

Abstract
The process of base excision repair (BER) recognizes and repairs small lesions or inappropriate bases on DNA through either a short-patch or long-patch pathway. The enzymes involved in BER have been well-characterized on DNA substrates and, somewhat surprisingly, many of these enzymes, including several DNA glycosylases, AP endonuclease (APE), FEN1 endonuclease, and DNA ligases have been shown to have activity on DNA substrates within nucleosomes. DNA polymerase β (Pol β), however, exhibits drastically reduced or no activity on nucleosomal DNA. Interestingly, acetylation of Pol β, by the acetyltransferase, p300, inhibits its 5′ dRP-lyase activity and presumably pushes repair of DNA substrates through the long patch base excision repair (LP-BER) pathway. In addition to the major enzymes involved in BER, a chromatin architectural factor, HMGB1, was found to directly interact with and enhance the activity of APE1 and FEN1, and thus may aid in altering the structure of the nucleosome to be more accessible to BER factors. In this work, we investigated whether acetylation of Pol β, either alone or in conjunction with HMGB1, facilitates its activity on nucleosome substrates. We find acetylated Pol β exhibits enhanced strand displacement synthesis activity on DNA substrates, but, similar to the unmodified enzyme, has little or no activity on nucleosomes. Preincubation of DNA templates with HMGB1 has little or no stimulatory effect on Pol β and even is inhibitory at higher concentrations. In contrast, pre-incubation of nucleosomes with HMGB1 rescues Pol β gap-filling activity in nucleosomes, suggesting that this factor may help overcome the repressive effects of chromatin.
Keywords: DNA Polymerase beta, Base excision repair, nucleosomes, HMGB1, DNA Polymerase beta acetylation
Graphical abstract
Although the eukaryotic genome is packaged into higher order chromatin structures, DNA remains susceptible to damage by endogenous and exogenous insults, resulting in the incorporation of incorrect bases or bases damaged through oxidative, alkylating, or deaminating agents. These bases are frequently repaired by the base excision repair (BER) pathway, which corrects approximately 10,000 lesions/cell/day and minimally requires just four enzyme activities, including a DNA glycosylase, AP endonuclease, a DNA polymerase, and a DNA ligase(1).
BER initiates upon the recognition of a damaged or misincorporated base by a specific DNA glycosylase that cleaves the base-sugar glycosidic bond(2). This cleavage results in the release of the damaged base and generates an apurinic/apyrimidinic (AP) site in the DNA backbone(3, 4). A DNA glycosylase possessing bifunctional activity is able to cleave the DNA backbone forming an aldehyde residue, which is a substrate for AP endonuclease 3′–5′ exonuclease activity. However, if the DNA glycosylase does not have bifunctional activity, AP endonuclease cleaves the DNA backbone forming a DNA gap containing a 3′-OH and a 5′-deoxyribose phosphate (5′-dRP). BER can also be initiated by recognition of an abasic site by AP endonuclease. Base excision repair can proceed via a short-patch (SP-) pathway in which a DNA polymerase with dRP lyase activity inserts a single base into the DNA gap leaving a nick in the DNA backbone, or through a long-patch base excision repair pathway (LP-BER). In the LP-BER pathway a DNA polymerase inserts 2–13 bases, displacing a ssDNA flap, which is cleaved by flap endonuclease I (FEN1) leaving a nick in the DNA backbone. DNA nicks in both pathways are sealed by a DNA ligase(1, 3).
While both SP- and LP-BER pathways have been fully reconstituted in vitro on naked DNA substrates, the in vivo genomic DNA substrate for BER is chromatin. The basic repeating subunit of chromatin is the nucleosome, which consists of ~147 bp of DNA wrapped around a histone core octamer approximately 1.7 times(5). Due to the interaction of genomic DNA with histones, the activity of DNA-binding factors, including those involved in DNA repair processes such as nucleotide excision repair (NER), are significantly inhibited and therefore, require the activities of ATP-dependent chromatin remodeling complexes to allow for these processes to occur(6–10).
Repair proteins involved in the initial (DNA glycosylases and AP endonuclesases) and final (FEN1 and DNA ligase) steps of BER have been shown to have activity on nucleosomal substrates, yet Pol β activity has been shown by several groups to have significantly reduced or no activity on nucleosome substrates(2). For example, Pol β was found to be unable to fill DNA gaps located about two DNA helical turns from the nucleosome dyad, regardless of the orientation of the gap, while limited gap-filling activity was observed closer to the edge of the nucleosome(11, 12). Pederson and colleagues found a significant increase in the amount of polymerase was required in order to observe gap-filling activity on nucleosome substrates, indicating that histone-DNA interactions could be out-competed by enzyme, with approximately 100-fold more enzyme required for an inward-facing gap compared to an outward-facing gap(13). Rodriguez et al. also found significantly reduced Pol β enzyme activity on both outward- and inward-facing gaps(14). Activity on inward-facing gaps was even further reduced upon formaldehyde crosslinking of DNA to the histone surface, suggesting Pol β requires nucleosome DNA flexibility/mobility to access these sites(14). Interestingly, recent work indicates that lesions within nucleosomes are preferentially repaired via the SP-BER pathway, due to a significant restriction of Pol β’s strand-displacement over gap-filling activity(15).
Interestingly, Pol β was found to directly interact and co-localize with p300, a transcriptional coactivator possessing histone acetyltransferase activity(16). Acetylation of Pol β (acPol β) by p300 significantly reduced the enzyme’s ability to remove the 5′-dRP, attributable to the acetylation of lysine 72, a residue important for 5′-dRP activity, as the main acetylation target of p300(16). It has been suggested acPol β may function to regulate the BER pathway, as the inability to remove the 5′-dRP would force the reaction to proceed via LP-BER and result in incorporation of additional nucleotides and displacement of downstream dsDNA harboring the 5′-dRP.
