1190, 910, 831, 807, 761, 730, 704 cm−1. HRMS [EI+, M+] Calcd for C20H24N6O4: 412.1859; Found: 412.1860.
1-butyl-5-[3-(1-butyl-4-nitro-1H-pyrazol-5-yl)phenyl]-4-nitro-1H-pyrazole (1v)
Following general procedure A, 1 (169 mg, 1.00 mmol, 2.0 equiv), 3-bromo phenyl tosylate (164 mg, 0.500 mmol, 1.0 equiv), Pd(OAc)2 (5.6 mg, 0.002 mmol, 0.05 equiv), XPhos (23.5 mg, 0.05 mmol, 0.10 equiv), K2CO3 (415 mg, 3.00 mmol, 6.0 equiv), and anhydrous toluene (0.5 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 120 °C for 24 h. Chromatography on a silica gel column using 80/20 hexanes/EtOAc (Rf = 0.25 in 80% hexanes/20% EtOAc) yielded product 1v as a yellow oil (152 mg, 74% yield). The spectroscopic data is identical to that of the product obtained using 3-chlorophenyl tosylate.
Product Synthesis and Characterization (Scheme 9)
[3-(1-methyl-4-nitro-1H-pyrazol-5-yl)phenyl]methyl acetate (2w)
Following general procedure D, 2 (63.6 mg, 0.50 mmol, 1.0 equiv), 3-chloro benzyl acetate (101 mg, 0.550 mmol, 1.1 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 1.5 equiv), and anhydrous xylene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 80 °C for 24 h. Chromatography on a silica gel column using 65/35 hexanes/EtOAc (Rf = 0.25 in 65% hexanes/35% EtOAc) yielded product 2w as a white solid (114 mg, 82% yield). mp = 102–105 °C. 1H NMR (CDCl3): 8.20 (s, 1H), 7.55-7.54 (m, 2H), 7.40 (s, 1H), 7.37-7.35 (m, 1H), 5.18 (s, 2H), 3.75 (s, 3H), 2.13 (s, 3H). 13C NMR (CDCl3): δ 170.6, 140.9, 136.9, 136.3, 132.9, 130.0, 129.4, 129.3, 129.0, 126.8, 65.4, 38.0, 20.9. IR (neat): 1732, 1499, 1469, 1406, 1381, 1360, 1328, 1288, 1195, 1178, 1029, 893, 825, 800, 762, 682, 644, 621, 605, 582 cm−1. HRMS [TOF, ES+, M+H] Calcd for C13H14N3O4: 276.0984; Found: 276.0989.
1-methyl-5-{3-[(1-methyl-4-nitro-1H-pyrazol-5-yl)methyl]phenyl}-4-nitro-1H-pyrazole (2x)
Following general procedure D, 2 (63.6 mg, 0.50 mmol, 2.0 equiv), 3-chloro benzyl acetate (46.2 mg, 0.25 mmol, 1.0 equiv), Pd(OAc)2 (5.6 mg, 0.025 mmol, 0.10 equiv), XPhos (35.3 mg, 0.075 mmol, 0.30 equiv), Cs2CO3 (244 mg, 0.750 mmol, 3.0 equiv), and anhydrous xylene (0.5 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 80 °C for 24 h. Chromatography on a silica gel column using 100% diethyl ether (Rf = 0.30 in 100% diethyl ether) yielded product 2x as an off white solid (60.0 mg, 70% yield). mp = 70–72 °C. 1H NMR (CDCl3): 8.19 (s, 1H), 8.14 (s, 1H), 7.51 (t, J = 7.8 Hz, 1H), 7.34-7.29 (m, 2H), 7.21-7.20 (m, 1H), 4.57 (s, 2H), 3.84 (s, 3H), 3.73 (s, 3H). 13C NMR (CDCl3): δ 140.6, 139.8, 136.3, 136.2, 135.7, 133.2, 132.9, 130.0, 129.7, 129.5, 128.5, 127.2, 38.1, 37.7, 29.8. IR (neat): 1552, 1497, 1432, 1404, 1317, 1193, 870, 832, 786, 772, 764, 752 cm−1. HRMS [EI+, M+] Calcd for C15H14N6O4: 342.1077; Found: 342.1075.
