

Abstract
Gene editing is moving rapidly from a highly useful laboratory-based tool towards human clinical application. In vivo gene editing has been documented in mouse models of Duchenne Muscular Dystrophy, where skeletal and cardiac muscle editing produces internally-truncated dystrophin. A recent report now demonstrates that editing the dystrophin gene in the heart improves cardiac function, paving the path to in vivo application of cardiac gene correction.
Keywords: cardiomyopathy, CRISPR, gene editing, dystrophin, mdx
Mutations in the gene encoding dystrophin cause Duchenne Muscular Dystrophy (DMD) and its associated cardiomyopathy. Large deletions, which span multiple exons, are the most common cause of DMD, and these deletions disrupt dystrophin’s reading frame and ablate its expression. Dystrophin binds actin at its N-terminus and with its C-terminus, dystrophin connects to the plasma membrane of myocytes. The central portion of dystrophin is composed of multiple spectrin repeat domains, and mutations that interrupt the internal spectrin repeat domains and leave intact the N- and C-terminus result in a milder form of disease called Becker Muscular Dystrophy1. A major therapeutic approach for DMD aims to restore the reading frame by converting DMD-associated frame-disrupting mutations into those that produce internally deleted but more functional BMD-associated dystrophins. In 2016, the FDA granted approval for eteplirsen, which uses antisense technology to restore dystrophin’s reading frame2. Antisense treatments act on RNA and therefore requires regular repeated dosing to induce the expected molecular effect. In contrast, gene-editing corrects DNA, and if directed to the correct cell type, it can afford a permanent treatment with a single application.
Gene editing using CRISPR/Cas9 induces double stranded breaks in DNA, which typically rejoin at predictable sites using nonhomologous end joining (NHEJ)3. Alternatively, when CRISPR/Cas9 is carried out in the presence of a corrective template, homology directed repair (HDR) can generate very specific base pair changes, such as that required to fully correct a premature stop codon or restore reading frame.
Because of the devastating progressive nature of muscle degeneration in DMD, proof-of-principle gene editing experiments focused more on skeletal muscle correction in mouse models of DMD4–6. Dystrophin is encoded by 79 exons, and the mdx mouse has a premature stop codon in exon 23 (Figure). Removing exon 23, either with exon skipping or gene editing, allows exons 22 and 24 to join and maintains the reading frame, thus improving skeletal muscle dysfunction. The effectiveness of gene editing has also been shown using the mdx4cv mouse, which has a mutation in exon 537. In this case, the gene editing strategy used guide RNAs to remove a 45 KB region encompassing exons 52 and 53, which joins exons 51 to 54 to re-establish an open reading frame. In both mdx and mdx4cv, systemic adeno-associated viral (AAV) delivery into 6 week or 11 week animals was used to document that genetic correction was also seen cardiomyocytes5, 7.
Figure.
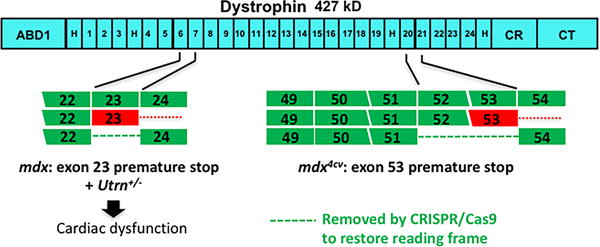
In vivo gene editing to restore dystrophin’s reading frame has been applied to both the mdx (left) and mdx4cv models (right), which have mutations either in exon 23 or 53, respectively. El Rafaey et al. now used mdx mice in combination with heterozygous loss of utrophin (Utrn+/−) to demonstrate improved cardiac function after dystrophin gene editing.
As a multinucleate syncytium, skeletal muscle benefit occurs even if only a subset of myonuclei have been corrected. Furthermore, correcting the regenerative cells within skeletal muscle, the satellite cells, is also thought to provide longer term benefit since the progeny of corrected satellite cells will also express dystrophin6. The mono/dinucleate nature of cardiomyocytes and the absence of an obvious correctable progenitor pool suggest that the target of gene editing in the heart is the cardiomyocyte itself.