In addition, evidence suggests the chromatin architectural factor high-mobility group protein B1 (HMGB1) contributes to the efficiency of the BER pathway(17, 18). HMGB1 contains two DNA binding domains (the A and B box domains), which have affinity for binding distorted DNAs in a non-sequence specific manner,(19, 20). HMGB1 can further distort DNA by inducing a bend through the intercalation of three residues into the DNA minor groove, which can enhance transcription by increasing the ability of transcription factors to interact with DNA and the activity of ATP-dependent nucleosome remodeling complexes(20, 21). Moreover, the association of HMGB proteins HMG-D and HMG-Z with nucleosomes has been shown to increase accessibility of DNA at sites near the dyad and near the periphery of the nucleosome core region(22)Wu, 2004 #34]. The ability of HMGB1 to interact with distorted DNA has implicated its activity in several DNA repair pathways, and co-immunoprecipitation studies have shown HMGB1 can directly interact with APE1, FEN1, and Pol β, and localize to sites of DNA damage in HeLa cells. In addition, BER was stimulated in the presence of HMGB1, particularly by enhancing the activities of APE1 (when APE1 is limiting) and FEN1(17). However, whether HMGB1 affects the activity of Pol β on naked DNA or nucleosome substrates is not known.
We asked whether the acetylation of Pol β was able to stimulate gap-filling and strand displacement activity on nucleosome substrates. In addition, due to the ability of HMGB1 to enhance BER activities on DNA substrates, it is possible that HMGB1 alone, or in cooperation with acPol β, would rescue Pol β activity on nucleosome substrates. We find that acetylation of Pol β stimulates gap-filling and strand-displacement synthesis on naked DNA templates, but not nucleosome substrates. However, HMGB1 stimulates both Pol β and acPol β gap-filling activity on nucleosomes, depending on the position of the DNA gap.
Materials and Methods
Construction of Gapped DNA Substrates
Oligonucleotides (Table S1) were designed to generate four 154 bp DNA templates containing the X. borealis 5S nucleosome positioning sequence with 2-nt gaps at positions −11/−12, −5/−6, +49/+50, and +54/+55 with respect to the predicted location of the nucleosome dyad base pair. The oligonucleotide to be extended by Pol β was 32P-radiolabeled with polynucleotide kinase at the 5′ end. Top-strand oligonucleotides were combined in molar ratios with the 154 bp DNA bottom strand in annealing buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 50 mM NaCl), heated to 95 °C for 10 min, then the reaction was allowed to slowly cool to room temperature by turning off the heating block. Annealed templates were gel purified on 6% PAGE containing 0.04% SDS and stored in TE (10 mM Tris, pH 8.0, 1 mM EDTA).
Nucleosome Reconstitution and Purification
Purified gapped DNAs (~0.1 ug) were combined with ~2.5 μg each of histones H2A/H2B and (H3/H4)2 purified from chicken erythrocyte nuclei(23), in addition to 5 μg linearized pBS+ plasmid as carrier DNA in a total volume of 200 μl of buffer containing 10 mM Tris-HCl, pH 8.0, 1 mM EDTA, and 2 M NaCl, and nucleosome reconstitution performed via standard salt dialysis against buffers containing 10 mM Tris, pH 8.0, 1 mM EDTA and 1.2, 1.0, 0.8, 0.6 and 0 M NaCl, with a final overnight dialysis against buffer without NaCl, as described(24). Amounts of individual histone preps required for optimal reconstitution were empirically determined. Nucleosomes were purified away from carrier DNA and excess histones through 7–20% sucrose gradients by ultracentrifugation in a Beckman Coulter Optima L-90 ultracentrifuge using a SW 41 Ti swinging bucket rotor spun at 34k rpm for 18 h at 4 °C. Gradients were fractionated into 600 μL aliquots and samples of each fraction run on a 0.7% agarose 0.5× TBE nucleoprotein gel, ethidium stained, photographed, then the gel was dried and exposed to a phosphorimage screen overnight to identify fractions containing purified nucleosomes free of carrier DNA.
Acetylation of DNA Polymerase β
DNA pol β was a kind gift from Dr. Samuel H. Wilson, National Institute of Environmental Health Sciences and was expressed and purified as described{Beard, 1995 #31}. Pol β was acetylated using the catalytic domain of p300 (purchased from Active Motif, 31205). In vitro acetylation reactions were carried out in 20 μl histone acetyltransferase buffer (50 mM Tris-HCl, pH 8.0, 10% glycerol, 150 mM NaCl, 1 mM DTT, 1 mM PMSF and 10 mM sodium butyrate) by incubating pol β (2 μg, 1 μM), p300 (100 ng, 1 pM) and acetyl CoA (10 μM) in a 1:1:10 molar ratio at 37 °C for 30 min(16). Based on a standard dilution curve of acetlyl-3H coenzyme A (Perkin Elmer Life Sciences) we found our method to acetylate approximately 83±4% of Pol β. A survey of reaction conditions showed these ratios to be optimal for maximal Pol β acetylation (Fig. S1). For autoradiography experiments, 14C acetyl-CoA (Perkin Elmer Life Sciences) was used. Acetylated Pol β was analyzed by separation on a 4–15% SDS-polyacrylamide gel (BioRad). The gels were subjected to Coomassie stain, dried in vacuum, and then subjected to autoradiography (Fig. S1).
DNA Polymerase β Gap-filling and Strand Displacement Synthesis Assay
Gapped DNA or nucleosome substrates (1 fmol) were incubated in 1× Pol β reaction buffer (50 mM Tris-HCl, pH 7.5, 2 mM DTT, 25 μg/mL BSA, 2 mM ATP, 5 mM MgCl2, 1 mM dNTPs, 30 mM NaCl) and 25, 50, 100, or 200 fmol Pol β or acPol β. Reactions were carried out at 37 °C for 10 min, stopped with formamide/loading dye solution, denatured at 95 °C for 5 min, then samples were separated on a 15% denaturing PAGE sequencing gel containing 8 M urea for approximately 1 h at 80 W. Gels were dried using a Bio-Rad Model 583 gel dryer and exposed to a phosphorimage screen overnight and imaged using a Molecular Dynamics Storm 820 molecular imager. The gels are easily capable of resolving 1 nt differences in DNA strand lengths.
HMGB1 Nucleosome Binding
HMGB1 was a kind gift of Dr. W. M. Scovell, Bowling Green State University and was purified as described(25). Incubation of HMGB1 with labeled gapped DNAs did not lead to degradation or extension of templates (results not shown). Serial dilutions of HMGB1 were incubated with 2 fmol 154 bp nucleosomes in 1× binding buffer (5% glycerol, 2× BSA, 0.1 mM EDTA, 10 mM Tris pH 8.0, and 50 mM NaCl) for 30 min at 4 °C in a 20 μL reaction volume. Binding reactions were also carried out with 217 bp ‘601’ nucleosomes(26). Binding reactions were run at 4 °C on a 6% native PAGE containing 0.5× TBE for 3 h at 100 V and phosphorimages were obtained as described above.