Product Synthesis and Characterization (Scheme 10)
2-(2-nitrophenyl)naphthalene (8a)
Following general procedure E, nitrobenzene (616 mg, 5.00 mmol, 10 equiv), 2-naphthyl tosylate (149 mg, 0.500 mmol, 1.0 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), K3PO4 (159 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous xylene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 140 °C for 24 h. Chromatography on a silica gel column using 97/3 hexanes/EtOAc (Rf = 0.12 in 97% hexanes/3% EtOAc) yielded product 8a as a yellow solid (78 mg, 63% yield). 1H NMR (CDCl3): 7.94-7.85 (m, 4H), 7.82 (s, 1H), 7.66 (td, J = 7.6, 1.4 Hz, 1H), 7.56-7.50 (m, 4H), 7.41 (dd, J = 8.5, 1.9 Hz, 1H). 13C NMR (CDCl3): δ 149.3, 136.4, 134.9, 133.2, 132.8, 132.4, 132.3, 128.3, 128.2, 128.1, 127.7, 126.9, 126.5, 125.7, 124.2. The spectroscopic data is consistent with that previously reported in the literature.23
2-methoxy-7-(2-nitrophenyl)naphthalene (8d)
Following general procedure E, nitrobenzene (616 mg, 5.00 mmol, 10 equiv), 7-methoxynaphthyl tosylate (164 mg, 0.500 mmol, 1.0 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), K3PO4 (159 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous xylene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 140 °C for 24 h. Chromatography on a silica gel column using 93/7 hexanes/EtOAc (Rf = 0.31 in 93% hexanes/7% EtOAc) yielded product 8d as a yellow oil (80 mg, 58% yield). 1H NMR (CDCl3): 7.90 (dd, J = 8.1, 1.3 Hz, 1H), 7.81 (d, J = 8.4 Hz, 1H), 7.77 (d, J = 9.0 Hz, 1H), 7.70 (d, J = 1.7 Hz, 1H), 7.65 (td, J = 7.6, 1.4 Hz, 1H), 7.55-7.49 (m, 2H), 7.27 (dd, J = 8.4, 1.8 Hz, 1H), 7.19 (dd, J = 8.9, 2.5 Hz, 1H), 7.15 (d, J = 2.6 Hz, 1H), 3.93 (s, 3H). 13C{1H} NMR (CDCl3): δ 158.1, 149.3, 136.4, 135.4, 134.4, 132.3, 132.1, 129.2, 128.3, 128.09, 128.06, 125.8, 124.1, 123.4, 119.4, 106.0, 55.3. HRMS [TOF, ES+, M+H] Calcd for C17H13NO3 280.0974; Found: 280.0971. IR (thin film, CH2Cl2): 3002, 1630, 1610, 1519, 1460, 1391, 1350, 1237, 1212, 1174, 1125, 1024, 838, 742 cm−1.
1,2-dimethoxy-4-(2-nitrophenyl)benzene (8p)
Following general procedure E, nitrobenzene (616 mg, 5.00 mmol, 10 equiv), 3,4-dimethoxyphenyl tosylate (154 mg, 0.500 mmol, 1.0 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), K3PO4 (159 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous xylene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 140 °C for 24 h. Chromatography on a silica gel column using 88/12 hexanes/EtOAc (Rf = 0.24 in 88% hexanes/12% EtOAc) yielded product 8p as a yellow solid (64 mg, 49% yield). 1H NMR (CDCl3): δ 7.80 (dd, J = 8.3, 1.4 Hz, 1H), 7.60 (td, J = 7.6, 1.3 Hz, 1H), 7.48-7.43 (m, 2H), 6.93-6.87 (m, 2H), 6.83 (d, J = 1.9 Hz, 1H), 3.92 (s, 3H), 3.88 (s, 3H). 13C{1H} NMR (CDCl3): δ 149.5, 149.1, 149.0, 135.8, 132.0, 131.8, 129.7, 127.8, 123.8, 120.3, 111.2, 111.1, 55.90, 55.86. The spectroscopic data is consistent with that previously reported in the literature.24
1,2,3-trimethoxy-5-(2-nitrophenyl)benzene (8f)
Following general procedure E, nitrobenzene (616 mg, 5.00 mmol, 10 equiv), 3,4,5-trimethoxyphenyl tosylate (169 mg, 0.500 mmol, 1.0 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), K3PO4 (159 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous toluene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 140 °C for 24 h. Chromatography on a silica gel column using 80/20 hexanes/EtOAc (Rf = 0.30 in 80% hexanes/20% EtOAc) yielded product 8f as a yellow solid (63 mg, 44% yield). 1H NMR (CDCl3): δ 7.80 (d, J = 8.0 Hz, 1H), 7.61 (t, J = 7.6 Hz, 1H), 7.50-7.45 (m, 2H), 6.52 (s, 2H), 3.89 (s, 3H), 3.86 (s, 6H). 13C NMR (CDCl3): δ 153.3, 149.4, 137.9, 135.9, 132.6, 132.0, 131.6, 128.1, 123.7, 105.0, 60.8, 56.1. HRMS [TOF, ES+, M+H] Calcd for C15H15NO5 290.1028; Found: 290.1030.