The percent of total cardiomyocytes needed for functional physiological improvement can be estimated from studies of female carriers of dystrophin gene mutations. As an X-linked recessive disorder, women with dystrophin mutations have ~ 50% of cardiomyocytes expressing dystrophin. Cardiomyopathy may occur in female carriers of dystrophin mutations, but largely this cardiomyopathy arises later in life and is considerably more mild that what occurs in their sons. X inactivation that deactivates a greater percentage of functional X-chromosomes can lead to earlier cardiomyopathy in female DMD carriers8. In the mdx mouse model, female mice have largely normal hearts suggesting that 50% cardiomyocyte expression is adequate to prevent cardiomyopathy or associated with only mild impairment9. However, cardiomyopathy in mdx mice is quite mild necessitating other stressors as surrogates for cardiac dysfunction. Studies in mice suggest that low level dystrophin production, as low as 5%, can contribute to improved skeletal and cardiac muscle function in the mdx mouse10, 11.
El Rafaey and colleagues now studied gene editing in the context of mdx mice lacking one copy of utrophin12. Utrophin is highly related to dystrophin and mdx mice lacking one copy of utrophin (mdx/Utrn+/−) develop cardiomyopathy more amenable for assessing therapeutic intervention13. The authors used Cas9 derived from Staphlyococcal aureus bacteria (saCas9) because its small size is more readily accommodated by AAV. The saCas9 and guide RNAs targeting exon 23 were packaged in AAV serotype rh.74. Neonatal mice were injected systemically with 1012 vector genomes (v.g.). Mice were assessed at ten weeks of age and found to have detectable dystrophin in ~40% of cardiomyocytes, which correlated to 23% of normal dystrophin levels on immunoblotting. To determine whether this level of correction was physiologically relevant, papillary muscle contractility was examined. Length-dependent activation studies identified increased force at 100% of optimal muscle length. Force was also measured with repetitive stimulation, and with higher frequencies, force was improved in treated compared to untreated hearts. These findings suggest that this degree of molecular correction was sufficient to improve cardiac function. The authors also conducted systemic delivery into 16 week animals using tail vein injection, and found that delivery later in life could restore dystrophin expression.
The editing strategy used by El Rafaey et al. was designed to remove 23 KB of sequence using NHEJ12. Deep sequencing of the resulting products indicated that NHEJ produced the expected outcome in 76% of the sequences. Approximately 14% of the deletions induced by this gene editing strategy produced larger or otherwise unexpected deletions. This type of “on-site” error rate is to be expected, and still produces a useful outcome in the majority of sites where it acted. Remote off-target mutations, away from the edited sites, are also known to occur with CRISPR/Cas9. Most commonly specific sites having related sequences to the guide RNAs are examined for off-target errors, but mutations can occur in regions unrelated to these predicted sites. Despite these concerns, there has been rapid advancement in applying gene editing to preclinical models, and the questions of how and when to apply these findings to human gene editing are now arising.
Translating gene editing into human cardiac therapy will require optimized efficacy with improved on-site fidelity and reduced off-target events. Viral delivery of Cas9 with guide RNAs is expected to the primary modality since this approach can restrict Cas9 expression using tissue specific promoters. AAV is a preferred virus for its safety, although it can still be expected that an immune response may occur to the virus and/or the protein being delivered. Immune response may arise to AAV itself, newly recognized dystrophin epitopes, and/or gene editing machinery. In humans who received viral delivery of mini-dystrophin genes, an anti-dystrophin response may occur14.
Even with these considerations and concerns, in vivo correction of dystrophin gene mutations in humans is a plausible application of therapeutic gene editing. Human dystrophin mutations are hemizygous, and therefore only one copy of the gene requires correction. This is distinct from the many heterozygous cardiomyopathy mutations where correcting the mutant allele must occur in the absence of changing the intact allele. Second, the repetitive nature of dystrophin is ideal for generating internal deletions. There is ample human genetic data indicating the functional nature of internally truncated dystrophin, and this human genetic data is highly complemented by experimental studies using transgenes and viral gene delivery. Much of dystrophin’s function can be achieved with internally deleted proteins, provided the N- and C-termini remain intact. Some internally truncated dystrophins may have support better physiological function than others. The biophysical demands on cardiomyocytes and skeletal myofibers differ, and the degree of correction sufficient for skeletal muscle function may differ from cardiac muscle function.
Thinking ahead to human clinical trials, there will be choices to be made by the pioneers who agree to participate in these studies. At this point, more is known about viral gene delivery in which mini-dystrophin is delivered, as this approach has been tested for several decades. Virally-mediated gene correction introduces several new variables but could afford a more long term correction. Thorough preclinical studies are needed to better delineate efficacy and safety of these approaches.