Identification of Acetylated Lysine Residues
Five hundred nanograms of purified recombinant Pol β was in vitro acetylated as described above. Twenty microliters of the reaction containing Pol β was precipitated with tricholoroacetic acid at 4°C overnight. 8M urea in 100mM Tris-HCl (pH 8.8) was added to TCA-precipitatant. The sample was further reduced and alkylated with TCEP and iodoacetamide (CAM modified). Endoproteinase Lys-C (0.2ug/ul in water) was added to the sample and incubated at 37°C with shaking overnight, followed by addition of trypsin (0.1ug/ul in water Promega Gold MS Grade). Digested peptides were filtered and injected onto the C18 column. Peptides were eluted with a linear gradient from 3 to 40% acetonitrile (in water with 0.1% FA) developed over 120 min at room temperature at a flow rate of 300 nL/min, and effluent was electro-sprayed into the mass spectrometer. Tandem mass spectra were collected in a data-dependent manner with an LTQ-Orbitrap Velos mass spectrometer running XCalibur 2.2 SP1 using a top-fifteen MS/MS method, a dynamic repeat count of one, and a repeat duration of 30 seconds. Enzyme specificity was set to trypsin, with up to two missed cleavages permitted. High-scoring peptide identifications are those with cross-correlation (Xcorr) values of ≥1.5, delta CN values of ≥0.10, and precursor accuracy measurements within ±3 ppm in at least one injection. A mass accuracy of ±10 ppm was used for precursor ions and a mass accuracy of 0.8 Da was used for product ions. Carboxamidomethyl cysteine was specified as a fixed modification, with oxidized methionine and acetylation of lysine residues allowed for dynamic modifications. Acetylated proteins were classified according to gene ontology (GO) annotations by Uniprot (http://www.ebi.ac.uk/uniprot/).
Results
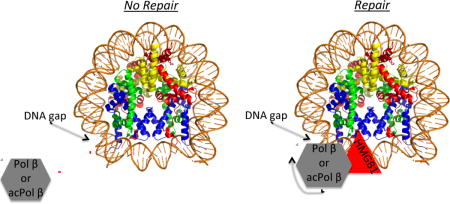
We examined the lysine residues on Pol β that were modified after in vitro acetylation by the p300 histone acetyl transferase (HAT) using mass spectrometry. We found twelve lysine sites to be acetylated on Pol β (K5, K35, K47, K67, K72, K81, K113, K141, K206, K209, K220, K230 (Fig S3, Table S2). Out of these twelve sites, K72 and K81 were identified in previous studies(16, 27, 28). Significantly, out of the 24 peptides recognized to contain an acetylated lysine residues, 16 peptides identified acetylated lysine residues 72 and 81 (Fig S2 and S3; Table S2). Of the previously identified sites, K72 was reported to be the main target for p300 modification and the acetylation of this site impaired its dRP lyase activity(29). In order to determine how acetylation affects the activity of Pol β on nucleosome substrates, DNA templates were prepared by incorporating 2-nt gaps within a 154 bp DNA fragment containing the X. borealis 5S nucleosome positioning element(30). Substrates were assembled such that after reconstitution with core histones, the single 2-nt gaps would be located either V or 1 turn from the nucleosome dyad (at positions −5/−6 or −11/−12, respectively) or near the edge of the nucleosome, 5 or 5 ½ turns from the dyad (at positions +49/+50 or +54/+55, respectively) (Fig. 1). Two positions were chosen in each location so that gaps would either face outward, away from the histone octamer (−11/−12 and +49/+50) or inward, toward the histone octamer (− 5/−6 and +54/+55). In addition, prior to annealing, a single 32P-end label was incorporated onto the 5′-end of the oligonucleotide strand expected to be extended by Pol β.
Fig. 1. Construction of gapped DNA substrates.

Four 154 bp DNA templates were prepared, each containing a 2-nt gap at different positions. Positions were chosen to place gaps at ½, 1, 5, and 5 ½ helical turns from the nucleosome dyad (−5/−6, −11/−12, −49/−50, −54/−55, respectively) and with gaps either facing away (−11/−12, +49/+50) or toward (−5/−6, +54/+55) the histone octamer when the templates are assembled into nucleosomes. Labeled DNA strands extended by Pol β (66-mer, 71-mer, 58-mer, and 63-mer) are indicated by red numbers and the position of the 32P radiolabels are indicated by the red stars. Top strand oligonucleotides were annealed with a 154 bp bottom strand oligonucleotide as described. The positions of the 2-nt gap are indicated by the purple arrows in the linear scheme (left) and the model of a nucleosome core (right)(40).
Once annealed, the naked gapped DNAs were incubated with either Pol β or acetylated Pol β (see Methods) at various concentrations for 10 min at 37 °C with all four dNTPs to allow both gap-filling and strand displacement synthesis. Reactions were stopped with addition of formamide dye and samples were denatured and separated on a 15% denaturing PAGE containing 8 M urea. When present at lower concentrations, Pol β rapidly incorporates 2-nt, filling the gap, and pausing where the downstream strand begins (Fig. 2A, lane 2, red line). At higher concentrations of enzyme, strand displacement synthesis is observed as indicated by the slower migrating bands corresponding to extended DNA products (Fig. 2A, lanes 3–5, red bracket). On average, acetylation of the enzyme caused a stimulation of strand extension activity but had little effect on gap-filling (Fig. 2A and see below). The influence of acetylation varied among the four templates, being least obvious for the −11/−12 template, indicating some variation among the templates. We show detailed analysis for the −5/−6 template (Fig. 2 B and C), but behavior of the other naked DNA substrates was similar. Acetylation of Pol β does not alter the total fraction of templates extended by the enzyme, which increases in an enzyme-dependent manner (Fig. 2B). However, acetylation does increase strand displacement synthesis, as evidenced by the relative decrease in 2-nt extended products, which are more efficiently extended into longer products (Fig. 2A, compare lanes 1–5 vs 6–10). This effect is somewhat dependent on template and ranges from the largest increase in strand-displacement synthesis for the −5/−6 template to smaller effects on the +49/+50 and +54/+55 templates, to little or no effect on the −11/−12 template (Fig. 2A). A quantitative scan analysis shows that the longest strand displacement products appear at a rate ~4-fold greater with the acetylated enzyme compared to the unacetylated Pol β on the −5/−6 template (Fig. 2C).