5-(2-nitrophenyl)-2H-1,3-benzodioxole (8e)
Following general procedure E, nitrobenzene (616 mg, 5.00 mmol, 10 equiv), sesimol tosylate (146 mg, 0.500 mmol, 1.0 equiv), Pd(OAc)2 (11.2 mg, 0.050 mmol, 0.10 equiv), XPhos (71.5 mg, 0.150 mmol, 0.30 equiv), K3PO4 (159 mg, 0.750 mmol, 1.5 equiv), CsOPiv (129 mg, 0.550 mmol, 1.1 equiv), and anhydrous xylene (1.0 mL) were combined in a 20 mL scintillation vial. The reaction mixture was allowed to stir at 140 °C for 24 h. Chromatography on a silica gel column using 97/3 hexanes/EtOAc (Rf = 0.26 in 97% hexanes/3% EtOAc) yielded product 8e as a dark orange solid (60.4 mg, 50% yield). 1H NMR (CDCl3): δ 7.80 (dd, J = 8.1, 1.3 Hz, 1H), 7.59 (td, J = 7.6, 1.3 Hz, 1H), 7.45 (td, J = 7.8, 1.4 Hz, 1H), 7.42 (dd, J = 7.8, 1.4 Hz, 1H), 6.86 (d, J = 7.8 Hz, 1H), 6.80-6.76 (m, 2H), 6.02 (s, 2H). The spectroscopic data is consistent with that previously reported in the literature.25
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIGMS, R15107892) and St. Olaf College. The authors also thank Emily Reeves for isolating a few catalysis products.
Footnotes
ASSOCIATED CONTENT
Supporting Information available. NMR spectra of all isolated products and new compounds are included in the supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a) Glasnov TN, Kappe CO. Adv. Synth. Catal. 2010;352:3089–3097. and references therein. [Google Scholar]; b) Ono N, editor. The Nitro Group in Organic Synthesis. Wiley-VCH; New York: 2001. [Google Scholar]; c) Hanan EJ, van Abbema A, Barrett K, Blair WS, Blaney J, Chang C, Eigenbrot C, Flynn S, Gibbons P, Hurley CA, Kenny JR, II, Kulagowski J, Lee L, Magnuson SR, Morris C, Murray J, Pastor RM, Rawson T, Siu M, Ultsch M, Zhou A, Sampath D, Lyssikatos JP. J. Med. Chem. 2012;55:10090. doi: 10.1021/jm3012239. [DOI] [PubMed] [Google Scholar]
- 2.Caron L, Campeau L-C, Fagnou K. Org. Lett. 2008;10:4533–4536. doi: 10.1021/ol801839w. [DOI] [PubMed] [Google Scholar]
- 3.a) Kelly SM, Lipshutz BH. Org. Lett. 2014;16:198–101. doi: 10.1021/ol403079x. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sanz R, Escribano J, Pedrosa MR, Aguado R, Arnaiz FJ. Adv. Synth. Catal. 2007;349:713–718. [Google Scholar]
- 4.a) Jung H, Bae S, Jang H-L, Joo JM. Bull. Korean. Chem. Soc. 2014;35:3009–3014. [Google Scholar]; b) Iaroshenko VO, Gevorgyan A, Davydova O, Villinger A, Langer P. J. Org. Chem. 2014;79:2906–2915. doi: 10.1021/jo4025418. [DOI] [PubMed] [Google Scholar]; c) Iaroshenko VO, Gevorgyan A, Mkrtchyan S, Arakelyan K, Grigoryan T, Yedoyan J, Villinger A, Langer P. J. Org. Chem. 2015;80:2103–2119. doi: 10.1021/jo5025927. [DOI] [PubMed] [Google Scholar]
- 5.References for Nitrobenzene arylation using aryl halides: Wang C, Yu Y-B, Fan S, Zhang X. Org. Lett. 2013;15:5004–5007. doi: 10.1021/ol4023326.Iaroshenko VO, Gevorgyan A, Mkrtchyan S, Grigoryan T, Movsisyan E, Villinger A, Langer P. Chem. Cat. Chem. 2015;7:316–324.