Acknowledgments
NIH HL061322, HL128075
Footnotes
Disclosure: Dr. McNally serves as a consultant to Exonics.
References
- 1.Gao QQ, McNally EM. The dystrophin complex: Structure, function, and implications for therapy. Comprehensive Physiology. 2015;5:1223–1239. doi: 10.1002/cphy.c140048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Niks EH, Aartsma-Rus A. Exon skipping: A first in class strategy for duchenne muscular dystrophy. Expert opinion on biological therapy. 2017;17:225–236. doi: 10.1080/14712598.2017.1271872. [DOI] [PubMed] [Google Scholar]
- 3.Doetschman T, Georgieva T. Gene editing with crispr/cas9 rna-directed nuclease. Circulation research. 2017;120:876–894. doi: 10.1161/CIRCRESAHA.116.309727. [DOI] [PubMed] [Google Scholar]
- 4.Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, Bhattacharyya S, Shelton JM, Bassel-Duby R, Olson EN. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science (New York, NY) 2016;351:400–403. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Castellanos Rivera RM, Madhavan S, Pan X, Ran FA, Yan WX, Asokan A, Zhang F, Duan D, Gersbach CA. In vivo genome editing improves muscle function in a mouse model of duchenne muscular dystrophy. Science (New York, NY) 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tabebordbar M, Zhu K, Cheng JKW, Chew WL, Widrick JJ, Yan WX, Maesner C, Wu EY, Xiao R, Ran FA, Cong L, Zhang F, Vandenberghe LH, Church GM, Wagers AJ. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science (New York, NY) 2016;351:407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bengtsson NE, Hall JK, Odom GL, Phelps MP, Andrus CR, Hawkins RD, Hauschka SD, Chamberlain JR, Chamberlain JS. Muscle-specific crispr/cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for duchenne muscular dystrophy. Nature communications. 2017;8:14454. doi: 10.1038/ncomms14454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Viggiano E, Picillo E, Cirillo A, Politano L. Comparison of x-chromosome inactivation in duchenne muscle/myocardium-manifesting carriers, non-manifesting carriers and related daughters. Clinical genetics. 2013;84:265–270. doi: 10.1111/cge.12048. [DOI] [PubMed] [Google Scholar]
- 9.Yue Y, Skimming JW, Liu M, Strawn T, Duan D. Full-length dystrophin expression in half of the heart cells ameliorates beta-isoproterenol-induced cardiomyopathy in mdx mice. Human molecular genetics. 2004;13:1669–1675. doi: 10.1093/hmg/ddh174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li D, Yue Y, Duan D. Preservation of muscle force in mdx3cv mice correlates with low-level expression of a near full-length dystrophin protein. The American journal of pathology. 2008;172:1332–1341. doi: 10.2353/ajpath.2008.071042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Putten M, van der Pijl EM, Hulsker M, Verhaart IE, Nadarajah VD, van der Weerd L, Aartsma-Rus A. Low dystrophin levels in heart can delay heart failure in mdx mice. Journal of molecular and cellular cardiology. 2014;69:17–23. doi: 10.1016/j.yjmcc.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 12.El Refaey M, Xu L, Gao Y, Canan BD, Adesanya TA, Warner SC, Akagi K, Symer DE, Mohler PJ, Ma J, Janssen PM, Han R. In vivo genome editing restores dystrophin expression and cardiac function in dystrophic mice. Circulation research. 2017 doi: 10.1161/CIRCRESAHA.117.310996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rafael-Fortney JA, Chimanji NS, Schill KE, Martin CD, Murray JD, Ganguly R, Stangland JE, Tran T, Xu Y, Canan BD, Mays TA, Delfin DA, Janssen PM, Raman SV. Early treatment with lisinopril and spironolactone preserves cardiac and skeletal muscle in duchenne muscular dystrophy mice. Circulation. 2011;124:582–588. doi: 10.1161/CIRCULATIONAHA.111.031716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S, Bowles D, Gray S, Li C, Galloway G, Malik V, Coley B, Clark KR, Li J, Xiao X, Samulski J, McPhee SW, Samulski RJ, Walker CM. Dystrophin immunity in duchenne’s muscular dystrophy. The New England journal of medicine. 2010;363:1429–1437. doi: 10.1056/NEJMoa1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]