Fig. 2. Acetylation stimulates Pol β activity on naked DNA templates.

Templates containing 2 nt gaps were incubated with 25, 50, 100 or 200 fmol of Pol β or acetylated Pol β and products analyzed. A. Products of Pol β activity on the indicated templates were run on 15% denaturing PAGE and visualized by phosphorimagery. The unextended template, products of gap-filling (extended by 2 nt), and strand-displacement products are indicated by the black arrow, red arrow and red bracket, respectively. B. Fraction of the −5/−6 template extended by Pol β or acetylated Pol β was quantified with respect to total radioactivity in each lane in Fig. 2A and plotted. Total extension = (total products > template/total lane signal). C. Scans of lanes for the −5/−6 template shown in Fig. 2A, with Pol β and acetylated Pol β represented by blue and black traces, respectively. The fraction of total products extended into longest strand-displacement products (areas above red dotted line) is indicated on the right for each. Horizontal arrow indicates direction of migration along the gel. Vertical axis is arbitrary units signifying band intensity.
We next determined whether the stimulation in activity of acPol β on naked DNA also occurs on nucleosome substrates, which have previously been found to be very poor substrates for Pol β(11–14). The 2-nt gap substrates were reconstituted into nucleosomes and purified via sucrose gradients. The DNA gap-containing nucleosomes were incubated with Pol β and acPol β at the same concentrations used on naked DNA substrates. In agreement with data reported by others, no Pol β gap-filling or strand displacement activity was detected on nucleosomes substrates with gaps at −5/−6, −11/−12, and +49/+50, indicating assembly of these substrates into a nucleosome completely inhibits Pol β activity at the concentration of enzyme used, regardless of the rotational orientation of the gap with respect to the histone surface (Fig. 3A–C). Of note, we found that acetylation of Pol β did not rescue gap-filling or stand displacement synthesis activities on these nucleosome substrates to detectable levels (Fig. 3A–C). We did observe, however, that both Pol β and acPol β show approximately equivalent levels of trace activity on the +54/+55 nucleosome substrates (Fig. 3D), showing that even when the gap is located near the edge of the nucleosome where spontaneous unwrapping of the DNA occurs much more frequently, Pol β activity is not altered by acetylation of the enzyme.
Fig. 3. Acetylation does not enhance activity of DNA Polymerase β on nucleosome substrates.

The ability of Pol β or acetylated Pol β to extend DNA templates assembled into nucleosomes was assessed. Increasing concentrations of Pol β or acPol β polymerase (0, 25, 50, 100, and 200 fmol) were incubated with nucleosomes (1 fmol) containing the −5/−6, −11/−12, +49/+50 or +54/+55 templates (A–D, respectively) and products analyzed as in Fig. 2. Products of Pol β extension (25 fmol) on naked templates is shown in lanes marked “DNA + Pol β.”
Due to the fact that HMGB1 co-immunoprecipitates with Pol β and has been shown to enhance the activity of APE1 and FEN1(18,31), and HMGB proteins have been shown to increase accessibility of DNA sites near the edge of a nucleosome core(32), we asked whether HMGB1 enhanced Pol β and acPol β activity on DNA and nucleosome substrates. The naked gapped DNA substrates (Fig. 1) were incubated with increasing concentrations of HMGB1 for 30 min at 4 °C before addition of Pol β or acPol β for 10 min at 37 °C. As before, reactions were stopped with formamide dye and products analyzed on a 15% denaturing gel. As shown in Fig. 4, the presence of HMGB1 either modestly stimulated or had no effect on the DNA synthesis activity of Pol β on the DNA templates compared to gap-filling reactions that lacked HMGB1. A stimulation of about ~1.4-fold in total extended products was observed for the −5/−6 template while the other templates did not exhibit significant stimulation (Fig. 4A–E). In addition, higher concentrations of HMGB1 were found to be inhibitory to Pol β (Fig. 4 A–E). Previous work suggests that HMGB1 and Pol β compete for the same substrate and that inhibition caused by this competition may be relieved in the presence of FEN1(18). In our experiments HMGB1 caused an increase in strand-displacement synthesis but only on the −5/−6 template (Fig. 4A, compare lanes 2–4; and Fig. 4F). Also, slightly greater amount of gap-filling was observed at the +49/+50 site, but again with inhibition at the higher concentrations of HMGB1 (Fig. 4B). Similar results were obtained with acPol β with some accentuation of extension products over 2-nt gap filling (results not shown).
Fig. 4. HMGB1 modestly stimulates Pol β DNA activity only on the −5/−6 gapped DNA template. A.
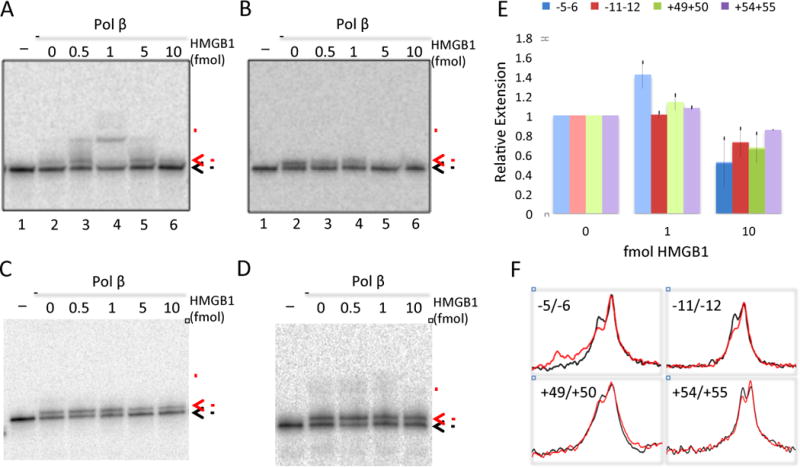
A–D. The −5/−6, −11/−12, +49/+50 or +54/+55 DNA templates were incubated without (Lanes 1) or with 25 fmol of Pol β in the absence (lanes 2) or presence of increasing amounts of HMGB1 (Lanes 3–6). (Note that in D, the 5 fmol HMGB1 lane was omitted). Products were analyzed as in Fig. 1. E. The fraction of templates extended by Pol β was determined by quantification of total extension products/total lane density, normalized to that found in the absence of HMGB1 for each experiment and the results averaged and plotted for 1 and 10 fmol HMGB1. Bars indicate +/− standard error, N= 2–4. F. Scans of the 0 and 0.5 fmol HMGB1 lanes in the gels shown in A–D (black and red traces, respectively), showing the increase in extension products observed only for the −5/−6 template.