- 6.For representative reviews on transition metal-catalyzed C–H arylation see: Alberico D, Scott ME, Lautens M. Chem. Rev. 2007;107:174–238. doi: 10.1021/cr0509760.Kakiuchi F, Kochi T. Synthesis. 2008:3013–3039.McGlacken GP, Bateman LM. Chem. Soc. Rev. 2009;38:2447–2464. doi: 10.1039/b805701j.Ackermann L, Vicente R, Kapdi AR. Angew. Chem., Int. Ed. 2009;48:9792–9826. doi: 10.1002/anie.200902996.Chiusoli GP, Catellani M, Costa M, Motti E, Della Ca’ N, Maestri G. Coord. Chem. Rev. 2010;254:456–469.Kuhl N, Hopkinson MN, Wencel-Delord J, Glorius F. Angew. Chem., Int. Ed. 2012;51:10236–10254. doi: 10.1002/anie.201203269.Rousseaux S, Liegault B, Fagnou K. In: Modern Tools for The Synthesis of Complex Bioactive Molecules. Cossy J, Arseniyadis S, editors. Wiley-VCH; 2012. pp. 1–32.Rossi R, Bellina F, Lessi M, Manzini C. Adv. Synth. & Catal. 2014;356:17–117.Segawa Y, Maekawa T, Itami K. Angew. Chem. Int. Ed. 2015;54:66–81. doi: 10.1002/anie.201403729.
- 7.For representative reviews on Pd-catalyzed C–H arylation see: Chen X, Engle KM, Wang D-H, Yu J-Q. Angew. Chem., Int. Ed. 2009;48:5094–5115. doi: 10.1002/anie.200806273.Lyons TW, Sanford MS. Chem. Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e.Daugulis O. Top. Curr. Chem. 2010;292:57–84. doi: 10.1007/128_2009_10.Gorelsky SI, Lapointe D, Fagnou K. J. Org. Chem. 2012;77:658–668. doi: 10.1021/jo202342q.
- 8.For representative reviews on Ni-catalyzed arylation see: Yamaguchi J, Muto K, Itami K. Eur. J. Org. Chem. 2013:19–30.Tasker SZ, Standley EA, Jamison TF. Nature. 2014;509:299–309. doi: 10.1038/nature13274.Cai X, Xie B. Arkivoc. 2015:184–211.Khan MS, Haque A, Al-Suti MK, Raithby PR. J. Organomet. Chem. 2015;793:114–133.Johnson SA. Dalton Trans. 2015;44:10905–10913. doi: 10.1039/c5dt00032g.Ananikov VP. ACS Catal. 2015;5:1964–1971.Hyster TK. Catal. Lett. 2015;145:458–467.
- 9.Bae S, Jang H-L, Jung H, Joo JM. J. Org. Chem. 2015;80:690–697. doi: 10.1021/jo5025317. [DOI] [PubMed] [Google Scholar]
- 10.a) Antoft-Finch A, Blackburn T, Snieckus V. J. Am. Chem. Soc. 2009;131:17750–17752. doi: 10.1021/ja907700e. [DOI] [PubMed] [Google Scholar]; b) Yu D-G, Li B-J, Shi Z-J. Acc. Chem. Res. 2010;43:1486–1495. doi: 10.1021/ar100082d. [DOI] [PubMed] [Google Scholar]; c) Rosen BM, Quasdorf KW, Wilson DA, Zhang N, Resmerita A-M, Garg NK, Percec V. Chem. Rev. 2011;111:1346–1416. doi: 10.1021/cr100259t. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Li B-J, Yu D-G, Sun C-L, Shi Z-J. Chem. Eur. J. 2011;17:1728–1759. doi: 10.1002/chem.201002273. [DOI] [PubMed] [Google Scholar]; e) Tobisu M, Chatani N. Top. Organomet. Chem. 2013;44:37–54. [Google Scholar]; f) Tobiso M, Chatani N. Acc. Chem. Res. 2015;48:1717–1726. doi: 10.1021/acs.accounts.5b00051. [DOI] [PubMed] [Google Scholar]; g) Mesganaw T, Garg NK. Org. Process Res. Dev. 2013;17:29–39. [Google Scholar]; h) Zeng H, Qiu Z, Dominguez-Huerta A, Hearne Z, Chen Z, Li C-J. ACS Catalysis. 2017;7:510–519. [Google Scholar]
- 11.a) So CM, Kwong FY. Chem. Soc. Rev. 2011;40:4963–4972. doi: 10.1039/c1cs15114b. [DOI] [PubMed] [Google Scholar]; b) Kozhushkov SI, Potukuchi HK, Ackermann L. Catal. Sci. Technol. 2013;3:562–571. [Google Scholar]