As a first step to determining whether HMGB1 enhanced the ability of Pol β to employ DNA templates assembled into nucleosomes, we assessed binding of HMGB1 to nucleosomes reconstituted with the gap templates. Incubation of increasing amounts of HMGB1 with sucrose gradient-purified 154 bp gap-containing nucleosomes resulted in formation of a stable, well-defined complex on the gel, in agreement with previously published data(25) (Fig. 5A, left). HMGB1 bound to gapped nucleosomes with an affinity nearly identical to that observed for (ungapped) control nucleosomes. (Fig 5A, right). The control nucleosomes contained longer linker DNA lengths and bound HMGB1 in an apparent 2:1 stoichiometry(33).
Fig. 5. HMGB1 rescues activity of Pol β on nucleosomes. A.
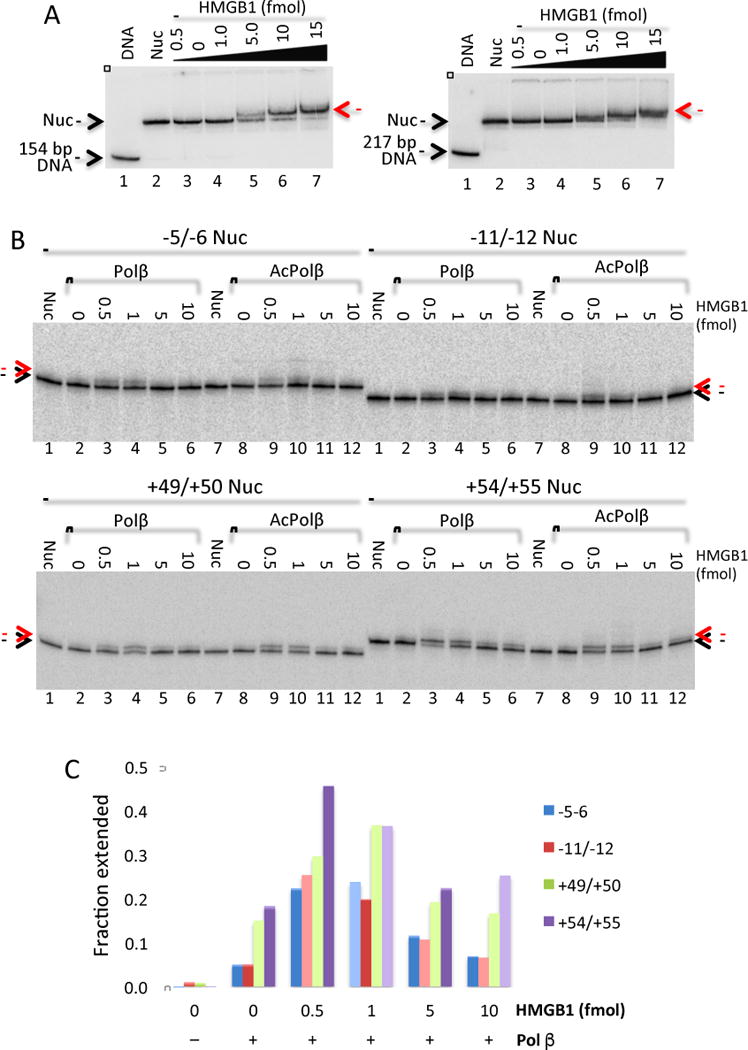
HMGB1 binds gapped nucleosomes with high affinity. Left: Radio-labeled nucleosomes containing the 154 bp −11/−12 gapped template were incubated with increasing amounts of HMGB1 in 1× binding buffer (5% glycerol, 2× BSA, 0.1 mM ETA, 10 mM Tris pH 8.0, and 50 mM NaCl) for 30 min at 4 °C. Binding reactions were run at 4 °C on a 6% native PAGE containing 0.5× TBE for 3 h at 100 V. Right: Binding reactions with nucleosomes containing a 217 bp 601 DNA fragment without gaps. B. HMGB1 partially rescues Pol β and acPol β activity on nucleosomes. Nucleosomes containing the indicated gap templates were incubated with increasing concentrations of HMGB1 (as indicated) for 30 minutes before addition of 50 fmol Pol β (lanes 2–6) or acPol β (8–12) for 10 min. Nucleosome substrates incubated in the absence of polymerase are indicted (Nuc). Extension products were analyzed on a 15% denaturing PAGE sequencing gels and the dried gels visualized by phosphorimagery. The unextended labeled oligos and 2 nt extension products are indicated by black and red arrows, respectively. Note that the size of labeled oligos within each substrate is different (see Fig. 1). C. The fraction of template extended by Pol β and acPol β was quantified, averaged, and plotted. The average standard error for all determinations was ±0.03, N=2.
We next tested whether incubation of nucleosomes with HMGB1 had any effect on Pol β or acPol β polymerase activity. Pol β and acPol β activity was undetectable on nucleosomes in the absence of HMGB1 for the −5/−6, −11/−12 and +49/+50 templates, with a trace level of activity detected on the +54/+55 template, with nearly identical extents of extension observed for Pol β vs. acPol β, consistent with prior results (Fig. 5B, lanes 2 and 7). Of note, addition of increasing amounts of HMGB1 to nucleosomes rescued polymerase activity. The stimulation of activity was, again, nearly identical extent for Pol β and acPol β. The stimulatory effect of HMGB1 was detectable but minimal on −5/−6 and −11/−12 nucleosome substrates, increasing the fraction of template extended from ~5% to ~20% at 0.5 and 1.0 fmol of HMGB1 (Fig. 5C), indicating positions near the dyad still remain largely inhibitory to Pol β and acPol β activity. However, the +49/+50 and +54/+55 nucleosome substrates showed significant stimulation of Pol β and acPol β activity, from ~15 to 30–50% in the presence of this factor, to levels greater than or equal to that observed with DNA substrates at the same concentration of polymerase (Fig. 5C). In addition, all nucleosome substrates show an inhibition in Pol β and acPol β activity at higher HMGB1 concentrations, similar to that observed with naked DNA substrates (Fig. 5C). We note that HMGB1 was able to stimulate gap-filling activity on nucleosomes, but not strand displacement activity.
Discussion
We investigated whether p300 acetylation and HMGB1 affect the activity of Pol β on DNA and nucleosome substrates. While p300 has been shown to acetylate Pol β both in vitro and in vivo(16), it is not currently clear if other acetyltransferases play a redundant role in acetylation of Pol β. Interestingly, when the efficiency of LP-BER was analyzed using a plasmid reporter construct(34) in HCT116 cells lacking p300 and was compared to wild-type HCT116 cells, no significant changes in the efficiency of repair in cells lacking p300 was observed(35). In this work, nucleosomes were reconstituted with DNA templates containing 2-nt gaps such that the gaps were positioned either near the dyad or the edge of the nucleosomes, and either facing inward, toward the histone octamer, or outward, away from the histone octamer. We find that acetylation of Pol β stimulates strand displacement synthesis activity on naked DNA substrates, but is unable to rescue polymerase gap-filling or strand-displacement activity on nucleosomes. However, in contrast to naked DNA templates, we discovered that preincubation of nucleosomes with HMGB1 does rescue Pol β and acPol β gap-filling activity, with the extent of stimulation by HMGB1 dependent on the distance of the gap from the nucleosome dyad as well as the concentration of HMGB1. Interestingly, rescue of Pol β and acPol β activity appears independent of the rotational orientation of the gap with respect to the nucleosome surface.
In particular, we find Pol β gap-filling activity occurs readily on naked DNA templates and at higher concentrations, the enzyme exhibits strand displacement synthesis downstream from the gap (Fig. 2). In addition, acetylated Pol β shows an even greater propensity for strand displacement activity at lower concentrations of enzyme (Fig. 2), possibly due to the inability of the acetylated form of the enzyme to remove the 5′-dRP, thus blocking the short-patch gap filling pathway, and necessarily resulting in the requirement for LP-BER and formation of a ssDNA flap.
Additionally, our data support that nucleosomes are not efficient substrates for Pol β gapfilling or strand displacement synthesis. While Pol β exhibits a complete lack of activity on −5/− 6, −11/−12, and +49/+50 nucleosomes, we do find trace amounts of activity on +54/+55 nucleosomes (Fig. 3). This is likely due to the fact that this position is closest to the edge of the nucleosome, where the transient DNA unwrapping from the nucleosome surface creates partially accessible DNA regions. In addition, the orientation of the gap at position +54/+55 is inwardfacing, suggesting the inward orientation of the gap is more compatible with synthesis compared to the outward-facing gap at +49/+50, in agreement with the results of Smerdon and co-workers(14). The 2-nt gap at position +49/+50 is outward-facing and only 5 bp further into the nucleosome than the gap at +54/+55. However, the complete lack of gap-filling activity observed at +49/+50 suggests the transient exposure of DNA in this region is not sufficient enough to allow even trace levels of Pol β activity. Interestingly, acetylation of Pol β does not overcome inhibition of activity at −5/−6, −11/−12, and +49/+50 nucleosomes, or significantly stimulate activity at the +54/+55 site (Fig. 3), indicating that acetylation does not alter the way in which the enzyme interacts with the nucleosome. Moreover, acetylation of Pol β does not lead to an increase in strand-displacement synthesis with nucleosome templates, consistent with an expected restriction for interaction with the larger segment of DNA required for incorporation of multiple nucleotides.
Due to the fact that HMGB1 was found to interact with enzymes involved in the BER pathway, including Pol β, and also was shown to enhance the activity of APE1 and FEN1 enzymes on DNA substrates(18, 31), it was reasonable to hypothesize that HMGB1 would also enhance the activity of Pol β. However, when DNA substrates were pre-incubated with HMGB1, little or no stimulation of Pol β or acPol β was observed compared to reactions lacking HMGB1. We observed a ~1.4-fold increase in overall extension products only for the −5/−6 template, while there was no stimulation for the other three gapped sites (Fig. 4). Indeed, higher concentrations of HMGB1 were mildly inhibitory to Pol β and acPol β activity perhaps due to binding of multiple HMGB1 proteins to the DNA templates. Interestingly the modest stimulation for the −5/−6 substrate, suggests some sequence specificity to HMGB1 binding the templates. In general, however, the lack of stimulation and strand displacement synthesis at low HMGB1 concentrations may be due to the fact that Pol β can readily interact with both strands of DNA with high affinity and induce a 90° bend such that the gapped or nicked DNA is exposed to the enzyme(36), potentially preventing the interaction of HMGB1 with the gapped DNA region. However, this does not seem to be the case on nucleosome substrates.
We discovered that, in contrast to the effect on naked DNA templates, exposure of nucleosomes to HMGB1 prior to addition of Pol β or acPol β results in a rescue of polymerase activity on these substrates. At gaps positioned near the nucleosome dyad, where previously no activity on nucleosome substrates was observed, HMGB1 was able to induce a low but detectable amount of Pol β and acPol β gap-filling activity, but not strand displacement synthesis activity (Fig. 5B, upper panel, and 5C). The observed gap-filling activity was inhibited at higher HMGB1 concentrations, similar to that observed on naked DNA substrates. At the two sites closer to the nucleosome edge, much greater HMGB1-dependent gap-filling activity, but not strand displacement activity, was observed (Fig. 5B, lower panel, and 5C). Importantly, our results align well with recent results by Meas and Smerdon showing that nucleosomes strongly bias BER toward gap filling synthesis and short-patch repair vs strand-displacement synthesis and long-patch repair(15). We also note that Pol β stimulation was observed at the lowest amount of HMGB1 (0.5 fmol), in the presence of 1 fmol of naked DNA or nucleosome template (Fig. 5B). Moreover, binding experiments indicated that approximately 10 times this amount of HMGB1 was required to observe partial nucleosome binding by gel-shift experiments (Fig. 5A). While further experimentation is required to ascertain actual binding in solution to gapped substrates, these results may imply either preferential binding of HMGB1 to gapped DNAs in nucleosomes or cooperative interactions between HMGB1 and Pol β for binding nucleosome substrates.