- 12.a) Nervig CS, Waller PJ, Kalyani D. Org. Lett. 2012;14:4838–4841. doi: 10.1021/ol302166n. [DOI] [PubMed] [Google Scholar]; b) Wang J, Ferguson D, Kalyani D. Tetrahedron. 2013;69:5780–5790. [Google Scholar]; c) Ferguson D, Rudolph SR, Kalyani D. ACS Catal. 2014;4:2395–2401. doi: 10.1021/cs500587b. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Steinberg DF, Turk MC, Kalyani D. Tetrahedron. 73:2017. 2196–2209. doi: 10.1016/j.tet.2017.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.For representative reports on Pd-catalyzed direct arylation using tosylates see: Ackermann L, Althammer A, Born R. Angew. Chem. Int. Ed. 2006;45:2619–2622. doi: 10.1002/anie.200504450.Ackermann L, Althammer A, Fenner S. Angew. Chem. Int. Ed. 2009;48:201–204. doi: 10.1002/anie.200804517.Ackermann L, Barfusser S, Pospech J. Org. Lett. 2010;12:724–726. doi: 10.1021/ol9028034.Ackermann L, Fenner S. Chem. Commun. 2011;47:430–432. doi: 10.1039/c0cc02360d.Fan S, Yang J, Zhang X. Org. Lett. 2011;13:4374–4377. doi: 10.1021/ol201706t.Dai F, Gui Q, Liu J, Yang Z, Chen X, Guo R, Tan Z. Chem. Commun. 2013;49:4634–4636. doi: 10.1039/c3cc41066h.
- 14.For representative reports on Pd-catalyzed direct arylation using mesylates see: So CM, Lau CP, Kwong FY. Chem. Eur. J. 2011;17:761–765. doi: 10.1002/chem.201002354.Chang JWW, Chia EY, Chai CLL, Seayad J. Org. Biomol. Chem. 2012;10:2289–2299. doi: 10.1039/c2ob06840k.Lee DS, Choy PY, So CM, Wang J, Lau CP, Kwong FY. RSC Adv. 2012;2:9179–9182.
- 15.The site-selectivity of pyrazole arylations is assigned based on analogy to arylations affording products 11b, 2h, 2c, 3i, and 5j (vide infra, Schemes 2 and 3). The 1H NMR spectra of products 1b, 2h, 2c, 3i, and 5j isolated under reaction conditions depicted in Schemes 2 and 3 is consistent with previously reported spectral data for these products (ref. 4b).
- 16.The mass balance of the reaction is poor at 120 °C.
- 17.Majority of arylations in Scheme 2–4 and 6 were conducted at 120 °C since conversion of azole to products is near complete at this temperature. The azole and the products have very similar retention factors on TLC thereby facilitating purification with higher conversions of the azole to the product. Only trace amounts of diarylation products were observed by GC/MS analysis of the crude reaction mixtures for reactions in Schemes 2 and 3 (vide infra). Mass balance was higher with azoles 2, 3, 5, 6 and 7 with less than quantitative yield being accounted by remaining azole. For reactions using azole 1, near complete conversion of 1 is observed in most cases but mass balance is poor at 120 °C.
- 18.CsOPiv is necessary for higher conversions using very electron rich (e.g., p-methoxyphenyl tosylate) or electron deficient (m-trifluoromethylphenyl and heteroaryl electrophiles).
- 19.The site-selectivity for pyrazole benzylations was established by comparison of spectral data of the isolated products with those previously reported in the literature (ref. 9).
- 20.Use of CsOPiv leads to higher yields of 4q and 6t. The benzylation of 2 using benzyl tosylate affords 2q in lower yield (40%) than obtained using benzyl acetate.
- 21.For an example of C-H arylation for which aryl tosylates react faster than aryl chlorides see reference 13e. This selectivity is opposite to that observed for arylations depicted in Scheme 8.
- 22.Ehara T, Hitomi Y, Konischi K, Masuya K. PCT. Int. Appl. 2006125621. 2006 Nov 30 [Google Scholar]
- 23.Song B, Knauber T, Gooβen LJ. Angew. Chem. Int. Ed. 2013;52:2954–2958. doi: 10.1002/anie.201208025. [DOI] [PubMed] [Google Scholar]
- 24.Gauchot V, Lee A-L. Chem. Commun. 2016;52:10163–10166. doi: 10.1039/c6cc05078f. [DOI] [PubMed] [Google Scholar]
- 25.Creech GS, Kwon O. Chem. Sci. 2013;4:2670–2674. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.