HMGB1 may be able to induce Pol β and acPol β activity on nucleosome substrates by stably binding to the nucleosome via interaction of the long, acidic C-terminal tail with linker DNA and pre-bending nucleosomal DNA. Indeed previous work has shown that pre-incubation of nucleosomes with HMGB1 stably alters nucleosome structure in a manner that drastically increases the binding of estrogen receptor to cognate sites within nucleosome DNA(25). The HMGB protein HMG-D has been shown to increase accessibility of DNA in spatially juxtaposed regions near the edge of the nucleosome and in the vicinity of the nucleosome dyad(22, 32). To further address this point we undertook a FRET study of the DNA end-to-end distance in two different nucleosomes in the absence and in the presence of HMGB1 (Fig. S4). If HMGB1 induces a stable structural alteration to the nucleosome, a likely mechanism would be promoting unwrapping of DNA from the nucleosome’s edge. However, we found that HMGB1 did not detectably alter DNA end-to-end distance suggesting that either this protein exposes internal nucleosome sites by a mechanism other than DNA unwrapping or that unwrapping is accompanied by distortions in the linker DNA that compensate for changes in relative end position due to the unwinding. The former possibility is more consistent with nuclease studies of Travers and colleagues, who found that sites near the dyad, including SHL 0 are increased in accessibility by HMGB1, which would be somewhat inconsistent with a simple unwinding mechanism. Alternatively, it is possible that HMGB1 does not stably alter nucleosome structure but induces more facile transition to an ‘open’ state upon incursion of a DNA binding factor. Thus in this latter mechanism HMGBl-stimulation of Pol β gap-filling activity would require an HMGBl-nucleosome-Pol β ternary complex.
While HMGB1 was shown to stimulate the activities of both APE1 and FEN1 on DNA substrates, it has not been determined if this is also true for nucleosome substrates(31). FEN1 has been shown to have efficient enzyme activity on nucleosomes(37, 38), thus HMGB1 may play a particularly important role in enhancing the nucleosome activity of APE1, which shows reduced activity on nucleosomes compared to naked DNA(13, 39). In addition, the ability of HMGB1 to enhance the activities of individual enzymes within the BER pathway may have an overall effect of increasing the total efficiency of the pathway on nucleosome substrates. Additionally, HMGB1 may eliminate or reduce the need to continually recruit ATP-dependent chromatin remodeling complexes during BER, especially for repair of lesions that are located near the edge of the nucleosome. As we have shown, HMGB1 shows only mild rescue of Pol β and acPol β gap-filling activity of gaps positioned near the nucleosome dyad. Therefore, while HMGB1 may enhance BER activity near the edge of the nucleosome, additional factors may still be required for efficient repair of lesions near the nucleosome dyad.
We have also shown that HMGB1 reduces strand displacement synthesis activity induced by acPol β on naked DNA substrates suggesting that it may inhibit the requirement of acPol β to pass through the LP-BER pathway. If this is the case, however, it could be potentially detrimental to the cell as the 5′-dRP moiety is not removed from the 5′-end of the gap and therefore, leaves an unligatable product. Alternatively, HMGB1 may bind to the DNA or nucleosomes in such a manner as to block efficient processive tracking of Pol β and acPol β on DNA and nucleosome substrates, as DNA substrates show reduced strand displacement activity compared to the absence of HMGB1 while nucleosome substrates show no strand displacement activity. It would be interesting to investigate whether increased concentrations of Pol β or acPol β are required for strand displacement synthesis in the presence of HMGB1.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by the National Institutes of Health [GM052426] to J.J.H, and [GM098328] to L.B. AJB was supported by NIH Training Grant [T32GM068411].
Footnotes
References
- 1.Wallace SS. Base excision repair: a critical player in many games. DNA Repair (Amst) 2014;19:14–26. doi: 10.1016/j.dnarep.2014.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balliano AJ, Hayes JJ. Base excision repair in chromatin: Insights from reconstituted systems. DNA repair. 2015;36:77–85. doi: 10.1016/j.dnarep.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Memisoglu A, Samson L. Base excision repair in yeast and mammals. Mutat Res. 2000;451:39–51. doi: 10.1016/s0027-5107(00)00039-7. [DOI] [PubMed] [Google Scholar]
- 4.Stivers JT, Drohat AC. Uracil DNA glycosylase: insights from a master catalyst. Arch Biochem Biophys. 2001;396:1–9. doi: 10.1006/abbi.2001.2605. [DOI] [PubMed] [Google Scholar]
- 5.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 6.Van Holde KE. Chromatin. Springer-Verlag New York, Inc 1989 [Google Scholar]
- 7.Ura K, Hayes JJ. Nucleotide excision repair and chromatin remodeling. Eur J Biochem. 2002;269:2288–2293. doi: 10.1046/j.1432-1033.2002.02888.x. [DOI] [PubMed] [Google Scholar]
- 8.Citterio E, Van Den Boom V, Schnitzler G, Kanaar R, Bonte E, Kingston RE, Hoeijmakers JH, Vermeulen W. ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol Cell Biol. 2000;20:7643–7653. doi: 10.1128/mcb.20.20.7643-7653.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gong F, Fahy D, Smerdon MJ. Rad4-Rad23 interaction with SWI/SNF links ATP-dependent chromatin remodeling with nucleotide excision repair. Nat Struct Mol Biol. 2006;13:902–907. doi: 10.1038/nsmb1152. [DOI] [PubMed] [Google Scholar]
- 10.Gong F, Fahy D, Liu H, Wang W, Smerdon MJ. Role of the mammalian SWI/SNF chromatin remodeling complex in the cellular response to UV damage. Cell Cycle. 2008;7:1067–1074. doi: 10.4161/cc.7.8.5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nilsen H, Lindahl T, Verreault A. DNA base excision repair of uracil residues in reconstituted nucleosome core particles. EMBO J. 2002;21:5943–5952. doi: 10.1093/emboj/cdf581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beard BC, Wilson SH, Smerdon MJ. Suppressed catalytic activity of base excision repair enzymes on rotationally positioned uracil in nucleosomes. Proc Natl Acad Sci U S A. 2003;100:7465–7470. doi: 10.1073/pnas.1330328100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Odell ID, Barbour JE, Murphy DL, Della-Maria JA, Sweasy JB, Tomkinson AE, Wallace SS, Pederson DS. Nucleosome disruption by DNA ligase III-XRCC1 promotes efficient base excision repair. Mol Cell Biol. 2011;31:4623–4632. doi: 10.1128/MCB.05715-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodriguez Y, Smerdon MJ. The structural location of DNA lesions in nucleosome core particles determines accessibility by base excision repair enzymes. J Biol Chem. 2013;288:13863–13875. doi: 10.1074/jbc.M112.441444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meas R, Smerdon MJ. Nucleosomes determine their own patch size in base excision repair. Scientific Reports. 2016;6:27122. doi: 10.1038/srep27122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hasan S, El-Andaloussi N, Hardeland U, Hassa PO, Burki C, Imhof R, Schar P, Hottiger MO. Acetylation regulates the DNA end-trimming activity of DNA polymerase beta. Mol Cell. 2002;10:1213–1222. doi: 10.1016/s1097-2765(02)00745-1. [DOI] [PubMed] [Google Scholar]
- 17.Prasad R, Liu Y, Deterding LJ, Poltoratsky VP, Kedar PS, Horton JK, Kanno S, Asagoshi K, Hou EW, Khodyreva SN, Lavrik OI, Tomer KB, Yasui A, Wilson SH. HMGB1 is a cofactor in mammalian base excision repair. Mol Cell. 2007;27:829–841. doi: 10.1016/j.molcel.2007.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Prasad R, Wilson SH. HMGB1: roles in base excision repair and related function. Biochimica et biophysica acta. 2010;1799:119–130. doi: 10.1016/j.bbagrm.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lange SS, Vasquez KM. HMGB1: the jack-of-all-trades protein is a master DNA repair mechanic. Molecular carcinogenesis. 2009;48:571–580. doi: 10.1002/mc.20544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stros M. HMGB proteins: interactions with DNA and chromatin. Biochimica et biophysica acta. 2010;1799:101–113. doi: 10.1016/j.bbagrm.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 21.Travers AA. Priming the nucleosome: a role for HMGB proteins? EMBO reports. 2003;4:131–136. doi: 10.1038/sj.embor.embor741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ragab A, Travers AA. HMG-D and histone H1 alter the local accessibility of nucleosomal DNA. Nucleic acids research. 2003;31 doi: 10.1093/nar/gkg923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simon RH, Felsenfeld G. A new procedure for purifying histone pairs H2A + H2B and H3 + H4 from chromatin using hydroxylapatite. Nucleic acids research. 1979;6:689–696. doi: 10.1093/nar/6.2.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee KM, Hayes JJ. In Vitro Reconstitution and Analysis of Mononucleosomes Containing Defined DNAs and Proteins. Methods: A companion to Methods in Enzymology. 1997;12:2–9. doi: 10.1006/meth.1997.0441. [DOI] [PubMed] [Google Scholar]
- 25.Joshi SR, Sarpong YC, Peterson RC, Scovell WM. Nucleosome dynamics: HMGB1 relaxes canonical nucleosome structure to facilitate estrogen receptor binding. Nucleic acids research. 2012;40:10161–10171. doi: 10.1093/nar/gks815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fang H, Clark DJ, Hayes JJ. DNA and nucleosomes direct distinct folding of a linker histone H1 C-terminal domain. Nucleic acids research. 2011;40:1475–1484. doi: 10.1093/nar/gkr866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mertins P, Qiao J, J P, Udeshi N, Clauser K, Mani D, Burgess M, Gillette M, Jaffe J, Carr S. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat Methods. 2013;10 doi: 10.1038/nmeth.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weinert B, Schölz C, Wagner S, Iesmantavicius V, Su D, Daniel J, Choudhary C. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell reports. 2013;4:842–851. doi: 10.1016/j.celrep.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 29.Hassan AH, Prochasson P, Neely KE, Galasinski SC, Chandy M, Carrozza MJ, Workman JL. Function and selectivity of bromodomains in anchoring chromatin-modifying complexes to promoter nucleosomes. Cell. 2002;111:369–379. doi: 10.1016/s0092-8674(02)01005-x. [DOI] [PubMed] [Google Scholar]
- 30.Cole HA, Tabor-Godwin JM, Hayes JJ. Uracil DNA glycosylase activity on nucleosomal DNA depends on rotational orientation of targets. J Biol Chem. 2010;285:2876–2885. doi: 10.1074/jbc.M109.073544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prasad A, Wallace SS, Pederson DS. Initiation of base excision repair of oxidative lesions in nucleosomes by the human, bifunctional DNA glycosylase NTH1. Mol Cell Biol. 2007;27:8442–8453. doi: 10.1128/MCB.00791-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu C, Travers A. A ‘one-pot’ assay for the accessibility of DNA in a nucleosome core particle. Nucleic acids research. 2004;32:e122. doi: 10.1093/nar/gnh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stros M, Shick VV, Belyavsky AV, Mirzabekov AD. Interaction of high mobility group proteins HMG 1 and HMG 2 with nucleosomes studied by gel electrophoresis. Molecular biology reports. 1985;10:221–226. doi: 10.1007/BF00775979. [DOI] [PubMed] [Google Scholar]
- 34.Sattler U, Frit P, Salles B, Calsou P. Long-patch DNA repair synthesis during base excision repair in mammalian cells. EMBO reports. 2003;4:363–367. doi: 10.1038/sj.embor.embor796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piekna-Przybylska D, Bambara RA, Balakrishnan L. Acetylation regulates DNA repair mechanisms in human cells. Cell Cycle. 2016:1–12. doi: 10.1080/15384101.2016.1176815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sawaya MR, Prasad R, Wilson SH, Kraut J, Pelletier H. Crystal structures of human DNA polymerase beta complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry. 1997;36:11205–11215. doi: 10.1021/bi9703812. [DOI] [PubMed] [Google Scholar]
- 37.Huggins CF, Chafin DR, Aoyagi S, Henricksen LA, Bambara RA, Hayes JJ. Flap endonuclease 1 efficiently cleaves base excision repair and DNA replication intermediates assembled into nucleosomes. Mol Cell. 2002;10:1201–1211. doi: 10.1016/s1097-2765(02)00736-0. [DOI] [PubMed] [Google Scholar]
- 38.Jagannathan I, Pepenella S, Hayes JJ. Activity of FEN1 endonuclease on nucleosome substrates is dependent upon DNA sequence but not flap orientation. J Biol Chem. 2011;286:17521–17529. doi: 10.1074/jbc.M111.229658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hinz JM. Impact of abasic site orientation within nucleosomes on human APE1 endonuclease activity. Mutat Res. 2014;766–767:19–24. doi: 10.1016/j.mrfmmm.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 40.Vasudevan D, Chua EYD, Davey CA. Crystal Structures of Nucleosome Core Particles Containing the ‘601’ Strong Positioning Sequence. J Mol Biol. 2010;403:1–10. doi: 10.1016/j.jmb.2010.08.039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.